
Instability Challenges and Stabilization Strategies of Pharmaceutical Proteins

Abstract
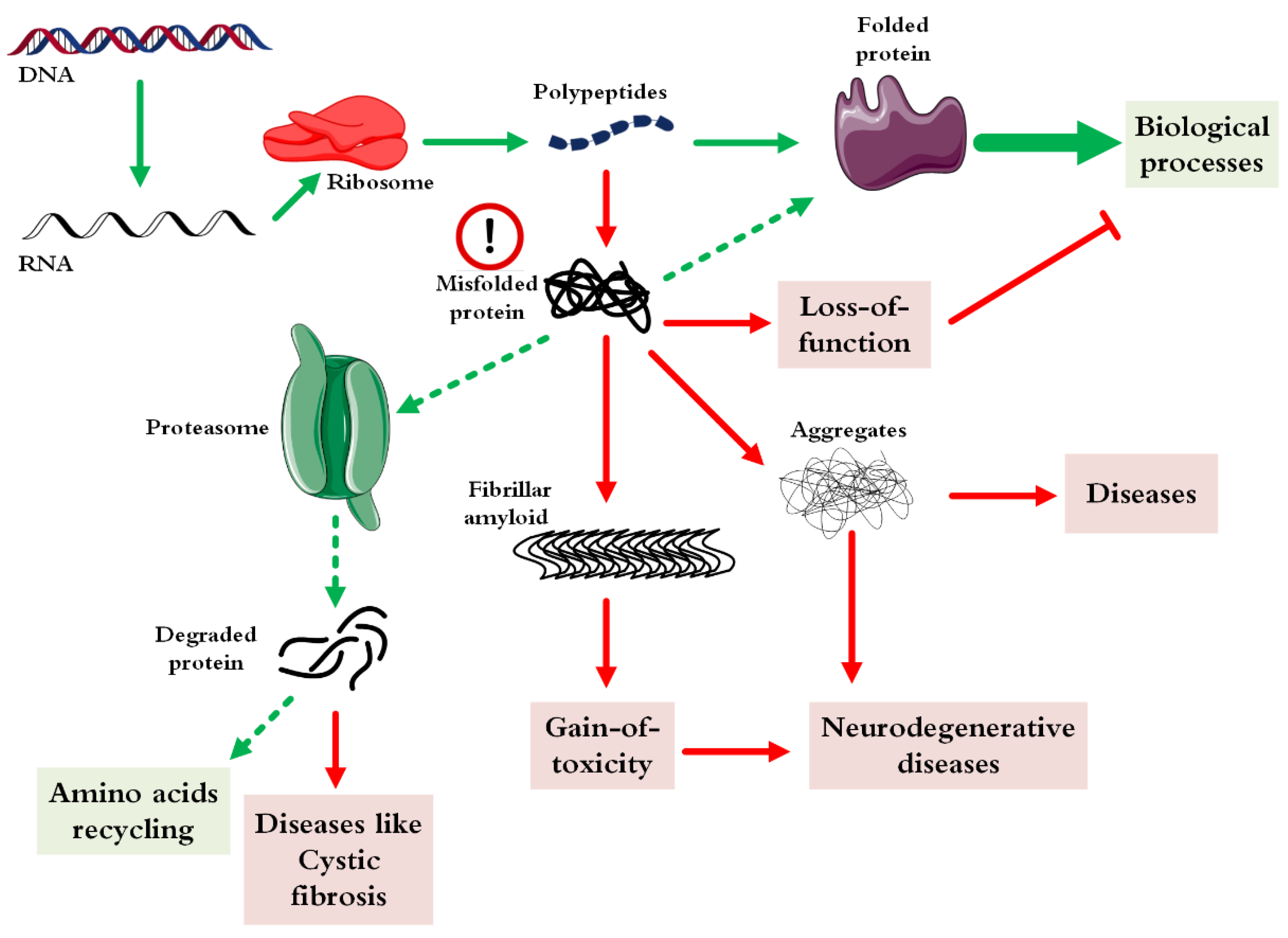
:1. Introduction
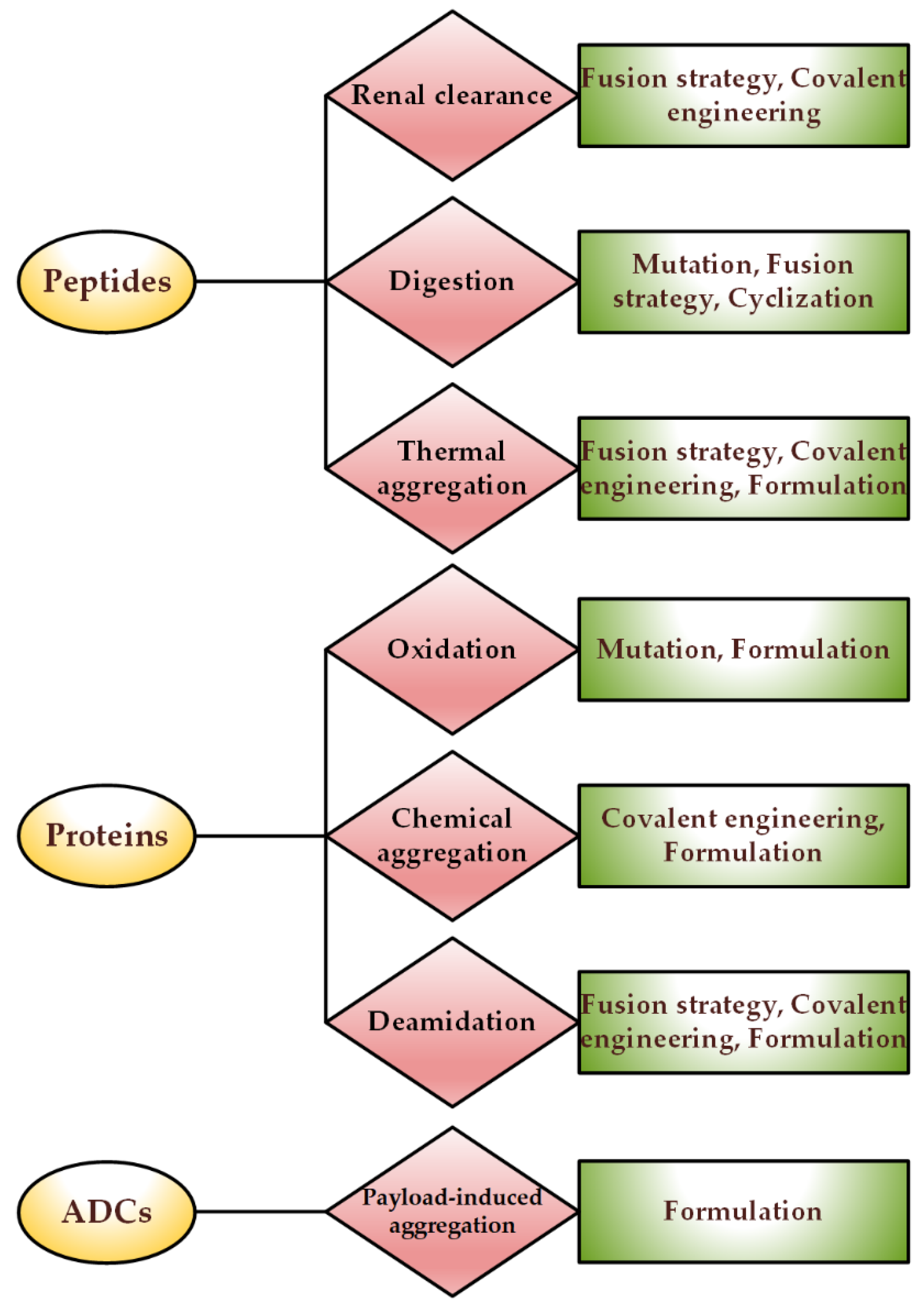
2. Stability Issues of Different Protein-Based Drugs
3. Causes of Instability
3.1. Physical Instability
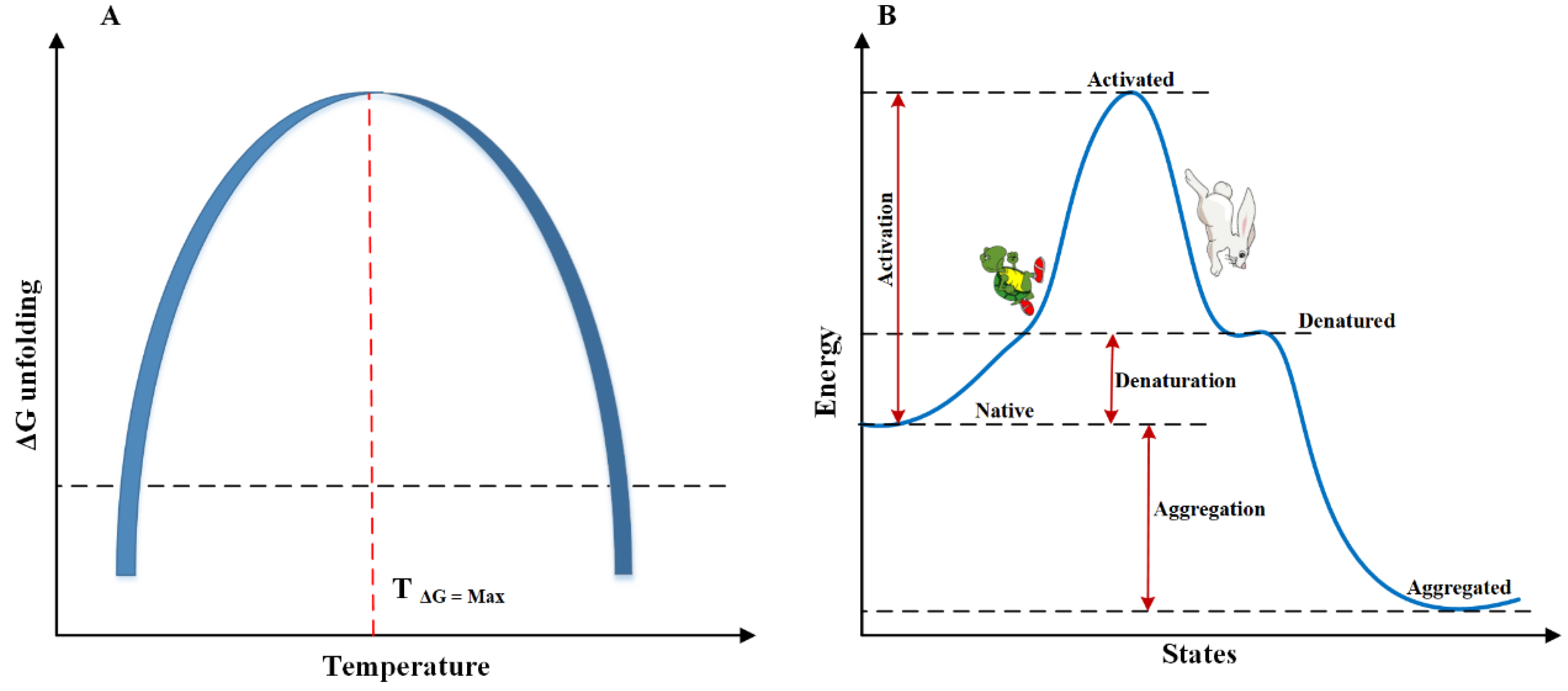
3.1.1. Temperature-Induced Instability
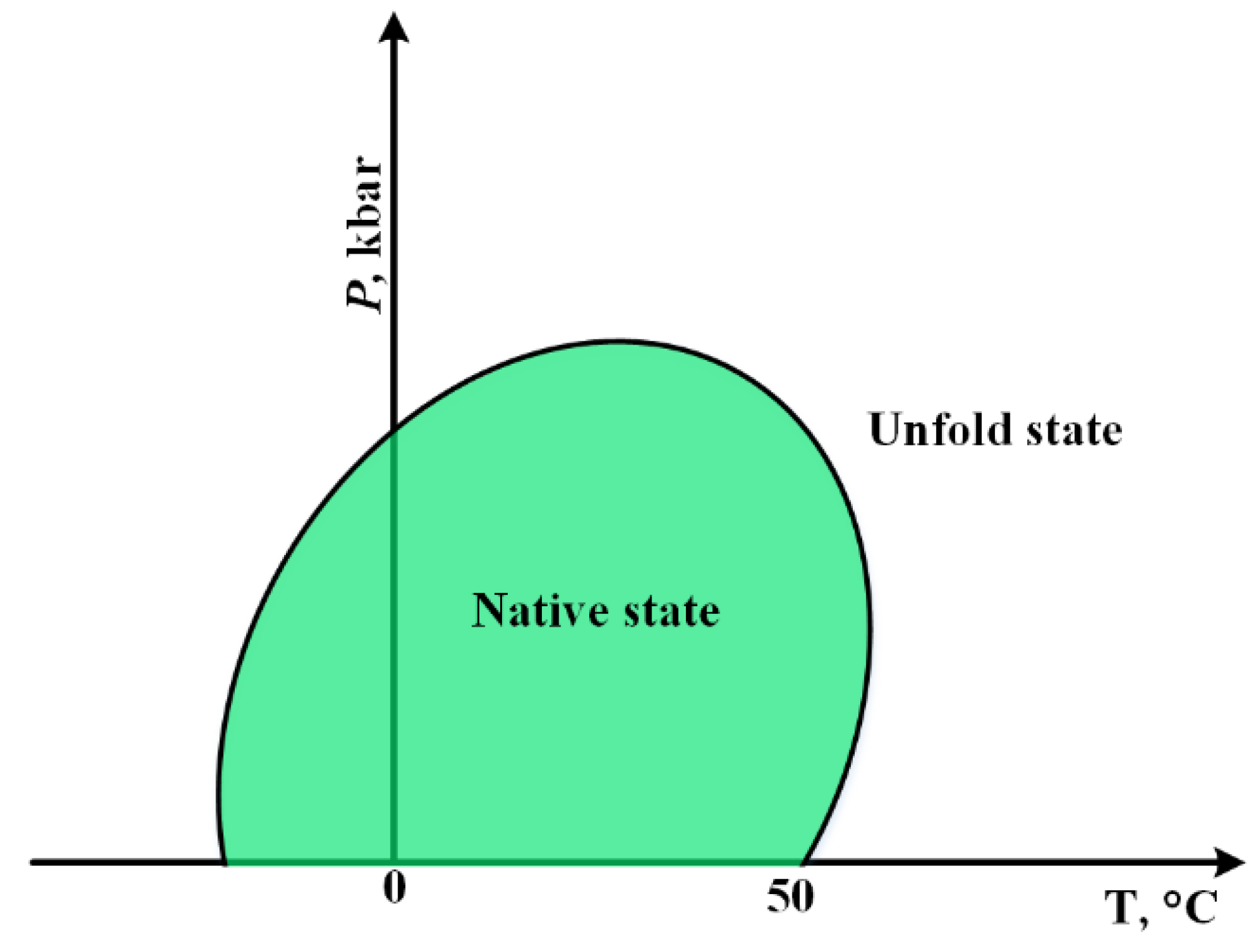
3.1.2. Cold Denaturation
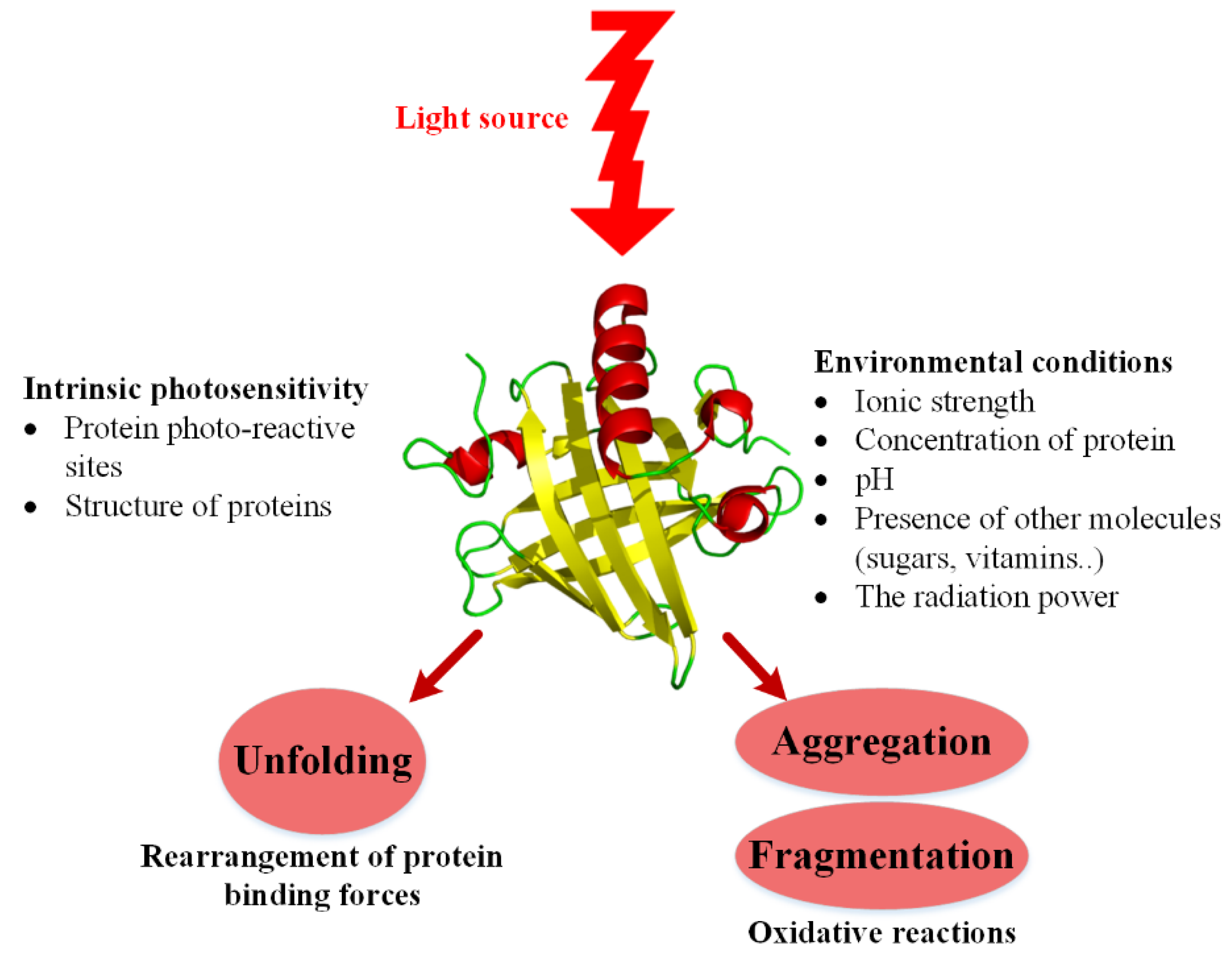
3.1.3. Photo-Induced Instability
3.1.4. Agitation-Induced Instability
3.2. Chemical Instability
3.2.1. Hydrolysis
3.2.2. Oxidation
3.2.3. Disulfide Exchanges
3.2.4. Deamidation
3.2.5. Conjugation-Induced Instability
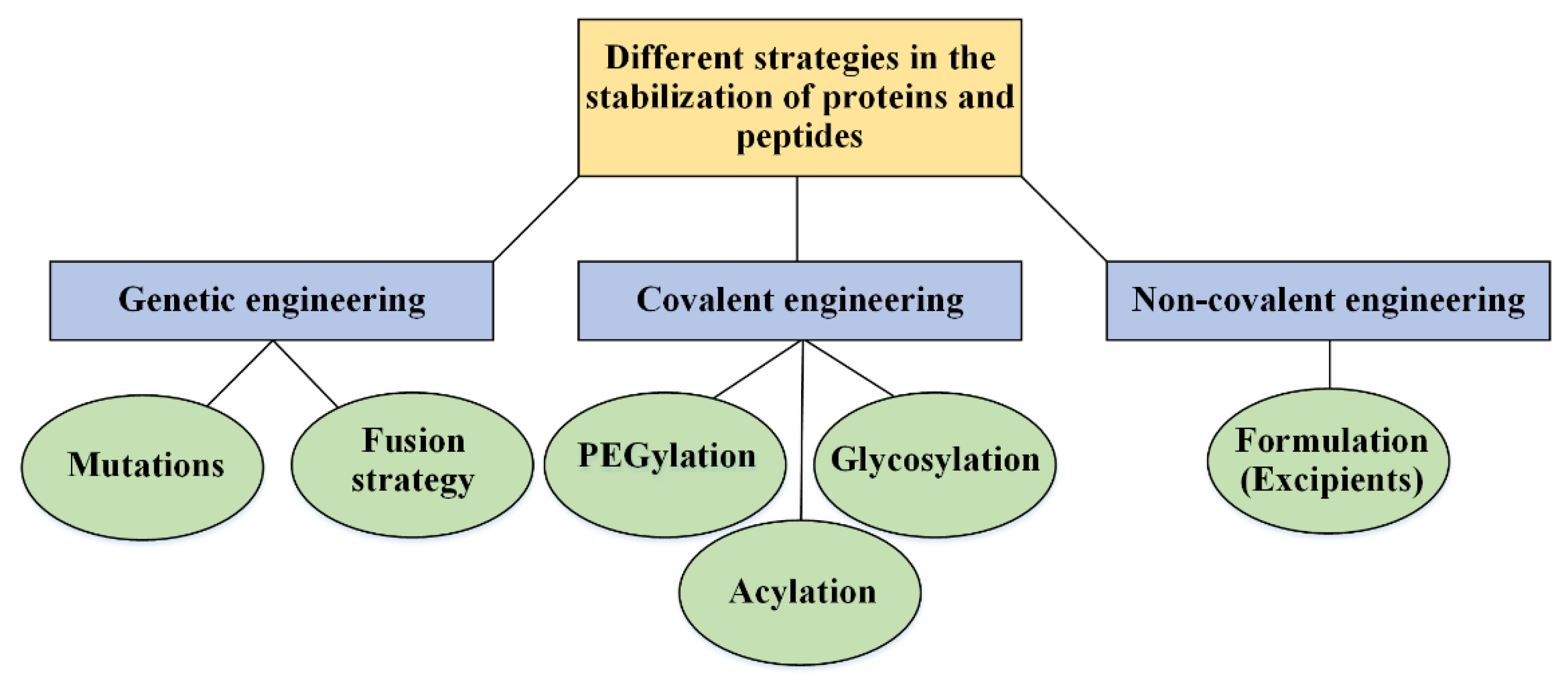
4. Strategies in the Stabilization
4.1. Genetic Engineering: Protein Analogs
4.1.1. Site-Directed Mutagenesis
4.1.2. Fusion Strategies
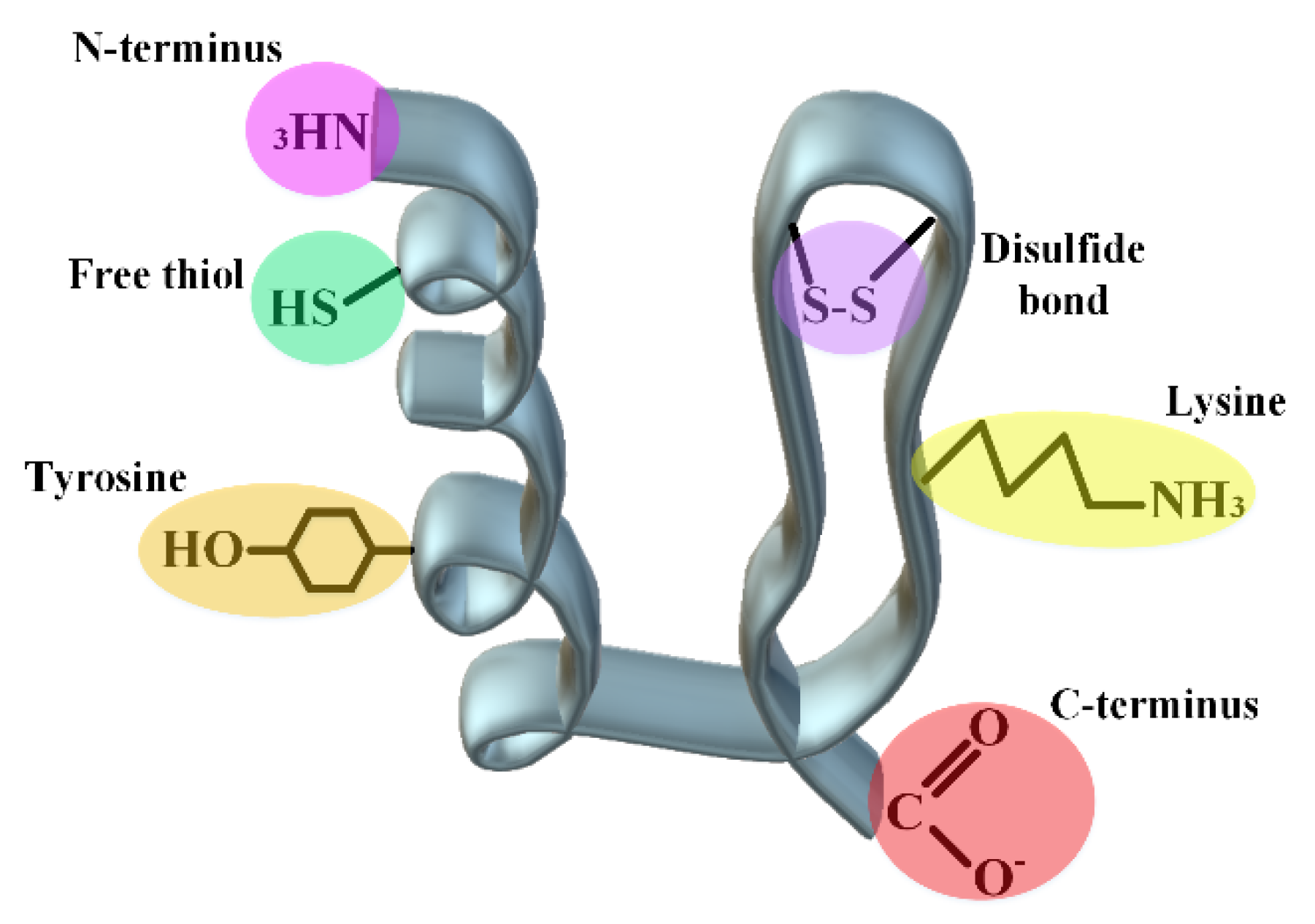
4.2. Covalent Engineering
4.2.1. Protein–Polymer Conjugates
4.2.2. Linker Chemistry
4.2.3. Acylation
4.2.4. Cyclization
4.2.5. Nanoparticles; Double-Blade Sword
4.3. Non-Covalent Engineering of Therapeutic Proteins and Peptides; Solvent Engineering Pathways
4.3.1. Formulation: Solvent Engineering Pathways
4.3.2. Choice of Container
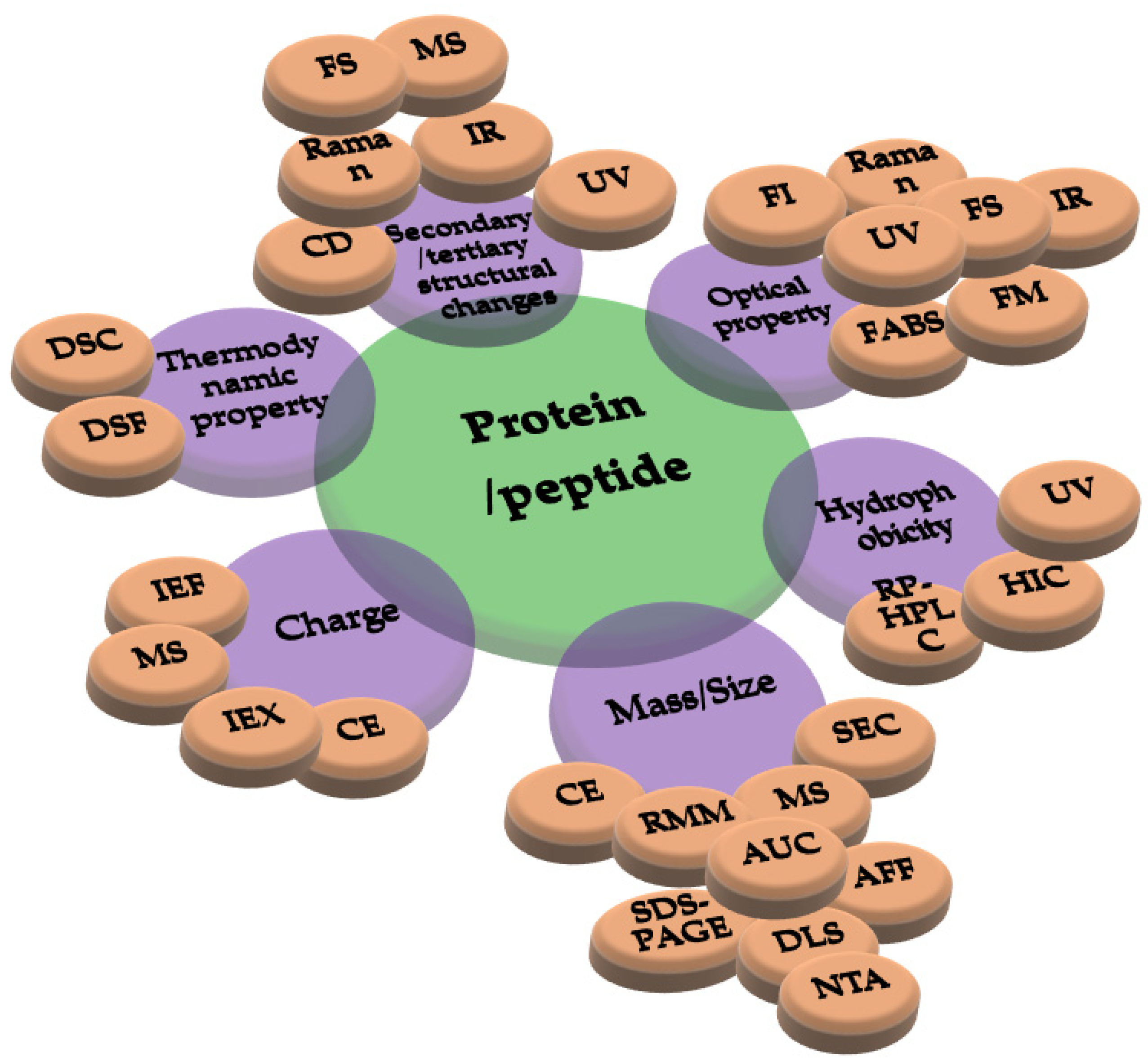
5. Different Approaches in Characterizing Protein Drugs and Induced Structural Changes
6. Conclusions and the Future of Protein and Peptide Drugs
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akbarian, M. Insulin therapy: A valuable legacy and its future perspective. Int. J. Biol. Macromol. 2021, 181, 1224–1230. [Google Scholar] [CrossRef]
- Akbarian, M.; Khani, A.; Eghbalpour, S.; Uversky, V.N. Bioactive Peptides: Synthesis, Sources, Applications, and Proposed Mechanisms of Action. Int. J. Mol. Sci. 2022, 23, 1445. [Google Scholar] [CrossRef]
- Karst, D.J.; Steinebach, F.; Morbidelli, M. Continuous integrated manufacturing of therapeutic proteins. Curr. Opin. Biotechnol. 2018, 53, 76–84. [Google Scholar] [CrossRef]
- Radhakrishnan, D.; Wells, E.A.; Robinson, A.S. Strategies to enhance productivity and modify product quality in therapeutic proteins. Curr. Opin. Chem. Eng. 2018, 22, 81–88. [Google Scholar] [CrossRef]
- Hong, M.S.; Severson, K.A.; Jiang, M.; Lu, A.E.; Love, J.C.; Braatz, R.D. Challenges and opportunities in biopharmaceutical manufacturing control. Comput. Chem. Eng. 2018, 110, 106–114. [Google Scholar] [CrossRef]
- Burnett, M.J.; Burnett, A.C. Therapeutic recombinant protein production in plants: Challenges and opportunities. Plants People Planet 2020, 2, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Zaman, R.; Islam, R.A.; Ibnat, N.; Othman, I.; Zaini, A.; Lee, C.Y.; Chowdhury, E.H. Current strategies in extending half-lives of therapeutic proteins. J. Control. Release 2019, 301, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Protein misfolding diseases. Annu. Rev. Biochem. 2017, 86, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.P.; Hewitt, E.W. Cellular proteostasis: Degradation of misfolded proteins by lysosomes. Essays Biochem. 2016, 60, 173–180. [Google Scholar] [PubMed] [Green Version]
- Butreddy, A.; Janga, K.Y.; Ajjarapu, S.; Sarabu, S.; Dudhipala, N. Instability of therapeutic proteins—An overview of stresses, stabilization mechanisms and analytical techniques involved in Lyophilized proteins. Int. J. Biol. Macromol. 2021, 167, 309–325. [Google Scholar] [CrossRef]
- Emami, F.; Vatanara, A.; Park, E.J.; Na, D.H. Drying technologies for the stability and bioavailability of biopharmaceuticals. Pharmaceutics 2018, 10, 131. [Google Scholar] [CrossRef] [Green Version]
- Braun, A.C.; Gutmann, M.; Lühmann, T.; Meinel, L. Bioorthogonal strategies for site-directed decoration of biomaterials with therapeutic proteins. J. Control. Release 2018, 273, 68–85. [Google Scholar] [CrossRef]
- Duivelshof, B.L.; Murisier, A.; Camperi, J.; Fekete, S.; Beck, A.; Guillarme, D.; D’Atri, V. Therapeutic Fc-fusion proteins: Current analytical strategies. J. Sep. Sci. 2021, 44, 35–62. [Google Scholar] [CrossRef]
- Akbarian, M.; Ghasemi, Y.; Uversky, V.N.; Yousefi, R. Chemical modifications of insulin: Finding a compromise between stability and pharmaceutical performance. Int. J. Pharm. 2018, 547, 450–468. [Google Scholar] [CrossRef] [PubMed]
- Falconer, R.J. Advances in liquid formulations of parenteral therapeutic proteins. Biotechnol. Adv. 2019, 37, 107412. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, C. Packing in the proteins. Nat. Rev. Chem. 2021, 5, 670. [Google Scholar] [CrossRef]
- Lundahl, M.L.; Fogli, S.; Colavita, P.E.; Scanlan, E.M. Aggregation of protein therapeutics enhances their immunogenicity: Causes and mitigation strategies. RSC Chem. Biol. 2021, 2, 1004–1020. [Google Scholar] [CrossRef]
- Paraskevopoulou, V.; Falcone, F.H. Polyionic tags as enhancers of protein solubility in recombinant protein expression. Microorganisms 2018, 6, 47. [Google Scholar] [CrossRef] [Green Version]
- Ekladious, I.; Colson, Y.L.; Grinstaff, M.W. Polymer–drug conjugate therapeutics: Advances, insights and prospects. Nat. Rev. Drug Discov. 2019, 18, 273–294. [Google Scholar] [CrossRef]
- Burcham, C.L.; Florence, A.J.; Johnson, M.D. Continuous manufacturing in pharmaceutical process development and manufacturing. Annu. Rev. Chem. Biomol. Eng. 2018, 9, 253–281. [Google Scholar] [CrossRef]
- Akbarian, M.; Yousefi, R.; Farjadian, F.; Uversky, V.N. Insulin fibrillation: Toward strategies for attenuating the process. Chem. Commun. 2020, 56, 11354–11373. [Google Scholar] [CrossRef]
- Younesi, F.S.; Pazhang, M.; Najavand, S.; Rahimizadeh, P.; Akbarian, M.; Mohammadian, M.; Khajeh, K. Deleting the Ig-like domain of Alicyclobacillus acidocaldarius endoglucanase Cel9A causes a simultaneous increase in the activity and stability. Mol. Biotechnol. 2016, 58, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.; Stenzel, M.H. All wrapped up: Stabilization of enzymes within single enzyme nanoparticles. J. Am. Chem. Soc. 2019, 141, 2754–2769. [Google Scholar] [CrossRef]
- Dimitrov, D.S. Therapeutic proteins. Methods Mol. Biol. 2012, 899, 1–26. [Google Scholar] [PubMed]
- Guo, J.; Carta, G. Unfolding and aggregation of monoclonal antibodies on cation exchange columns: Effects of resin type, load buffer, and protein stability. J. Chromatogr. A 2015, 1388, 184–194. [Google Scholar] [CrossRef]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–drug conjugates for cancer. The Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody–drug conjugates: The last decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef]
- Sau, S.; Alsaab, H.O.; Kashaw, S.K.; Tatiparti, K.; Iyer, A.K. Advances in antibody–drug conjugates: A new era of targeted cancer therapy. Drug Discov. Today 2017, 22, 1547–1556. [Google Scholar] [CrossRef]
- Duerr, C.; Friess, W. Antibody-drug conjugates-stability and formulation. Eur. J. Pharm. Biopharm. 2019, 139, 168–176. [Google Scholar] [CrossRef]
- Eustáquio, A.S.; Janso, J.E.; Ratnayake, A.S.; O’Donnell, C.J.; Koehn, F.E. Spliceostatin hemiketal biosynthesis in Burkholderia spp. is catalyzed by an iron/α-ketoglutarate–dependent dioxygenase. Proc. Natl. Acad. Sci. USA 2014, 111, E3376–E3385. [Google Scholar] [CrossRef]
- Coleman, R.G.; Sharp, K.A. Shape and evolution of thermostable protein structure. Proteins Struct. Funct. Bioinform. 2010, 78, 420–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delahaije, R.J.; Wierenga, P.A.; Giuseppin, M.L.; Gruppen, H. Comparison of heat-induced aggregation of globular proteins. J. Agric. Food Chem. 2015, 63, 5257–5265. [Google Scholar] [CrossRef] [PubMed]
- Wakankar, A.; Chen, Y.; Gokarn, Y.; Jacobson, F.S. Analytical methods for physicochemical characterization of antibody drug conjugates. MAbs 2011, 3, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumey, L.N.; Li, F.; Rago, B.; Han, X.; Loganzo, F.; Musto, S.; Graziani, E.I.; Puthenveetil, S.; Casavant, J.; Marquette, K.; et al. Site selection: A case study in the identification of optimal cysteine engineered antibody drug conjugates. AAPS J. 2017, 19, 1123–1135. [Google Scholar] [CrossRef]
- Pauling, L. Protein interactions. Aggregation of globular proteins. Discuss. Faraday Soc. 1953, 13, 170–176. [Google Scholar] [CrossRef]
- March, D.; Bianco, V.; Franzese, G. Protein unfolding and aggregation near a hydrophobic interface. Polymers 2021, 13, 156. [Google Scholar] [CrossRef]
- Jha, S.K.; Udgaonkar, J.B. Free energy barriers in protein folding and unfolding reactions. Curr. Sci. 2010, 457–475. [Google Scholar]
- Wang, W.; Nema, S.; Teagarden, D. Protein aggregation—Pathways and influencing factors. Int. J. Pharm. 2010, 390, 89–99. [Google Scholar] [CrossRef]
- Mahler, H.-C.; Friess, W.; Grauschopf, U.; Kiese, S. Protein aggregation: Pathways, induction factors and analysis. J. Pharm. Sci. 2009, 98, 2909–2934. [Google Scholar] [CrossRef]
- Zhang, J.; Peng, X.; Jonas, A.; Jonas, J. NMR study of the cold, heat, and pressure unfolding of ribonuclease A. Biochemistry 1995, 34, 8631–8641. [Google Scholar] [CrossRef]
