
Discovery of 1-Benzhydryl-Piperazine-Based HDAC Inhibitors with Anti-Breast Cancer Activity: Synthesis, Molecular Modeling, In Vitro and In Vivo Biological Evaluation

 , , , , , , ,
, , , , , , , 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemistry—General Procedures
2.2. Alkylation of 1-Benzhydryl Piperazine (General Procedure A)
2.3. Synthesis of Hydroxamic Acid Derivatives (General Procedure B)
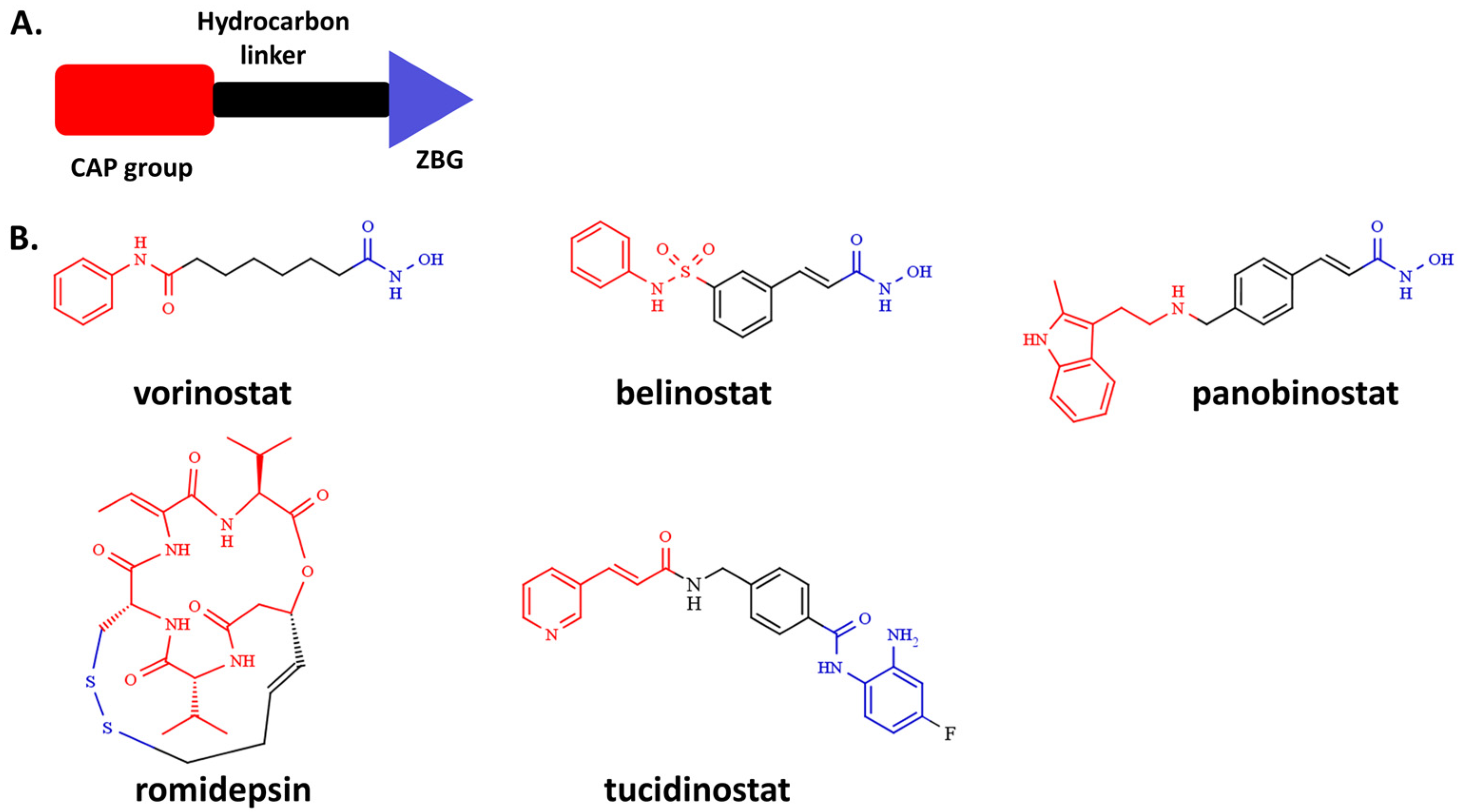
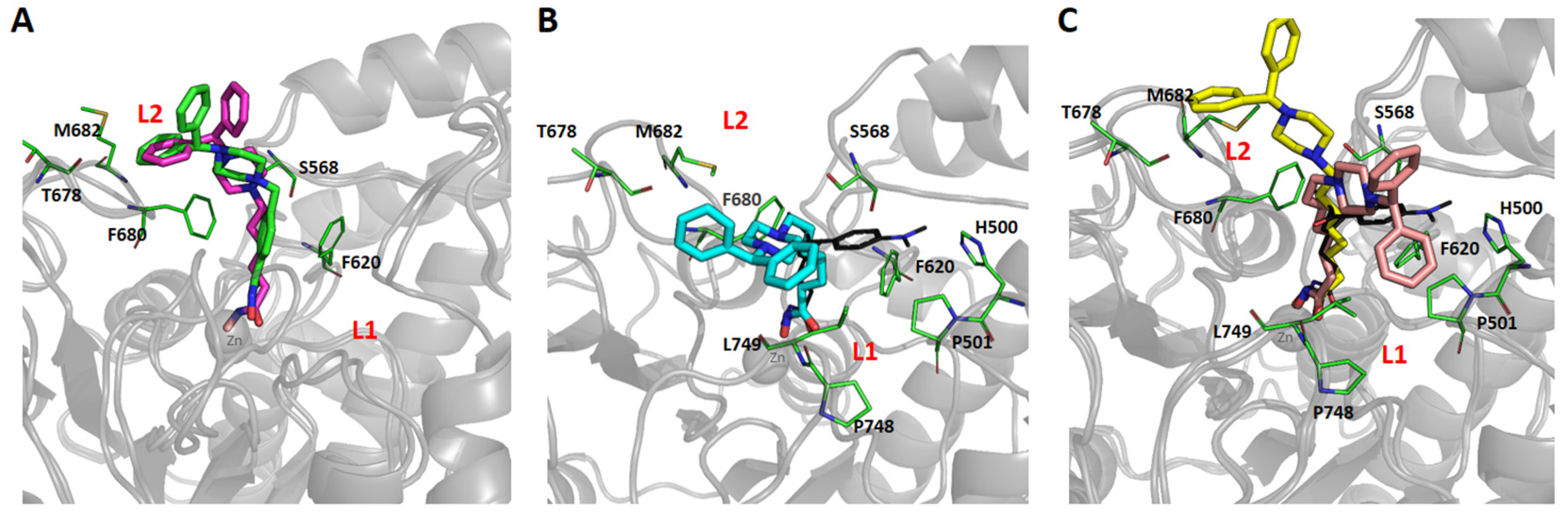
2.4. Molecular Docking
2.5. HDAC Enzymes Assay
2.6. Breast Cancer Cell Cultures
2.6.1. Cell Viability (MTT) Assay
2.6.2. Apoptosis Assay
2.6.3. Cell Cycle Analysis
2.6.4. Detection of Mitochondrial Membrane Potential
2.6.5. Generation and Analysis of Tumor Spheres
2.6.6. Bicameral Motility and Invasion Assay
2.6.7. Wound Healing Assay
2.7. Animal Study in the Zebrafish (Danio rerio) Model
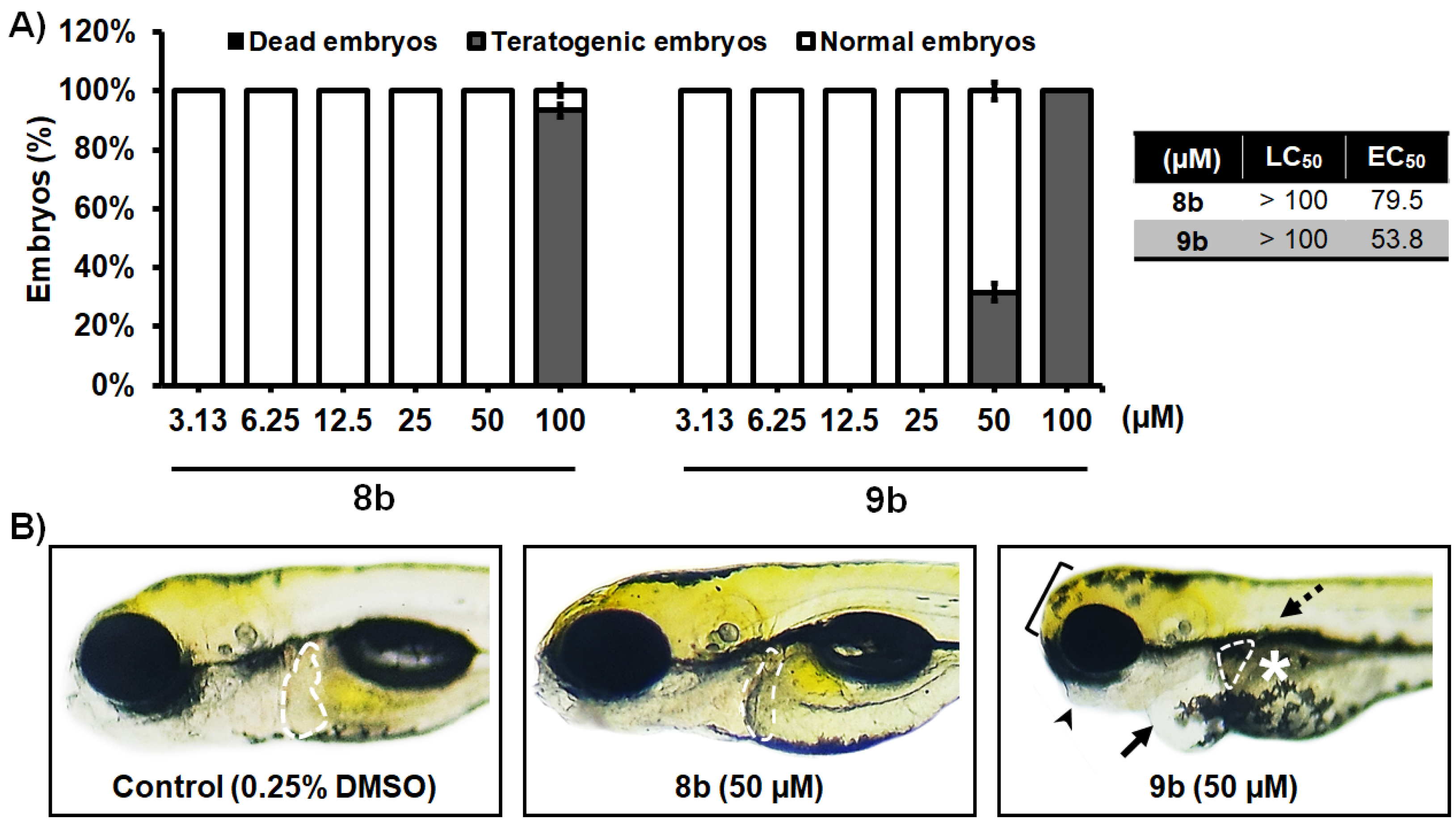
2.7.1. Acute and Developmental Toxicity Assessment
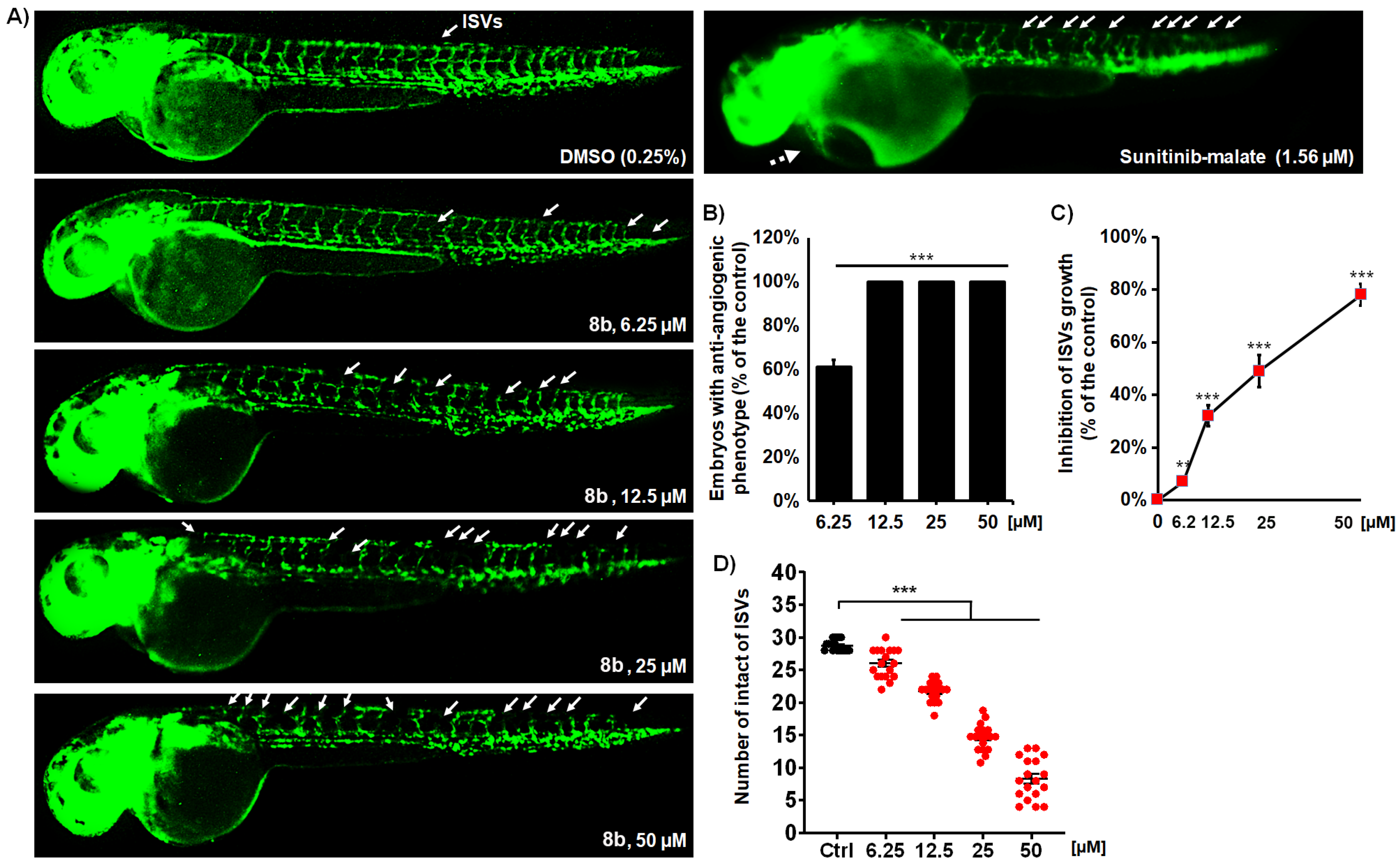
2.7.2. Anti-Angiogenic Activity Assessment
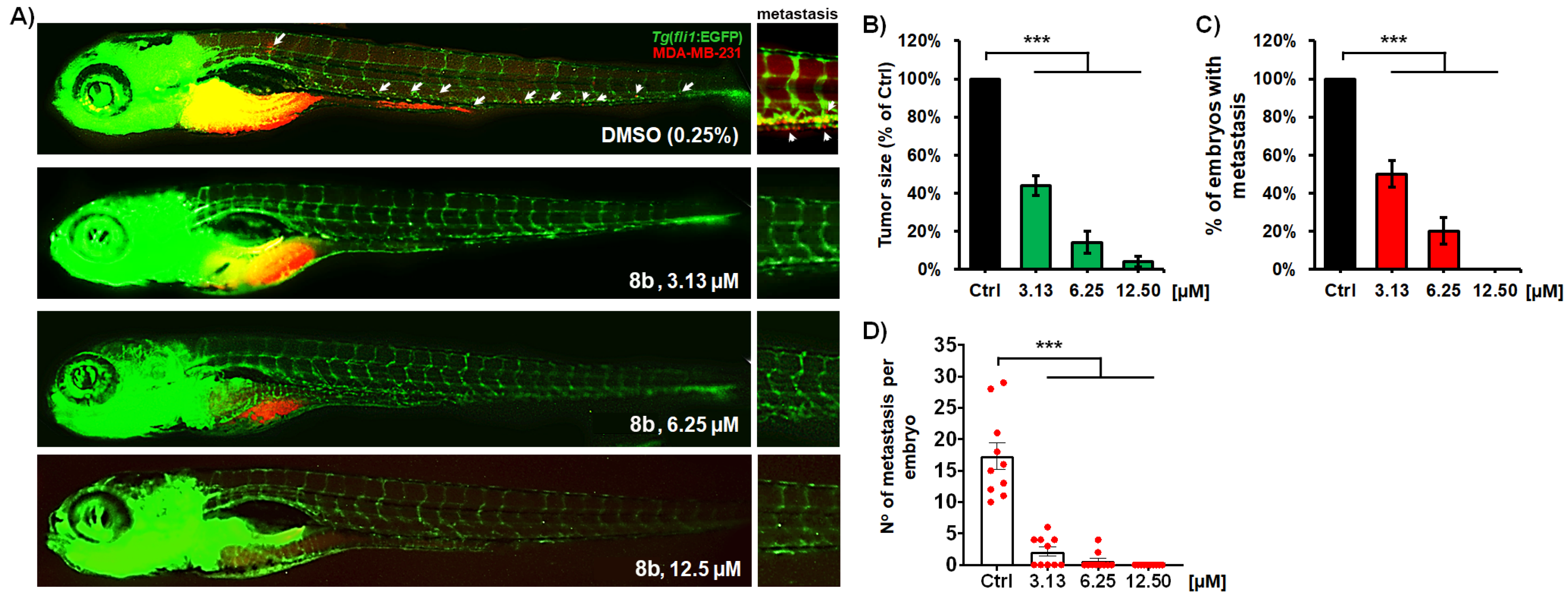
2.7.3. Anti-Cancer Activity Evaluation in Human and Zebrafish Cell-Derived Xenografts (CDX)
Cell Line Culture Preparation
2.7.4. Zebrafish Xenografts and Treatment Efficacy Assessment
2.8. Statistical Analysis
3. Results and Discussion
3.1. Design and Synthesis of HDAC Inhibitors
3.2. Biology
3.2.1. Analysis of HDAC Inhibitory Profiles
3.2.2. Examination of Cytotoxic Effects of Synthesized Compounds
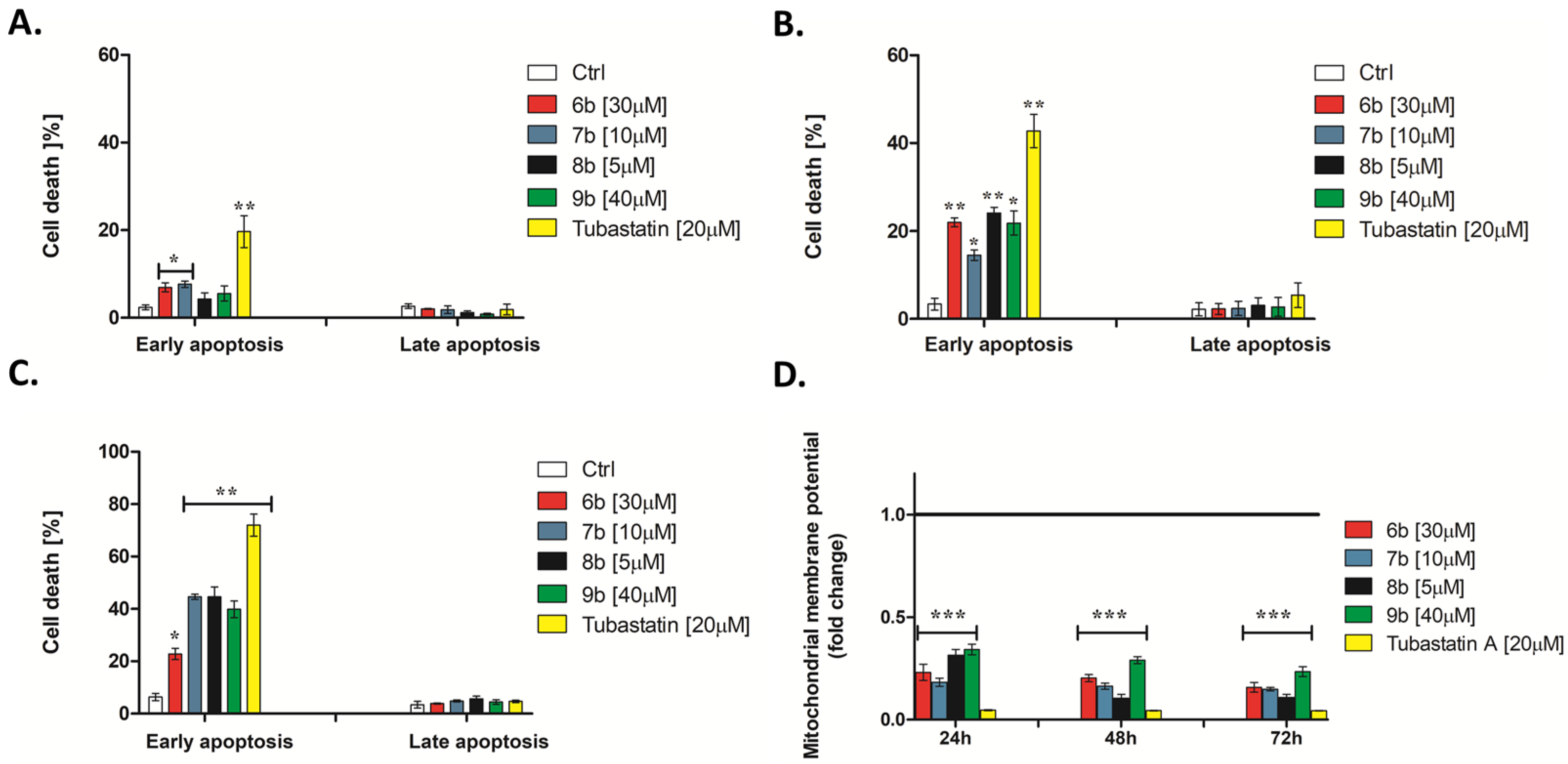
3.2.3. 1-Benzhydryl-Piperazine-Based HDAC Inhibitors Induce Translocation of Cell Membrane Phosphatidylserine
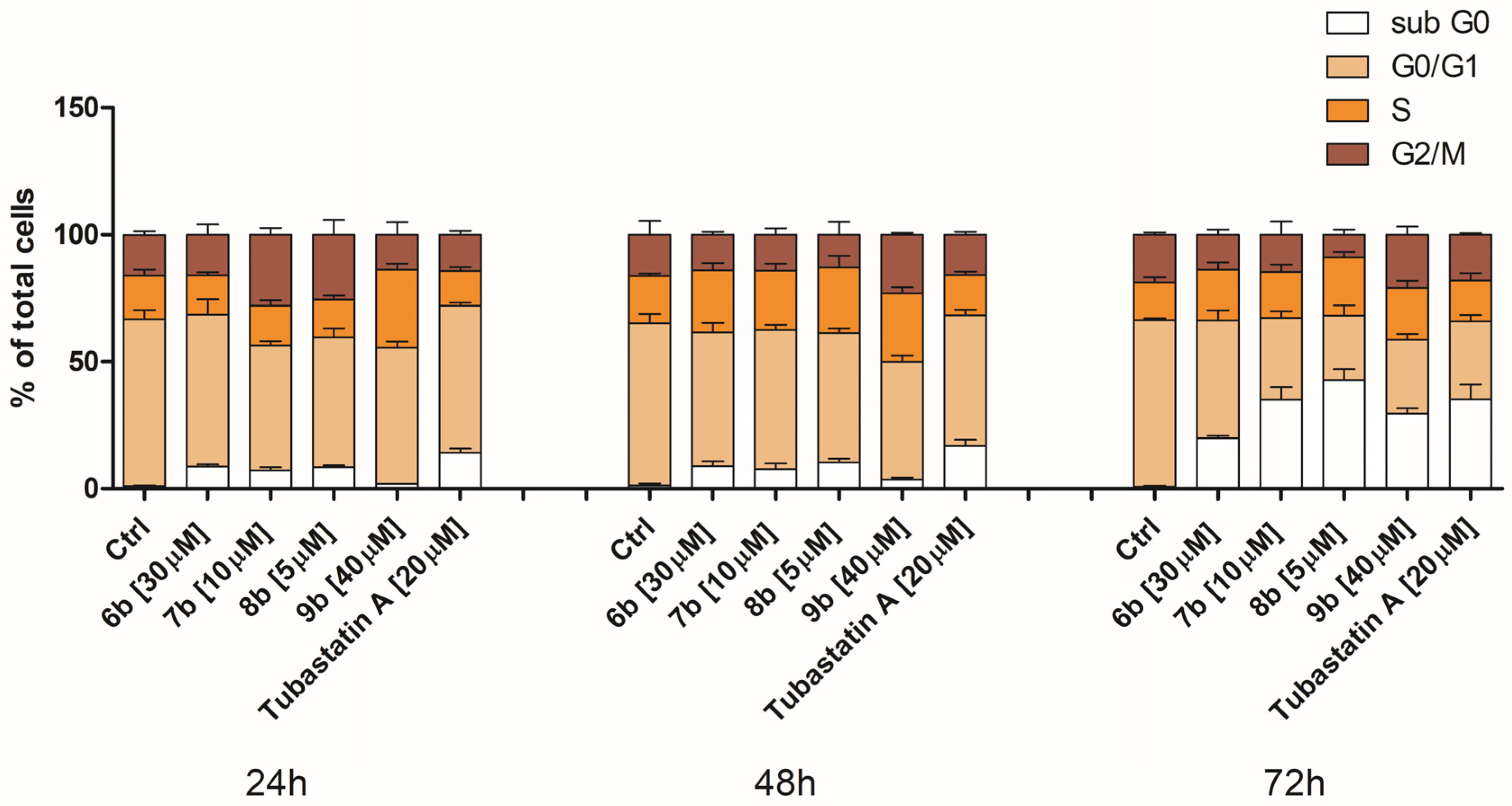
3.2.4. Treatment of MDA-MB-231 Cells with Inhibitors Induces Changes in the Cell Cycle
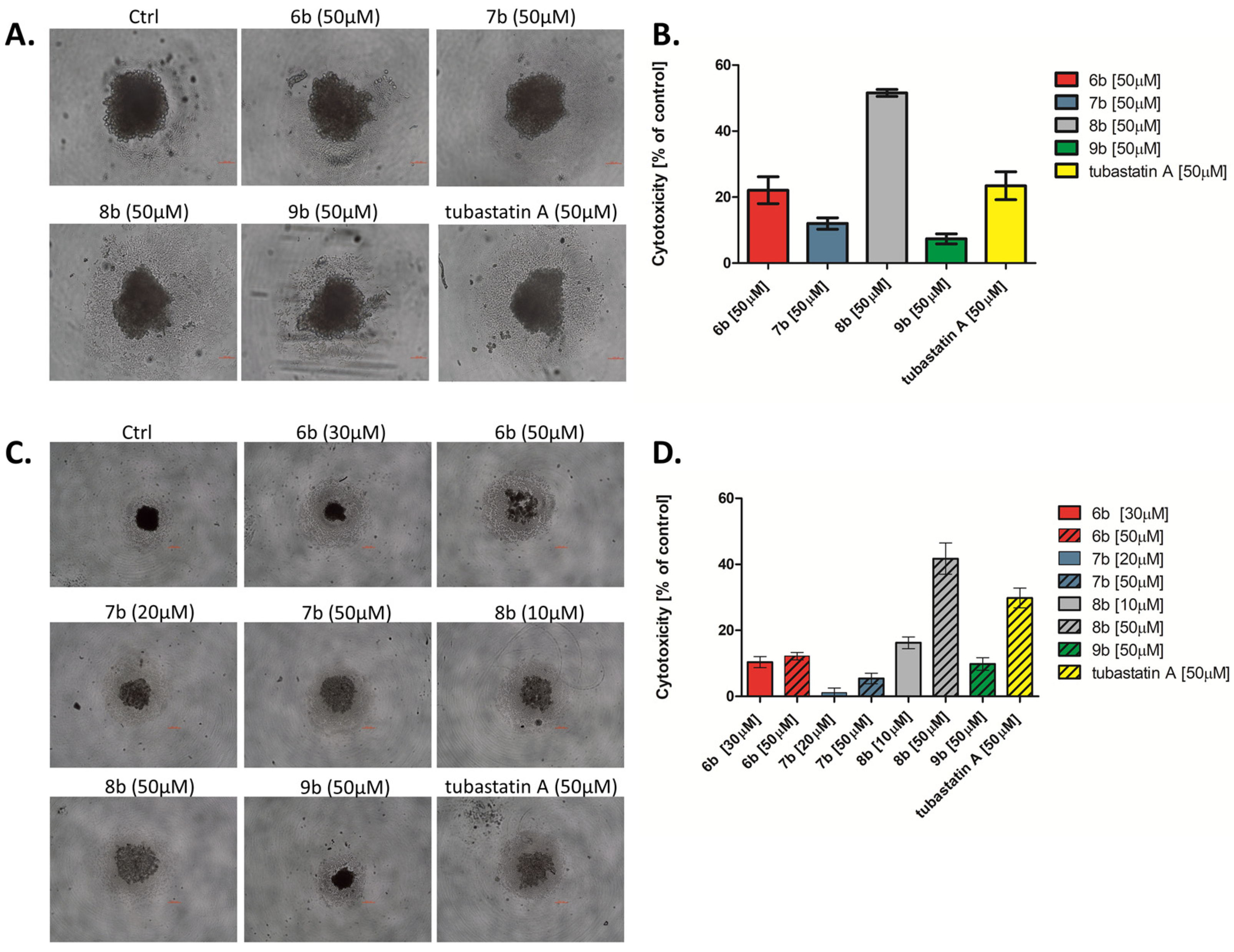
3.2.5. Analyses of the Efficacy of Compounds in a 3D Cancer Model
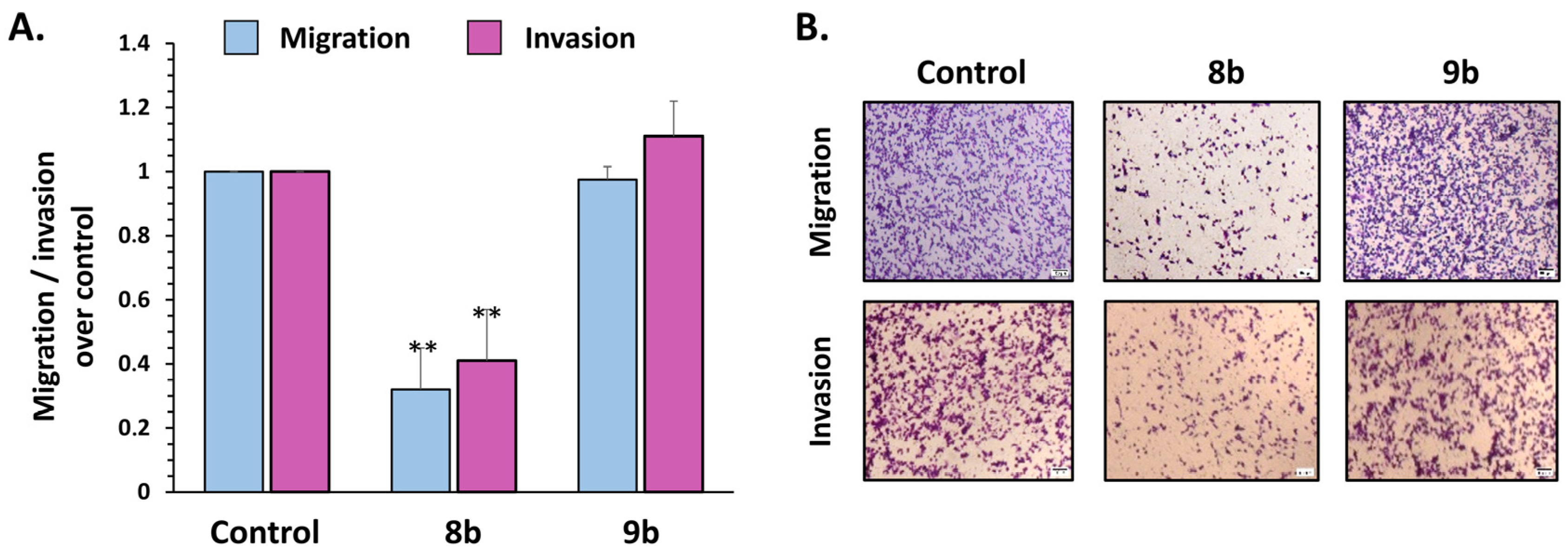
3.2.6. Compound 8b Decreases Migration and Invasion of Triple-Negative Breast Cancer MDA-MB-231 Cell Line In Vitro
3.2.7. Toxicity Assessment In Vivo Using the Zebrafish Model
3.2.8. Anti-Angiogenic Effects of 8b and 9b on Developing Zebrafish
