
Augmented Oral Bioavailability and Prokinetic Activity of Levosulpiride Delivered in Nanostructured Lipid Carriers
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Selection of Lipids and Surfactants
2.3. Experimental Design and Optimization of LSP-NLCs
2.4. Preparation of LSP-NLCs
2.5. Physicochemical Characterization of LSP-NLCs
2.6. Solid State Characterization of LSP-NLCs
2.7. In Vitro Drug Release from LSP-NLCs
2.8. In Vitro Lipid Digestion Study
2.9. In Vivo Pharmacokinetics
2.9.1. Animals
2.9.2. Oral Administration and Blood Collection
2.9.3. LSP Quantification and Pharmacokinetics Parameters
2.10. In Vivo Prokinetic Studies
2.10.1. Gastric Emptying and Intestinal Transit
2.10.2. Fecal Excretion
2.11. Statistical Analysis
3. Results and Discussion
3.1. Preliminary Studies for the Selection of Ingredients
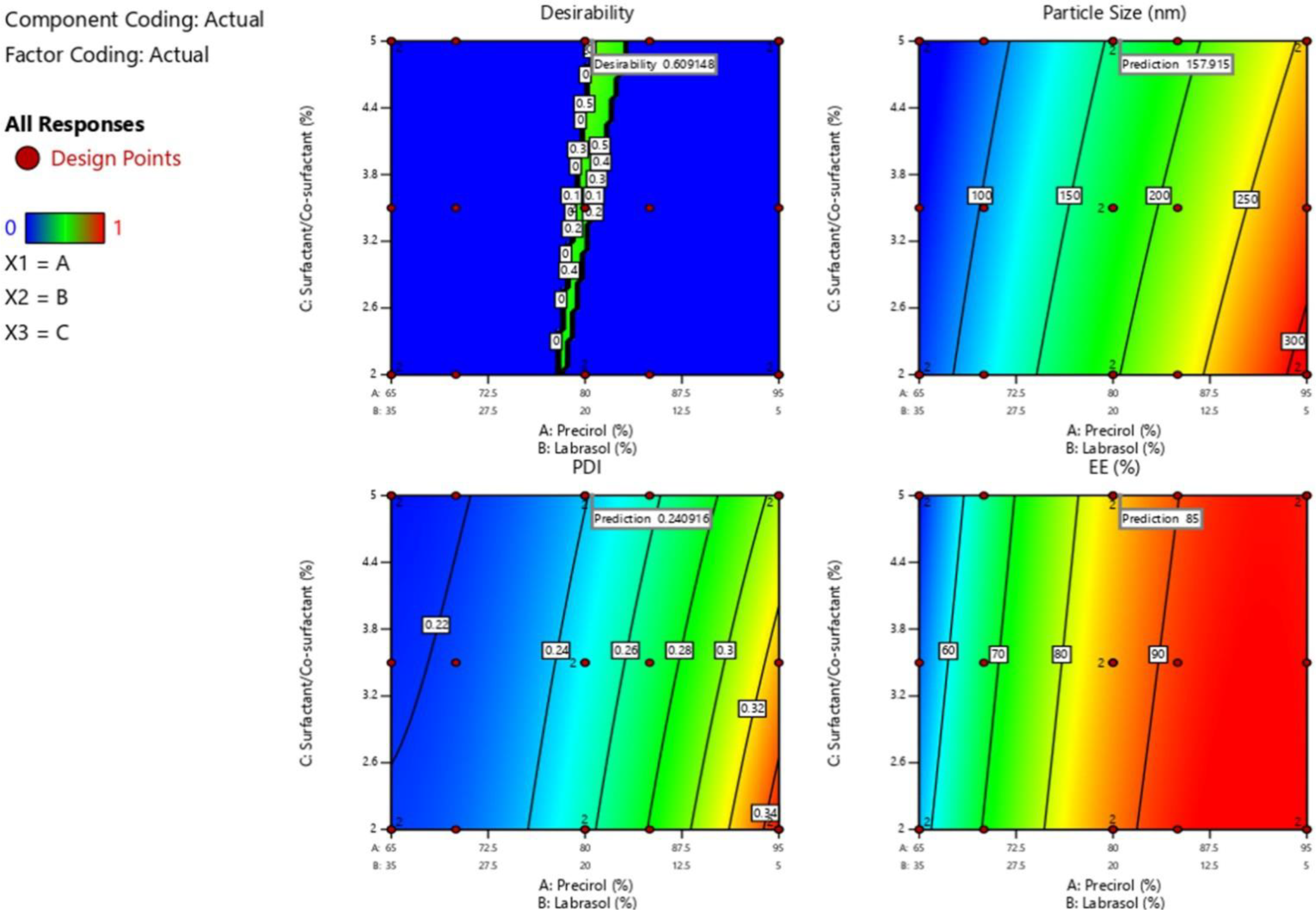
3.2. D-Optimal Combined Mixture Design
3.3. Effects of Independent Variables on Particle Size, PDI and Entrapment Efficiency
3.4. Optimization of LSP-NLCs
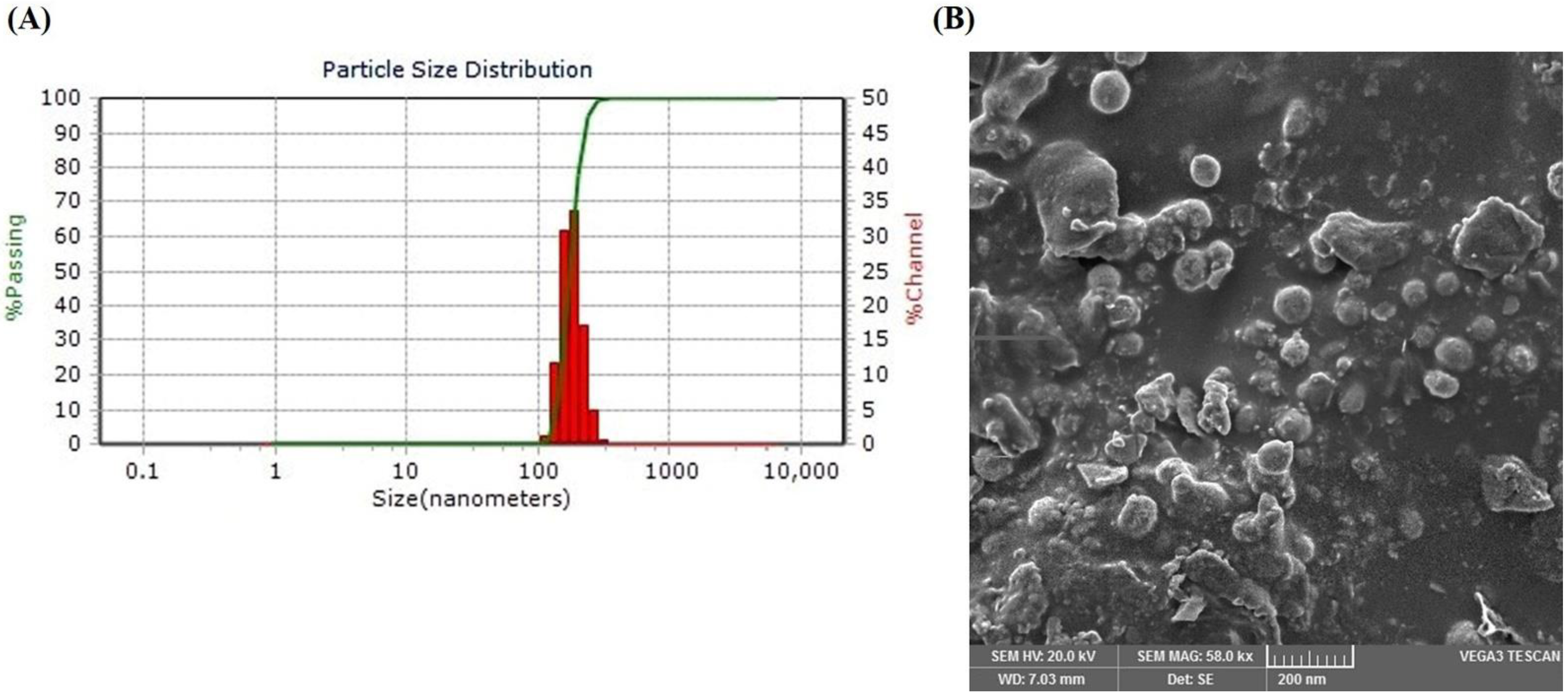
3.5. Physicochemical Properties of Optimized LSP-NLCs
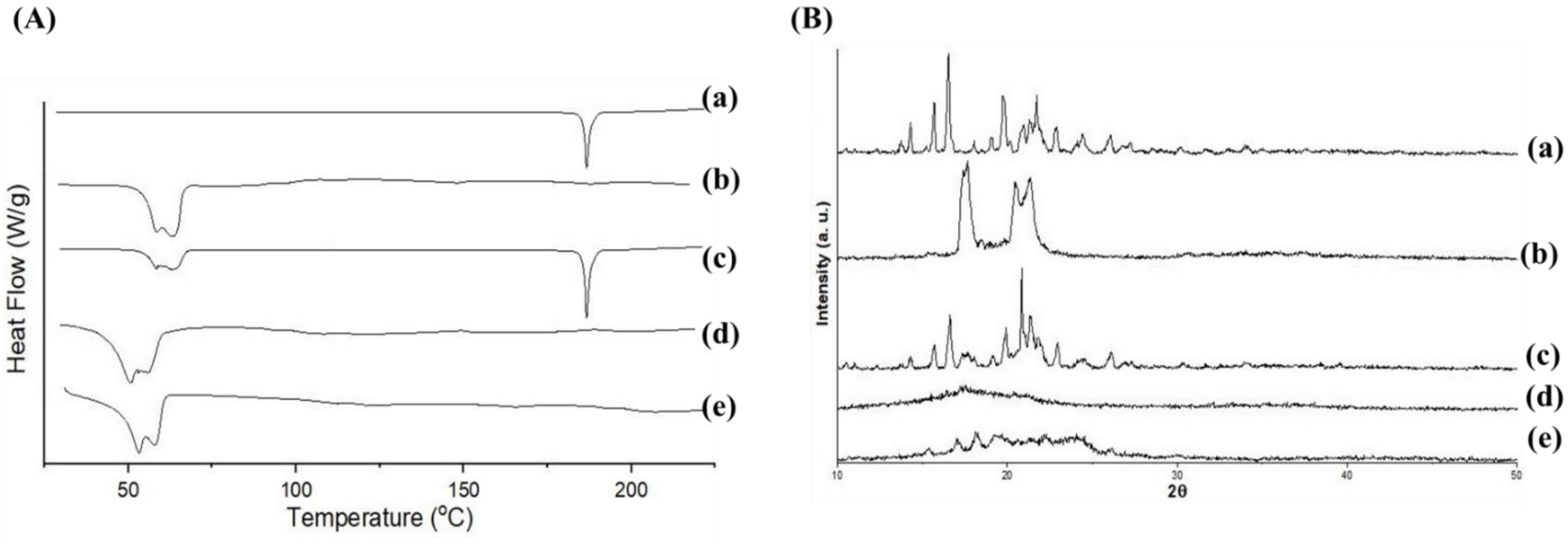
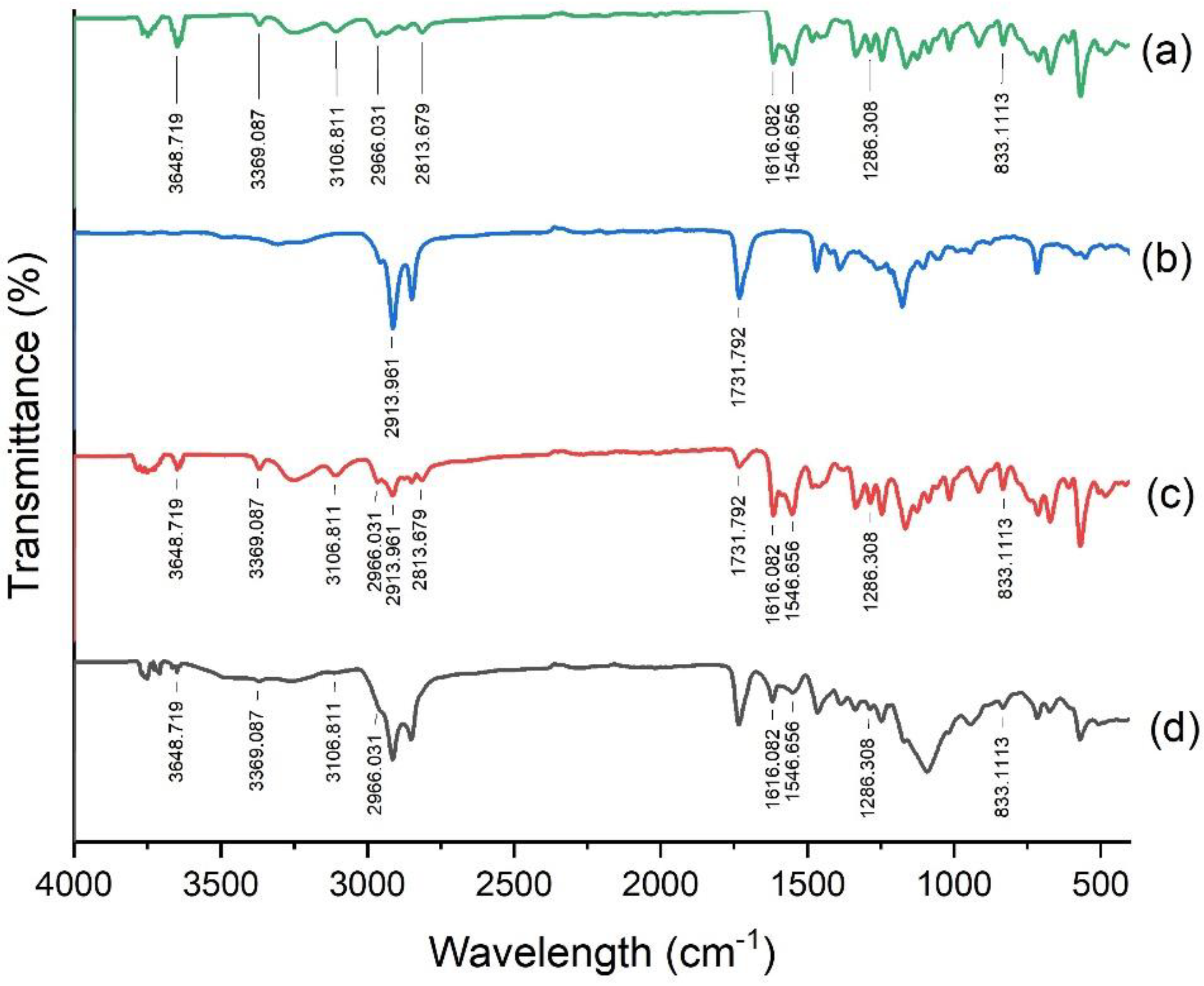
3.6. DSC, PXRD and FTIR Analysis
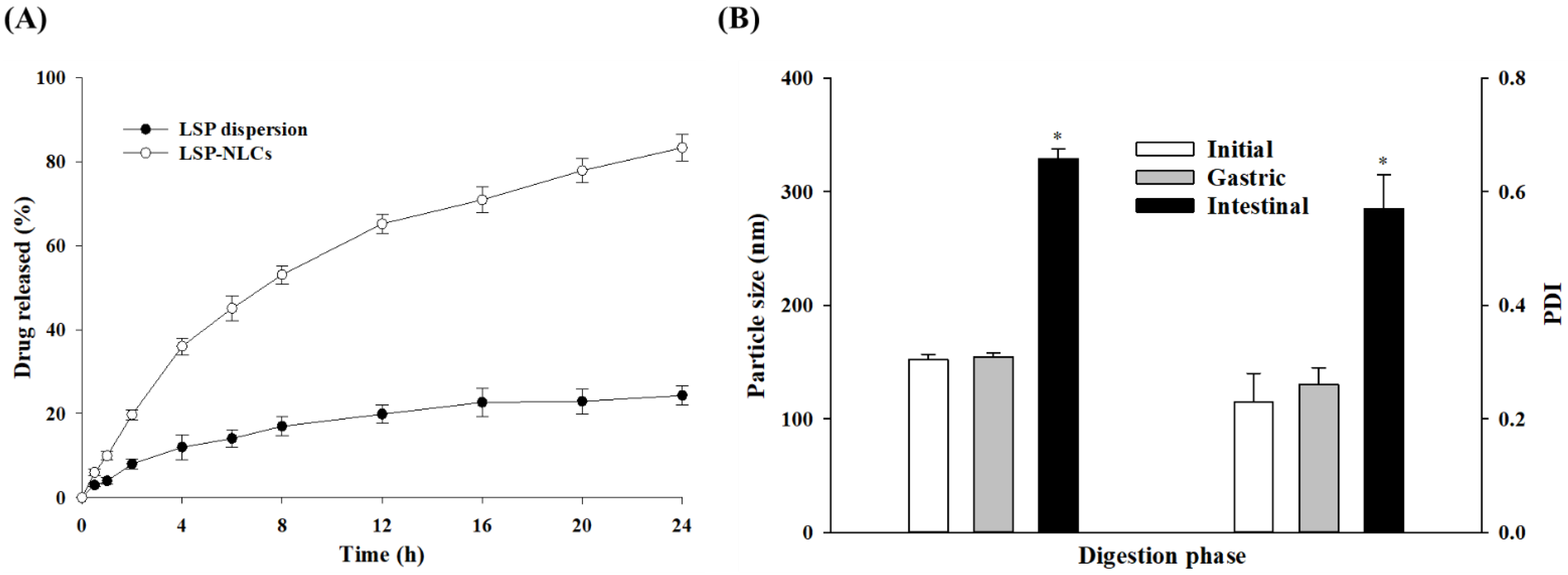
3.7. In Vitro Drug Release
3.8. Lipid Digestion in Gastric and Intestinal Fluids
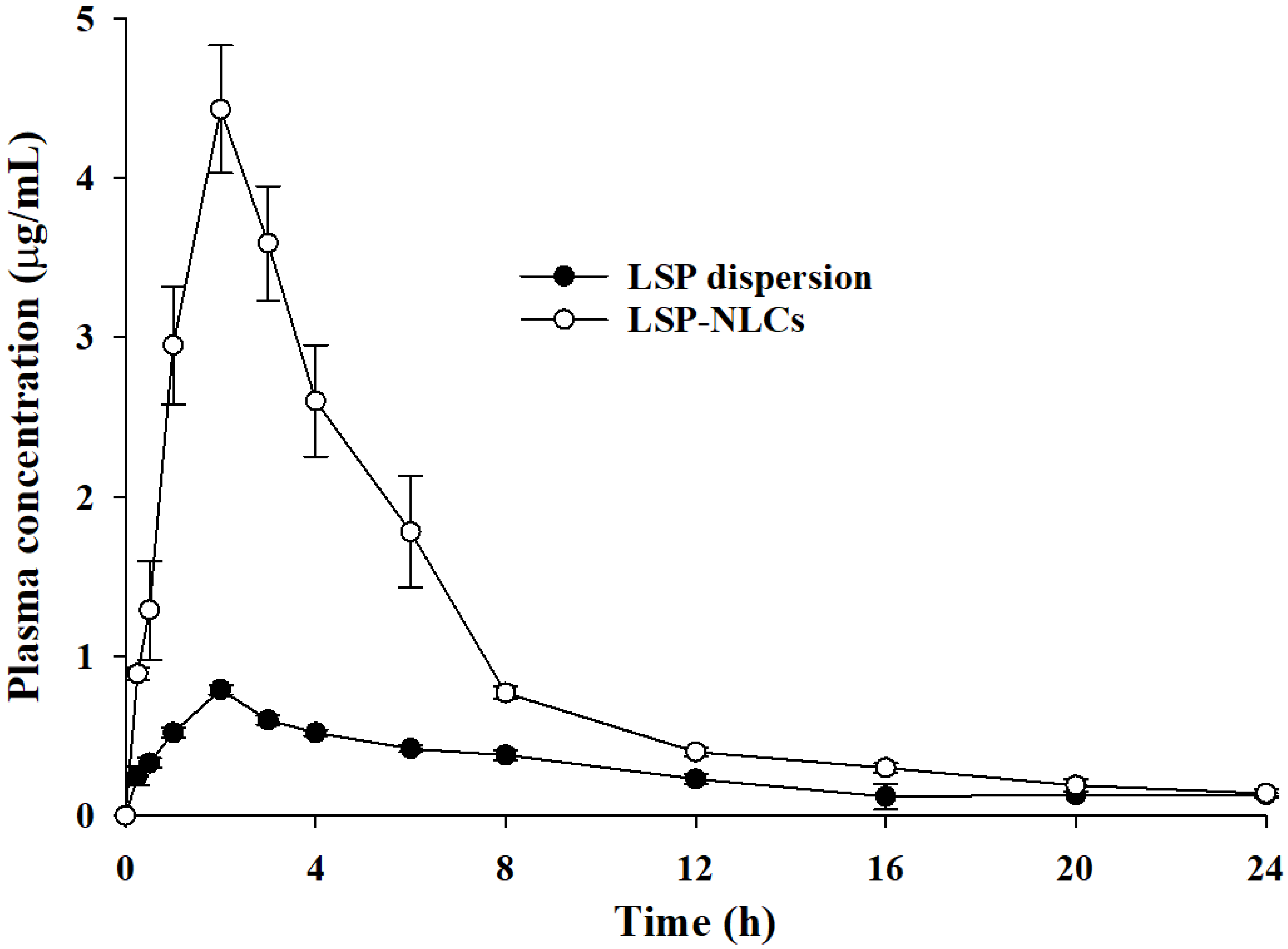
3.9. Pharmacokinetics of LSP-NLCs
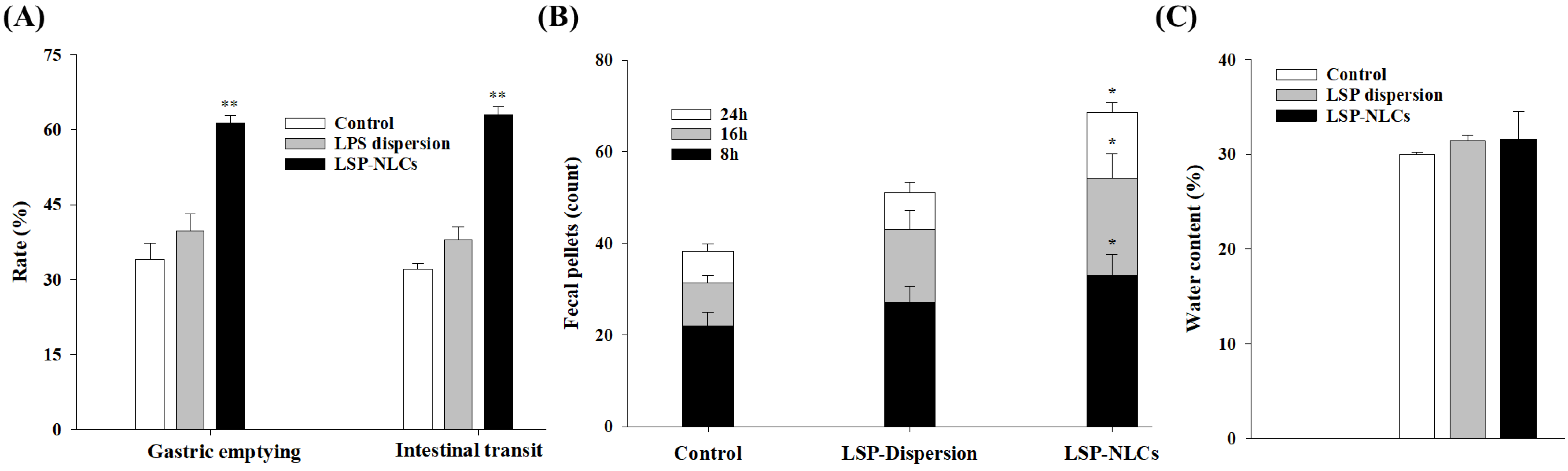
3.10. Prokinetic Activity of LSP-NLCs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Tack, J.; Talley, N.J.; Camilleri, M.; Holtmann, G.; Hu, P.; Malagelada, J.-R.; Stanghellini, V. Functional Gastroduodenal Disorders. Gastroenterol. 2006, 130, 1466–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhram, M.; Anturlikar, S.D.; Rafiq, M.; Patki, P.S.; Rangesh, P. Gastrokinetic and Gastroprotective Activity of Gasex. Int. J. Phytomedicine 2011, 3, 557–566. [Google Scholar]
- Mansi, C.; Borro, P.; Giacomini, M.; Biagini, R.; Mele, M.; Pandolfo, N.; Savarino, V. Comparative effects of levosulpiride and cisapride on gastric emptying and symptoms in patients with functional dyspepsia and gastroparesis. Aliment. Pharmacol. Ther. 2000, 14, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Serra, J. Levosulpiride in the management of functional dyspepsia and delayed gastric emptying. Gastroenterol. Hepatol. 2010, 33, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Khaleeq, N.; Din, F.-U.; Khan, A.S.; Rabia, S.; Dar, J.; Khan, G.M. Development of levosulpiride-loaded solid lipid nanoparticles and their in vitro and in vivo comparison with commercial product. J. Microencapsul. 2020, 37, 160–169. [Google Scholar] [CrossRef]
