
Synthesis of β-Cyclodextrin-Decorated Dendritic Compounds Based on EDTA Core: A New Class of PAMAM Dendrimer Analogs

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
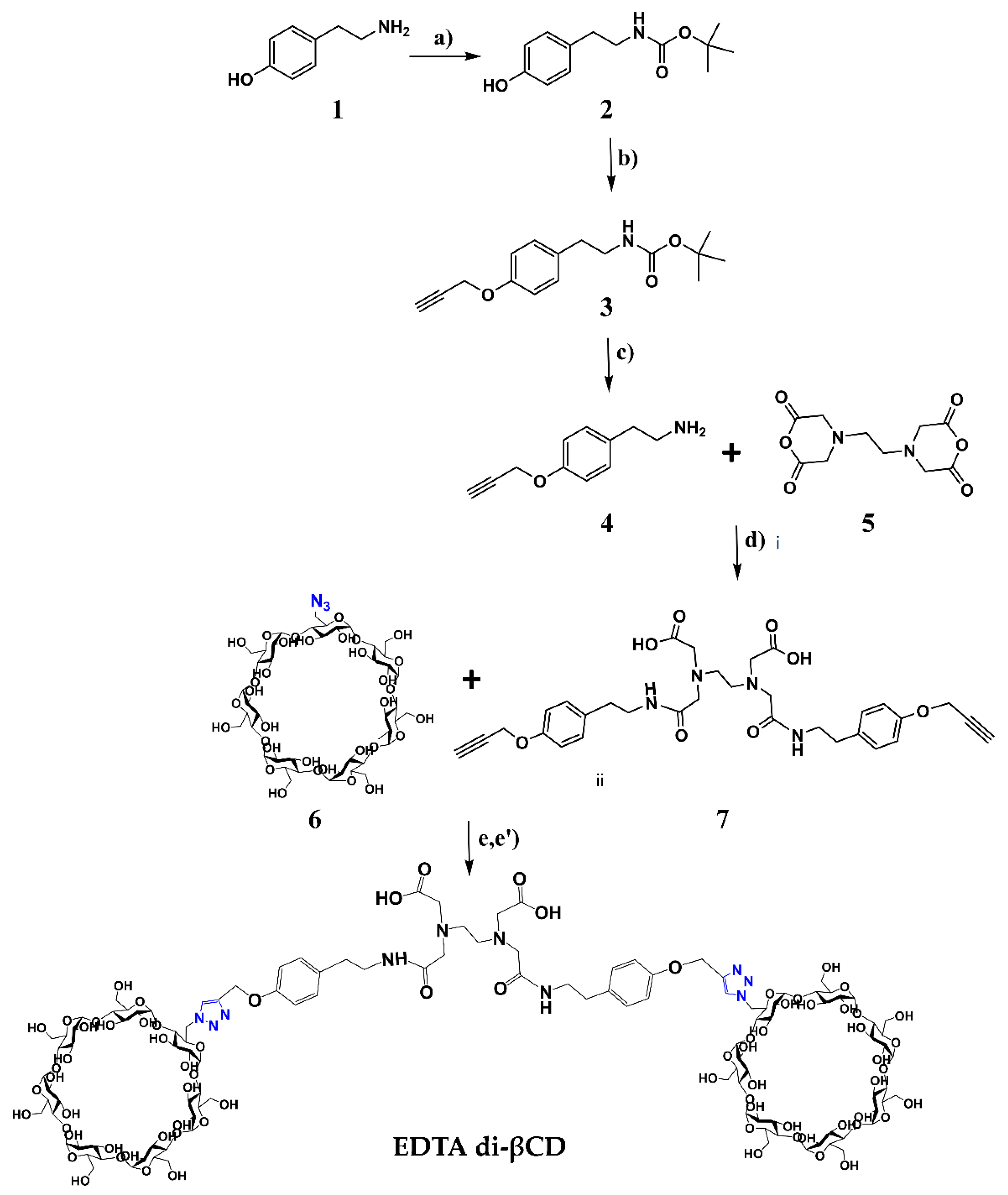
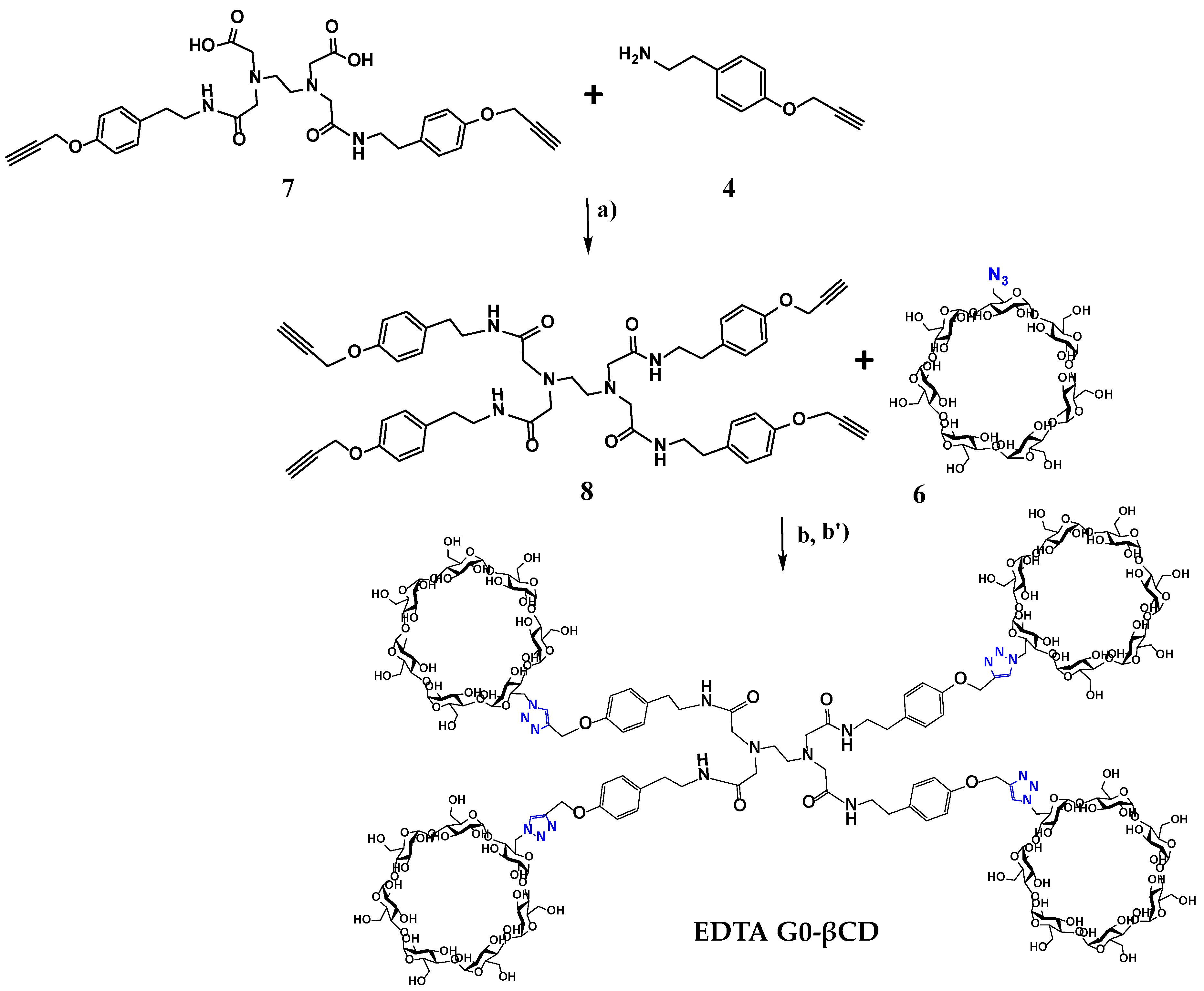
2.1. Synthesis
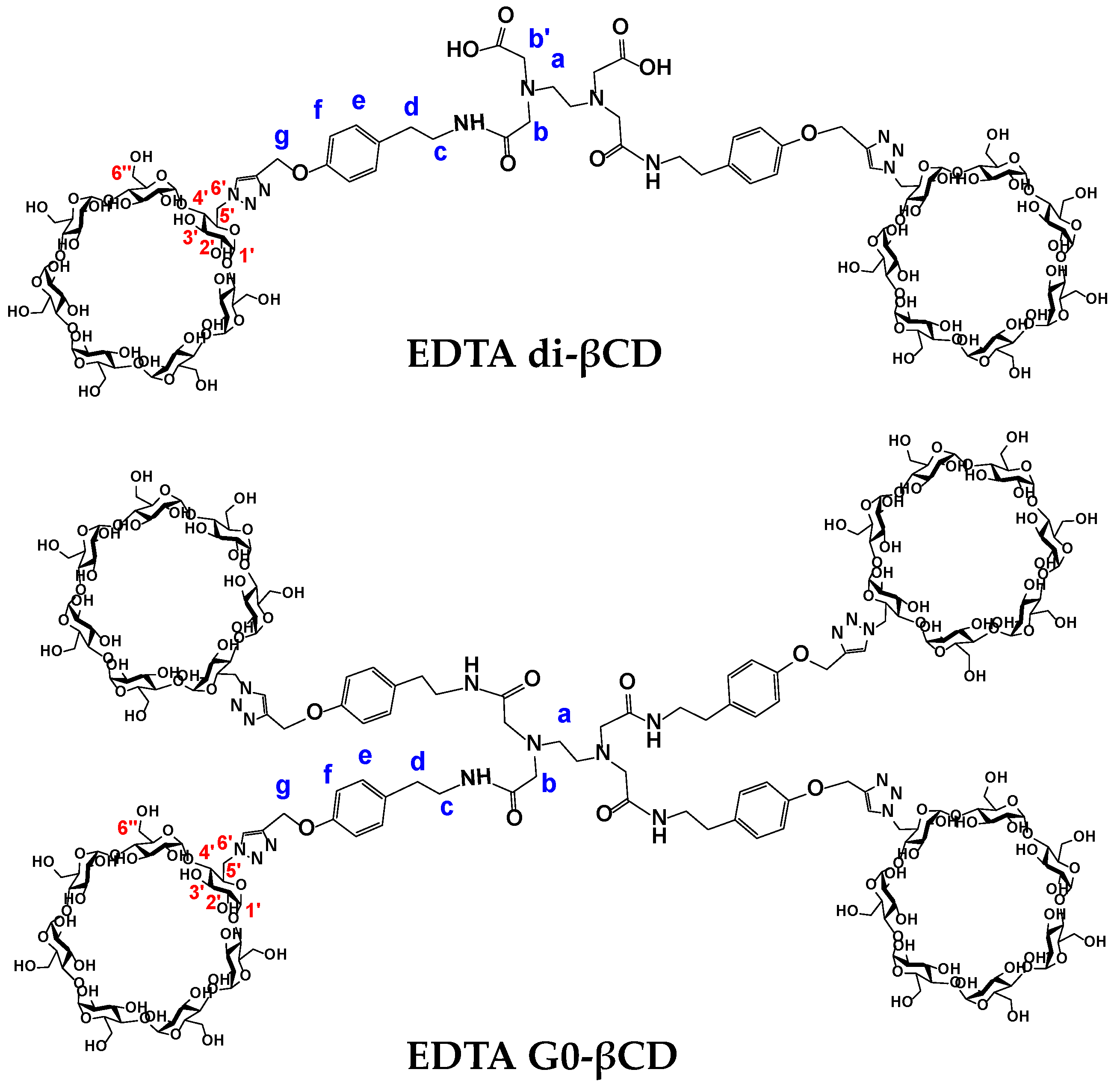
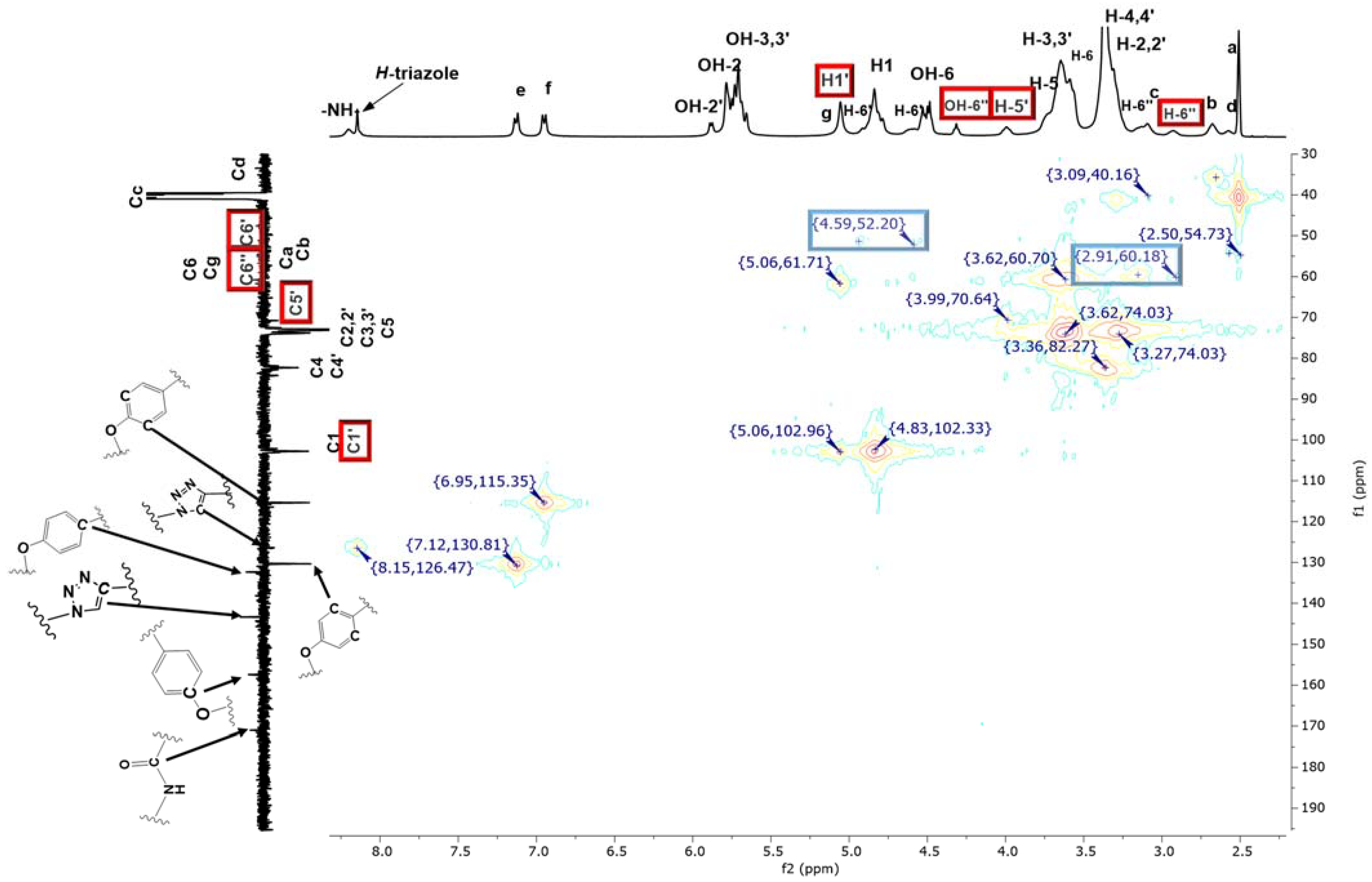
2.2. Characterization
2.3. Determination of Water Solubility for EDTA di-βCD and EDTA G0-βCD
3. Materials and Methods
3.1. General Notes
3.2. Synthetic Procedures
3.2.1. Synthesis of tert-butyl (4-hydroxyphenethyl)carbamate (2)
3.2.2. Synthesis of tert-butyl (4-(prop-2-yn-1-yloxy)phenyl)carbamate (3)
3.2.3. Synthesis of 2-(4-(prop-2-yn-1-yloxy)phenyl)ethan-1-amine (4)
3.2.4. Synthesis of Disubstituted EDTA Alkyne (7)
3.2.5. Synthesis of Tetrasubstituted EDTA G0-Alkyne (8)
3.2.6. Synthesis of Dendritic EDTA di-βCD
3.2.7. Synthesis of EDTA G0-βCD Dendrimer
3.3. Determination of Water Solubility for EDTA di-βCD Dendritic and EDTA G0-βCD Dendrimer
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abbasi, E.; Aval, S.F.; Akbarzadeh, A.; Milani, M.; Nasrabadi, H.T.; Joo, S.W.; Hanifehpour, Y.; Nejati-Koshki, K.; Pashaei-Asl, R. Dendrimers: Synthesis, Applications, and Properties. Nanoscale Res. Lett. 2014, 9, 247. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.-P.; Ficker, M.; Christensen, J.B.; Trohopoulos, P.N.; Moghimi, S.M. Dendrimers in Medicine: Therapeutic Concepts and Pharmaceutical Challenges. Bioconjug. Chem. 2015, 26, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.K.; Çaǧın, T.; Wang, G.; Goddard, W.A. Structure of PAMAM Dendrimers: Generations 1 Through 11. Macromolecules 2004, 37, 6236–6254. [Google Scholar] [CrossRef]
- Mittal, P.; Saharan, A.; Verma, R.; Altalbawy, F.M.A.; Alfaidi, M.A.; Batiha, G.E.-S.; Akter, W.; Gautam, K.R.; Uddin, S.; Rahman, S. Dendrimers: A New Race of Pharmaceutical Nanocarriers. BioMed Res. Int. 2021, 2021, 8844030. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lei, L.; Voets, I.K.; Stuart, M.A.; Velders, A.H. Dendrimicelles with pH-Controlled Aggregation Number of Core-Dendrimers and Stability. Soft Matter 2020, 16, 7893–7897. [Google Scholar] [CrossRef] [PubMed]
