
Optimization of a Novel Mandelamide-Derived Pyrrolopyrimidine Series of PERK Inhibitors
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Biochemical Assays
2.2. PERK Crystallization and Structure Determination
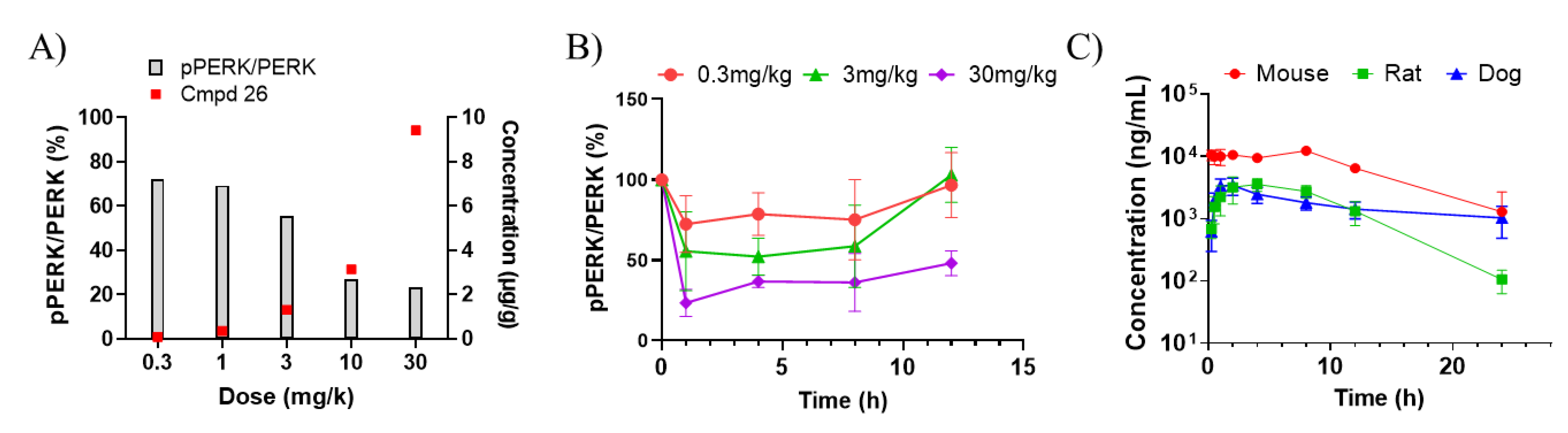
2.3. Pharmacokinetic and Pharmacodynamic Analysis in Plasma and Pancreas
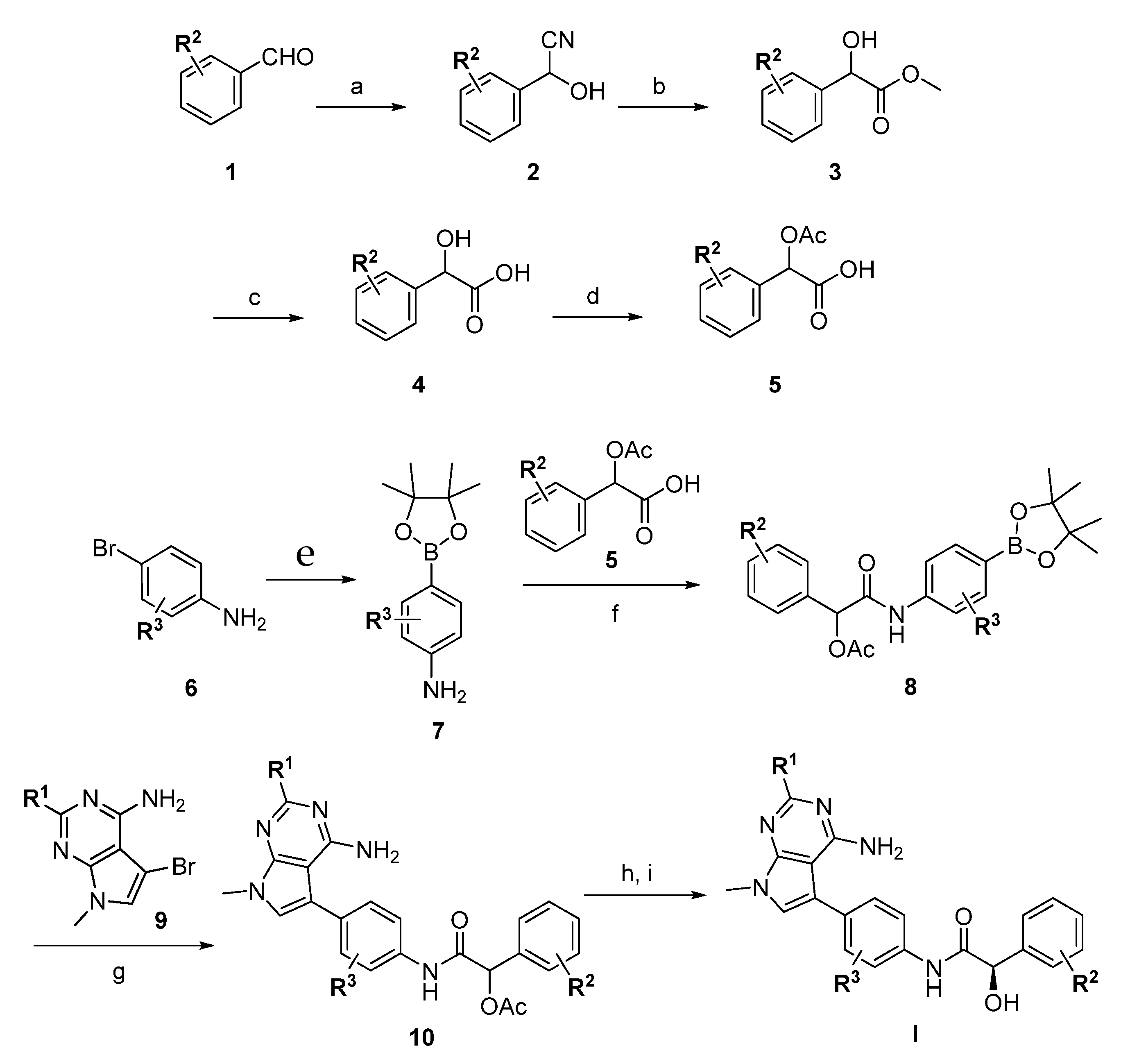
2.4. Biochemical and Cell-Based Characterization of Pyrrolopyrimidine Analogs
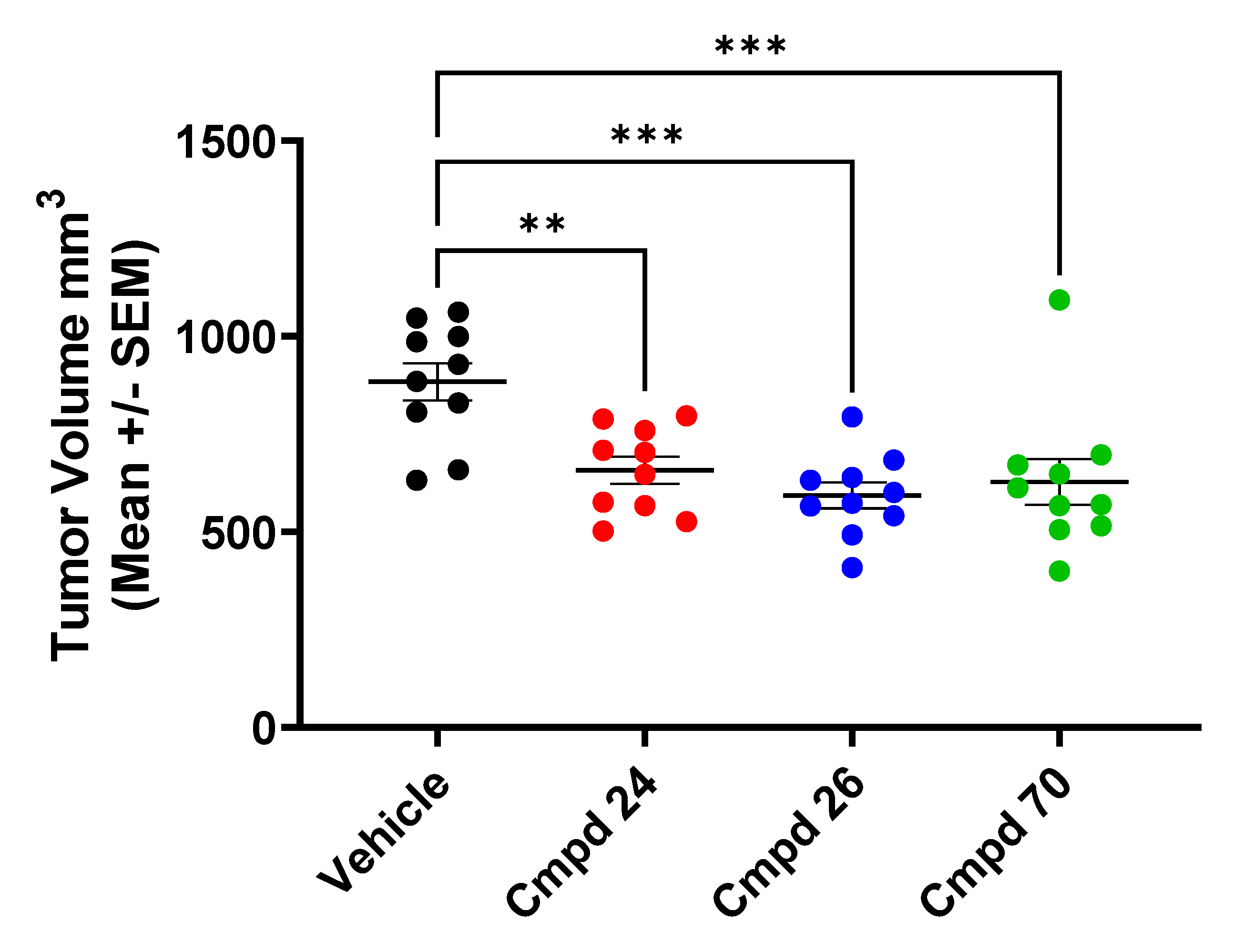
2.5. In Vivo Tumor Xenograft Studies
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.J.; Hendershot, L.M. UPR activation alters chemosensitivity of tumor cells. Cancer Biol. Ther. 2006, 5, 736–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Hendershot, L.M. The role of the unfolded protein response in tumour development: Friend or foe? Nat. Rev. Cancer 2004, 4, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Avril, T.; Vauléon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef] [PubMed]
- Calvo, V.; Surguladze, D.; Li, A.-H.; Surman, M.D.; Malibhatla, S.; Bandaru, M.; Jonnalagadda, S.K.; Adarasandi, R.; Velmala, M.; Singireddi, D.R.P.; et al. Discovery of 2-amino-3-amido-5-aryl-pyridines as highly potent, orally bioavailable, and efficacious PERK kinase inhibitors. Bioorg. Med. Chem. Lett. 2021, 43, 128058. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.-Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a Novel PERK Kinase Inhibitor with Antitumor and Antiangiogenic Activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakes, S.A.; Papa, F.R. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.B.; Landa-Galván, H.V.; Alonso, L.C. Living Dangerously: Protective and Harmful ER Stress Responses in Pancreatic β-Cells. Diabetes 2021, 70, 2431–2443. [Google Scholar] [CrossRef] [PubMed]
- Brozzi, F.; Eizirik, D.L. ER stress and the decline and fall of pancreatic beta cells in type 1 diabetes. Upsala J. Med. Sci. 2016, 121, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marré, M.L.; James, E.A.; Piganelli, J.D. β cell ER stress and the implications for immunogenicity in type 1 diabetes. Front. Cell Dev. Biol. 2015, 3, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marhfour, I.; Lopez, X.M.; Lefkaditis, D.; Salmon, I.; Allagnat, F.; Richardson, S.J.; Morgan, N.G.; Eizirik, D.L. Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 2012, 55, 2417–2420. [Google Scholar] [CrossRef] [PubMed]
- Tersey, S.A.; Nishiki, Y.; Templin, A.T.; Cabrera, S.M.; Stull, N.D.; Colvin, S.C.; Evans-Molina, C.; Rickus, J.L.; Maier, B.; Mirmira, R.G. Islet β-Cell Endoplasmic Reticulum Stress Precedes the Onset of Type 1 Diabetes in the Nonobese Diabetic Mouse Model. Diabetes 2012, 61, 