- Xu, M.; Zhou, Y.; Ni, Y.; He, X.; Li, H.; Sattar, H.; Chen, H.; Li, W. Tolerability and Pharmacokinetic Comparison of Oral, Intramuscular, and Intravenous Administration of Levosulpiride After Single and Multiple Dosing in Healthy Chinese Volunteers. Clin. Ther. 2015, 37, 2458–2467. [Google Scholar] [CrossRef]
- Kim, D.S.; Choi, J.S.; Kim, D.W.; Kim, K.S.; Seo, Y.G.; Cho, K.H.; Kim, J.O.; Yong, C.S.; Youn, Y.S.; Lim, S.-J.; et al. Comparison of solvent wetted and kneaded l-sulpiride loaded solid dispersions: Powder characterization and in vivo evaluation. Int. J. Pharm. 2016, 511, 351–358. [Google Scholar]
- Kim, D.S.; Kim, D.W.; Kim, K.S.; Choi, J.S.; Seo, Y.G.; Youn, Y.S.; Oh, K.T.; Yong, C.S.; Kim, J.O.; Jin, S.G.; et al. Development of a novel l-sulpiride-loaded quaternary microcapsule: Effect of TPGS as an absorption enhancer on physicochemical characterization and oral bioavailability. Colloids Surf. B Biointerfaces 2016, 147, 250–257. [Google Scholar] [CrossRef]
- Ibrahim, W.M.; AlOmrani, A.H.; Yassin, A.E.B. Novel sulpiride-loaded solid lipid nanoparticles with enhanced intestinal permeability. Int. J. Nanomedicine 2014, 9, 129–144. [Google Scholar]
- Poorani, G.; Uppuluri, S.; Uppuluri, K.B. Formulation, characterization, in vitro and in vivo evaluation of castor oil based self-nano emulsifying levosulpiride delivery systems. J. Microencapsul. 2016, 33, 535–543. [Google Scholar]
- Khan, N.; Shah, F.A.; Rana, I.; Ansari, M.M.; Din, F.u.; Rizvi, S.Z.H.; Aman, W.; Lee, G.-Y.; Lee, E.-S.; Kim, J.-K.; et al. Nanostructured lipid carriers-mediated brain delivery of carbamazepine for improved in vivo anticonvulsant and anxiolytic activity. Int. J. Pharm. 2020, 577, 119033. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.Z.H.; Shah, F.A.; Khan, N.; Muhammad, I.; Ali, K.H.; Ansari, M.M.; Din, F.u.; Qureshi, O.S.; Kim, K.-W.; Choe, Y.-H.; et al. Simvastatin-loaded solid lipid nanoparticles for enhanced anti-hyperlipidemic activity in hyperlipidemia animal model. Int. J. Pharm. 2019, 560, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Trevaskis, N.L.; Charman, W.N.; Porter, C.J. Lipid-based delivery systems and intestinal lymphatic drug transport: A mechanistic update. Adv. Drug Deliv. Rev. 2008, 60, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Ali, Z.; Sajjad Khan, A.; Ullah, K.; Jamshaid, H.; Zeb, A.; Imran, M.; Sarwar, S.; Choi, H.-G.; ud Din, F. Preparation, Pharmacokinetics, and Antitumor Potential of Miltefosine-Loaded Nanostructured Lipid Carriers. Int. J. Nanomedicine 2021, 16, 3255–3273. [Google Scholar] [CrossRef]