- Matveev, V.V.; Markelov, D.A.; Dvinskikh, S.V.; Shishkin, A.N.; Tyutyukin, K.V.; Penkova, A.V.; Tatarinova, E.A.; Ignat’eva, G.M.; Milenin, S.A. Investigation of Melts of Polybutylcarbosilane Dendrimers by 1H NMR Spectroscopy. Sci. Rep. 2017, 7, 13710. [Google Scholar] [CrossRef] [Green Version]
- Leiro, V.; Garcia, J.P.; Tomás, H.; Pêgo, A.P. The Present and the Future of Degradable Dendrimers and Derivatives in Theranostics. Bioconjug. Chem. 2015, 26, 1182–1197. [Google Scholar] [CrossRef] [Green Version]
- Sadekar, S.; Ghandehari, H. Transepithelial Transport and Toxicity of PAMAM Dendrimers: Implications for Oral Drug Delivery. Adv. Drug Delivery Rev. 2012, 64, 571–588. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Katsumi, H.; Sakane, T.; Yamamoto, A. Effects of Polyamidoamine (PAMAM) Dendrimers on the Nasal Absorption of Poorly Absorbable Drugs in Rats. Int. J. Pharm. 2010, 393, 245–253. [Google Scholar] [CrossRef]
- Cui, T.; Li, S.; Chen, S.; Liang, Y.; Sun, H.; Wang, L. “Stealth” Dendrimers with Encapsulation of Indocyanine Green for Photothermal and Photodynamic Therapy of Cancer. Int. J. Pharm. 2021, 600, 120502. [Google Scholar] [CrossRef]
- Kim, H.; Choi, B.; Lim, H.; Min, H.; Oh, J.H.; Choi, S.; Cho, J.G.; Park, J.-S.; Lee, S.J. Polyamidoamine Dendrimer-Conjugated Triamcinolone Acetonide Attenuates Nerve Injury-Induced Spinal Cord Microglia Activation and Mechanical Allodynia. Mol. Pain 2017, 13, 1744806917697006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorzkiewicz, M.; Janaszewska, A.; Ficker, M.; Svenningsen, S.W.; Christensen, J.B.; Klajnert-Maculewicz, B. Pyrrolidone-modified PAMAM Dendrimers Enhance Anti-Inflammatory Potential of Indomethacin in vitro. Colloids Surf. B 