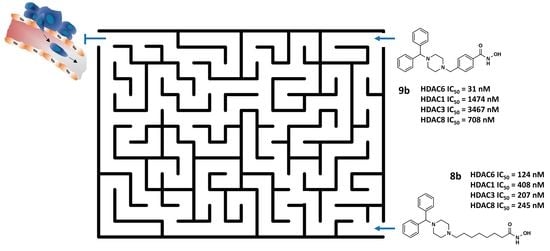
3.2.9. Compound 8b Inhibits MDA-MB-231 Breast Tumor Development and Successfully Prevents Tumor Cell Metastasis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Experimental Section—Structural Characterization of Synthesized Compounds
References
- Wolff, A.C.; Hammond, M.E.H.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.S.; Bilous, M.; Fitzgibbons, P. American Society of Clinical Oncology; College of American Pathologists. Recommendations for Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Update. J. Clin. Oncol. 2013, 31, 3997–4013. [Google Scholar] [CrossRef] [PubMed]
- Kadamkulam Syriac, A.; Nandu, N.S.; Leone, J.P. Central Nervous System Metastases from Triple-Negative Breast Cancer: Current Treatments and Future Prospective. Breast Cancer 2022, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cobleigh, M.; Yardley, D.A.; Brufsky, A.M.; Rugo, H.S.; Swain, S.M.; Kaufman, P.A.; Tripathy, D.; Hurvitz, S.A.; O’Shaughnessy, J.; Mason, G. Baseline Characteristics, Treatment Patterns, and Outcomes in Patients with HER2-Positive Metastatic Breast Cancer by Hormone Receptor Status from SystHERs. Clin. Cancer Res. 2020, 26, 1105–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchini, G.; De Angelis, C.; Licata, L.; Gianni, L. Treatment Landscape of Triple-Negative Breast Cancer—Expanded Options, Evolving Needs. Nat. Rev. Clin. Oncol. 2022, 19, 91–113. [Google Scholar] [CrossRef]
- Nephew, K.P.; Huang, T.H.-M. Epigenetic Gene Silencing in Cancer Initiation and Progression. Cancer Lett. 2003, 190, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Kim, M.-Y. Cancer Epigenetics: Past, Present and Future. Semin. Cancer Biol. 2022, 83, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Dang, F.; Wei, W. Targeting the Acetylation Signaling Pathway in Cancer Therapy. Semin. Cancer Biol. 2022, 85, 209–218. [Google Scholar] [CrossRef]
- Carew, J.S.; Giles, F.J.; Nawrocki, S.T. Histone Deacetylase Inhibitors: Mechanisms of Cell Death and Promise in Combination Cancer Therapy. Cancer Lett. 2008, 269, 7–17. [Google Scholar] [CrossRef]
