
Intranasal Administration of N-acetyl-L-cysteine Combined with Cell-Penetrating Peptide-Modified Polymer Nanomicelles as a Potential Therapeutic Approach for Amyotrophic Lateral Sclerosis
 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Radioisotopes and Chemicals
2.2. Synthesis of PEG-PCL-Tat Micelles
2.3. Preparation of the NAC Containing PEG-PCL-Tat Micelles Solution
2.4. Physicochemical Characterization of the NAC Containing PEG-PCL-Tat Micelles Solution
2.5. Animals
2.5.1. G93A Mice
2.5.2. ddY Mice
2.6. Intranasal Administration
2.7. Drug Administration and Survival Analyses
2.8. Pharmacokinetics Following Intranasal Administration of [14C]-NAC/PEG-PCL-Tat Micelles in ddY Mice
2.8.1. Preparation of [14C]-NAC/PEG-PCL-Tat
2.8.2. Blood Sampling after the Intranasal Administration
2.9. Collection of CSF, Plasma, and Tissues Following an Intranasal Administration to ddY Mice
2.10. Determination of [14C] Radioactivity
2.11. Statistical Analysis
3. Results
3.1. Physicochemical Characterization of NAC/PEG-PCL-Tat Micelles
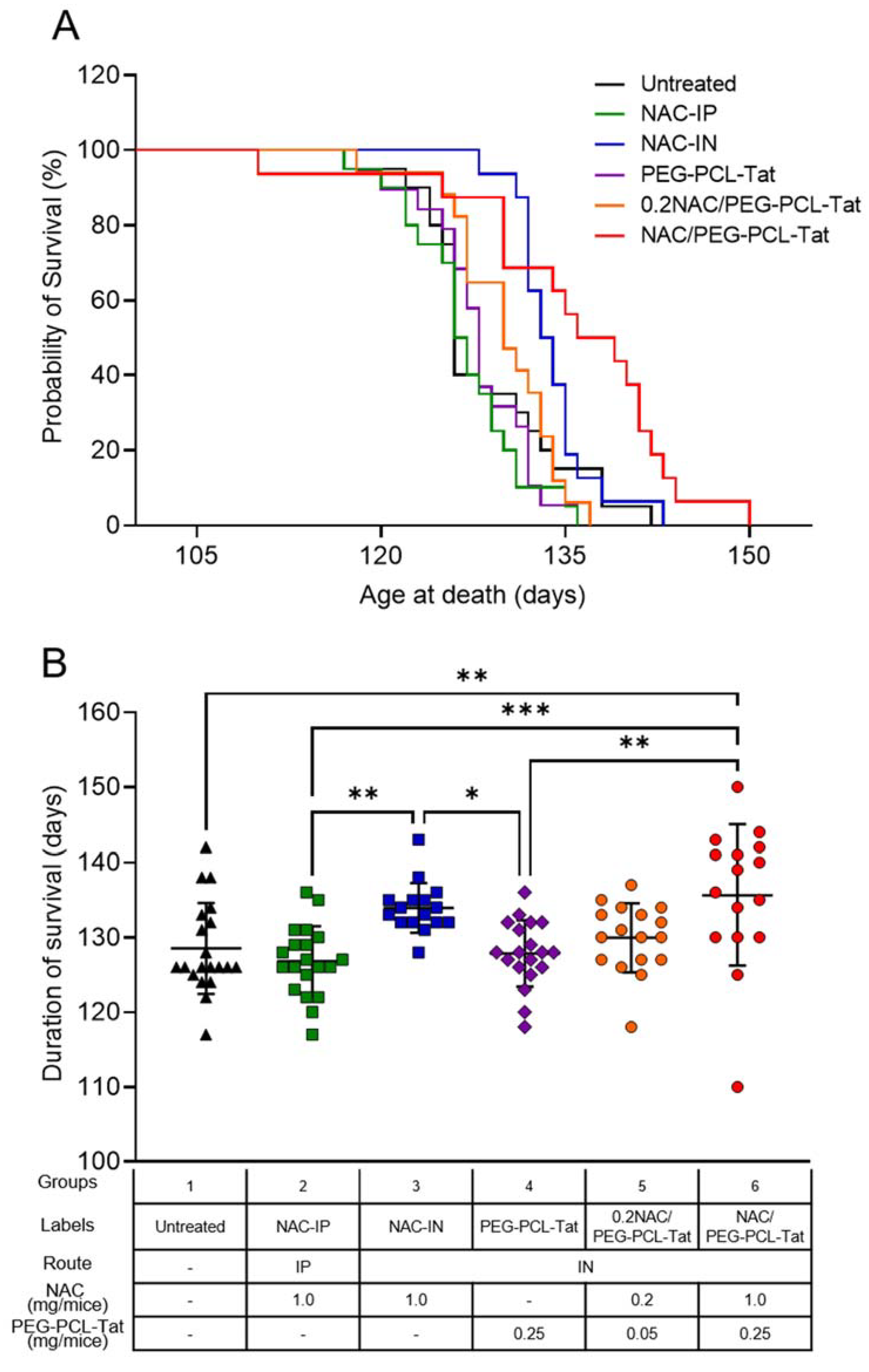
3.2. Effect of NAC on Survival Duration of G93A Mice
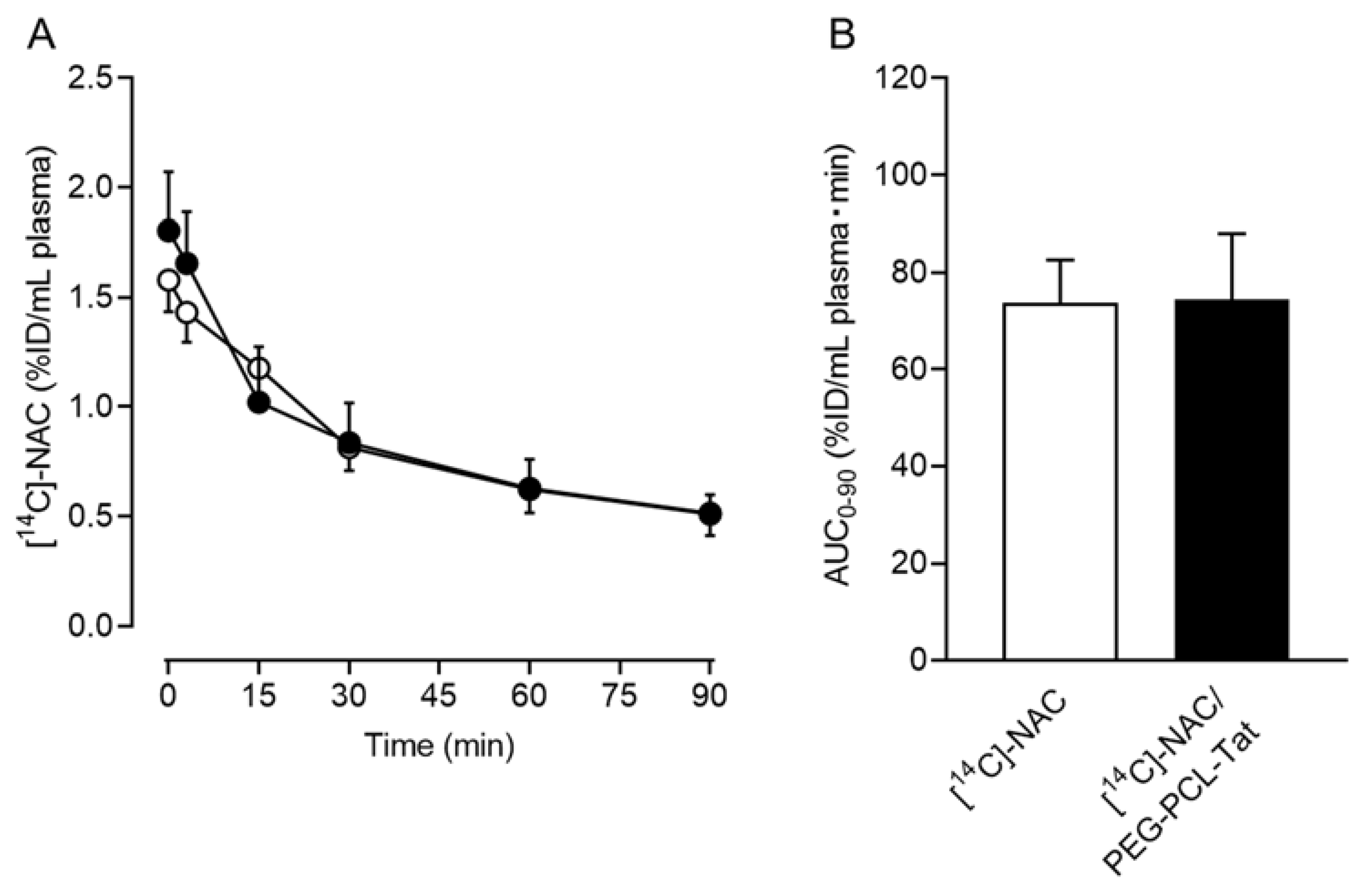
3.3. Plasma Time-Dependent Concentration of [14C]-NAC/PEG-PCL-Tat Micelles after a Single Intranasal Administration
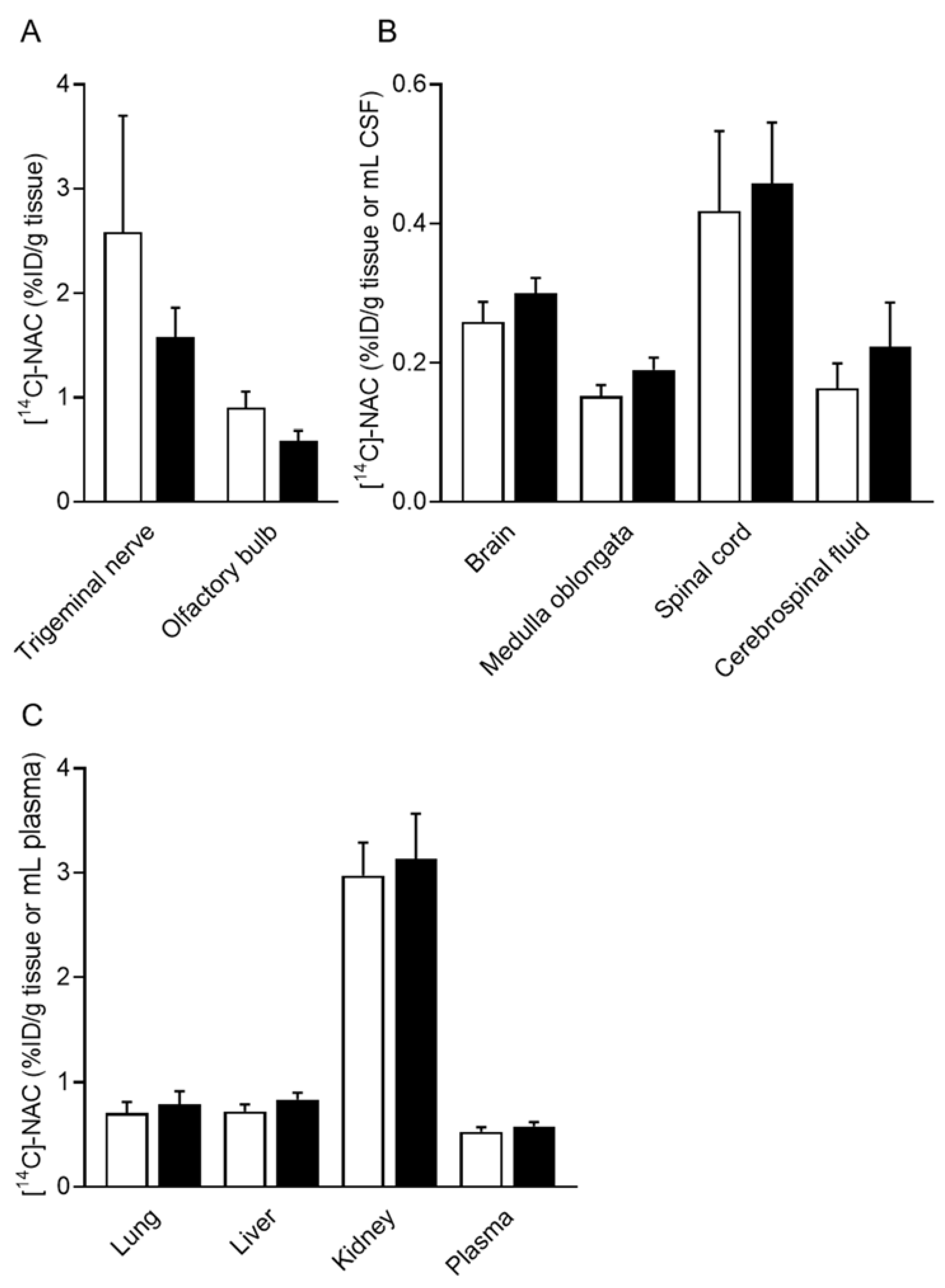
3.4. Drug Distribution in Tissue after a Single Intranasal Administration of [14C]-NAC/PEG-PCL-Tat Micelles
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Bonafede, R.; Mariotti, R. ALS Pathogenesis and Therapeutic Approaches: The Role of Mesenchymal Stem Cells and Extracellular Vesicles. Front. Cell. Neurosci. 2017, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Beretta, S.; Sala, G.; Mattavelli, L.; Ceresa, C.; Casciati, A.; Ferri, A.; Carrì, M.T.; Ferrarese, C. Mitochondrial Dysfunction Due to Mutant Copper/Zinc Superoxide Dismutase Associated with Amyotrophic Lateral Sclerosis Is Reversed by N-Acetylcysteine. Neurobiol. Dis. 2003, 13, 213–221. [Google Scholar] [CrossRef]