2019, 181, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Mendes, P.L.; Pan, J.; Torchilin, V.P. Dendrimers as Nanocarriers for Nucleic Acid and Drug Delivery in Cancer Therapy. Molecules 2017, 22, 1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noriega-Luna, B.; Godínez, L.A.; Rodríguez, F.J.; Rodríguez, A.; Zaldívar-Lelo de Larrea, G.; Sosa-Ferreyra, C.F.; Mercado-Curiel, R.F.; Manríquez, J.; Bustos, E. Applications of Dendrimers in Drug Delivery Agents, Diagnosis, Therapy, and Detection. J. Nanomater. 2014, 2014, 39. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Ravizzini, G.; Choyke, P.L.; Kobayashi, H. Dendrimers in Medical Nanotechnology. IEEE Eng. Med. Biol. Mag. 2009, 28, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Gorzkiewicz, M.; Kopeć, O.; Janaszewska, A.; Konopka, M.; Pędziwiatr-Werbicka, E.; Tarasenko, I.I.; Bezrodnyi, V.V.; Neelov, I.M.; Klajnert-Maculewicz, B. Poly(Lysine) Dendrimers Form Complexes with siRNA and Provide its Efficient Uptake by Myeloid Cells: Model Studies for Therapeutic Nucleic Acid Delivery. Int. J. Mol. Sci. 2020, 21, 3138. [Google Scholar] [CrossRef]
- Idris, A.O.; Mamba, B.; Feleni, U. Poly (Propylene Imine) Dendrimer: A Potential Nanomaterial for Electrochemical Application. Mater. Chem. Phys. 2020, 244, 122641. [Google Scholar] [CrossRef]
- Singh, V.; Sahebkar, A.; Kesharwani, P. Poly (Propylene Imine) Dendrimer as an Emerging Polymeric Nanocarrier for Anticancer Drug and Gene Delivery. Eur. Polym. J. 2021, 158, 110683. [Google Scholar] [CrossRef]
- Cangiotti, M.; Staneva, D.; Ottaviani, M.F.; Vasileva-Tonkova, E.; Grabchev, I. Synthesis and Characterization of Fluorescent PAMAM Dendrimer Modified with 1,8-Naphthalimide Units and its Cu(II) Complex Designed for Specific Biomedical Application. J. Photochem. Photobiol. A 2021, 415, 113312. [Google Scholar] [CrossRef]
- Jose, J.; Charyulu, R.N. Prolonged Drug Delivery System of an Antifungal Drug by Association with Polyamidoamine Dendrimers. Int. J. Pharm. Investig. 2016, 6, 123–127. [Google Scholar] [CrossRef]