- Zeb, A.; Qureshi, O.S.; Kim, H.S.; Kim, M.S.; Kang, J.H.; Park, J.S.; Kim, J.K. High payload itraconazole-incorporated lipid nanoparticles with modulated release property for oral and parenteral administration. J. Pharm. Pharmacol. 2017, 69, 955–966. [Google Scholar] [CrossRef]
- Kasongo, W.A.; Pardeike, J.; Müller, R.H.; Walker, R.B. Selection and Characterization of Suitable Lipid Excipients for use in the Manufacture of Didanosine-Loaded Solid Lipid Nanoparticles and Nanostructured Lipid Carriers. J. Pharm. Sci. 2011, 100, 5185–5196. [Google Scholar] [CrossRef]
- Jazuli, I.; Annu; Nabi, B.; Moolakkadath, T.; Alam, T.; Baboota, S.; Ali, J. Optimization of Nanostructured Lipid Carriers of Lurasidone Hydrochloride Using Box-Behnken Design for Brain Targeting: In Vitro and In Vivo Studies. J. Pharm. Sci. 2019, 108, 3082–3090. [Google Scholar] [CrossRef]
- Cirri, M.; Maestrini, L.; Maestrelli, F.; Mennini, N.; Mura, P.; Ghelardini, C.; Di Cesare Mannelli, L. Design, characterization and in vivo evaluation of nanostructured lipid carriers (NLC) as a new drug delivery system for hydrochlorothiazide oral administration in pediatric therapy. Drug Deliv. 2018, 25, 1910–1921. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Ansari, M.M.; Arif, S.T.; Raza, A.; Choi, H.-I.; Lim, C.-W.; Noh, H.-Y.; Noh, J.-S.; Akram, S.; Nawaz, H.A.; et al. Eplerenone nanocrystals engineered by controlled crystallization for enhanced oral bioavailability. Drug Deliv. 2021, 28, 2510–2524. [Google Scholar] [CrossRef]
- Feng, J.; Huang, M.; Chai, Z.; Li, C.; Huang, W.; Cui, L.; Li, Y. The influence of oil composition on the transformation, bioaccessibility, and intestinal absorption of curcumin in nanostructured lipid carriers. Food Funct. 2020, 11, 5223–5239. [Google Scholar] [CrossRef]
- Mulè, F.; Amato, A.; Serio, R. Gastric emptying, small intestinal transit and fecal output in dystrophic (mdx) mice. J. Physiol. Sci. 2010, 60, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Doktorovová, S.; Araújo, J.; Garcia, M.L.; Rakovský, E.; Souto, E.B. Formulating fluticasone propionate in novel PEG-containing nanostructured lipid carriers (PEG-NLC). Colloids Surf. B Biointerfaces 2010, 75, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Soni, K.; Rizwanullah, M.; Kohli, K. Development and optimization of sulforaphane-loaded nanostructured lipid carriers by the Box-Behnken design for improved oral efficacy against cancer: In vitro, ex vivo and in vivo assessments. Artif. Cells Nanomed. Biotechnol. 2018, 46, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hejri, A.; Khosravi, A.; Gharanjig, K.; Hejazi, M. Optimisation of the formulation of β-carotene loaded nanostructured lipid carriers prepared by solvent diffusion method. Food Chem. 2013, 141, 117–123. [Google Scholar] [CrossRef]