- Kosa, T.; Maruyama, T.; Otagiri, M. Species differences of serum albumins: II. Chemical and thermal stability. Pharm. Res. 1998, 15, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Makin, O.S.; Serpell, L.C. Structures for amyloid fibrils. FEBS J. 2005, 272, 5950–5961. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int. J. Pharm. 1999, 185, 129–188. [Google Scholar] [CrossRef]
- Bischof, J.C.; He, X. Thermal stability of proteins. Ann. N. Y. Acad. Sci. 2006, 1066, 12–33. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, F.G. Denaturation of Proteins by Urea and Related Substances. Nature 1930, 126, 328–330. [Google Scholar] [CrossRef]
- Clark, J.H. The temperature coefficient of the urea denaturation of egg albumin. J. Gen. Physiol. 1945, 28, 539–545. [Google Scholar] [CrossRef]
- Ervin, J.; Larios, E.; Osváth, S.; Schulten, K.; Gruebele, M. What causes hyperfluorescence: Folding intermediates or conformationally flexible native states? Biophys. J. 2002, 83, 473–483. [Google Scholar] [CrossRef] [Green Version]
- Privalov, P.L. Cold denaturation of protein. Crit. Rev. Biochem. Mol. Biol. 1990, 25, 281–306. [Google Scholar] [CrossRef]
- Menon, S.; Sengupta, N. The cold thermal response of an amyloid oligomer differs from typical globular protein cold denaturation. J. Phys. Chem. Lett. 2019, 10, 2453–2457. [Google Scholar] [CrossRef]
- Sabelko, J.; Ervin, J.; Gruebele, M. Cold-denatured ensemble of apomyoglobin: Implications for the early steps of folding. J. Phys. Chem. B 1998, 102, 1806–1819. [Google Scholar] [CrossRef]
- Babu, C.R.; Hilser, V.J.; Wand, A.J. Direct access to the cooperative substructure of proteins and the protein ensemble via cold denaturation. Nat. Struct. Mol. Biol. 2004, 11, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S. High-pressure techniques. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1978; Volume 49, pp. 14–24. [Google Scholar]
- Marqués, M.I.; Borreguero, J.M.; Stanley, H.E.; Dokholyan, N.V. Possible mechanism for cold denaturation of proteins at high pressure. Phys. Rev. Lett. 2003, 91, 138103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, R.; Winter, R. Cold-and Pressure-Induced Dissociation of Protein Aggregates and Amyloid Fibrils. Angew. Chem. Int. Ed. 2008, 47, 6518–6521. [Google Scholar] [CrossRef]
- Bensasson, R.; Land, E.; Truscott, T. Pulse Radiolysis and Flash Photolysis: Contributions to the Chemistry of Biology and Medicine; Pergamon Press: Oxford, UK, 1983. [Google Scholar]
- Cockrell, G.M.; Wolfe, M.S.; Wolfe, J.L.; Schöneich, C. Photoinduced aggregation of a model antibody–drug conjugate. Mol. Pharm. 2015, 12, 1784–1797. [Google Scholar] [CrossRef] [PubMed]
- Pattison, D.I.; Rahmanto, A.S.; Davies, M.J. Photo-oxidation of proteins. Photochem. Photobiol. Sci. 2012, 11, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Truscott, R.J. Photo-oxidation of proteins and its role in cataractogenesis. J. Photochem. Photobiol. B Biol. 2001, 63, 114–125. [Google Scholar] [CrossRef]
- Herbert, J.M.; Coons, M.P. The hydrated electron. Annu. Rev. Phys. Chem 2017, 68, 447–472. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, P. The Chemical Basis of Radiation Biology; Taylor & Francis: Oxfordshire, UK, 1987. [Google Scholar]
- Davies, M.; Dean, R.; Davies, D. Radical-Mediated Protein Oxidation: From Chemistry to Medicine; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Afonso, S.; Enriquez de Salamanca, R.; Batlle, A.d.C. The photodynamic and non-photodynamic actions of porphyrins. Braz. J. Med. Biol. Res. 1999, 32, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Manzocco, L. Photo-induced modification of food protein structure and functionality. Food Eng. Rev. 2015, 7, 346–356. [Google Scholar] [CrossRef]
- Chen, X.; Ding, G.; Gao, Q.; Sun, J.; Zhang, Q.; Du, L.; Qiu, Z.; Wang, C.; Zheng, F.; Sun, B.; et al. A human anti-c-Met Fab fragment conjugated with doxorubicin as targeted chemotherapy for hepatocellular carcinoma. PLoS ONE 2013, 8, e63093. [Google Scholar] [CrossRef] [Green Version]
- Trail, P.A.; Willner, D.; Lasch, S.J.; Henderson, A.J.; Hofstead, S.; Casazza, A.M.; Firestone, R.A.; Hellström, I.; Hellström, K.E. Cure of xenografted human carcinomas by BR96-doxorubicin immunoconjugates. Science 1993, 261, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Nawara, K.; Krysinski, P.; Blanchard, G. Photoinduced reactivity of doxorubicin: Catalysis and degradation. J. Phys. Chem. A 2012, 116, 4330–4337. [Google Scholar] [CrossRef] [PubMed]
- Ducry, L.; Stump, B. Antibody− drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconjugate Chem. 2010, 21, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.F.; Kendrick, B.S.; Chang, B.S.; Manning, M.C.; Randolph, T.W. Inhibition of stress-induced aggregation of protein therapeutics. In Methods Enzymol; Elsevier: Amsterdam, The Netherlands, 1999; Volume 309, pp. 236–255. [Google Scholar]
- Mahler, H.-C.; Müller, R.; Frieβ, W.; Delille, A.; Matheus, S. Induction and analysis of aggregates in a liquid IgG1-antibody formulation. Eur. J. Pharm. Biopharm. 2005, 59, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Kiese, S.; Papppenberger, A.; Friess, W.; Mahler, H.-C. Shaken, not stirred: Mechanical stress testing of an IgG1 antibody. J. Pharm. Sci. 2008, 97, 4347–4366. [Google Scholar] [CrossRef]
- Ishikawa, T.; Kobayashi, N.; Osawa, C.; Sawa, E.; Wakamatsu, K. Prevention of stirring-induced microparticle formation in monoclonal antibody solutions. Biol. Pharm. Bull. 2010, 33, 1043–1046. [Google Scholar] [CrossRef] [Green Version]
- Randolph, T.W.; Schiltz, E.; Sederstrom, D.; Steinmann, D.; Mozziconacci, O.; Schöneich, C.; Freund, E.; Ricci, M.S.; Carpenter, J.F.; Lengsfeld, C.S. Do not drop: Mechanical shock in vials causes cavitation, protein aggregation, and particle formation. J. Pharm. Sci. 2015, 104, 602–611. [Google Scholar] [CrossRef] [Green Version]
- Lentz, Y.; Anchordoquy, T.; Lengsfeld, C. DNA acts as a nucleation site for transient cavitation in the ultrasonic nebulizer. J. Pharm. Sci. 2006, 95, 607–619. [Google Scholar] [CrossRef]
- Wiesbauer, J.; Cardinale, M.; Nidetzky, B. Shaking and stirring: Comparison of controlled laboratory stress conditions applied to the human growth hormone. Process Biochem. 2013, 48, 33–40. [Google Scholar] [CrossRef]
- Biddlecombe, J.G.; Smith, G.; Uddin, S.; Mulot, S.; Spencer, D.; Gee, C.; Fish, B.C.; Bracewell, D.G. Factors influencing antibody stability at solid–liquid interfaces in a high shear environment. Biotechnol. Prog. 2009, 25, 1499–1507. [Google Scholar] [CrossRef]
- Dunstan, D.E.; Hamilton-Brown, P.; Asimakis, P.; Ducker, W.; Bertolini, J. Shear flow promotes amyloid-β fibrilization. Protein Eng. Des. Sel. 2009, 22, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Hamilton-Brown, P.; Bekard, I.; Ducker, W.A.; Dunstan, D.E. How does shear affect Aβ fibrillogenesis? J. Phys. Chem. B 2008, 112, 16249–16252. [Google Scholar] [CrossRef] [PubMed]
- Hill, E.K.; Krebs, B.; Goodall, D.G.; Howlett, G.J.; Dunstan, D.E. Shear flow induces amyloid fibril formation. Biomacromolecules 2006, 7, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Kiyoshi, M.; Tada, M.; Shibata, H.; Aoyama, M.; Ishii-Watabe, A. Characterization of aggregated antibody-silicone oil complexes: From perspectives of morphology, 3D image, and Fcγ receptor activation. J. Pharm. Sci. 2021, 110, 1189–1196. [Google Scholar] [CrossRef]
- Torisu, T.; Maruno, T.; Hamaji, Y.; Ohkubo, T.; Uchiyama, S. Synergistic effect of cavitation and agitation on protein aggregation. J. Pharm. Sci. 2017, 106, 521–529. [Google Scholar] [CrossRef]
- Bam, N.B.; Cleland, J.L.; Yang, J.; Manning, M.C.; Carpenter, J.F.; Kelley, R.F.; Randolph, T.W. Tween protects recombinant human growth hormone against agitation-induced damage via hydrophobic interactions. J. Pharm. Sci. 1998, 87, 1554–1559. [Google Scholar] [CrossRef]
- Yano, Y.F.; Uruga, T.; Tanida, H.; Toyokawa, H.; Terada, Y.; Takagaki, M.; Yamada, H. Driving force behind adsorption-induced protein unfolding: A time-resolved X-ray reflectivity study on lysozyme adsorbed at an air/water interface. Langmuir 2009, 25, 32–35. [Google Scholar] [CrossRef]
- Yano, Y.F.; Kobayashi, Y.; Ina, T.; Nitta, K.; Uruga, T. Hofmeister anion effects on protein adsorption at an air–water interface. Langmuir 2016, 32, 9892–9898. [Google Scholar] [CrossRef]
- Miller, R.; Fainerman, V.; Makievski, A.; Krägel, J.; Grigoriev, D.O.; Kazakov, V.; Sinyachenko, O. Dynamics of protein and mixed protein/surfactant adsorption layers at the water/fluid interface. Adv. Colloid Interface Sci. 2000, 86, 39–82. [Google Scholar] [CrossRef]
- Bos, M.A.; Van Vliet, T. Interfacial rheological properties of adsorbed protein layers and surfactants: A review. Adv. Colloid Interface Sci. 2001, 91, 437–471. [Google Scholar] [CrossRef]
- Dickinson, E. Adsorbed protein layers at fluid interfaces: Interactions, structure and surface rheology. Colloids Surf. B Biointerfaces 1999, 15, 161–176. [Google Scholar] [CrossRef]
- Hong, T.; Iwashita, K.; Han, J.; Nishinami, S.; Handa, A.; Shiraki, K. Aggregation of hen egg white proteins with additives during agitation. LWT 2021, 146, 111378. [Google Scholar] [CrossRef]