- Villamagna, I.J.; Gordon, T.N.; Hurtig, M.B.; Beier, F.; Gillies, E.R. Poly(Ester Amide) Particles for Controlled Delivery of Celecoxib. J. Biomed. Mater. Res. Part A 2019, 107, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Lancelot, A.; González-Pastor, R.; Clavería-Gimeno, R.; Romero, P.; Abian, O.; Martín-Duque, P.; Serrano, J.L.; Sierra, T. Cationic Poly(Ester Amide) Dendrimers: Alluring Materials for Biomedical Applications. J. Mater. Chem. B 2018, 6, 3956–3968. [Google Scholar] [CrossRef] [PubMed]
- Caminade, A.-M.; Maraval, V.; Laurent, R.; Turrin, C.-O.; Sutra, P.; Leclaire, J.; Griffe, L.; Marchand, P.; Baudoin-Dehoux, C.; Rebout, C.; et al. Phosphorus Dendrimers: From Synthesis to Applications. Comptes Rendus Chim. 2003, 6, 791–801. [Google Scholar] [CrossRef]
- Mignani, S.; Shi, X.; Ceña, V.; Shcharbin, D.; Bryszewska, M.; Majoral, J.-P. In vivo Therapeutic Applications of Phosphorus Dendrimers: State of the Art. Drug Discov. Today 2021, 26, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Z.; Ding, L.; Huang, A.Y.-T.; Kao, C.-L.; Peng, L. Poly(Amidoamine) Dendrimers: Covalent and Supramolecular Synthesis. Mater. Today Chem. 2019, 13, 34–48. [Google Scholar] [CrossRef]
- Kaur, D.; Jain, K.; Mehra, N.K.; Kesharwani, P.; Jain, N.K. A Review on Comparative Study of PPI and PAMAM Dendrimers. J. Nanopart. Res. 2016, 18, 146. [Google Scholar] [CrossRef]
- Boas, U.; Heegaard, P.M.H. Dendrimers in Drug Research. Chem. Soc. Rev. 2004, 33, 43–63. [Google Scholar] [CrossRef]
- Sebestik, J.; Niederhafner, P.; Jezek, J. Peptide and Glycopeptide Dendrimers and Analogous Dendrimeric Structures and their Biomedical Applications. Amino Acids 2011, 40, 301–370. [Google Scholar] [CrossRef]
- de Araújo, R.V.; da Silva Santos, S.; Igne Ferreira, E.; Giarolla, J. New Advances in General Biomedical Applications of PAMAM Dendrimers. Molecules 2018, 23, 2849. [Google Scholar] [CrossRef] [Green Version]