- Liu, J.; Gong, T.; Wang, C.; Zhong, Z.; Zhang, Z. Solid lipid nanoparticles loaded with insulin by sodium cholate-phosphatidylcholine-based mixed micelles: Preparation and characterization. Int. J. Pharm. 2007, 340, 153–162. [Google Scholar] [CrossRef]
- Moghddam, S.M.M.; Ahad, A.; Aqil, M.; Imam, S.S.; Sultana, Y. Optimization of nanostructured lipid carriers for topical delivery of nimesulide using Box–Behnken design approach. Artif. Cells Nanomed. Biotechnol. 2017, 45, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Azhar Shekoufeh Bahari, L.; Hamishehkar, H. The impact of variables on particle size of solid lipid nanoparticles and nanostructured lipid carriers. A comparative literature review. Adv. Pharm. Bull. 2016, 6, 143–151. [Google Scholar] [CrossRef]
- Rahman, Z.; Zidan, A.S.; Khan, M.A. Non-destructive methods of characterization of risperidone solid lipid nanoparticles. Eur. J. Pharm. Biopharm. 2010, 76, 127–137. [Google Scholar] [CrossRef]
- Safwat, S.; Ishak, R.A.H.; Hathout, R.M.; Mortada, N.D. Nanostructured lipid carriers loaded with simvastatin: Effect of PEG/glycerides on characterization, stability, cellular uptake efficiency and in vitro cytotoxicity. Drug Dev. Ind. Pharm. 2017, 43, 1112–1125. [Google Scholar] [CrossRef]
- Singh, A.; Neupane, Y.R.; Mangla, B.; Kohli, K. Nanostructured Lipid Carriers for Oral Bioavailability Enhancement of Exemestane: Formulation Design, In Vitro, Ex Vivo, and In Vivo Studies. J. Pharm. Sci. 2019, 108, 3382–3395. [Google Scholar] [CrossRef]
- Estella-Hermoso de Mendoza, A.; Campanero, M.A.; Mollinedo, F.; Blanco-Prieto, M.J. Lipid nanomedicines for anticancer drug therapy. J. Biomed. Nanotechnol. 2009, 5, 323–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubab, S.; Naeem, K.; Rana, I.; Khan, N.; Afridi, M.; Ullah, I.; Shah, F.A.; Sarwar, S.; Din, F.u.; Choi, H.-I.; et al. Enhanced neuroprotective and antidepressant activity of curcumin-loaded nanostructured lipid carriers in lipopolysaccharide-induced depression and anxiety rat model. Int. J. Pharm. 2021, 603, 120670. [Google Scholar] [CrossRef] [PubMed]