- Salvatore, S.; Vandenplas, Y. Hydrolyzed proteins in allergy. In Protein in Neonatal and Infant Nutrition: Recent Updates; Karger Publishers: Basel, Switzerland, 2016; Volume 86, pp. 11–27. [Google Scholar]
- Kahne, D.; Still, W.C. Hydrolysis of a peptide bond in neutral water. J. Am. Chem. Soc. 1988, 110, 7529–7534. [Google Scholar] [CrossRef]
- Almela, L.; Sánchez-Muñoz, B.; Fernández-López, J.A.; Roca, M.J.; Rabe, V. Liquid chromatograpic–mass spectrometric analysis of phenolics and free radical scavenging activity of rosemary extract from different raw material. J. Chromatogr. A 2006, 1120, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Cordoba, A.J.; Shyong, B.-J.; Breen, D.; Harris, R.J. Non-enzymatic hinge region fragmentation of antibodies in solution. J. Chromatogr. B 2005, 818, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Dillon, T.M.; Bondarenko, P.V.; Rehder, D.S.; Pipes, G.D.; Kleemann, G.R.; Ricci, M.S. Optimization of a reversed-phase high-performance liquid chromatography/mass spectrometry method for characterizing recombinant antibody heterogeneity and stability. J. Chromatogr. A 2006, 1120, 112–120. [Google Scholar] [CrossRef]
- Smith, R.M.; Hansen, D.E. The pH-rate profile for the hydrolysis of a peptide bond. J. Am. Chem. Soc. 1998, 120, 8910–8913. [Google Scholar] [CrossRef]
- Pan, B.; Ricci, M.S.; Trout, B.L. A molecular mechanism of hydrolysis of peptide bonds at neutral pH using a model compound. J. Phys. Chem. B 2011, 115, 5958–5970. [Google Scholar] [CrossRef]
- Tsabouri, S.; Douros, K.; Priftis, K.N. Cow’s milk allergenicity. Endocr. Metab. Immune Disord.-Drug Targets 2014, 14, 16–26. [Google Scholar] [CrossRef]
- Banks, D.D.; Hambly, D.M.; Scavezze, J.L.; Siska, C.C.; Stackhouse, N.L.; Gadgil, H.S. The effect of sucrose hydrolysis on the stability of protein therapeutics during accelerated formulation studies. J. Pharm. Sci. 2009, 98, 4501–4510. [Google Scholar] [CrossRef]
- Torosantucci, R.; Schöneich, C.; Jiskoot, W. Oxidation of therapeutic proteins and peptides: Structural and biological consequences. Pharm. Res. 2014, 31, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Yates, Z.; Balland, A.; Kleemann, G.R. Human IgG1 hinge fragmentation as the result of H2O2-mediated radical cleavage. J. Biol. Chem. 2009, 284, 35390–35402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, R.; Madurawe, R.D.; Bush, K.D.; Lumpkin, J.A. Conditions Promoting Metal-Catalyzed Oxidations during Immobilized Cu—Iminodiacetic Acid Metal Affinity Chromatography. Biotechnol. Prog. 1995, 11, 643–650. [Google Scholar] [CrossRef]
- Men, L.; Wang, Y. The oxidation of yeast alcohol dehydrogenase-1 by hydrogen peroxide in vitro. J. Proteome Res. 2007, 6, 216–225. [Google Scholar] [CrossRef]
- Vogt, W. Oxidation of methionyl residues in proteins: Tools, targets, and reversal. Free. Radic. Biol. Med. 1995, 18, 93–105. [Google Scholar] [CrossRef]
- Li, S.; Schöneich, C.; Borchardt, R.T. Chemical instability of protein pharmaceuticals: Mechanisms of oxidation and strategies for stabilization. Biotechnol. Bioeng. 1995, 48, 490–500. [Google Scholar] [CrossRef]
- Tada, M.; Ishii-Watabe, A.; Suzuki, T.; Kawasaki, N. Development of a cell-based assay measuring the activation of FcγRIIa for the characterization of therapeutic monoclonal antibodies. PLoS ONE 2014, 9, e95787. [Google Scholar] [CrossRef] [Green Version]
- Bertolotti-Ciarlet, A.; Wang, W.; Lownes, R.; Pristatsky, P.; Fang, Y.; McKelvey, T.; Li, Y.; Li, Y.; Drummond, J.; Prueksaritanont, T.; et al. Impact of methionine oxidation on the binding of human IgG1 to FcRn and Fcγ receptors. Mol. Immunol. 2009, 46, 1878–1882. [Google Scholar] [CrossRef]
- Amici, A.; Levine, R.; Tsai, L.; Stadtman, E. Conversion of amino acid residues in proteins and amino acid homopolymers to carbonyl derivatives by metal-catalyzed oxidation reactions. J. Biol. Chem. 1989, 264, 3341–3346. [Google Scholar] [CrossRef]
- Turyan, I.; Khatwani, N.; Sosic, Z.; Jayawickreme, S.; Mandler, D. A novel approach for oxidation analysis of therapeutic proteins. Anal. Biochem. 2016, 494, 108–113. [Google Scholar] [CrossRef]
- Moussa, E.M.; Panchal, J.P.; Moorthy, B.S.; Blum, J.S.; Joubert, M.K.; Narhi, L.O.; Topp, E.M. Immunogenicity of therapeutic protein aggregates. J. Pharm. Sci. 2016, 105, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Varkhede, N.; Bommana, R.; Schöneich, C.; Forrest, M.L. Proteolysis and oxidation of therapeutic proteins after intradermal or subcutaneous administration. J. Pharm. Sci. 2020, 109, 191–205. [Google Scholar] [CrossRef] [Green Version]
- Buecheler, J.W.; Winzer, M.; Weber, C.; Gieseler, H. Oxidation-induced destabilization of model antibody-drug conjugates. J. Pharm. Sci. 2019, 108, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; May, K. Disulfide bond structures of IgG molecules: Structural variations, chemical modifications and possible impacts to stability and biological function. MAbs 2012, 4, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Chumsae, C.; Gaza-Bulseco, G.; Hurkmans, K.; Radziejewski, C.H. Ranking the susceptibility of disulfide bonds in human IgG1 antibodies by reduction, differential alkylation, and LC−MS analysis. Anal. Chem. 2010, 82, 5219–5226. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.T.-Y. Deamidation: A source of microheterogeneity in pharmaceutical proteins. Trends Biotechnol. 1992, 10, 364–369. [Google Scholar] [CrossRef]
- Charache SFox, J.; McCurdy, P.; Kazazian, H., Jr.; Winslow, R.; Hathaway, P.; van Beneden, R.; Jessop, M. Postsynthetic deamidation of hemoglobin Providence (Beta 82 Lys replaced by Asn, Asp) and its effect on oxygen transport. J. Clin. Invest. 1977, 59, 652–658. [Google Scholar] [CrossRef]
- Gráf, L.; Hajós, G.; Patthy, A.; Cseh, G. The influence of deamidation on the biological activity of porcine adrenocorticotropic hormone (ACTH). Horm. Metab. Res. 1973, 5, 142–143. [Google Scholar] [CrossRef]
- Johnson, B.A.; Shirokawa, J.M.; Aswad, D.W. Deamidation of calmodulin at neutral and alkaline pH: Quantitative relationships between ammonia loss and the susceptibility of calmodulin to modification by protein carboxyl methyltransferase. Arch. Biochem. Biophys. 1989, 268, 276–286. [Google Scholar] [CrossRef]
- Zeunik, R.; Ryuzoji, A.F.; Peariso, A.; Wang, X.; Lannan, M.; Spindler, L.J.; Knierman, M.; Copeland, V.; Patel, C.; Wen, Y. Investigation of immune responses to oxidation, deamidation, and isomerization in therapeutic antibodies using preclinical immunogenicity risk assessment assays. J. Pharm. Sci. 2022. [Google Scholar] [CrossRef]
- Srinivasan, C.; Ma, Y.; Liu, Y.; Wang, Y.; Hengst, L.; Liu, X.; Toth, R.; Rodriguez, J.; Mohammad, A.; Bandaranayake, B.M.; et al. Quality attributes and evaluation of pharmaceutical glass containers for parenterals. Int. J. Pharm. 2019, 568, 118510. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.; Lowenson, J.; Clarke, S.; Wolkow, C.; Wang, R.; Cotter, R.; Reardon, I.; Zürcher-Neely, H.; Heinrikson, R.; Ball, M. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar] [CrossRef]
- Takata, T.; Oxford, J.T.; Demeler, B.; Lampi, K.J. Deamidation destabilizes and triggers aggregation of a lens protein, βA3-crystallin. Protein Sci. 2008, 17, 1565–1575. [Google Scholar] [CrossRef]
- Shimizu, T.; Fukuda, H.; Murayama, S.; Izumiyama, N.; Shirasawa, T. Isoaspartate formation at position 23 of amyloid beta peptide enhanced fibril formation and deposited onto senile plaques and vascular amyloids in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Hao, P.; Adav, S.S.; Gallart-Palau, X.; Sze, S.K. Recent advances in mass spectrometric analysis of protein deamidation. Mass Spectrom. Rev. 2017, 36, 677–692. [Google Scholar] [CrossRef]
- Milczek, E.M. Commercial applications for enzyme-mediated protein conjugation: New developments in enzymatic processes to deliver functionalized proteins on the commercial scale. Chem. Rev. 2018, 118, 119–141. [Google Scholar] [CrossRef]
- Ciepluch, K.; Radulescu, A.; Hoffmann, I.; Raba, A.; Allgaier, J.; Richter, D.; Biehl, R. Influence of PEGylation on domain dynamics of phosphoglycerate kinase: PEG acts like entropic spring for the protein. Bioconjugate Chem. 2018, 29, 1950–1960. [Google Scholar] [CrossRef]
- Jeyachandran, Y.L.; Mielczarski, E.; Rai, B.; Mielczarski, J.A. Quantitative and qualitative evaluation of adsorption/desorption of bovine serum albumin on hydrophilic and hydrophobic surfaces. Langmuir 2009, 25, 11614–11620. [Google Scholar] [CrossRef]
- Mohammadpanah, H.; Rastegar, H.; Ramazani, M.R.; Jaafari, M.R. Effects of different buffers and pH on the stability of recombinant human growth hormone. Biosci. Biotechnol. Res. Asia 2013, 10, 193–203. [Google Scholar] [CrossRef]
- Kumar, S.; Nussinov, R. Salt bridge stability in monomeric proteins. J. Mol. Biol. 1999, 293, 1241–1255. [Google Scholar] [CrossRef]
- Chakravarty, S.; Varadarajan, R. Elucidation of factors responsible for enhanced thermal stability of proteins: A structural genomics based study. Biochemistry 2002, 41, 8152–8161. [Google Scholar] [CrossRef] [PubMed]