- Dias, A.P.; da Silva Santos, S.; da Silva, J.V.; Parise-Filho, R.; Igne Ferreira, E.; El Seoud, O.; Giarolla, J. Dendrimers in the Context of Nanomedicine. Int. J. Pharm. 2020, 573, 118814. [Google Scholar] [CrossRef]
- Nwe, K.; Milenic, D.E.; Ray, G.L.; Kim, Y.-S.; Brechbiel, M.W. Preparation of Cystamine Core Dendrimer and Antibody–Dendrimer Conjugates for MRI Angiography. Mol. Pharm. 2012, 9, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Nayak, S.K. Dendrimers: A Review on Synthetic Approaches. J. Appl. Pharm. Sci. 2015, 5, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Kharwade, R.; More, S.; Warokar, A.; Agrawal, P.; Mahajan, N. Starburst Pamam Dendrimers: Synthetic Approaches, Surface Modifications, and Biomedical Applications. Arab. J. Chem. 2020, 13, 6009–6039. [Google Scholar] [CrossRef]
- Peterson, J.; Allikmaa, V.; Subbi, J.; Pehk, T.; Lopp, M. Structural Deviations in Poly(Amidoamine) Dendrimers: A MALDI-TOF MS Analysis. Eur. Polym. J. 2003, 39, 33–42. [Google Scholar] [CrossRef]
- Sánchez-Navarro, M.; Rojo, J. Chapter 5—Synthetic Strategies to Create Dendrimers: Advantages and Drawbacks. In Nanobiotechnology: Inorganic Nanoparticles vs Organic Nanoparticles, 1st ed.; de la Fuente, J.M., Grazu, V., Eds.; Elsevier: Oxford, UK, 2012; Volume 4, pp. 143–156. [Google Scholar] [CrossRef]
- Shaunak, S. Perspective: Dendrimer Drugs for Infection and Inflammation. Biochem. Biophys. Res. Commun. 2015, 468, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.P.; Huang, B.; Choi, S.K.; Silpe, J.E.; Kotlyar, A.; Desai, A.M.; Zong, H.; Gam, J.; Joice, M.; Baker, J.R., Jr. Polyvalent Dendrimer-Methotrexate as a Folate Receptor-Targeted Cancer Therapeutic. Mol. Pharm. 2012, 9, 2669–2676. [Google Scholar] [CrossRef] [Green Version]
- Fox, L.J.; Richardson, R.M.; Briscoe, W.H. PAMAM Dendrimer-Cell Membrane Interactions. Adv. Colloid Interface Sci. 2018, 257, 1–18. [Google Scholar] [CrossRef]