- Arakawa, M.; Ito, Y. N-Acetylcysteine and Neurodegenerative Diseases: Basic and Clinical Pharmacology. Cerebellum 2007, 6, 308–314. [Google Scholar] [CrossRef]
- Jaarsma, D.; Guchelaar, H.J.; Haasdijk, E.; De Jong, J.M.B.V.; Holstege, J.C. The Antioxidant N-Acetylcysteine Does Not Delay Disease Onset and Death in a Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. Ann. Neurol. 1998, 44, 293. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The Blood–Brain Barrier: Bottleneck in Brain Drug Development. Neurorx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- McLellan, L.I.; Lewis, A.D.; Hall, D.J.; Ansell, J.D.; Wolf, C.R. Uptake and Distribution of N-Acetylcysteine in Mice: Tissue-Specific Effects on Glutathione Concentrations. Carcinogenesis 1995, 16, 2099–2106. [Google Scholar] [CrossRef]
- Ogawa, K.; Kato, N.; Kawakami, S. Recent Strategies for Targeted Brain Drug Delivery. Chem. Pharm. Bull 2020, 68, 567–582. [Google Scholar] [CrossRef]
- Agrawal, M.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Chougule, M.B.; Shoyele, S.A.; Alexander, A. Nose-To-Brain Drug Delivery: An Update on Clinical Challenges and Progress Towards Approval of Anti-Alzheimer Drugs. J. Control. Release 2018, 281, 139–177. [Google Scholar] [CrossRef]
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of Intranasal Drug Delivery Directly to the Brain. Life Sci. 2018, 195, 44–52. [Google Scholar] [CrossRef]
- Kumar, N.N.; Lochhead, J.J.; Pizzo, M.E.; Nehra, G.; Boroumand, S.; Greene, G.; Thorne, R.G. Delivery of Immunoglobulin G Antibodies to the Rat Nervous System Following Intranasal Administration: Distribution, Dose–Response, and Mechanisms of Delivery. J. Control. Release 2018, 286, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Davis, T.P. Perivascular and Perineural Pathways Involved in Brain Delivery and Distribution of Drugs after Intranasal Administration. Pharmaceutics 2019, 11, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lochhead, J.J.; Wolak, D.J.; Pizzo, M.E.; Thorne, R.G. Rapid Transport within Cerebral Perivascular Spaces Underlies Widespread Tracer Distribution in the Brain after Intranasal Administration. J. Cereb. Blood Flow Metab. 2015, 35, 371–381. [Google Scholar] [CrossRef]