- Kimchi-Sarfaty, C.; Schiller, T.; Hamasaki-Katagiri, N.; Khan, M.A.; Yanover, C.; Sauna, Z.E. Building better drugs: Developing and regulating engineered therapeutic proteins. Trends Pharmacol. Sci. 2013, 34, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Stolnik, S.; Shakesheff, K. Formulations for delivery of therapeutic proteins. Biotechnol. Lett. 2009, 31, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, M.; Yousefi, R.; Moosavi-Movahedi, A.A.; Ahmad, A.; Uversky, V.N. Modulating insulin fibrillation using engineered B-chains with mutated C-termini. Biophys. J. 2019, 117, 1626–1641. [Google Scholar] [CrossRef]
- Arakawa, T.; Prestrelski, S.J.; Narhi, L.O.; Boone, T.C.; Kenney, W.C. Cysteine 17 of recombinant human granulocyte-colony stimulating factor is partially solvent-exposed. J. Protein Chem. 1993, 12, 525–531. [Google Scholar] [CrossRef]
- Culajay, J.F.; Blaber, S.I.; Khurana, A.; Blaber, M. Thermodynamic characterization of mutants of human fibroblast growth factor 1 with an increased physiological half-life. Biochemistry 2000, 39, 7153–7158. [Google Scholar] [CrossRef]
- Luo, P.; Hayes, R.J.; Chan, C.; Stark, D.M.; Hwang, M.Y.; Jacinto, J.M.; Juvvadi, P.; Chung, H.S.; Kundu, A.; Ary, M.L.; et al. Development of a cytokine analog with enhanced stability using computational ultrahigh throughput screening. Protein Sci. 2002, 11, 1218–1226. [Google Scholar] [CrossRef] [Green Version]
- Marshall, S.A.; Lazar, G.A.; Chirino, A.J.; Desjarlais, J.R. Rational design and engineering of therapeutic proteins. Drug Discov. Today 2003, 8, 212–221. [Google Scholar] [CrossRef]
- Pipe, S.W.; Kaufman, R.J. Characterization of a genetically engineered inactivation-resistant coagulation factor VIIIa. Proc. Natl. Acad. Sci. USA 1997, 94, 11851–11856. [Google Scholar] [CrossRef] [Green Version]
- Sytkowski, A.J.; Lunn, E.D.; Risinger, M.A.; Davis, K.L. An erythropoietin fusion protein comprised of identical repeating domains exhibits enhanced biological properties. J. Biol. Chem. 1999, 274, 24773–24778. [Google Scholar] [CrossRef] [Green Version]
- Strohl, W.R. Fusion proteins for half-life extension of biologics as a strategy to make biobetters. Biodrugs 2015, 29, 215–239. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.A.; George, J.K.; Smith, I.J.; Bhakta, V.; Sheffield, W.P. A barbourin-albumin fusion protein that is slowly cleared in vivo retains the ability to inhibit platelet aggregation in vitro. Thromb. Haemost. 2001, 86, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Syed, S.; Schuyler, P.D.; Kulczycky, M.; Sheffield, W.P. Potent antithrombin activity and delayed clearance from the circulation characterize recombinant hirudin genetically fused to albumin. Blood J. Am. Soc. Hematol. 1997, 89, 3243–3252. [Google Scholar] [CrossRef] [Green Version]
- Dennis, M.S.; Zhang, M.; Meng, Y.G.; Kadkhodayan, M.; Kirchhofer, D.; Combs, D.; Damico, L.A. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J. Biol. Chem. 2002, 277, 35035–35043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbarian, M.; Yousefi, R. Human αB-crystallin as fusion protein and molecular chaperone increases the expression and folding efficiency of recombinant insulin. PLoS ONE 2018, 13, e0206169. [Google Scholar] [CrossRef] [Green Version]
- Khosravi, F.; Upadhyay, M.; Kumar, A.; Shahsavani, M.B.; Akbarian, M.; Najafi, H.; Tamaddon, A.M.; Yousefi, R. A novel method for the chaperone aided and efficient production of human proinsulin in the prokaryotic system. J. Biotechnol. 2022, 346, 35–46. [Google Scholar] [CrossRef]
- Ko, J.H.; Maynard, H.D. A guide to maximizing the therapeutic potential of protein–polymer conjugates by rational design. Chem. Soc. Rev. 2018, 47, 8998–9014. [Google Scholar] [CrossRef]
- Pelegri-O’Day, E.M.; Lin, E.-W.; Maynard, H.D. Therapeutic protein–polymer conjugates: Advancing beyond PEGylation. J. Am. Chem. Soc. 2014, 136, 14323–14332. [Google Scholar] [CrossRef]
- Rosen, C.B.; Francis, M.B. Targeting the N terminus for site-selective protein modification. Nat. Chem. Biol. 2017, 13, 697–705. [Google Scholar] [CrossRef]
- Kuan, S.L.; Wang, T.; Weil, T. Site-Selective Disulfide Modification of Proteins: Expanding Diversity beyond the Proteome. Chem.–A Eur. J. 2016, 22, 17112–17129. [Google Scholar] [CrossRef]
- Kinstler, O.; Molineux, G.; Treuheit, M.; Ladd, D.; Gegg, C. Mono-N-terminal poly (ethylene glycol)–protein conjugates. Adv. Drug Deliv. Rev. 2002, 54, 477–485. [Google Scholar] [CrossRef]
- Qi, Y.; Amiram, M.; Gao, W.; McCafferty, D.G.; Chilkoti, A. Sortase-catalyzed initiator attachment enables high yield growth of a stealth polymer from the C terminus of a protein. Macromol. Rapid Commun. 2013, 34, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Stephanopoulos, N.; Francis, M.B. Choosing an effective protein bioconjugation strategy. Nat. Chem. Biol. 2011, 7, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Xiao, D.; Xie, F.; Liu, L.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Antibody–drug conjugates: Recent advances in linker chemistry. Acta Pharm. Sin. B 2021, 11, 3889–3907. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Nakajima, T.; Miyazaki, K.; Ohuchi, Y.; Ikegami, T.; Choyke, P.L.; Kobayashi, H. Short PEG-linkers improve the performance of targeted, activatable monoclonal antibody-indocyanine green optical imaging probes. Bioconjugate Chem. 2013, 24, 811–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostova, V.; Désos, P.; Starck, J.-B.; Kotschy, A. The chemistry behind ADCs. Pharmaceuticals 2021, 14, 442. [Google Scholar] [CrossRef] [PubMed]
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody–drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881. [Google Scholar] [CrossRef]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abaza, Y.; Fathi, A. Monoclonal Antibodies in Acute Myeloid Leukemia—Are We There Yet? Cancer J. 2022, 28, 37–42. [Google Scholar] [CrossRef]
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef] [Green Version]
- LoRusso, P.M.; Weiss, D.; Guardino, E.; Girish, S.; Sliwkowski, M.X. Trastuzumab emtansine: A unique antibody-drug conjugate in development for human epidermal growth factor receptor 2–positive cancer. Clin. Cancer Res. 2011, 17, 6437–6447. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, H.D.; Dubowchik, G.M.; Mastalerz, H.; Willner, D.; Hofstead, S.J.; Firestone, R.A.; Lasch, S.J.; Trail, P.A. Monoclonal antibody conjugates of doxorubicin prepared with branched peptide linkers: Inhibition of aggregation by methoxytriethyleneglycol chains. J. Med. Chem. 2002, 45, 4336–4343. [Google Scholar] [CrossRef] [PubMed]
- Finbloom, D.S.; Abeles, D.; Rifai, A.; Plotz, P.H. The specificity of uptake of model immune complexes and other protein aggregates by the murine reticuloendothelial system. J. Immunol. 1980, 125, 1060–1065. [Google Scholar]
- Chari, R.V.; Miller, M.L.; Widdison, W.C. Antibody–drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735. [Google Scholar] [CrossRef]
- Sunna, A.; Care, A.; Bergquist, P.L. Peptides and Peptide-Based Biomaterials and Their Biomedical Applications; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Resh, M. Fatty acylation of proteins: The long and the short of it. Prog. Lipid Res. 2016, 63, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Diallo, I.; Seve, M.; Cunin, V.; Minassian, F.; Poisson, J.-F.; Michelland, S.; Bourgoin-Voillard, S. Current trends in protein acetylation analysis. Expert Rev. Proteomic 2019, 16, 139–159. [Google Scholar] [CrossRef]
- Driessen, H.; De Jong, W.; Tesser, G.; Bloemendal, H. The mechanism of N-terminal acetylation of protein. Crit. Rev. Biochem. 1985, 18, 281–325. [Google Scholar] [CrossRef]
- Lee, K.K.; Workman, J. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef]
- Silva, O.; Alves, E.S.F.; De La Fuente-Núñez, C.; Ribeiro, S.; Mandal, S.M.; Gaspar, D.; Veiga, A.S.; Castanho, M.; de Andrade, C.A.S.; Nascimento, J.M.; et al. Structural studies of a lipid-binding peptide from tunicate hemocytes with anti-biofilm activity. Sci. Rep. 2016, 6, 27128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bulaj, G. Converting peptides into drug leads by lipidation. Curr. Med. Chem. 2012, 19, 1602–1618. [Google Scholar] [CrossRef] [PubMed]
- Grimsey, E.; Collis, D.W.; Mikut, R.; Hilpert, K. The effect of lipidation and glycosylation on short cationic antimicrobial peptides. Biochim. Biophys. Acta-Biomembr. 2020, 1862, 183195. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Dritselis, A.; Kirkpatrick, P. Liraglutide. Nat. Rev. Drug Discov. 2010, 9, 267–268. [Google Scholar] [CrossRef]
- Wang, J.; Chow, D.; Heiati, H.; Shen, W.-C. Reversible lipidization for the oral delivery of salmon calcitonin. J. Control. Release 2003, 88, 369–380. [Google Scholar] [CrossRef]
- Wang, J.; Hogenkamp, D.J.; Tran, M.; Li, W.-Y.; Yoshimura, R.F.; Johnstone, T.B.; Shen, W.-C.; Gee, K.W. Reversible lipidization for the oral delivery of leu-enkephalin. J. Drug Target. 2006, 14, 127–136. [Google Scholar] [CrossRef]