818–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; McGrath, B.; Li, S.; Frank, A.; Zambito, F.; Reinert, J.; Gannon, M.; Ma, K.; McNaughton, K.; Cavener, D.R. The PERK Eukaryotic Initiation Factor 2α Kinase Is Required for the Development of the Skeletal System, Postnatal Growth, and the Function and Viability of the Pancreas. Mol. Cell. Biol. 2002, 22, 3864–3874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes Mellitus and Exocrine Pancreatic Dysfunction in Perk−/− Mice Reveals a Role for Translational Control in Secretory Cell Survival. Mol. Cell 2001, 7, 1153–1163. [Google Scholar] [CrossRef]
- Smith, A.L.; Andrews, K.L.; Beckmann, H.; Bellon, S.F.; Beltran, P.J.; Booker, S.; Chen, H.; Chung, Y.-A.; D’Angelo, N.D.; Dao, J.; et al. Discovery of 1H-Pyrazol-3(2H)-ones as Potent and Selective Inhibitors of Protein Kinase R-like Endoplasmic Reticulum Kinase (PERK). J. Med. Chem. 2015, 58, 1426–1441. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Aguilera, C.C.; Valero, T.; Lorente-Macías, Á; Baillache, D.J.; Croke, S.; Unciti-Broceta, A. Small Molecule Kinase Inhibitor Drugs (1995–2021): Medical Indication, Pharmacology, and Synthesis. J. Med. Chem. 2021, 65, 1047–1131. [Google Scholar] [CrossRef]