- Singh, T.; Kaur, P.; Singh, P.; Singh, S.; Munshi, A. Differential Molecular Mechanistic Behavior of HDACs in Cancer Progression. Med. Oncol. 2022, 39, 171. [Google Scholar] [CrossRef]
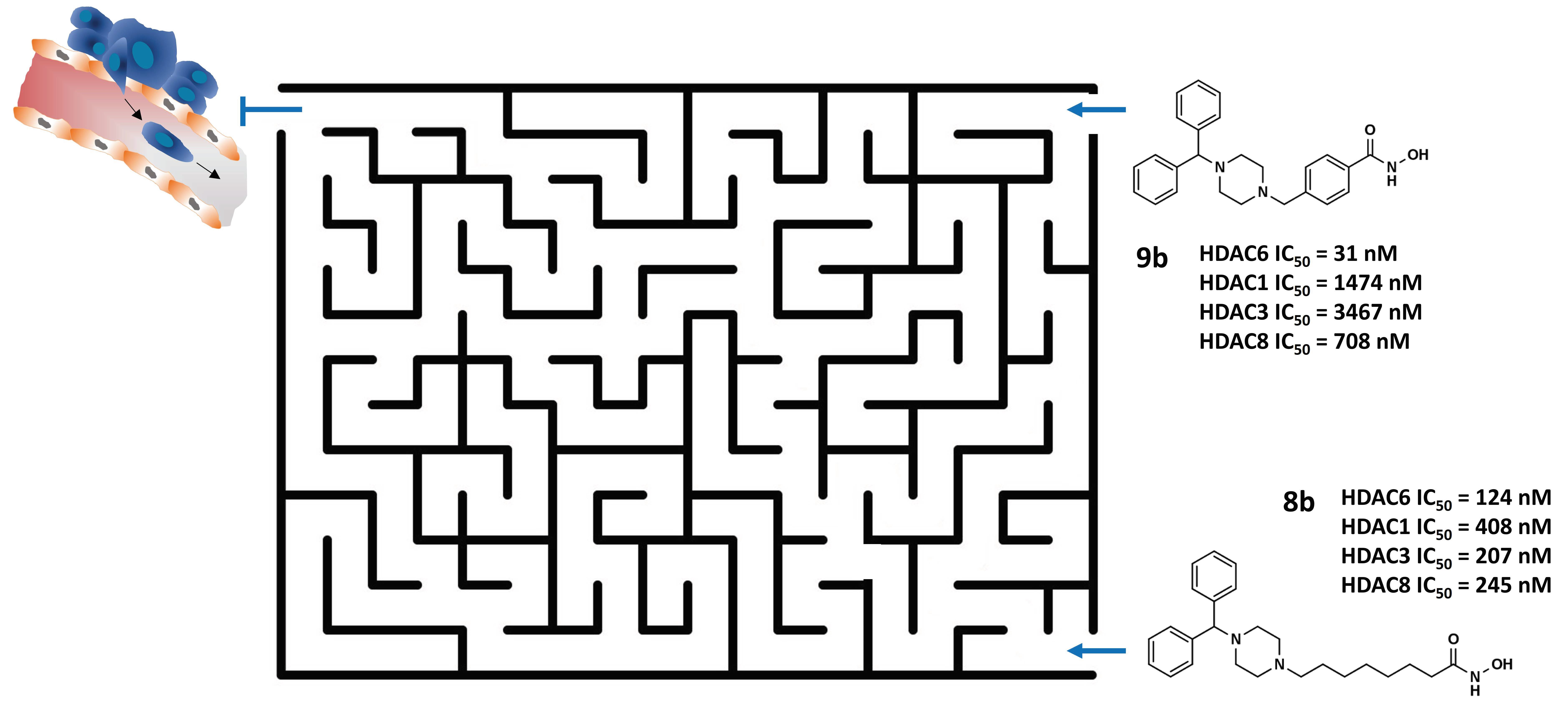
- Roche, J.; Bertrand, P. Inside HDACs with More Selective HDAC Inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a Histone Deacetylase Homologue Bound to the TSA and SAHA Inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Fernández, A.; Cabrero, J.R.; Serrador, J.M.; Sánchez-Madrid, F. HDAC6: A Key Regulator of Cytoskeleton, Cell Migration and Cell-Cell Interactions. Trends Cell Biol. 2008, 18, 291–297. [Google Scholar] [CrossRef]
- Aldana-Masangkay, G.I.; Sakamoto, K.M. The Role of HDAC6 in Cancer. J. Biomed. Biotechnol. 2011, 2011, 875824. [Google Scholar] [CrossRef] [Green Version]
- Deakin, N.O.; Turner, C.E. Paxillin Inhibits HDAC6 to Regulate Microtubule Acetylation, Golgi Structure, and Polarized Migration. J. Cell Biol. 2014, 206, 395–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arsenault, D.; Brochu-Gaudreau, K.; Charbonneau, M.; Dubois, C.M. HDAC6 Deacetylase Activity Is Required for Hypoxia-Induced Invadopodia Formation and Cell Invasion. PLoS ONE 2013, 8, e55529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.-F.; Yao, T.-P. HDAC6 Is a Microtubule-Associated Deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Kaluza, D.; Kroll, J.; Gesierich, S.; Yao, T.-P.; Boon, R.A.; Hergenreider, E.; Tjwa, M.; Rössig, L.; Seto, E.; Augustin, H.G. Class IIb HDAC6 Regulates Endothelial Cell Migration and Angiogenesis by Deacetylation of Cortactin. EMBO J. 2011, 30, 4142–4156. [Google Scholar] [CrossRef] [Green Version]
- Hai, Y.; Christianson, D.W. Histone Deacetylase 6 Structure and Molecular Basis of Catalysis and Inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef] [Green Version]
- Miyake, Y.; Keusch, J.J.; Wang, L.; Saito, M.; Hess, D.; Wang, X.; Melancon, B.J.; Helquist, P.; Gut, H.; Matthias, P. Structural Insights into HDAC6 Tubulin Deacetylation and Its Selective Inhibition. Nat. Chem. Biol. 2016, 12, 748–754. [Google Scholar] [CrossRef]