- Tanaka, K.; Fujita, T.; Yamamoto, Y.; Murakami, M.; Yamamoto, A.; Muranishi, S. Enhancement of intestinal transport of thyrotropin-releasing hormone via a carrier-mediated transport system by chemical modification with lauric acid. Biochim. Biophys. Acta-Biomembr. 1996, 1283, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Muranishi, S.; Sakai, A.; Yamada, K.; Murakami, M.; Takada, K.; Kiso, Y. Lipophilic peptides: Synthesis of lauroyl thyrotropin-releasing hormone and its biological activity. Pharm. Res. 1991, 8, 649–652. [Google Scholar] [CrossRef]
- Lin, W.-C.; Iversen, L.; Tu, H.-L.; Rhodes, C.; Christensen, S.M.; Iwig, J.S.; Hansen, S.D.; Huang, W.Y.; Groves, J.T. H-Ras forms dimers on membrane surfaces via a protein–protein interface. Proc. Natl. Acad. Sci. 2014, 111, 2996–3001. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Peng, D.; Schlebach, J.P.; Hadziselimovic, A.; Sanders, C.R. Modest effects of lipid modifications on the structure of caveolin-3. Biochemistry 2014, 53, 4320–4322. [Google Scholar] [CrossRef]
- Garst, E.H.; Lee, H.; Das, T.; Bhattacharya, S.; Percher, A.; Wiewiora, R.; Witte, I.P.; Li, Y.; Peng, T.; Im, W.; et al. Site-specific lipidation enhances IFITM3 membrane interactions and antiviral activity. ACS Chem. Biol. 2021, 16, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Kočevar, N.; Obermajer, N.; Štrukelj, B.; Kos, J.; Kreft, S. Improved acylation method enables efficient delivery of functional palmitoylated cystatin into epithelial cells. Chem. Biol. Drug Des. 2007, 69, 124–131. [Google Scholar] [CrossRef]
- Tang, Y.-Q.; Yuan, J.; Osapay, G.; Osapay, K.; Tran, D.; Miller, C.J.; Ouellette, A.J.; Selsted, M.E. A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated α-defensins. Science 1999, 286, 498–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craik, D.J.; Daly, N.L.; Bond, T.; Waine, C. Plant cyclotides: A unique family of cyclic and knotted proteins that defines the cyclic cystine knot structural motif. J. Mol. Biol. 1999, 294, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Colgrave, M.L.; Craik, D.J. Thermal, chemical, and enzymatic stability of the cyclotide kalata B1: The importance of the cyclic cystine knot. Biochemistry 2004, 43, 5965–5975. [Google Scholar] [CrossRef]
- Abdalla, M.A.; McGaw, L.J. Natural cyclic peptides as an attractive modality for therapeutics: A mini review. Molecules 2018, 23, 2080. [Google Scholar] [CrossRef] [Green Version]
- Luckett, S.; Garcia-Fernandez, R.; Barker, J.; Konarev, A.; Shewry, P.; Clarke, A.; Brady, R. High-resolution structure of a potent, cyclic proteinase inhibitor from sunflower seeds. J. Mol. Biol. 1999, 290, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Kondejewski, L.H.; Farmer, S.W.; Wishart, D.S.; Kay, C.M.; Hancock, R.E.W.; Hodges, R.S. Modulation of structure and antibacterial and hemolytic activity by ring size in cyclic gramicidin S analogs. J. Biol. Chem. 1996, 271, 25261–25268. [Google Scholar] [CrossRef] [Green Version]
- Gause, G.F.; Brazhnikova, M.G. Gramicidin S and its use in the treatment of infected wounds. Nature 1944, 154, 703. [Google Scholar] [CrossRef]
- Adrio, J.; Cuevas, C.; Manzanares, I.; Joullié, M.M. Total synthesis and biological evaluation of tamandarin B analogues. J. Org. Chem. 2007, 72, 5129–5138. [Google Scholar] [CrossRef]
- Nuijen, B.; Bouma, M.; Manada, C.; Jimeno, J.; Schellens, J.H.; Bult, A.; Beijnen, J. Pharmaceutical development of anticancer agents derived from marine sources. Anti-Cancer Drugs 2000, 11, 793–811. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Bu, X.; Wu, X.; Guo, Z. A chemical approach to generate molecular diversity based on the scaffold of cyclic decapeptide antibiotic tyrocidine A. J. Comb. Chem. 2003, 5, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Kitani, S.; Ueguchi, T.; Igarashi, Y.; Leetanasaksakul, K.; Thamchaipenet, A.; Nihira, T. Rakicidin F, a new antibacterial cyclic depsipeptide from a marine sponge-derived Streptomyces sp. J. Antibiot. 2018, 71, 139–141. [Google Scholar] [CrossRef]
- Alonso-Álvarez, S.; Pardal, E.; Sánchez-Nieto, D.; Navarro, M.; Caballero, M.D.; Mateos, M.V.; Martin, A. Plitidepsin: Design, development, and potential place in therapy. Drug Des. Dev. Ther. 2017, 11, 253. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Shao, C.-L.; Fu, X.-M.; Kong, C.-J.; She, Z.-G.; Wang, C.-Y. Lumazine peptides penilumamides B–D and the cyclic pentapeptide asperpeptide A from a gorgonian-derived Aspergillus sp. fungus. J. Nat. Prod. 2014, 77, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.L.; Crabtree, G. The mechanism of action of cyclosporin A and FK506. Immunol. Today 1992, 13, 136–142. [Google Scholar] [CrossRef]
- Ortiz-Lopez, F.J.; Monteiro, M.C.n.; Gonzalez-Menendez, V.; Tormo, J.R.; Genilloud, O.; Bills, G.F.; Vicente, F.; Zhang, C.; Roemer, T.; Singh, S.B. Cyclic colisporifungin and linear cavinafungins, antifungal lipopeptides isolated from Colispora cavincola. J. Nat. Prod. 2015, 78, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.D.; Zhang, X.; Mooberry, S.L.; Patterson, G.M.; Moore, R.E. Cryptophycin: A new antimicrotubule agent active against drug-resistant cells. Cancer Res. 1994, 54, 3779–3784. [Google Scholar]
- Campas-Moya, C. Romidepsin for the treatment of cutaneous T-cell lymphoma. Drugs Today 2009, 45, 787–795. [Google Scholar] [CrossRef]
- Bertino, E.M.; Otterson, G. Romidepsin: A novel histone deacetylase inhibitor for cancer. Expert Opin. Investig. Drugs 2011, 20, 1151–1158. [Google Scholar] [CrossRef]
- Youssef, D.T.; Shaala, L.A.; Mohamed, G.A.; Badr, J.M.; Bamanie, F.H.; Ibrahim, S.R. Theonellamide G, a potent antifungal and cytotoxic bicyclic glycopeptide from the Red Sea marine sponge Theonella swinhoei. Mar. Drugs 2014, 12, 1911–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidtko, A.; Lötsch, J.; Freynhagen, R.; Geisslinger, G. Ziconotide for treatment of severe chronic pain. Lancet 2010, 375, 1569–1577. [Google Scholar] [CrossRef]
- Suárez, Y.; González, L.; Cuadrado, A.; Berciano, M.; Lafarga, M.; Muñoz, A. Kahalalide F, a new marine-derived compound, induces oncosis in human prostate and breast cancer cells. Mol. Cancer Ther. 2003, 2, 863–872. [Google Scholar] [PubMed]
- De Hoog, M.; Mouton, J.W.; van den Anker, J. Vancomycin: Pharmacokinetics and administration regimens in neonates. Clin Pharm. 2004, 43, 417–440. [Google Scholar] [CrossRef]
- Gallay, P.A.; Lin, K. Profile of alisporivir and its potential in the treatment of hepatitis C. Drug Des. Dev. Ther. 2013, 7, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubinin, M.V.; Starinets, V.S.; Talanov, E.Y.; Mikheeva, I.B.; Belosludtseva, N.V.; Belosludtsev, K. Alisporivir improves mitochondrial function in skeletal muscle of mdx mice but suppresses mitochondrial dynamics and biogenesis. Int. J. Mol. Sci. 2021, 22, 9780. [Google Scholar] [CrossRef]
- VanderMolen, K.M.; McCulloch, W.; Pearce, C.J.; Oberlies, N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): A natural product recently approved for cutaneous T-cell lymphoma. J. Antibiot. 2011, 64, 525–531. [Google Scholar] [CrossRef]
- Heo, Y.-A. Voclosporin: First approval. Drugs 2021, 81, 605–610. [Google Scholar] [CrossRef]
- Feelders, R.A.; Yasothan, U.; Kirkpatrick, P. Pasireotide. Nat. Rev. Drug Discov. 2012, 11, 597–599. [Google Scholar] [CrossRef]
- McGivern, J.G. Ziconotide: A review of its pharmacology and use in the treatment of pain. Neuropsychiatr. Dis. Treat. 2007, 3, 69–85. [Google Scholar] [CrossRef] [Green Version]
- Claro, B.; Bastos, M.; Garcia-Fandino, R. Design and applications of cyclic peptides. In Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Elsevier: Amsterdam, The Netherlands, 2018; pp. 87–129. [Google Scholar]
- Kianpour, M.; Akbarian, M.; Uversky, V.N. Nanoparticles for Coronavirus Control. Nanomaterials 2022, 12, 1602. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Tang, P.S.; Chan, W.C. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisse, E.; Braet, F.; Luo, D.; De Zanger, R.; Jans, D.; Crabbe, E.; Vermoesen, A. Structure and function of sinusoidal lining cells in the liver. Toxicol. Pathol. 1996, 24, 100–111. [Google Scholar] [CrossRef]
- Yuan, F.; Dellian, M.; Fukumura, D.; Leunig, M.; Berk, D.A.; Torchilin, V.P.; Jain, R.K. Vascular permeability in a human tumor xenograft: Molecular size dependence and cutoff size. Cancer Res. 1995, 55, 3752–3756. [Google Scholar] [PubMed]
- Batista, P.; Castro, P.M.; Madureira, A.R.; Sarmento, B.; Pintado, M. Recent insights in the use of nanocarriers for the oral delivery of bioactive proteins and peptides. Peptides 2018, 101, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Fonte, P.; Nogueira, T.; Gehm, C.; Ferreira, D.; Sarmento, B. Chitosan-coated solid lipid nanoparticles enhance the oral absorption of insulin. Drug Deliv. Transl. Res. 2011, 1, 299–308. [Google Scholar] [CrossRef]