- Rana, I.; Khan, N.; Ansari, M.M.; Shah, F.A.; Din, F.u.; Sarwar, S.; Imran, M.; Qureshi, O.S.; Choi, H.-I.; Lee, C.-H.; et al. Solid lipid nanoparticles-mediated enhanced antidepressant activity of duloxetine in lipopolysaccharide-induced depressive model. Colloids Surf. B Biointerfaces 2020, 194, 111209. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; ud Din, F.; Ali, Z.; Bibi, M.; Zahid, F.; Zeb, A.; Rehman, M.u.; Khan, G.M. Development, in vitro and in vivo evaluation of miltefosine loaded nanostructured lipid carriers for the treatment of Cutaneous Leishmaniasis. Int. J. Pharm. 2021, 593, 120109. [Google Scholar] [CrossRef]
- Gul, M.; Shah, F.A.; Sahar, N.-u.; Malik, I.; Din, F.u.; Khan, S.A.; Aman, W.; Choi, H.-I.; Lim, C.-W.; Noh, H.-Y.; et al. Formulation optimization, in vitro and in vivo evaluation of agomelatine-loaded nanostructured lipid carriers for augmented antidepressant effects. Colloids Surf. B Biointerfaces 2022, 216, 112537. [Google Scholar] [PubMed]
- Qureshi, O.S.; Kim, H.-S.; Zeb, A.; Choi, J.-S.; Kim, H.-S.; Kwon, J.-E.; Kim, M.-S.; Kang, J.-H.; Ryou, C.; Park, J.-S.; et al. Sustained release docetaxel-incorporated lipid nanoparticles with improved pharmacokinetics for oral and parenteral administration. J. Microencapsul. 2017, 34, 250–261. [Google Scholar] [CrossRef]
- Mura, P.; Maestrelli, F.; D’Ambrosio, M.; Luceri, C.; Cirri, M. Evaluation and Comparison of Solid Lipid Nanoparticles (SLNs) and Nanostructured Lipid Carriers (NLCs) as Vectors to Develop Hydrochlorothiazide Effective and Safe Pediatric Oral Liquid Formulations. Pharmaceutics 2021, 13, 437. [Google Scholar] [CrossRef]
- Mircioiu, C.; Voicu, V.; Anuta, V.; Tudose, A.; Celia, C.; Paolino, D.; Fresta, M.; Sandulovici, R.; Mircioiu, I. Mathematical Modeling of Release Kinetics from Supramolecular Drug Delivery Systems. Pharmaceutics 2019, 11, 140. [Google Scholar] [CrossRef] [Green Version]
- Ghate, V.M.; Lewis, S.A.; Prabhu, P.; Dubey, A.; Patel, N. Nanostructured lipid carriers for the topical delivery of tretinoin. Eur. J. Pharm. Biopharm. 2016, 108, 253–261. [Google Scholar] [CrossRef]
- Espinosa-Olivares, M.A.; Delgado-Buenrostro, N.L.; Chirino, Y.I.; Trejo-Márquez, M.A.; Pascual-Bustamante, S.; Ganem-Rondero, A. Nanostructured lipid carriers loaded with curcuminoids: Physicochemical characterization, in vitro release, ex vivo skin penetration, stability and antioxidant activity. Eur. J. Pharm. Sci. 2020, 155, 105533. [Google Scholar] [CrossRef]
- van Aken, G.A.; Bomhof, E.; Zoet, F.D.; Verbeek, M.; Oosterveld, A. Differences in in vitro gastric behaviour between homogenized milk and emulsions stabilised by Tween 80, whey protein, or whey protein and caseinate. Food Hydrocoll. 2011, 25, 781–788. [Google Scholar] [CrossRef]