- Sixto-López, Y.; Gómez-Vidal, J.A.; de Pedro, N.; Bello, M.; Rosales-Hernández, M.C.; Correa-Basurto, J. Hydroxamic Acid Derivatives as HDAC1, HDAC6 and HDAC8 Inhibitors with Antiproliferative Activity in Cancer Cell Lines. Sci. Rep. 2020, 10, 10462. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-L.; Tu, H.-J.; Pan, S.-L.; Liou, J.-P.; Yang, C.-R. Anti-Metastatic Activity of MPT0G211, a Novel HDAC6 Inhibitor, in Human Breast Cancer Cells in Vitro and in Vivo. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1866, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Niu, X.; Quan, J.; Li, Y.; Yuan, L.; Wang, J.; Ma, C.; Ma, E. Discovery of Specific HDAC6 Inhibitor with Anti-Metastatic Effects in Pancreatic Cancer Cells through Virtual Screening and Biological Evaluation. Bioorg. Chem. 2020, 97, 103679. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Quan, J.; Song, H.; Li, D.; Ma, E.; Wang, Y.; Ma, C. Novel Pyrrolo[2,1-c][1,4]Benzodiazepine-3,11-Dione (PBD) Derivatives as Selective HDAC6 Inhibitors to Suppress Tumor Metastasis and Invasion in Vitro and in Vivo. Bioorg. Chem. 2021, 114, 105081. [Google Scholar] [CrossRef] [PubMed]
- Depetter, Y.; Geurs, S.; De Vreese, R.; Goethals, S.; Vandoorn, E.; Laevens, A.; Steenbrugge, J.; Meyer, E.; de Tullio, P.; Bracke, M. Selective Pharmacological Inhibitors of HDAC6 Reveal Biochemical Activity but Functional Tolerance in Cancer Models. Int. J. Cancer 2019, 145, 735–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Emerging Role of Histone Deacetylase Inhibitors as Anti-Breast-Cancer Agents. Drug Discov. Today 2019, 24, 685–702. [Google Scholar] [CrossRef]
- Halley, F.; Reinshagen, J.; Ellinger, B.; Wolf, M.; Niles, A.L.; Evans, N.J.; Kirkland, T.A.; Wagner, J.M.; Jung, M.; Gribbon, P. A Bioluminogenic HDAC Activity Assay: Validation and Screening. J. Biomol. Screen 2011, 16, 1227–1235. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Reers, M.; Smiley, S.T.; Mottola-Hartshorn, C.; Chen, A.; Lin, M.; Chen, L.B. Mitochondrial Membrane Potential Monitored by JC-1 Dye. Methods Enzymol. 1995, 260, 406–417. [Google Scholar] [CrossRef]
- Trivanović, D.; Jauković, A.; Krstić, J.; Nikolić, S.; Okić Djordjević, I.; Kukolj, T.; Obradović, H.; Mojsilović, S.; Ilić, V.; Santibanez, J.F. Inflammatory Cytokines Prime Adipose Tissue Mesenchymal Stem Cells to Enhance Malignancy of MCF-7 Breast Cancer Cells via Transforming Growth Factor-Β1. IUBMB Life 2016, 68, 190–200. [Google Scholar] [CrossRef]