- Shrestha, N.; Shahbazi, M.-A.; Araújo, F.; Zhang, H.; Mäkilä, E.M.; Kauppila, J.; Sarmento, B.; Salonen, J.J.; Hirvonen, J.T.; Santos, H.A. Chitosan-modified porous silicon microparticles for enhanced permeability of insulin across intestinal cell monolayers. Biomaterials 2014, 35, 7172–7179. [Google Scholar] [CrossRef]
- Song, M.; Li, L.; Zhang, Y.; Chen, K.; Wang, H.; Gong, R. Carboxymethyl-β-cyclodextrin grafted chitosan nanoparticles as oral delivery carrier of protein drugs. React. Funct. Polym. 2017, 117, 10–15. [Google Scholar] [CrossRef]
- Lee, S.H.; Zhang, Z.; Feng, S.-S. Nanoparticles of poly(lactide)—Tocopheryl polyethylene glycol succinate (PLA-TPGS) copolymers for protein drug delivery. Biomaterials 2007, 28, 2041–2050. [Google Scholar] [CrossRef]
- Masuda, K.; Takegami, K.; An, H.; Kumano, F.; Chiba, K.; Andersson, G.; Schmid, T.; Thonar, E. Recombinant osteogenic protein-1 upregulates extracellular matrix metabolism by rabbit annulus fibrosus and nucleus pulposus cells cultured in alginate beads. J. Orthop. Res. 2003, 21, 922–930. [Google Scholar] [CrossRef]
- Arab, A.; Behravan, J.; Razazan, A.; Gholizadeh, Z.; Nikpoor, A.R.; Barati, N.; Mosaffa, F.; Badiee, A.; Jaafari, M.R. A nano-liposome vaccine carrying E75, a HER-2/neu-derived peptide, exhibits significant antitumour activity in mice. J. Drug Target. 2018, 26, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Varypataki, E.M.; Benne, N.; Bouwstra, J.; Jiskoot, W.; Ossendorp, F. Efficient Eradication of Established Tumors in Mice with Cationic Liposome-Based Synthetic Long-Peptide VaccinesLiposomal Peptide Vaccines for Cancer Immunotherapy. Cancer Immunol. Res. 2017, 5, 222–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salade, L.; Wauthoz, N.; Deleu, M.; Vermeersch, M.; De Vriese, C.; Amighi, K.; Goole, J. Development of coated liposomes loaded with ghrelin for nose-to-brain delivery for the treatment of cachexia. Int. J. Nanomed. 2017, 12, 8531. [Google Scholar] [CrossRef] [PubMed]
- Ramezanzade, L.; Hosseini, S.F.; Nikkhah, M. Biopolymer-coated nanoliposomes as carriers of rainbow trout skin-derived antioxidant peptides. Food Chem. 2017, 234, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Mura, P.; Mennini, N.; Nativi, C.; Richichi, B. In situ mucoadhesive-thermosensitive liposomal gel as a novel vehicle for nasal extended delivery of opiorphin. Eur. J. Pharm. Biopharm. 2018, 122, 54–61. [Google Scholar] [CrossRef]
- Ganda, I.S.; Zhong, Q.; Hali, M.; Albuquerque, R.L.; Padilha, F.F.; da Rocha, S.R.; Whittum-Hudson, J.A. Dendrimer-conjugated peptide vaccine enhances clearance of Chlamydia trachomatis genital infection. Int. J. Pharm. 2017, 527, 79–91. [Google Scholar] [CrossRef]
- Sudhakar, S.; Chandran, S.V.; Selvamurugan, N.; Nazeer, R.A. Biodistribution and pharmacokinetics of thiolated chitosan nanoparticles for oral delivery of insulin in vivo. Int. J. Biol. Macromol. 2020, 150, 281–288. [Google Scholar] [CrossRef]
- Yamamoto, H.; Kuno, Y.; Sugimoto, S.; Takeuchi, H.; Kawashima, Y. Surface-modified PLGA nanosphere with chitosan improved pulmonary delivery of calcitonin by mucoadhesion and opening of the intercellular tight junctions. J. Control. Release 2005, 102, 373–381. [Google Scholar] [CrossRef]
- Kim, J.-H.; Kim, Y.-S.; Park, K.; Kang, E.; Lee, S.; Nam, H.Y.; Kim, K.; Park, J.H.; Chi, D.Y.; Park, R.-W.; et al. Self-assembled glycol chitosan nanoparticles for the sustained and prolonged delivery of antiangiogenic small peptide drugs in cancer therapy. Biomaterials 2008, 29, 1920–1930. [Google Scholar] [CrossRef]
- Rayaprolu, B.M.; Strawser, J.J.; Anyarambhatla, G. Excipients in parenteral formulations: Selection considerations and effective utilization with small molecules and biologics. Drug Dev. Ind. Pharm. 2018, 44, 1565–1571. [Google Scholar] [CrossRef]
- Rubin, J.; Linden, L.; Coco, W.M.; Bommarius, A.S.; Behrens, S. Salt-induced aggregation of a monoclonal human immunoglobulin G1. J. Pharm. Sci. 2013, 102, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Guo, M.; Liu, D.; Liu, X.; Labuza, T. Maillard-reaction-induced modification and aggregation of proteins and hardening of texture in protein bar model systems. J. Food Sci. 2013, 78, C437–C444. [Google Scholar] [CrossRef] [PubMed]
- Izutsu, K.-i. Stabilization of therapeutic proteins by chemical and physical methods. In Therapeutic Proteins; Springer: Berlin/Heidelberg, Germany, 2005; pp. 287–292. [Google Scholar]
- Manning, M.C.; Chou, D.K.; Murphy, B.M.; Payne, R.W.; Katayama, D.S. Stability of protein pharmaceuticals: An update. Pharm. Res. 2010, 27, 544–575. [Google Scholar] [CrossRef] [PubMed]
- Joshi, O.; McGuire, J.; Wang, D. Adsorption and function of recombinant factor VIII at solid–water interfaces in the presence of Tween-80. J. Pharm. Sci. 2008, 97, 4741–4755. [Google Scholar] [CrossRef] [PubMed]
- Chi, E.Y.; Weickmann, J.; Carpenter, J.F.; Manning, M.C.; Randolph, T.W. Heterogeneous nucleation-controlled particulate formation of recombinant human platelet-activating factor acetylhydrolase in pharmaceutical formulation. J. Pharm. Sci. 2005, 94, 256–274. [Google Scholar] [CrossRef]
- Allmendinger, A.; Lebouc, V.; Bonati, L.; Woehr, A.; Kishore, R.S.; Abstiens, K. Glass leachables as a nucleation factor for free fatty acid particle formation in biopharmaceutical formulations. J. Pharm. Sci. 2021, 110, 785–795. [Google Scholar] [CrossRef]
- Sharma, B. Immunogenicity of therapeutic proteins. Part 2: Impact of container closures. Biotechnol. Adv. 2007, 25, 318–324. [Google Scholar] [CrossRef]
- Yoneda, S.; Torisu, T.; Uchiyama, S. Development of syringes and vials for delivery of biologics: Current challenges and innovative solutions. Expert Opin. Drug Deliv. 2021, 18, 459–470. [Google Scholar] [CrossRef]
- Ditter, D.; Nieto, A.; Mahler, H.-C.; Roehl, H.; Wahl, M.; Huwyler, J.; Allmendinger, A. Evaluation of glass delamination risk in pharmaceutical 10 mL/10R vials. J. Pharm. Sci. 2018, 107, 624–637. [Google Scholar] [CrossRef]
- Jenke, D.R.; Jene, J.M.; Poss, M.; Story, J.; Tsilipetros, T.; Odufu, A.; Terbush, W. Accumulation of extractables in buffer solutions from a polyolefin plastic container. Int. J. Pharm. 2005, 297, 120–133. [Google Scholar] [CrossRef]
- Stan, M.S.; Cinteză, L.O.; Petrescu, L.; Mernea, M.A.; Călborean, O.; Mihailescu, D.F.; Sima, C.; Dinischiotu, A. Dynamic analysis of the interactions between Si/SiO2 quantum dots and biomolecules for improving applications based on nano-bio interfaces. Sci. Rep. 2018, 8, 5289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchman, J.T.; Elmer, W.H.; Ma, C.; Landy, K.M.; White, J.C.; Haynes, C.L. Chitosan-Coated mesoporous silica nanoparticle treatment of Citrullus lanatus (watermelon): Enhanced fungal disease suppression and modulated expression of stress-related genes. ACS Sustain. Chem. Eng. 2019, 7, 19649–19659. [Google Scholar] [CrossRef]
- Patten, P.; Schellekens, H. The immunogenicity of biopharmaceuticals. Lessons learned and consequences for protein drug development. Dev. Biol. 2003, 112, 81–97. [Google Scholar]
- Ross, P.L.; Wolfe, J. Physical and chemical stability of antibody drug conjugates: Current status. J. Pharm. Sci. 2016, 105, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Vendruscolo, M.; Dobson, C. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, J.J.B. Characterization and QC of biopharmaceuticals by MS-based ‘multi-attribute method’: Advantages and challenges. Bioanalysis 2017, 9, 499–502. [Google Scholar] [CrossRef] [Green Version]
- Parr, M.K.; Montacir, O.; Montacir, H. Physicochemical characterization of biopharmaceuticals. J. Pharm. Biomed. Anal. 2016, 130, 366–389. [Google Scholar] [CrossRef]
- Rogstad, S.; Yan, H.; Wang, X.; Powers, D.; Brorson, K.; Damdinsuren, B.; Lee, S. Multi-attribute method for quality control of therapeutic proteins. Anal. Chem. 2019, 91, 14170–14177. [Google Scholar] [CrossRef]
- Huang, L.-J.; Chiang, C.-W.; Chen, S.-L.; Wei, S.-Y.; Chen, S.-H. Complete mapping of disulfide linkages for etanercept products by multi-enzyme digestion coupled with LC-MS/MS using multi-fragmentations including CID and ETD. J. Food Drug Anal. 2019, 27, 531–541. [Google Scholar] [CrossRef]
- Kuo, C.-M.; Wei, S.-Y.; Du, S.-H.; Lin, J.-L.; Chu, C.-H.; Chen, C.-H.; Tai, J.-H.; Chen, S.-H. Comprehensive workflow for mapping disulfide linkages including free thiols and error checking by on-line UV-induced precolumn reduction and spiked control. Anal. Chem. 2020, 93, 1544–1552. [Google Scholar] [CrossRef]
- Said, N.; Gahoual, R.; Kuhn, L.; Beck, A.; François, Y.-N.; Leize-Wagner, E. Structural characterization of antibody drug conjugate by a combination of intact, middle-up and bottom-up techniques using sheathless capillary electrophoresis–Tandem mass spectrometry as nanoESI infusion platform and separation method. Anal. Chim. Acta 2016, 918, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, H.E.; Mohamed, A.A.; Al-Ghobashy, M.A.; Fathalla, F.A.; Abbas, S.S. Stability assessment of antibody-drug conjugate Trastuzumab emtansine in comparison to parent monoclonal antibody using orthogonal testing protocol. J. Pharm. Biomed. Anal. 2018, 150, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Wagh, A.; Song, H.; Zeng, M.; Tao, L.; Das, T.K. Challenges and new frontiers in analytical characterization of antibody-drug conjugates. MAbs 2018, 10, 222–243. [Google Scholar] [CrossRef]