- Xing, L.; Klug-Mcleod, J.; Rai, B.; Lunney, E.A. Kinase hinge binding scaffolds and their hydrogen bond patterns. Bioorg. Med. Chem. 2015, 23, 6520–6527. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.; Waterman, D.G.; Parkhurst, J.M.; Brewster, A.S.; Gildea, R.J.; Gerstel, M.; Fuentes-Montero, L.; Vollmar, M.; Michels-Clark, T.; Young, I.D.; et al. DIALS: Implementation and evaluation of a new integration package. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74 Pt 2, 85–97. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69 Pt 7, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40 Pt 4, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 Pt 4, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67 Pt 4, 355–367. [Google Scholar] [CrossRef] [Green Version]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Noble, M.E.M. Presenting your structures: TheCCP4mgmolecular-graphics software. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67 Pt 4, 386–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, M.A.; Biggs, W.H., III; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule–kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef] [PubMed]








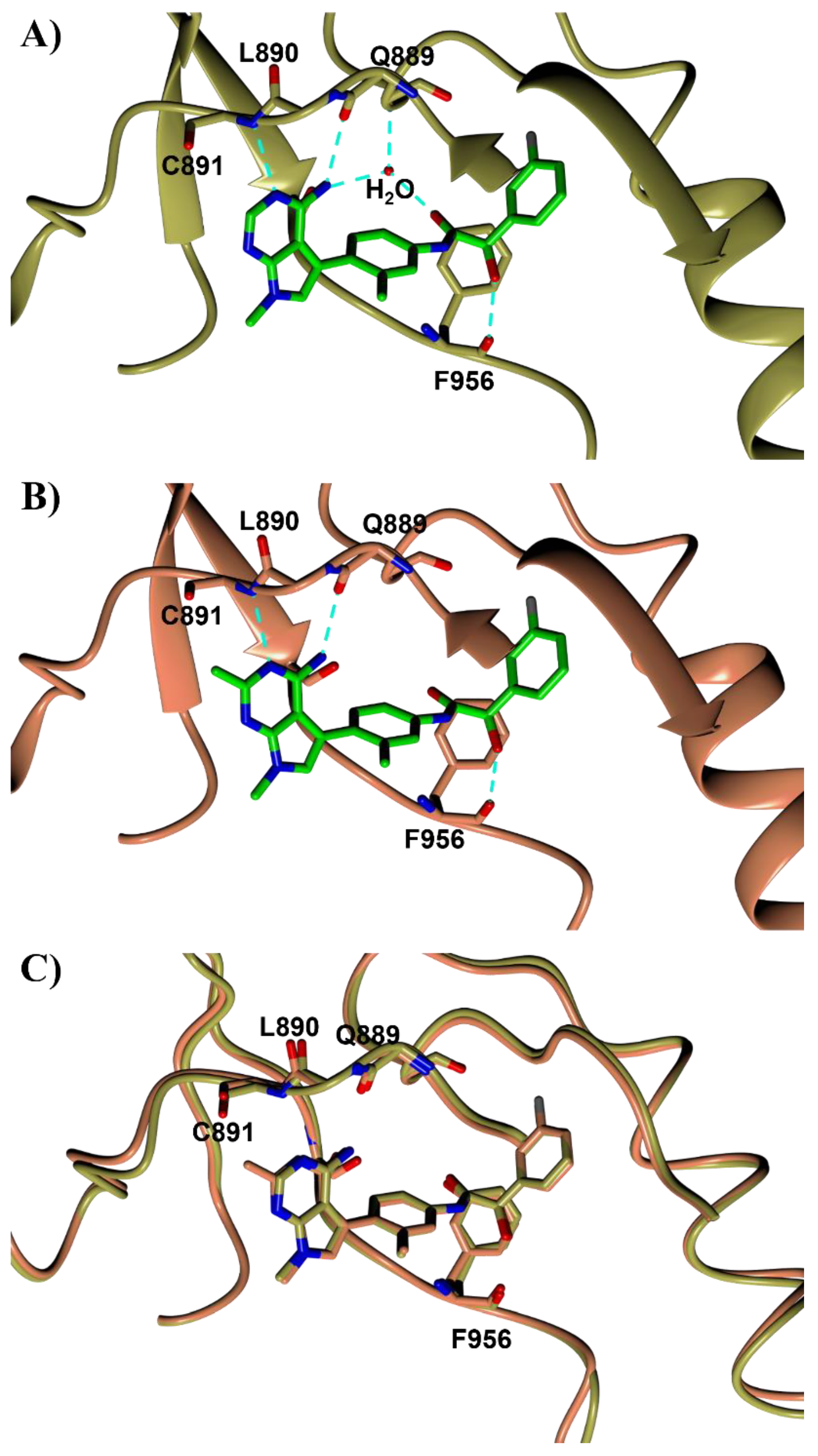{kind=link}
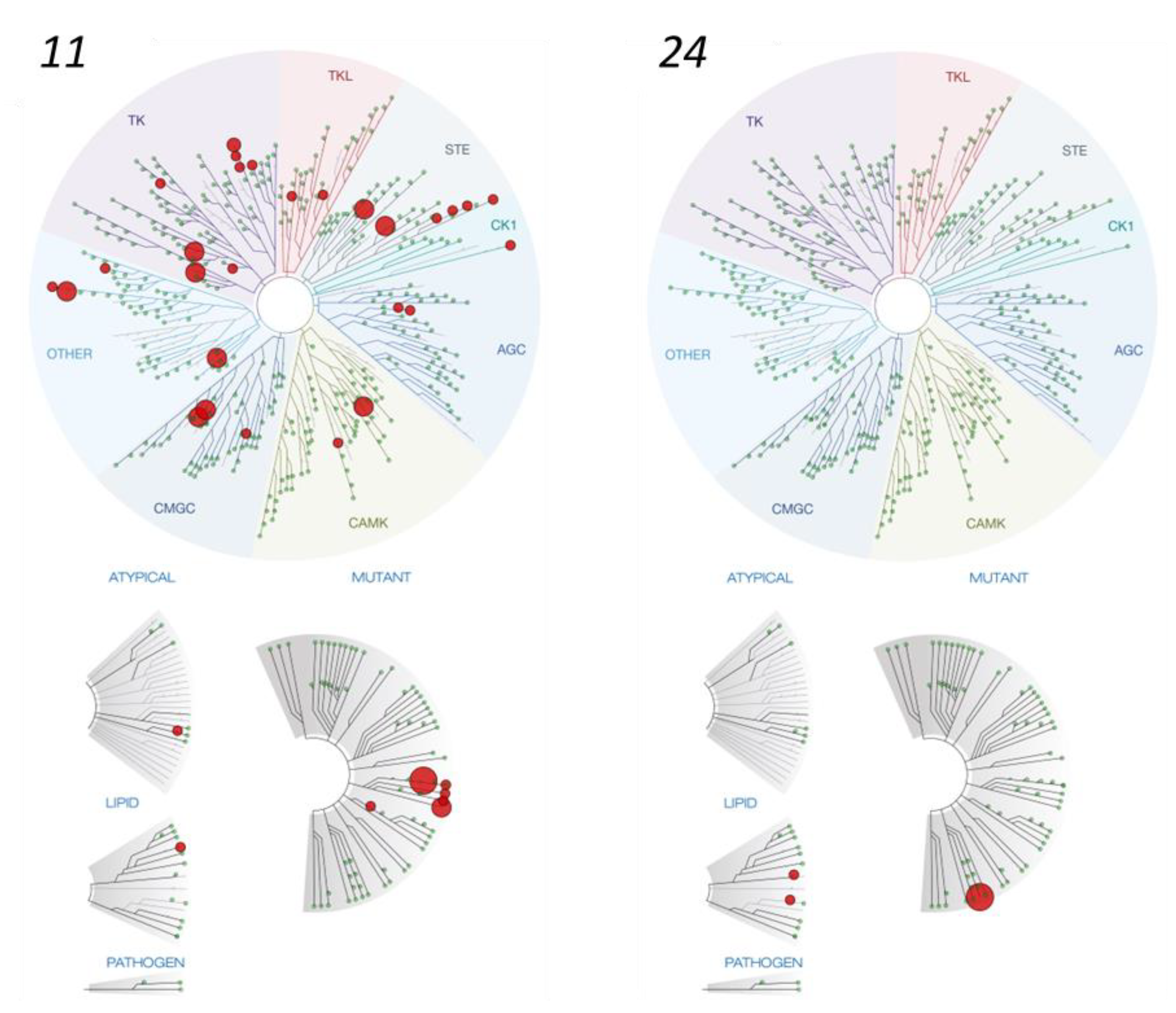{kind=link}
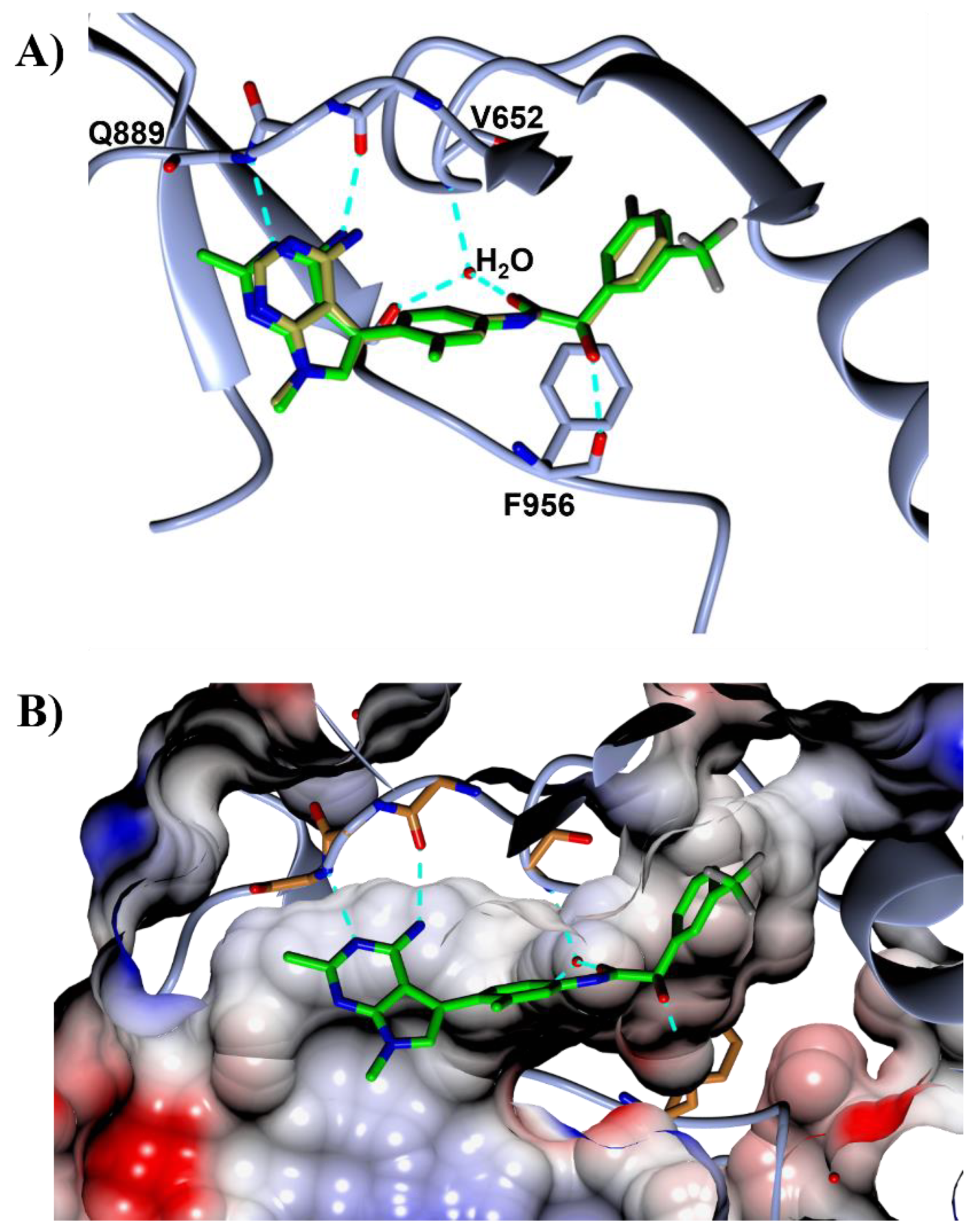{kind=link}
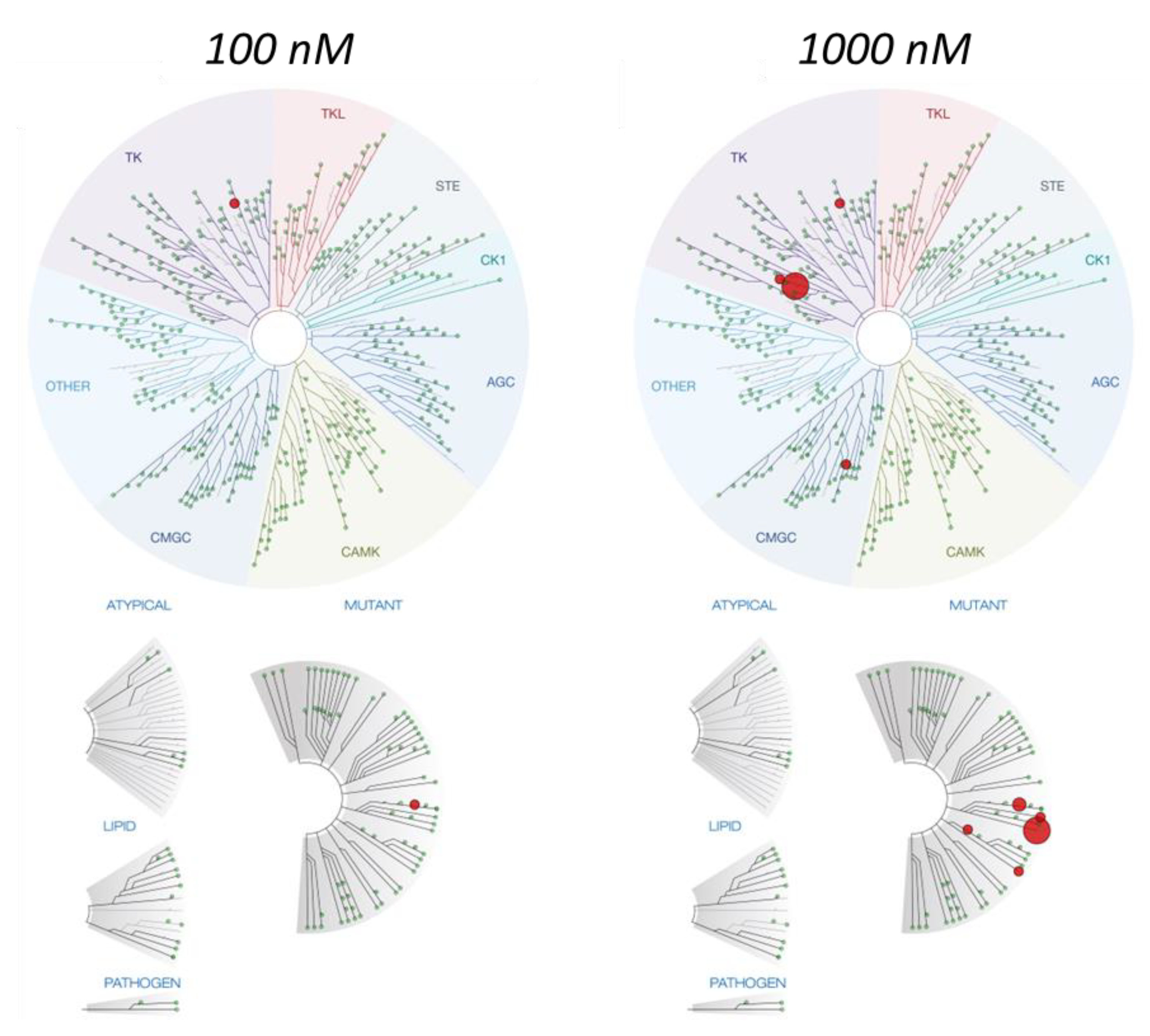{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Carna Cat# | Assay Buffer |
|---|---|---|