- OECD. Test No. 236: Fish Embryo Acute Toxicity (FET) Test; Organisation for Economic Co-Operation and Development: Paris, France, 2013. [Google Scholar]
- Delasoie, J.; Pavic, A.; Voutier, N.; Vojnovic, S.; Crochet, A.; Nikodinovic-Runic, J.; Zobi, F. Identification of Novel Potent and Non-Toxic Anticancer, Anti-Angiogenic and Antimetastatic Rhenium Complexes against Colorectal Carcinoma. Eur. J. Med. Chem. 2020, 204, 112583. [Google Scholar] [CrossRef] [PubMed]
- Tulotta, C.; Stefanescu, C.; Beletkaia, E.; Bussmann, J.; Tarbashevich, K.; Schmidt, T.; Snaar-Jagalska, B.E. Inhibition of Signaling between Human CXCR4 and Zebrafish Ligands by the Small Molecule IT1t Impairs the Formation of Triple-Negative Breast Cancer Early Metastases in a Zebrafish Xenograft Model. Dis. Model Mech. 2016, 9, 141–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruzic, D.; Petkovic, M.; Agbaba, D.; Ganesan, A.; Nikolic, K. Combined Ligand and Fragment-Based Drug Design of Selective Histone Deacetylase—6 Inhibitors. Mol. Inform. 2019, 38, 1800083. [Google Scholar] [CrossRef] [PubMed]
- Molecular Descriptors Calculation—Dragon—Talete Srl. Available online: http://www.talete.mi.it/products/dragon_description.htm (accessed on 30 October 2022).
- Krukowski, K.; Ma, J.; Golonzhka, O.; Laumet, G.O.; Gutti, T.; van Duzer, J.H.; Mazitschek, R.; Jarpe, M.B.; Heijnen, C.J.; Kavelaars, A. HDAC6 Inhibition Effectively Reverses Chemotherapy-Induced Peripheral Neuropathy. Pain 2017, 158, 1126–1137. [Google Scholar] [CrossRef]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational Design and Simple Chemistry Yield a Superior, Neuroprotective HDAC6 Inhibitor, Tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [Green Version]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.-C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C. Preclinical Activity, Pharmacodynamic, and Pharmacokinetic Properties of a Selective HDAC6 Inhibitor, ACY-1215, in Combination with Bortezomib in Multiple Myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef]