- Filipe, V.; Hawe, A.; Carpenter, J.F.; Jiskoot, W. Analytical approaches to assess the degradation of therapeutic proteins. TrAC Trends Anal. Chem. 2013, 49, 118–125. [Google Scholar] [CrossRef]
- Laganowsky, A.; Clemmer, D.E.; Russell, D.H. Variable-temperature native mass spectrometry for studies of protein folding, stabilities, assembly, and molecular interactions. Annu. Rev. Biophys. 2022, 51, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-C.; Huang, L.-J.; Hsu, L.-S.; Huang, S.-T.; Lo, W.-T.; Wang, T.-F.; Sun, W.-T.; Wei, W.-Y.; Lee, Y.-S.; Chuang, S.-H.; et al. Selective and predicable amine conjugation sites by kinetic characterization under excess reagents. Sci. Rep. 2021, 11, 21222. [Google Scholar] [CrossRef]
- Matsuda, Y. Current approaches for the purification of antibody–drug conjugates. J. Sep. Sci. 2022, 45, 27–37. [Google Scholar] [CrossRef]
- Stoll, D.R.; Carr, P.W. Two-dimensional liquid chromatography: A state of the art tutorial. Anal. Chem. 2017, 89, 519–531. [Google Scholar] [CrossRef]
- Kumar, R.; Guttman, A.; Rathore, A.S. Applications of capillary electrophoresis for biopharmaceutical product characterization. Electrophoresis 2022, 43, 143–166. [Google Scholar] [CrossRef]
- Cho, E.; Mayhugh, B.M.; Srinivasan, J.M.; Sacha, G.A.; Nail, S.L.; Topp, E.M. Stability of antibody drug conjugate formulations evaluated using solid-state hydrogen-deuterium exchange mass spectrometry. J. Pharm. Sci. 2021, 110, 2379–2385. [Google Scholar] [CrossRef]
- Xu, G.; Chance, M.R. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem. Rev. 2007, 107, 3514–3543. [Google Scholar] [CrossRef] [PubMed]
- Maleknia, S.D.; Downard, K.M. Advances in radical probe mass spectrometry for protein footprinting in chemical biology applications. Chem. Soc. Rev. 2014, 43, 3244–3258. [Google Scholar] [CrossRef] [PubMed]
- Jhan, S.-Y.; Huang, L.-J.; Wang, T.-F.; Chou, H.-H.; Chen, S.-H. Dimethyl labeling coupled with mass spectrometry for topographical characterization of primary amines on monoclonal antibodies. Anal. Chem. 2017, 89, 4255–4263. [Google Scholar] [CrossRef] [PubMed]
- Ravisankar, P.; Gowthami, S.; Rao, G. A review on analytical method development. Indian J. Res. Pharm. Biotechnol. 2014, 2, 1183. [Google Scholar]
- Zölls, S.; Tantipolphan, R.; Wiggenhorn, M.; Winter, G.; Jiskoot, W.; Friess, W.; Hawe, A. Particles in therapeutic protein formulations, Part 1: Overview of analytical methods. J. Pharm. Sci. 2012, 101, 914–935. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H. Characterization of glycosylation in monoclonal antibodies and its importance in therapeutic antibody development. Crit. Rev. Biotechnol. 2021, 41, 300–315. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Partner Type | Marketed Examples | The Effects of the Partner | |
|---|---|---|---|
| Non-peptide partners | Fc | Enbrel®, Eloctate® and Alprolix® (FDA approved). | Enlargement to a size greater than 70 kDa (the critical size to escape renal excretion). |
| Albumin | Tanzeum® (FDA approved). | Increases blood circulation | |
| Transferrin | - | Improve oral administration, no FDA-approved cases, | |
| Carboxyl terminal peptide | Elonva® (produced by Merck and Co., Rahway, NJ, USA, but not FDA approved). LagovaTM (in phase 3 clinical trials). | 31 residues with four O-glycosylation sites | |
| Recombinant polypeptide partners | XTEN | - | Increases blood circulation |
| Elastin-like polypeptides | GlymeraTM, VasomeraTM | Contains V-P-G-x-G, the sequence where x is any residue except proline. | |
| Proline-alanine-serine | XL-Protein GmbH (preclinical state). | The repetition of Pro, Ala and Ser residues in 100–200 copies. Delaying the renal clearance | |
| Glycine-Serine rich peptides | - | As a linker and a partner for medicinal peptides |
| Factors Must Be Consider | Example | Description |
|---|---|---|
| Identity | PEG and PEG analogs | Increases the hydrodynamic radius, decreasing immunogenicity |
| Stimuli-responsive polymers | Thermo responsive: p(NIPAAm) pH-responsive: poly(acrylic acid) and p(DMAEMA) | |
| Biomimetic polymers | Trehalose Heparin-mimicking polymers: poly(styrene sulfonate) and poly(vinyl sulfonate) | |
| Degradable polymers | Cyclic ketene acetals copolymerized with vinyl monomers: degradable under basic pH Hydroxyethyl starch: a-amylase sensitive | |
| Toxicity | Hydroxyethyl starch | Plasma volume expander |
| Molecular weight | PEGylated proteins | Decreasing renal filtration |
| Polymer architecture | Branched, brush and linear polymers are accessible to be conjugate with proteins. | Stimulating the immune system |
| Polymer solubility | - | Soluble in the range between 100 and 500 mg/mL |
| Lipidopeptide/Protein | Lipid | Attachment Strategy | Effects | Ref. |
|---|---|---|---|---|
| Liraglutide | Palmitic acid | γ-glutamic acid | Extension of the half-life, reducing renal clearance | [171] |
| Salmon calcitonin | N-palmitoylated | Cys-1 and Cys-7 of the peptide | Extension of the half-life, reducing renal clearance | [172] |
| Opioid peptide leu-enkephalin | 3,4 bis(decylthiomethyl)- 2,5-furandione 16 | N-terminus binding | Reducing receptor binding, increasing protease stability | [173] |
| Thyrotropin-releasing hormone | lauric acid | N-terminus binding | Peptide penetration across the small intestine | [174,175] |
| H-Ras | 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine | maleimide-functionalized phospholipids/ S-palmitoylation at Cys181 and Cys184 | Dimerization | [176] |
| Caveolin-3 | octanethiol | cysteine S-fatty acylation | As a small protein model in the study of lipidation of proteins | [177] |
| Interferon-induced transmembrane protein 3 (IFITM3) | maleimide palmitate | maleimide-functionalized phospholipids/ S-palmitoylation at Cys72 and Cys105 | Enhancing the conserved amphipathic domain | [178] |
| Cathepsin-B inhibitor | Palmitic acid | Palmitoylation of the amino groups (ε-lysyl amino groups) | Increasing the inhibitory effect on cathepsin-B | [179] |
| Name | Source | Application | Ref. |
|---|---|---|---|
| Sunflower trypsin inhibitor 1 (SFTI-1) | Helianthus annuus | Trypsin inhibitor, angiogenic activity | [184] |
| Gramicidin S | Bacillus brevis | Antibiotic activity towards Gram-negative, Gram-positive and several pathogenic fungi | [185,186] |
| Plitidepsin | Aplidium albicans | In vitro anticancer activity | [187,188] |
| Tyrocidine | Bacillus brevis | Antibiotic action | [189] |
| Rakicidin F | Streptomyces | Antibacterial effect | [190] |
| Plitidepsin | Aplidium albicans | Antitumor, antiviral and immunosuppressive activities | [191] |
| Asperpeptide A cyclo(-Pro-Ala-Ala-Tyr-5-OHAA) | Aspergillus sp. XS-20090B15 | Antibacterial activity | [192] |
| Cyclosporin A | Tolypocladium inflatum | Calcineurin inhibitor, decreasing the function of lymphocytes | [193] |
| Depsilipopeptide colisporifungin | Colispora cavincola | Antifungal activity | [194] |
| Cryptophycin | Nostoc | Fungicide and anticancer | [195] |
| Romidepsin | Chromobacterium violaceum | Apoptotic activity, anticancer activity | [196,197] |
| Theonellamide G | Theonella swinhoei | Cytotoxic activity | [198] |
| Ziconotide | Conus magus | Analgesic agent; a strong pain killer. | [199] |
| Kahalalide F | Elysia rufescens | Antitumor | [200] |
| Vancomycin | Amycolatopsis orientalis | Antibacterial | [201] |
| Alisporivir | Chemically synthesized from ciclosporin. | Inhibits cyclophilin A, the potential effect on Alzheimer’s disease and hepatitis C | [202,203] |
| Istodax (Romidepsin) | Head–tail lactone cyclization which is stabilized by a pair of disulfide bonds. | Anticancer | [204] |
| Lupkynis (Voclosporin) | Head–tail cyclization | Calcineurin like activity | [205] |
| Vasostrict (Vasopressin) | Disulfide mediated cyclization | Used in Anti-diuretic hormone deficiency | |
| Signifor (Pasireotide) | Head–tail cyclization | Activating a broad spectrum of somatostatin receptors, reducing cortisol secretion | [206] |
| Prialt (Ziconotide) | Disulfide mediated cyclization | Pain killer | [207] |
| Nanoparticle | Protein Cargo | Delivery Rout | Ref. |
|---|---|---|---|
| Chitosan functionalized lipid nanoparticle (LN) | Insulin | Oral | [214] |
| Chitosan-modified mesoporous silica | Insulin | Suggested for oral delivery | [215] |
| Chitosan-modified carboxymethyl-β-cyclodextrin | BSA | Oral | [216] |
| Poly(lactide)-tocopheryl polyethyde glycol succinate | BSA | Oral | [217] |
| Alginate | Osteogenic protein-1 | Intranasal | [218] |
| DOPE-liposome | E75 peptide (HER-2/neu-369–377) | Intravenous | [219] |
| DOTAP-liposome | HSV-derivated E7 oncoprotein | Intravenous | [220] |
| Chitosan-coated liposomal system | Ghrelin (a peptide hormone that can regulate appetite and body weight changing) | Nose-to-brain delivery | [221] |
| Chitosan-covered liposomes | Rainbow trout skin-derived peptides | Suggested for oral delivery | [222] |
| PEGylated liposomes | Opiorphin | Intravenous | [223] |