| GCN2 | 05-153 | GCN2 (1 nM); DTT (2 mM); ATP (150 µM); GFP-eIF2a (100 nM; Invitrogen Cat# PV4809) |
| HRI | 05-154 | HRI (0.12 nM); DTT (2 mM); ATP (30 µM); GFP-eIF2a (100 nM; Invitrogen Cat# PV4809) |
| PERK | 05-155 | PERK (0.09 nM); DTT (2 mM); ATP (8 µM); GFP-eIF2a (100 nM; Invitrogen Cat# PV4809) |
| PKR | 05-156 | PKR (0.11 nM); DTT (2 mM); ATP (6 µM); GFP-eIF2a (100 nM; Invitrogen Cat# PV4809) |
 | PERK | p-eIF2α | |||
|---|---|---|---|---|---|
| Cmpd | R1 | L | Ar | IC50 (µM) | IC50 (µM) |
| 11 | H |  |  | 0.0022 | 0.0084 |
| 12 | H |  |  | 0.0078 | 0.057 |
| 13 | H |  |  | 0.0018 | 0.023 |
| 14 | H |  |  | 0.0052 | 0.041 |
| 15 | H |  |  | 0.126 | 1.93 |
| 16 | H | --- |  | 0.168 | 3.96 |
| 17 | H |  |  | 0.3 | 0.532 |
| 18 | H |  |  | 3.98 | >10.0 |
| 19 | H |  |  | 0.021 | 0.211 |
| 20 | H |  |  | 0.108 | 1.75 |
| 21 | H |  |  | 0.014 | 0.174 |
| 22 | H |  |  | 0.056 | 0.184 |
| 23 | H |  |  | 0.033 | 0.581 |
| 24 | Me |  |  | 0.02 | 0.051 |
| 25 | Me |  |  | 0.102 | 0.303 |
| 26 | Me |  |  | 0.0028 | 0.015 |
| 27 | Me |  |  | 0.343 | 2.05 |
| 28 | Me |  |  | 0.0018 | 0.016 |
| 29 | Me |  |  | 0.107 | 0.713 |
| 30 | Me |  |  | 0.014 | 0.083 |
| 31 | Me |  |  | 0.026 | 0.122 |
| 32 | Me |  |  | 0.003 | 0.0078 |
| 33 | Me |  |  | 0.003 | 0.014 |
| 34 | Me |  |  | 0.0059 | 0.021 |
| 35 | Me |  |  | 0.0059 | 0.017 |
| 36 | Me |  |  | 0.008 | 0.325 |
| 37 | Me |  |  | >1 | >10 |