- Aditya, N.P.; Shim, M.; Lee, I.; Lee, Y.; Im, M.-H.; Ko, S. Curcumin and genistein coloaded nanostructured lipid carriers: In vitro digestion and antiprostate cancer activity. J. Agric. Food Chem. 2013, 61, 1878–1883. [Google Scholar] [CrossRef] [PubMed]
- Cirri, M.; Mennini, N.; Maestrelli, F.; Mura, P.; Ghelardini, C.; Di Cesare Mannelli, L. Development and in vivo evaluation of an innovative “Hydrochlorothiazide-in Cyclodextrins-in Solid Lipid Nanoparticles” formulation with sustained release and enhanced oral bioavailability for potential hypertension treatment in pediatrics. Int. J. Pharm. 2017, 521, 73–83. [Google Scholar] [CrossRef]
- Noack, A.; Oidtmann, J.; Kutza, J.; Mäder, K. In vitro digestion of curcuminoid-loaded lipid nanoparticles. J. Nanopart. Res. 2012, 14, 1113. [Google Scholar] [CrossRef]
- Mayer, S.; Weiss, J.; McClements, D.J. Behavior of vitamin E acetate delivery systems under simulated gastrointestinal conditions: Lipid digestion and bioaccessibility of low-energy nanoemulsions. J. Colloid Interface Sci. 2013, 404, 215–222. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Pouton, C.W.; Cuine, J.F.; Charman, W.N. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv. Drug Deliv. Rev. 2008, 60, 673–691. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef]
- Walsh, K.R.; Zhang, Y.C.; Vodovotz, Y.; Schwartz, S.J.; Failla, M.L. Stability and Bioaccessibility of Isoflavones from Soy Bread during In Vitro Digestion. J. Agric. Food Chem. 2003, 51, 4603–4609. [Google Scholar] [CrossRef]
- Din, F.u.; Kim, D.W.; Choi, J.Y.; Thapa, R.K.; Mustapha, O.; Kim, D.S.; Oh, Y.-K.; Ku, S.K.; Youn, Y.S.; Oh, K.T.; et al. Irinotecan-loaded double-reversible thermogel with improved antitumor efficacy without initial burst effect and toxicity for intramuscular administration. Acta Biomater. 2017, 54, 239–248. [Google Scholar] [CrossRef]
- Zhuang, C.-Y.; Li, N.; Wang, M.; Zhang, X.-N.; Pan, W.-S.; Peng, J.-J.; Pan, Y.-S.; Tang, X. Preparation and characterization of vinpocetine loaded nanostructured lipid carriers (NLC) for improved oral bioavailability. Int. J. Pharm. 2010, 394, 179–185. [Google Scholar] [CrossRef]
- Dahan, A.; Hoffman, A. The effect of different lipid based formulations on the oral absorption of lipophilic drugs: The ability of in vitro lipolysis and consecutive ex vivo intestinal permeability data to predict in vivo bioavailability in rats. Eur. J. Pharm. Biopharm. 2007, 67, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Kalepu, S.; Manthina, M.; Padavala, V. Oral lipid-based drug delivery systems—An overview. Acta Pharm. Sin. B 2013, 3, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Merwid-Ląd, A.; Trocha, M.; Ksiądzyna, D.; Sozański, T.; Szeląg, A. Animal models for the gastrointestinal motility evaluation. Gastroenterol. Pol. 2009, 16, 201–206. [Google Scholar]