- Lochhead, J.J.; Kellohen, K.L.; Ronaldson, P.T.; Davis, T.P. Distribution of Insulin in Trigeminal Nerve and Brain after Intranasal Administration. Sci. Rep. 2019, 9, 2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.; Furubayashi, T.; Arai, M.; Inoue, D.; Kimura, S.; Kiriyama, A.; Kusamori, K.; Katsumi, H.; Yutani, R.; Sakane, T.; et al. Delivery of Oxytocin to the Brain for the Treatment of Autism Spectrum Disorder by Nasal Application. Mol. Pharm. 2018, 15, 1105–1111. [Google Scholar] [CrossRef]
- Stenslik, M.J.; Evans, A.; Pomerleau, F.; Weeks, R.; Huettl, P.; Foreman, E.; Turchan-Cholewo, J.; Andersen, A.; Cass, W.A.; Zhang, Z.; et al. Methodology and Effects of Repeated Intranasal Delivery of DNSP-11 in Awake Rhesus Macaques. J. Neurosci. Methods 2018, 303, 30–40. [Google Scholar] [CrossRef]
- Born, J.; Lange, T.; Kern, W.; McGregor, G.P.; Bickel, U.; Fehm, H.L. Sniffing Neuropeptides: A Transnasal Approach to the Human Brain. Nat. Neurosci. 2002, 5, 514–516. [Google Scholar] [CrossRef]
- Kamei, N.; Shingaki, T.; Kanayama, Y.; Tanaka, M.; Zochi, R.; Hasegawa, K.; Watanabe, Y.; Takeda-Morishita, M. Visualization and Quantitative Assessment of the Brain Distribution of Insulin Through Nose-To-Brain Delivery Based on the Cell-Penetrating Peptide Noncovalent Strategy. Mol. Pharm. 2016, 13, 1004–1011. [Google Scholar] [CrossRef]
- Lin, T.; Liu, E.; He, H.; Shin, M.C.; Moon, C.; Yang, V.C.; Huang, Y. Nose-To-Brain Delivery of Macromolecules Mediated by Cell-Penetrating Peptides. Acta Pharm. Sin. B 2016, 6, 352–358. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Sun, Y.Y.; Lin, X.; Baumann, J.M.; Dunn, R.S.; Lindquist, D.M.; Kuan, C.Y. Intranasal Delivery of Cell-Penetrating Anti-NF-ΚB Peptides (Tat-NBD) Alleviates Infection-Sensitized Hypoxic–Ischemic Brain Injury. Exp. Neurol. 2013, 247, 447–455. [Google Scholar] [CrossRef]