- Porter, N.J.; Wagner, F.F.; Christianson, D.W. Entropy as a Driver of Selectivity for Inhibitor Binding to Histone Deacetylase 6. Biochemistry 2018, 57, 3916–3924. [Google Scholar] [CrossRef]
- Porter, N.J.; Mahendran, A.; Breslow, R.; Christianson, D.W. Unusual Zinc-Binding Mode of HDAC6-Selective Hydroxamate Inhibitors. Proc. Natl. Acad. Sci. USA 2017, 114, 13459–13464. [Google Scholar] [CrossRef] [Green Version]
- Osko, J.D.; Porter, N.J.; Narayana Reddy, P.A.; Xiao, Y.-C.; Rokka, J.; Jung, M.; Hooker, J.M.; Salvino, J.M.; Christianson, D.W. Exploring Structural Determinants of Inhibitor Affinity and Selectivity in Complexes with Histone Deacetylase 6. J. Med. Chem. 2020, 63, 295–308. [Google Scholar] [CrossRef]
- Gaisina, I.N.; Tueckmantel, W.; Ugolkov, A.; Shen, S.; Hoffen, J.; Dubrovskyi, O.; Mazar, A.; Schoon, R.A.; Billadeau, D.; Kozikowski, A.P. Identification of HDAC6-Selective Inhibitors of Low Cancer Cell Cytotoxicity. ChemMedChem 2016, 11, 81–92. [Google Scholar] [CrossRef]
- Jan, R.; Chaudhry, G.-E.-S. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langhans, S.A. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharmacol. 2018, 9, 6. [Google Scholar] [CrossRef]
- Justus, C.R.; Leffler, N.; Ruiz-Echevarria, M.; Yang, L.V. In Vitro Cell Migration and Invasion Assays. J. Vis. Exp. 2014, 88, 51046. [Google Scholar] [CrossRef] [Green Version]
- Wolf, K.; Alexander, S.; Schacht, V.; Coussens, L.M.; von Andrian, U.H.; van Rheenen, J.; Deryugina, E.; Friedl, P. Collagen-Based Cell Migration Models in Vitro and in Vivo. Semin Cell Dev. Biol. 2009, 20, 931–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, T.Q.; Robinson, K.; Xu, L.; Pavlova, M.N.; Skapek, S.X.; Chen, E.Y. HDAC6 Promotes Growth, Migration/Invasion, and Self-Renewal of Rhabdomyosarcoma. Oncogene 2021, 40, 578–591. [Google Scholar] [CrossRef]
- Azuma, K.; Urano, T.; Horie-Inoue, K.; Hayashi, S.; Sakai, R.; Ouchi, Y.; Inoue, S. Association of Estrogen Receptor α and Histone Deacetylase 6 Causes Rapid Deacetylation of Tubulin in Breast Cancer Cells. Cancer Res. 2009, 69, 2935–2940. [Google Scholar] [CrossRef] [Green Version]