- Janaszewska, A.; Lazniewska, J.; Trzepiński, P.; Marcinkowska, M.; Klajnert-Maculewicz, B. Cytotoxicity of Dendrimers. Biomolecules 2019, 9, 330. [Google Scholar] [CrossRef] [Green Version]
- Chis, A.A.; Dobrea, C.; Morgovan, C.; Arseniu, A.M.; Rus, L.L.; Butuca, A.; Juncan, A.M.; Totan, M.; Vonica-Tincu, A.L.; Cormos, G.; et al. Applications and Limitations of Dendrimers in Biomedicine. Molecules 2020, 25, 3982. [Google Scholar] [CrossRef]
- Mishra, V.; Gupta, U.; Jain, N.K. Surface-Engineered Dendrimers: A Solution for Toxicity Issues. J. Biomater. Sci. Polym. Ed. 2009, 20, 141–166. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Q.; Chang, H.; Cheng, Y. Surface-Engineered Dendrimers in Gene Delivery. Chem. Rev. 2015, 115, 5274–5300. [Google Scholar] [CrossRef] [PubMed]
- González-Méndez, I.; Hameau, A.; Laurent, R.; Bijani, C.; Bourdon, V.; Caminade, A.-M.; Rivera, E.; Moineau-Chane Ching, K.I. β-Cyclodextrin PAMAM Dendrimer: How to Overcome the Tumbling Process for Getting Fully Available Host Cavities. Eur. J. Org. Chem. 2020, 2020, 1114–1121. [Google Scholar] [CrossRef]
- Sorroza-Martínez, K.; González-Méndez, I.; Martínez-Serrano, R.D.; Solano, J.D.; Ruiu, A.; Illescas, J.; Zhu, X.X.; Rivera, E. Efficient Modification of PAMAM G1 Dendrimer Surface with β-Cyclodextrin Units by CuAAC: Impact on the Water Solubility and Cytotoxicity. RSC Adv. 2020, 10, 25557–25566. [Google Scholar] [CrossRef] [PubMed]
- Przybyla, M.A.; Yilmaz, G.; Becer, C.R. Natural Cyclodextrins and their Derivatives for Polymer Synthesis. Polym. Chem. 2020, 11, 7582–7602. [Google Scholar] [CrossRef]
- Arima, H. Twenty Years of Research on Cyclodextrin Conjugates with PAMAM Dendrimers. Pharmaceutics 2021, 13, 697. [Google Scholar] [CrossRef]
- Qiu, J.; Kong, L.; Cao, X.; Li, A.; Tan, H.; Shi, X. Dendrimer-Entrapped Gold Nanoparticles Modified with β-Cyclodextrin for Enhanced Gene Delivery Applications. RSC Adv. 2016, 6, 25633–25640. [Google Scholar] [CrossRef]