- Samaridou, E.; Alonso, M.J. Nose-To-Brain Peptide Delivery—The Potential of Nanotechnology. Bioorg. Med. Chem. 2018, 26, 2888–2905. [Google Scholar] [CrossRef] [PubMed]
- Sonvico, F.; Clementino, A.; Buttini, F.; Colombo, G.; Pescina, S.; Stanisçuaski Guterres, S.; Raffin Pohlmann, A.; Nicoli, S. Surface-Modified Nanocarriers for Nose-To-Brain Delivery: From Bioadhesion to Targeting. Pharmaceutics 2018, 10, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanazawa, T.; Akiyama, F.; Kakizaki, S.; Takashima, Y.; Seta, Y. Delivery of SiRNA to the Brain Using a Combination of Nose-To-Brain Delivery and Cell-Penetrating Peptide-Modified Nano-Micelles. Biomaterials 2013, 34, 9220–9226. [Google Scholar] [CrossRef] [PubMed]
- Taki, H.; Kanazawa, T.; Akiyama, F.; Takashima, Y.; Okada, H. Intranasal Delivery of Camptothecin-Loaded Tat-Modified Nanomicells for Treatment of Intracranial Brain Tumors. Pharmaceuticals 2012, 5, 1092–1102. [Google Scholar] [CrossRef] [Green Version]
- Kanazawa, T.; Morisaki, K.; Suzuki, S.; Takashima, Y. Prolongation of Life in Rats with Malignant Glioma by Intranasal siRNA/Drug Codelivery to the Brain with Cell-Penetrating Peptide-Modified Micelles. Mol. Pharm. 2014, 11, 1471–1478. [Google Scholar] [CrossRef]
- Kanazawa, T.; Taki, H.; Okada, H. Nose-To-Brain Drug Delivery System with Ligand/Cell-Penetrating Peptide-Modified Polymeric Nano-Micelles for Intracerebral Gliomas. Eur. J. Pharm. Biopharm. 2020, 152, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T.; Kurano, T.; Ibaraki, H.; Takashima, Y.; Suzuki, T.; Seta, Y. Therapeutic Effects in a Transient Middle Cerebral Artery Occlusion Rat Model by Nose-To-Brain Delivery of Anti-TNF-alpha siRNA with Cell-Penetrating Peptide-Modified Polymer Micelles. Pharmaceutics 2019, 11, 478. [Google Scholar] [CrossRef] [Green Version]