| 38 | Me |  |  | 0.0019 | 0.014 |
| 39 | Me |  |  | 0.0026 | 0.011 |
| 40 | Me |  |  | 0.0029 | 0.0085 |
| 41 | Me |  |  | 0.0014 | 0.01 |
| 42 | Me |  |  | 0.0042 | 0.032 |
| 43 | Me |  |  | 0.052 | 0.562 |
| 44 | Me |  |  | 0.017 | 0.129 |
| 45 | Me |  |  | 0.019 | 0.165 |
| Cmpd No. |  | PERK IC50 (µM) | p-eIF2α IC50 (µM) | |
|---|---|---|---|---|
| Ar | R2 | |||
| 24 |  | 3-F | 0.020 | 0.051 |
| 46 |  | 3-F | 0.0022 | 0.041 |
| 47 |  | 3-CF3 | 0.0051 | 0.011 |
| 48 |  | 3-Cl | 0.0010 | 0.010 |
| 49 |  | 3-Me | 0.0011 | 0.017 |
| 50 |  | 3-Et | 0.0009 | 0.025 |
| 51 |  | 3-F, 5-CF3 | 0.0027 | 0.062 |
| 52 |  | 3-F, 5-Me | 0.0011 | 0.011 |
| 53 |  | 3-F | 0.017 | 0.056 |
| 54 |  | 3-CF3 | 0.0027 | 0.021 |
| 55 |  | 3-Cl | 0.0016 | 0.017 |
| 56 |  | 3-Me | 0.0041 | 0.018 |
| 57 |  | 3-Et | 0.059 | 0.014 |
| 58 |  | 3-F | 0.0085 | 0.204 |
| 59 |  | 3-F | 0.0040 | 0.041 |
| 60 |  | 3-F | 0.236 | 1.85 |
| 61 |  | 3-F | 0.023 | 0.435 |
| 62 |  | 3-F | 0.0020 | 0.126 |
| 63 |  | 3-CF3 | 0.0028 | 0.013 |
| 64 |  | 3-Br | 0.0015 | 0.014 |
| 65 |  | 3-F | 0.0019 | 0.030 |
| 66 |  | 3-F | 0.011 | 0.042 |
| 67~ |  | 3-F | 0.022 | 0.078 |
| 68~ |  | 3-F | 0.013 | 0.082 |
| 69 |  | 3-F | 0.0010 | 0.026 |
| 70 |  | 3-F | 0.0015 | 0.024 |
| 71~ |  | 3-F | 0.0010 | 0.034 |
| 72 |  | 3-F | 0.017 | 0.105 |
| 73 |  | 3-CF3 | 0.013 | 0.051 |
| 74 |  | 3-F | 0.0083 | 0.146 |