- Menjoge, A.R.; Kannan, R.M.; Tomalia, D.A. Dendrimer-based drug and imaging conjugates: Design considerations for nanomedical applications. Drug Discov. Today 2010, 15, 171–185. [Google Scholar] [CrossRef]
- Buschhaus, B.; Hampel, F.; Grimme, S.; Hirsch, A. Metal-Induced Chiral folding of Depsipeptide Dendrimers. Chem. Eur. J. 2005, 11, 3530–3540. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-Palma, M.T.; Apolonio, V.M.; González, J.; Martínez-Barrera, G.; Corona, D.; Cuevas-Yañez, E. Synthesis of EDTA Core Dendrimers Through a Consecutive Esterification-CuAAC Process. J. Macromol. Sci. Part A Pure Appl. Chem. 2017, 54, 908–914. [Google Scholar] [CrossRef]
- Hameau, A.; Fuchs, S.; Laurent, R.; Majoral, J.-P.; Caminade, A.-M. Synthesis of Dye/Fluorescent Functionalized Dendrons Based on Cyclotriphosphazene. Beilstein J. Org. Chem. 2011, 7, 1577–1583. [Google Scholar] [CrossRef]
- Cheng, M.H.Y.; Savoie, H.; Bryden, F.; Boyle, R.W. A Convenient Method for Multicolour Labelling of Proteins with BODIPY Fluorophores via Tyrosine Residues. Photochem. Photobiol. Sci. 2017, 16, 1260–1267. [Google Scholar] [CrossRef]
- Ting, C.-H.; Chen, J.-T.; Hsu, C.-S. Synthesis and Thermal and Photoluminescence Properties of Liquid Crystalline Polyacetylenes Containing 4-Alkanyloxyphenyl trans-4-Alkylcyclohexanoate Side Groups. Macromolecules 2002, 35, 1180–1189. [Google Scholar] [CrossRef]
- Li, Z.; Huang, R.; Xu, H.; Chen, J.; Zhan, Y.; Zhou, X.; Chen, H.; Jiang, B. Divinylsulfonamides as Specific Linkers for Stapling Disulfide Bonds in Peptides. Org. Lett. 2017, 19, 4972–4975. [Google Scholar] [CrossRef] [PubMed]
- Antelo, A.; Jover, A.; Galantini, L.; Meijide, F.; Alvarez Alcalde, M.; Viorel Pavel, N.; Vázquez Tato, J. Formation of Host-Guest and Sandwich Complexes by a β-Cyclodextrin Derivative. J. Incl. Phenom. Macrocycl. Chem. 2011, 69, 245–253. [Google Scholar] [CrossRef]