- MacRae, C.A.; Peterson, R.T. Zebrafish as Tools for Drug Discovery. Nat. Rev. Drug Discov. 2015, 14, 721–731. [Google Scholar] [CrossRef]
- Chakraborty, C.; Hsu, C.H.; Wen, Z.H.; Lin, C.S.; Agoramoorthy, G. Zebrafish: A Complete Animal Model for in Vivo Drug Discovery and Development. Curr. Drug Metab. 2009, 10, 116–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassar, S.; Adatto, I.; Freeman, J.L.; Gamse, J.T.; Iturria, I.; Lawrence, C.; Muriana, A.; Peterson, R.T.; Van Cruchten, S.; Zon, L.I. Use of Zebrafish in Drug Discovery Toxicology. Chem. Res. Toxicol. 2020, 33, 95–118. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-C.; Lin, C.-Y.; Tsai, H.-J. Zebrafish, an In Vivo Platform to Screen Drugs and Proteins for Biomedical Use. Pharmaceuticals 2021, 14, 500. [Google Scholar] [CrossRef]
- Cully, M. Zebrafish Earn Their Drug Discovery Stripes. Nat. Rev. Drug Discov. 2019, 18, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis in Cancer, Vascular, Rheumatoid and Other Disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Kerbel, R.S. Angiogenesis as a Therapeutic Target. Nature 2005, 438, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Pennesi, G.; Donatelli, F.; Cammarota, R.; De Flora, S.; Noonan, D.M. Cardiotoxicity of Anticancer Drugs: The Need for Cardio-Oncology and Cardio-Oncological Prevention. J. Natl. Cancer Inst. 2010, 102, 14–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fior, R.; Póvoa, V.; Mendes, R.V.; Carvalho, T.; Gomes, A.; Figueiredo, N.; Ferreira, M.G. Single-Cell Functional and Chemosensitive Profiling of Combinatorial Colorectal Therapy in Zebrafish Xenografts. Proc. Natl. Acad. Sci. USA 2017, 114, E8234–E8243. [Google Scholar] [CrossRef] [Green Version]
- White, R.; Rose, K.; Zon, L. Zebrafish Cancer: The State of the Art and the Path Forward. Nat. Rev. Cancer 2013, 13, 624–636. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % HDAC6 Inhibition at 5 μM | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | 2b | 3b | 4b | 5b | 6b | 7b | 8b | 9b |
| 12.2 ± 1.7 | 22.2 ± 1.1 | 3.1 ± 1.7 | 18.2 ± 1.7 | 95.6 ± 0.9 | 97.1 ± 0.8 | 97.7 ± 0.2 | 96.9 ± 0.3 | |
| Compound | HDAC Inhibition, IC50 ± SD (μM) | Selectivity Ratio | |||||