| 75 |  | 3-CF3 | 0.0068 | 0.095 |
| 76 |  | 3-F | 0.647 | >10 |
| Cmpd No. | Kinetic Aqueous Solubility (µM) | Caco-2 | Plasma Protein Binding | Hepatocyte Clearance (t1/2 min) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Papp (A→B) (10−6 cm/s) | Efflux Ratio | Human | Rat | Mouse | Dog | Human | Rat | Mouse | Dog | ||
| 26 | 19 | 21 | 2.2 | 99.8 ± 0.0 | 99.8 ± 0.0 | 99.9 ± 0.0 | 99.5 ± 0.0 | 194 | 166 | 118 | 130 |
| 28 | 35 | 23 | 1.9 | 99.8 ± 0.1 | 99.7 ± 0.0 | >99.9 | 98.3 ± 0.3 | 778 | 27 | 62 | 355 |
| 65 | 58 | 26 | 1.6 | 99.6 ± 0.1 | 98.9 ± 0.0 | 99.3 ± 0.2 | 98.7 ± 0.1 | 390 | 22 | 64 | 567 |
| 24 | 76 | 9 | 5.2 | 99.2 ± 0.2 | 99.1 ± 0.2 | 99.4 ± 0.0 | 97.0 ± 0.4 | 733 | 23 | 31 | >1000 |
| 41 | 24 | 20 | 2.5 | 99.6 ± 0.2 | 99.5 ± 0.2 | 99.9 ± 0.0 | 98.8 ± 0.3 | 68 | 16 | 24 | 232 |
| 39 | 10 | 16 | 2.0 | 99.9 ± 0.0 | 99.9 ± 0.0 | >99.9 | >99.9 | 277 | 252 | 320 | 223 |
| 38 | 19 | 19 | 1.9 | 99.0 ± 0.1 | 99.1 ± 0.3 | 99.2 ± 0.3 | 98.4 ± 0.1 | 897 | 121 | 805 | 460 |
| 48 | 14 | 24 | 1.6 | 99.9 ± 0.0 | 99.6 ± 0.0 | 99.8 ± 0.0 | 99.6 ± 0.1 | 556 | 25 | 53 | 215 |
| 66 | 61 | 24 | 1.5 | 99.3 ± 0.2 | 98.6 ± 0.1 | 98.8 ± 0.2 | 98.2 ± 0.2 | 358 | 13 | 167 | 39 |
| 70 | 66 | 29 | 1.7 | 99.4 ± 0.0 | 99.3 ± 0.1 | 99.7 ± 0.1 | 99.4 ± 0.1 | 360 | 15 | 45 | 268 |
| Compound | Cmax (ng/mL) | AUC0–last (h·ng/mL) | t1/2 (h) | F (%) |