- Zhong, N.; Byun, H.-S.; Bittman, R. An Improved Synthesis of 6-O-Monotosyl-6-Deoxy-β-Cyclodextrin. Tetrahedron Lett. 1998, 39, 2919–2920. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Y.; Hu, J.; Li, C.; Liu, S. Multi-Responsive Supramolecular Double Hydrophilic Diblock Copolymer Driven by Host-Guest Inclusion Complexation between β-Cyclodextrin and Adamantyl Moieties. Macromol. Chem. Phys. 2009, 210, 2125–2137. [Google Scholar] [CrossRef]
- Sorroza-Martínez, K.; González-Méndez, I.; Vonlanthen, M.; Moineau-Chane Ching, K.I.; Caminade, A.-M.; Illescas, J.; Rivera, E. First Class of Phosphorus Dendritic Compounds Containing β-Cyclodextrin Units in the Periphery Prepared by CuAAC. Molecules 2020, 25, 4034. [Google Scholar] [CrossRef]
- Jozwiakowski, M.J.; Connors, K.A. Aqueous solubility behavior of three cyclodextrins. Carbohydr. Res. 1985, 143, 51–59. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Méndez, I.; Loera-Loera, E.; Sorroza-Martínez, K.; Vonlanthen, M.; Cuétara-Guadarrama, F.; Bernad-Bernad, M.J.; Rivera, E.; Gracia-Mora, J. Synthesis of β-Cyclodextrin-Decorated Dendritic Compounds Based on EDTA Core: A New Class of PAMAM Dendrimer Analogs. Pharmaceutics 2022, 14, 2363. https://doi.org/10.3390/pharmaceutics14112363
González-Méndez I, Loera-Loera E, Sorroza-Martínez K, Vonlanthen M, Cuétara-Guadarrama F, Bernad-Bernad MJ, Rivera E, Gracia-Mora J. Synthesis of β-Cyclodextrin-Decorated Dendritic Compounds Based on EDTA Core: A New Class of PAMAM Dendrimer Analogs. Pharmaceutics. 2022; 14(11):2363. https://doi.org/10.3390/pharmaceutics14112363
Chicago/Turabian StyleGonzález-Méndez, Israel, Esteban Loera-Loera, Kendra Sorroza-Martínez, Mireille Vonlanthen, Fabián Cuétara-Guadarrama, María Josefa Bernad-Bernad, Ernesto Rivera, and Jesús Gracia-Mora. 2022. "Synthesis of β-Cyclodextrin-Decorated Dendritic Compounds Based on EDTA Core: A New Class of PAMAM Dendrimer Analogs" Pharmaceutics 14, no. 11: 2363. https://doi.org/10.3390/pharmaceutics14112363