- Kurano, T.; Kanazawa, T.; Ooba, A.; Masuyama, Y.; Maruhana, N.; Yamada, M.; Iioka, S.; Ibaraki, H.; Kosuge, Y.; Kondo, H.; et al. Nose-To-Brain/Spinal Cord Delivery Kinetics of Liposomes with Different Surface Properties. J. Control. Release 2022, 344, 225–234. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X. Motor Neuron Degeneration in Mice That Express a Human Cu, Zn Superoxide Dismutase Mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Tanaka, K.; Kanazawa, T.; Shibata, Y.; Suda, Y.; Fukuda, T.; Takashima, Y.; Okada, H. Development of Cell-Penetrating Peptide-Modified MPEG-PCL Diblock Copolymeric Nanoparticles for Systemic Gene Delivery. Int. J. Pharm. 2010, 396, 229–238. [Google Scholar] [CrossRef]
- Tokuda, E.; Ono, S.; Ishige, K.; Naganuma, A.; Ito, Y.; Suzuki, T. Metallothionein Proteins Expression, Copper and Zinc Concentrations, and Lipid Peroxidation Level in a Rodent Model for Amyotrophic Lateral Sclerosis. Toxicology 2007, 229, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T.; Fukuda, M.; Suzuki, N.; Suzuki, T. Novel Methods for Intranasal Administration under Inhalation Anesthesia to Evaluate Nose-To-Brain Drug Delivery. J. Vis. Exp. 2018, 141, 2–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, M.; Kanazawa, T.; Iioka, S.; Oguma, T.; Iwasa, R.; Masuoka, S.; Suzuki, N.; Kosuge, Y.; Suzuki, T. Quantitative Analysis of Inulin Distribution in the Brain Focused on Nose-To-Brain Route via Olfactory Epithelium by Reverse Esophageal Cannulation. J. Control. Release 2021, 332, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Cheroni, C.; Peviani, M.; Cascio, P.; DeBiasi, S.; Monti, C.; Bendotti, C. Accumulation of Human SOD1 and Ubiquitinated Deposits in the Spinal Cord of SOD1G93A Mice during Motor Neuron Disease Progression Correlates with a Decrease of Proteasome. Neurobiol. Dis. 2005, 18, 509–522. [Google Scholar] [CrossRef]
- Kosuge, Y.; Kaneko, E.; Nango, H.; Miyagishi, H.; Ishige, K.; Ito, Y. Bidens Pilosa Extract Administered after Symptom Onset Attenuates Glial Activation, Improves Motor Performance, and Prolongs Survival in a Mouse Model of Amyotrophic Lateral Sclerosis. Oxid. Med. Cell. Longev. 2020, 2020, 1020673. [Google Scholar] [CrossRef] [Green Version]