| Polyacrylate-coated superparamagnetic Fe3O4 | Elastin-like VPGVG pentapeptides | Subcutaneous injections | [223] |
| G4 OH-terminated PAMAM | AFPQFRSATLLL | Subcutaneous injections | [224] |
| Thiolated chitosan nanoparticles | Insulin | Subcutaneous injections | [225] |
| Chitosan-modified PLGA nanospheres | Elcatonin | Pulmonary | [226] |
| Chitosan nanoparticles | RGD peptide (Arg-Gly-Asp) | Intravenous | [227] |
| Excipient | Mechanism | General Comments |
|---|---|---|
| Buffering agents | Keeping the pH of protein solutions | Usually, the buffers work in the range of 3–10 for proteins. In certain conditions, some buffers may be decomposed, and their by-products destroy the protein structure. |
| Chelators and antioxidants | The roles of antioxidants and chelators are to prevent and/or remove oxidazing factors. | Some reducing agents such as glutathione and ascorbic acid in the presence of metals and enhancing oxidation stresses can have a negative/destructive role on protein structure, although these agents are used in the pharmaceutical sector of proteins. |
| Proteins | By interacting with therapeutic peptides, excipient proteins can increase the blood circulation time. | Nowadays, chaperones have been given a special look as a preservative for medicinal proteins. |
| Polymers | Maintaining the structure of proteins | The main examples of this group include polyvinyl alcohol, dextran and hydroxyethyl starch. |
| Amino acids | Buffering properties, preferential interactions, favored hydration, antioxidant effect and strong binding to protein regions | Glycine (buffering agent and bulking agent during lyophilization), arginine (solubilizing agent and works as chaperone) and histidine (antioxidant and buffering agent) |
| Sugars and carbohydrates | Forming a crystal network with preferential interactions | Sorbitol in lyophilization and liquid formulation conditions, has a stabilizing role, however, in freezing conditions, due to the formation of sorbitol crystals, it assumes a stabilizing role. |
| Salts | Tonicifying agent, reacting with the charged surfaces of proteins and take two stabilizing or destabilizing paths. | Among the cations and anions of the Hofmeister series, the latter has dual protective/destructive effects on proteins. |
| Antimicrobial preservatives | Preventing the growth of bacteria in solution-rich medicinal proteins | m-cresol, phenol and benzyl alcohol are among the popular. |
| Surfactants | Reducing the interface area of solution and air during purification (inner wall of purification column and dialysis bags, etc.). | In this group, polysorbate 20 (PS20) and polysorbate 80 (PS80) are the most used in protein drugs and especially antibodies. |
| Osmolytes | Generating favored hydration, preferential interactions, and polar interactions | Sorbitol, sucrose, glycine, and trehalose have been able to reach the pharmaceutical sector |
| Technique | Output | Destructive | QC Method | General Comments | |
|---|---|---|---|---|---|
| Conformational assessments | DSC | Thermal parameter (Tm, ΔG unfolding) | Yes | No | The best option for checking temperature-dependent parameters; not high throughput. |
| CD | Secondary and tertiary statures | No | No | Sensitive to the polarity of the solution. | |
| Fluorescence spectroscopy | Tertiary statures/ general view on the protein structure | Yes | No | Sensitive to small structural changes of proteins and peptides; the need for relatively small amounts of material; relatively high speed; takes a general view of structural changes and cannot go into details. | |
| DSF | Tm, aggregation onset | Yes | No | During the thermal unfolding of a protein, a dye (commonly Sypro Orange) is added to the sample, which, by connecting to the unfolded parts, leads to an increase in its fluorescence emission. | |
| UV | Tertiary structure | No | No | Makes a general view of structural changes. | |
| Raman | Secondary structure/ chemical characterization | Yes | No | It has a high overlap with FTIR, but on the contrary, a wide range of solvents can be used in Raman analysis to examine samples. | |
| Infrared | Secondary structure/chemical integrity | Yes | No | By examining the amine-I and II regions of proteins and their deconvolution, it is possible to reach the percentage of the secondary structure in the protein (in solution and solid states) either in the form of fold and/or aggregation/fibrils. ATR facility requires very small amounts of substance without the need to prepare KBR tablets. | |
| Oligomerization studies | AUC | Molecular weight/shape | No | Yes | Determining the size of particle aggregation. |
| DLS | Hydrodynamic size | No | High range of particle detection (between 1nm to 5 µM), reliable within a certain range of polydispersity | ||
| FAPS | Particles/Serum interactions | No | Extracting fluorescence data from the aggregated samples. One of its limitations is the use of dyes to identify aggregates, which may affect the protein structure. | ||
| AF4 | Hydrodynamic size | Yes | Yes | AF4 technique separates particles based on their diffusion coefficients. | |
| NTA | Hydrodynamic size | No | It can measure the size of particles, imaging and quantifying them. | ||
| RMM | Concentration/size/mass | Yes | No | In a microfluidic way, it can calculate particle size. | |
| SEC | Hydrodynamic size | Yes | Yes | Determine the molecular weight, aggregation rate, and interactions between proteins. | |
| SDS-PAGE (all types) | Molecular weight/ interactions between proteins | Yes | Yes | Covalent interactions between proteins and also protein digests; in reducing and non-reducing types, it can observe disulfide bonds. | |
| Optical microscopy | Size/morphology | No | No | Detecting large particles (larger than 1 µm). | |
| Native MS | Fragments/aggregates | Yes | No | With principles similar to MS, it investigates non-covalent interactions and post-translational changes in proteins. | |
| Light obscuration | Concentration/size | No | Yes | This technique, which is also known as Single Particle Optical Sensing (SPOS), is not very sensitive to small sizes (detecting sizes more than 1 µm). | |
| Fluorescence microscopy | Particles/amorphous and morphous aggregates | Yes | No | A fluorophore molecule is needed that can provide the output signal. Some fluorophore dyes change fluorescence intensity by interacting with proteins and being buried in their structure, which can be a pattern of protein folding and even aggregation. | |
| Flow imaging | Concentration/size/morphology | No | Yes | The basis is similar to optical microscopy, except that it can provide data qualitatively. | |
| CE-SDS | Molecular weight | Yes | Yes | Advantages such as quicker analysis, the facility for quantification, full automation, the need for low sample, and better resolution | |
| Electrical zone sensing | Concentration/size | Yes | No | By applying electric force and migration of the two-pole magnifier, it can achieve the size of the particles. | |
| Turbidity | Optical density > 360 nm | No | Yes | It is a low-cost, rough method that can be used to detect large particles. Its high speed and simplicity are its positive points. | |
| Chemical changes | RP-HPLC | Hydrophobicity | Yes | Yes | Sensitive to slight changes in surface hydrophobicity of proteins, requiring small amounts of samples. |
| cIEF | Charge | Yes | Yes | Similar to IEF, it can separate proteins based on their PI. Compared to its traditional sample, i.e., IEF, it requires much less material, and due to its capillary nature, a higher voltage can be applied to the sample, which leads to a reduction in the test time. This test is successful in the case of samples higher than 150,000 Daltons that dissolve well in aqueous solutions. | |
| IEX chromatography | Charge | Yes | Yes | Separating proteins by considering their charge. High speed and acceptable accuracy. | |
| MS | By the difference in weight molecular changes | Yes | No | Differentiation of diverse components with considering mass-to-charge ratio (m/z). | |
| LC-MS | By the difference in weight molecular changes | Yes | No | In cases where a protein complex is present, initial separation by HPLC enables MS to obtain more details of the sample by removing noise. | |
| Zeta potential | Charge | No | Yes | Measuring the particle charges; the types of solvents and even the percentage of ions in water strongly affect the data. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akbarian, M.; Chen, S.-H. Instability Challenges and Stabilization Strategies of Pharmaceutical Proteins. Pharmaceutics 2022, 14, 2533. https://doi.org/10.3390/pharmaceutics14112533
Akbarian M, Chen S-H. Instability Challenges and Stabilization Strategies of Pharmaceutical Proteins. Pharmaceutics. 2022; 14(11):2533. https://doi.org/10.3390/pharmaceutics14112533
Chicago/Turabian StyleAkbarian, Mohsen, and Shu-Hui Chen. 2022. "Instability Challenges and Stabilization Strategies of Pharmaceutical Proteins" Pharmaceutics 14, no. 11: 2533. https://doi.org/10.3390/pharmaceutics14112533