- Suchitra, A.D.; Dkhar, S.A.; Shewade, D.G.; Shashindran, C.H. Relative efficacy of some prokinetic drugs in morphine-induced gastrointestinal transit delay in mice. World J. Gastroenterol. 2003, 9, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.S.; Son, M. DA-9701: A New Multi-Acting Drug for the Treatment of Functional Dyspepsia. Biomol. Ther. 2013, 21, 181–189. [Google Scholar] [CrossRef]
- Badary, O.A.; Awad, A.S.; Sherief, M.A.; Hamada, F.M. In vitro and in vivo effects of ferulic acid on gastrointestinal motility: Inhibition of cisplatin-induced delay in gastric emptying in rats. World J. Gastroenterol. 2006, 12, 5363–5367. [Google Scholar] [CrossRef]
- Singh, H.; Bala, R.; Kaur, K. Efficacy and tolerability of levosulipride, domperidone and metoclopramide in patients with non-ulcer functional dyspepsia: A comparative analysis. J. Clin. Diagn. Res. 2015, 9, FC09–FC12. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variables | Lower Level | Upper Level | |
|---|---|---|---|
| Factor 1 (A+B) | A = solid lipid | 65% | 95% |
| B = liquid lipid | 5% | 35% | |
| Factor 2 | C = surfactant | 2% | 5% |
| Dependent variables | Constraints | ||
| Response 1 | Particle size (nm) | Minimum | |
| Response 2 | PDI | In range (0.2–0.25) | |
| Response 3 | Entrapment efficiency (%) | In range (85–95%) | |
| Factor 1 (A + B) | Factor 2 | Response 1 | Response 2 | Response 3 | ||
|---|---|---|---|---|---|---|
| Run | A: Precirol (%) | B: Labrasol (%) | C: Tween 80/Span 80 (%) | Particle Size (nm) | PDI | Entrapment Efficiency (%) |
| 1 | 65 | 35 | 2 | 74.2 ± 5.3 | 0.219 ± 0.011 | 57.0 ± 6.4 |
| 2 | 65 | 35 | 2 | 75.0 ± 3.7 | 0.220 ± 0.027 | 60.0 ± 4.9 |
| 3 | 65 | 35 | 5 | 65.4 ± 7.7 | 0.213 ± 0.020 | 49.0 ± 7.7 |
| 4 | 65 | 35 | 5 | 66.6 ± 8.3 | 0.215 ± 0.023 | 53.0 ± 5.6 |
| 5 | 65 | 35 | 3.5 | 67.4 ± 6.2 | 0.216 ± 0.032 | 54.0 ± 4.1 |
| 6 | 70 | 30 | 2 | 106.3 ± 4.6 | 0.234 ± 0.021 | 67.2 ± 7.5 |
| 7 | 70 | 30 | 5 | 78.1 ± 5.2 | 0.224 ± 0.024 | 61.0 ± 3.0 |
| 8 | 70 | 30 | 3.5 | 103.2 ± 4.1 | 0.228 ± 0.027 | 63.5 ± 5.2 |
| 9 | 80 | 20 | 5 | 106.7 ± 5.3 | 0.235 ± 0.031 | 84.5 ± 6.9 |
| 10 | 80 | 20 | 2 | 188.7 ± 6.8 | 0.249 ± 0.019 | 89.3 ± 5.2 |
| 11 | 80 | 20 | 3.5 | 165.3 ± 8.9 | 0.240 ± 0.029 | 87.9 ± 6.4 |
| 12 | 80 | 20 | 3.5 | 186.1 ± 9.3 | 0.244 ± 0.028 | 88.6 ± 3.7 |
| 13 | 80 | 20 | 5 | 123.4 ± 4.5 | 0.239 ± 0.011 | 85.0 ± 7.2 |