|---|---|---|---|---|
| 26 | 17,200 | 126,019 | 3.5 | 82 |
| 28 | 14,067 | 65,764 | 2.4 | 84 |
| 65 | 8150 | 40,105 | 2.2 | 129 |
| 24 | 13,100 | 37,495 | 2.1 | 77 |
| 41 | 12,467 | 26,010 | 2.7 | 40 |
| 39 | 14,300 | 86,444 | 2.9 | 78 |
| 38 | 8597 | 28,932 | 2.0 | 94 |
| 48 | 3017 | 10,889 | 2.1 | 70 |
| 66 | 9610 | 39,835 | 2.0 | 126 |
| 70 | 9723 | 32,258 | 2.0 | 117 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stokes, M.E.; Surman, M.D.; Calvo, V.; Surguladze, D.; Li, A.-H.; Gasparek, J.; Betzenhauser, M.; Zhu, G.; Du, H.; Rigby, A.C.; et al. Optimization of a Novel Mandelamide-Derived Pyrrolopyrimidine Series of PERK Inhibitors. Pharmaceutics 2022, 14, 2233. https://doi.org/10.3390/pharmaceutics14102233
Stokes ME, Surman MD, Calvo V, Surguladze D, Li A-H, Gasparek J, Betzenhauser M, Zhu G, Du H, Rigby AC, et al. Optimization of a Novel Mandelamide-Derived Pyrrolopyrimidine Series of PERK Inhibitors. Pharmaceutics. 2022; 14(10):2233. https://doi.org/10.3390/pharmaceutics14102233
Chicago/Turabian StyleStokes, Michael E., Matthew D. Surman, Veronica Calvo, David Surguladze, An-Hu Li, Jennifer Gasparek, Matthew Betzenhauser, Guangyu Zhu, Hongwen Du, Alan C. Rigby, and et al. 2022. "Optimization of a Novel Mandelamide-Derived Pyrrolopyrimidine Series of PERK Inhibitors" Pharmaceutics 14, no. 10: 2233. https://doi.org/10.3390/pharmaceutics14102233