- Ernsting, M.J.; Murakami, M.; Roy, A.; Li, S.D. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release 2013, 172, 782–794. [Google Scholar] [CrossRef] [Green Version]
- Ignatovich, I.A.; Dizhe, E.B.; Pavlotskaya, A.V.; Akifiev, B.N.; Burov, S.V.; Orlov, S.V.; Perevozchikov, A.P. Complexes of Plasmid DNA with Basic Domain 47–57 of the HIV-1 Tat Protein Are Transferred to Mammalian Cells by Endocytosis-Mediated Pathways. J. Biol. Chem. 2003, 278, 42625–42636. [Google Scholar] [CrossRef] [Green Version]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The Chemistry and Biological Activities of N-Acetylcysteine. Biochim. Biophys. Acta 2013, 1830, 4117–4129. [Google Scholar] [CrossRef]
- Borrajo, M.L.; Alonso, M.J. Using Nanotechnology to Deliver Biomolecules from Nose to Brain—Peptides, Proteins, Monoclonal Antibodies and RNA. Drug Deliv. Transl. Res. 2021, 12, 133–140. [Google Scholar] [CrossRef]
- Pires, P.C.; Santos, A.O. Nanosystems in nose-to-brain drug delivery: A review of non-clinical brain targeting studies. J. Control. Release 2018, 270, 89–100. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Dedeoglu, A.; Klivenyi, P.; Beal, M.F.; Bush, A.I. N-acetyl-L-cysteine Improves Survival and Preserves Motor Performance in an Animal Model of Familial Amyotrophic Lateral Sclerosis. Neuroreport 2000, 11, 2491–2493. [Google Scholar] [CrossRef] [PubMed]
- Hogg, M.C.; Halang, L.; Woods, I.; Coughlan, K.S.; Prehn, J.H.M. Riluzole Does Not Improve Lifespan or Motor Function in Three Als Mouse Models. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Wate, R.; Zhang, J.; Ohnishi, S.; Kaneko, S.; Ito, H.; Nakano, S.; Kusaka, H. Treatment with Edaravone, Initiated at Symptom Onset, Slows Motor Decline and Decreases SOD1 Deposition in ALS Mice. Exp. Neurol. 