|---|---|---|---|---|---|---|---|
| HDAC1 | HDAC3 | HDAC6 | HDAC8 | HDAC1/6 | HDAC3/6 | HDAC8/6 | |
| 6b | 4.730 ± 0.670 | 1.860 ± 0.090 | 0.186 ± 0.005 | 2.440 ± 0.510 | 25.4 | 10 | 13.1 |
| 7b | 0.619 ± 0.023 | 0.267 ± 0.019 | 0.096 ± 0.008 | 0.345 ± 0.017 | 6.4 | 2.8 | 3.6 |
| 8b | 0.408 ± 0.022 | 0.207 ± 0.014 | 0.124 ± 0.013 | 0.245 ± 0.002 | 3.3 | 1.7 | 2 |
| 9b | 1.474 ± 0.172 | 3.467 ± 0.121 | 0.031 ± 0.004 | 0.708 ± 0.045 | 47.5 | 111.8 | 23 |
| Valproic acid # | 660 | >1000 | >1000 | 103 | - | - | - |
| Sodium butyrate # | 8.3 | 4.8 | >1000 | 10.4 | - | - | - |
| Trichostatin A # | 0.0013 | 0.0015 | 0.0036 | 0.400 | 0.3 | 0.4 | 111.1 |
| Compound | IC50 ± SD (μM) | |
|---|---|---|
| MDA-MB-231 | MCF-7 | |
| 6b | 33.40 ± 2.79 | 84.05 ± 5.2 |
| 7b | 10.55 ± 1.95 | >100 |
| 8b | 5.42 ± 0.77 | 39.10 ± 2.7 |
| 9b | 38.21 ± 3.01 | 99.50 ± 0.7 |
| tubastatin A | 20.83 ± 2.84 | 93.31 ± 9.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruzic, D.; Ellinger, B.; Djokovic, N.; Santibanez, J.F.; Gul, S.; Beljkas, M.; Djuric, A.; Ganesan, A.; Pavic, A.; Srdic-Rajic, T.; et al. Discovery of 1-Benzhydryl-Piperazine-Based HDAC Inhibitors with Anti-Breast Cancer Activity: Synthesis, Molecular Modeling, In Vitro and In Vivo Biological Evaluation. Pharmaceutics 2022, 14, 2600. https://doi.org/10.3390/pharmaceutics14122600
Ruzic D, Ellinger B, Djokovic N, Santibanez JF, Gul S, Beljkas M, Djuric A, Ganesan A, Pavic A, Srdic-Rajic T, et al. Discovery of 1-Benzhydryl-Piperazine-Based HDAC Inhibitors with Anti-Breast Cancer Activity: Synthesis, Molecular Modeling, In Vitro and In Vivo Biological Evaluation. Pharmaceutics. 2022; 14(12):2600. https://doi.org/10.3390/pharmaceutics14122600
Chicago/Turabian StyleRuzic, Dusan, Bernhard Ellinger, Nemanja Djokovic, Juan F. Santibanez, Sheraz Gul, Milan Beljkas, Ana Djuric, Arasu Ganesan, Aleksandar Pavic, Tatjana Srdic-Rajic, and et al. 2022. "Discovery of 1-Benzhydryl-Piperazine-Based HDAC Inhibitors with Anti-Breast Cancer Activity: Synthesis, Molecular Modeling, In Vitro and In Vivo Biological Evaluation" Pharmaceutics 14, no. 12: 2600. https://doi.org/10.3390/pharmaceutics14122600