| 14 | 80 | 20 | 2 | 231.6 ± 8.1 | 0.253 ± 0.029 | 89.6 ± 8.8 |
| 15 | 85 | 15 | 3.5 | 234.6 ± 6.3 | 0.283 ± 0.014 | 90.2 ± 2.9 |
| 16 | 85 | 15 | 2 | 236.1 ± 9.4 | 0.286 ± 0.022 | 90.5 ± 5.9 |
| 17 | 85 | 15 | 5 | 232.5 ± 7.2 | 0.256 ± 0.016 | 90.0 ± 3.8 |
| 18 | 95 | 5 | 2 | 298.4 ± 6.1 | 0.342 ± 0.023 | 93.2 ± 2.6 |
| 19 | 95 | 5 | 5 | 247.0 ± 5.4 | 0.292 ± 0.019 | 92.1 ± 3.1 |
| 20 | 95 | 5 | 2 | 302.1 ± 9.3 | 0.351 ± 0.020 | 93.5 ± 2.9 |
| 21 | 95 | 5 | 3.5 | 387.2 ± 7.6 | 0.336 ± 0.016 | 93.0 ± 2.5 |
| 22 | 95 | 5 | 5 | 265.2 ± 8.3 | 0.314 ± 0.023 | 92.3 ± 3.4 |
| Response | F-Value | Model p-Value | Lack of Fit | Lack of Fit p-Value | R2 | Adjusted R2 | Predicted Mean Value | Obtained Value | Error (%) |
|---|---|---|---|---|---|---|---|---|---|
| Particle size (nm) | 103.83 | <0.0001 | 3.03 | 0.0756 | 0.9454 | 0.9363 | 157.9 | 152.0 ± 4.7 | 3.7 |
| PDI | 139.21 | <0.0001 | 1.50 | 0.3039 | 0.9775 | 0.9705 | 0.240 | 0.230 ± 0.05 | 4.2 |
| Entrapment efficiency (%) | 240.21 | <0.0001 | 3.58 | 0.0533 | 0.9869 | 0.9827 | 85.0 | 88.0 ± 2.4 | 3.5 |
| Kinetic Model | R2 | n |
|---|---|---|
| Zero order | 0.7686 | - |
| First order | 0.9809 | - |
| Higuchi | 0.9816 | - |
| Korsmeyer–Peppas | 0.9834 | 0.533 |
| Hixson–Crowell | 0.9464 | - |
| Parameters | LSP Dispersion | LSP-NLCs |
|---|---|---|
| AUC0→t (μg∙h/mL) | 7.14 ± 0.09 | 24.44 ± 2.52 ** |
| AUC0→∞ (μg∙h/mL) | 8.55 ± 0.07 | 25.98 ± 3.03 ** |
| Cmax (µg/mL) | 1.01 ± 0.02 | 4.43 ± 0.50 ** |
| Tmax (h) | 1.5 ± 0.00 | 2.00 ± 0.00 |
| t1/2 (h) | 7.97 ± 1.16 | 7.42 ± 1.09 |
| Kel | 0.09 ± 0.01 | 0.09 ± 0.01 |
| Frel (%) | --- | 339.18 ± 26.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arif, S.T.; Zaman, S.u.; Khan, M.A.; Tabish, T.A.; Sohail, M.F.; Arshad, R.; Kim, J.-K.; Zeb, A. Augmented Oral Bioavailability and Prokinetic Activity of Levosulpiride Delivered in Nanostructured Lipid Carriers. Pharmaceutics 2022, 14, 2347. https://doi.org/10.3390/pharmaceutics14112347
Arif ST, Zaman Su, Khan MA, Tabish TA, Sohail MF, Arshad R, Kim J-K, Zeb A. Augmented Oral Bioavailability and Prokinetic Activity of Levosulpiride Delivered in Nanostructured Lipid Carriers. Pharmaceutics. 2022; 14(11):2347. https://doi.org/10.3390/pharmaceutics14112347
Chicago/Turabian StyleArif, Sadia Tabassam, Shahiq uz Zaman, Muhammad Ayub Khan, Tanveer A. Tabish, Muhammad Farhan Sohail, Rabia Arshad, Jin-Ki Kim, and Alam Zeb. 2022. "Augmented Oral Bioavailability and Prokinetic Activity of Levosulpiride Delivered in Nanostructured Lipid Carriers" Pharmaceutics 14, no. 11: 2347. https://doi.org/10.3390/pharmaceutics14112347