2008, 213, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, M.A.; Hemley, S.J.; Najafi, E.; Vella, N.G.F.; Bilston, L.E.; Stoodley, M.A. The Ultrastructure of Spinal Cord Perivascular Spaces: Implications for the Circulation of Cerebrospinal Fluid. Sci. Rep. 2017, 7, 12924. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Groups | Labels | Route of Administration | Dosing Volume (µL) | NAC (mg/Mice) | PEG-PCL-Tat (mg/Mice) | Number of Animals |
|---|---|---|---|---|---|---|
| 1 | Untreated | - | - | - | - | 20 |
| 2 | NAC-IP | IP | 100 | 1.0 | - | 20 |
| 3 | NAC-IN | IN | 20 | 1.0 | - | 16 |
| 4 | PEG-PCL-Tat | IN | 20 | - | 0.25 | 19 |
| 5 | 0.2NAC/PEG-PCL-Tat | IN | 20 | 0.2 | 0.05 | 17 |
| 6 | NAC/PEG-PCL-Tat | IN | 20 | 1.0 | 0.25 | 16 |
| Nanocarrier | Mean Particle Size (nm) | PDI | Zeta Potential (mV) |
|---|---|---|---|
| PEG-PCL-Tat | 285 ± 6.1 | 0.496 ± 0.016 | +14.1 ± 0.12 |
| 0.2NAC/PEG-PCL-Tat | 270 ± 8.7 | 0.612 ± 0.104 | +12.9 ± 0.45 |
| NAC/PEG-PCL-Tat | 294 ± 7.2 | 0.541 ± 0.106 | +9.29 ± 0.52 **** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurano, T.; Kanazawa, T.; Iioka, S.; Kondo, H.; Kosuge, Y.; Suzuki, T. Intranasal Administration of N-acetyl-L-cysteine Combined with Cell-Penetrating Peptide-Modified Polymer Nanomicelles as a Potential Therapeutic Approach for Amyotrophic Lateral Sclerosis. Pharmaceutics 2022, 14, 2590. https://doi.org/10.3390/pharmaceutics14122590
Kurano T, Kanazawa T, Iioka S, Kondo H, Kosuge Y, Suzuki T. Intranasal Administration of N-acetyl-L-cysteine Combined with Cell-Penetrating Peptide-Modified Polymer Nanomicelles as a Potential Therapeutic Approach for Amyotrophic Lateral Sclerosis. Pharmaceutics. 2022; 14(12):2590. https://doi.org/10.3390/pharmaceutics14122590
Chicago/Turabian StyleKurano, Takumi, Takanori Kanazawa, Shingo Iioka, Hiromu Kondo, Yasuhiro Kosuge, and Toyofumi Suzuki. 2022. "Intranasal Administration of N-acetyl-L-cysteine Combined with Cell-Penetrating Peptide-Modified Polymer Nanomicelles as a Potential Therapeutic Approach for Amyotrophic Lateral Sclerosis" Pharmaceutics 14, no. 12: 2590. https://doi.org/10.3390/pharmaceutics14122590