
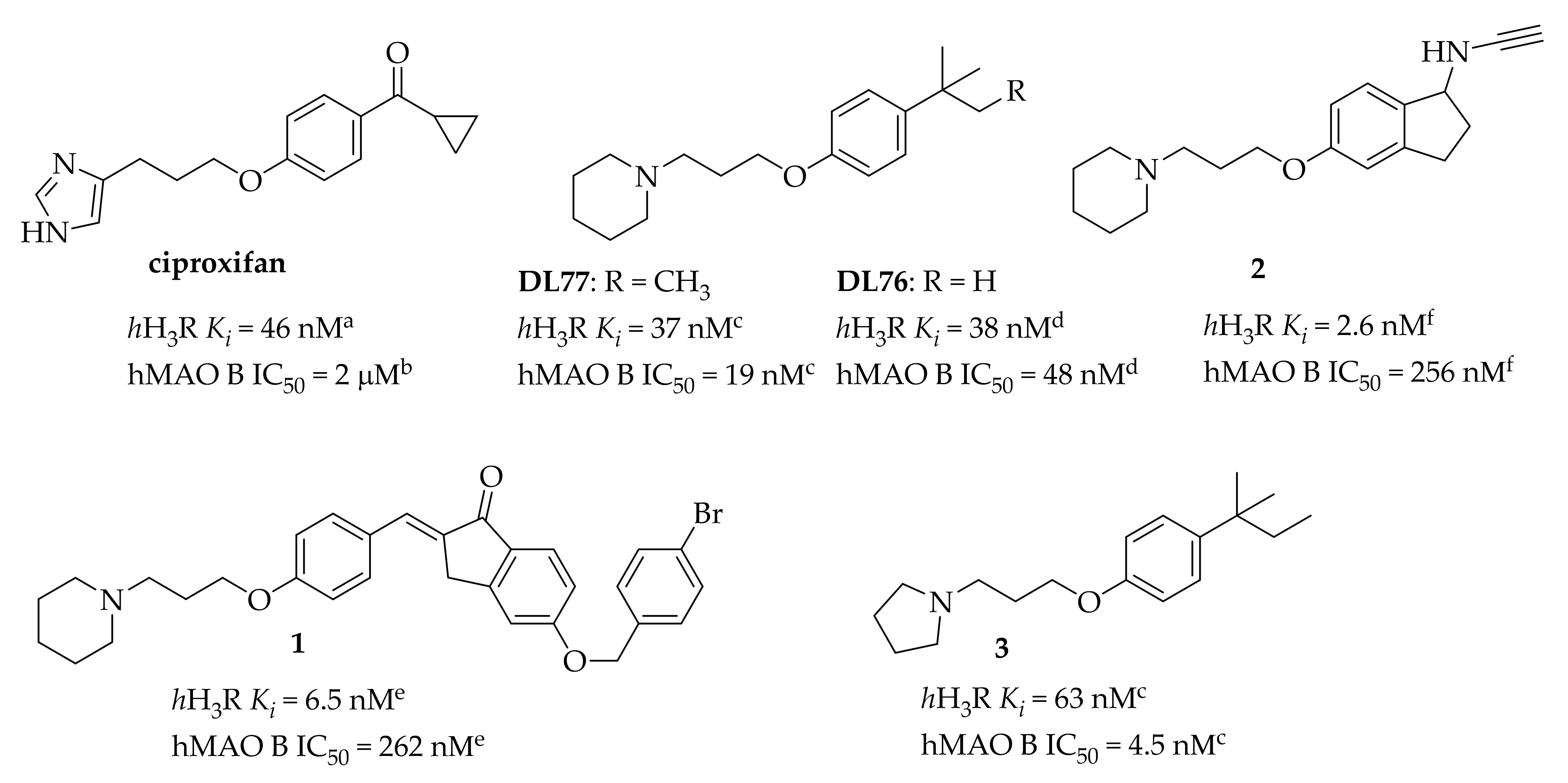
Dual Targeting Ligands—Histamine H3 Receptor Ligands with Monoamine Oxidase B Inhibitory Activity—In Vitro and In Vivo Evaluation
 , , ,
, , ,  , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
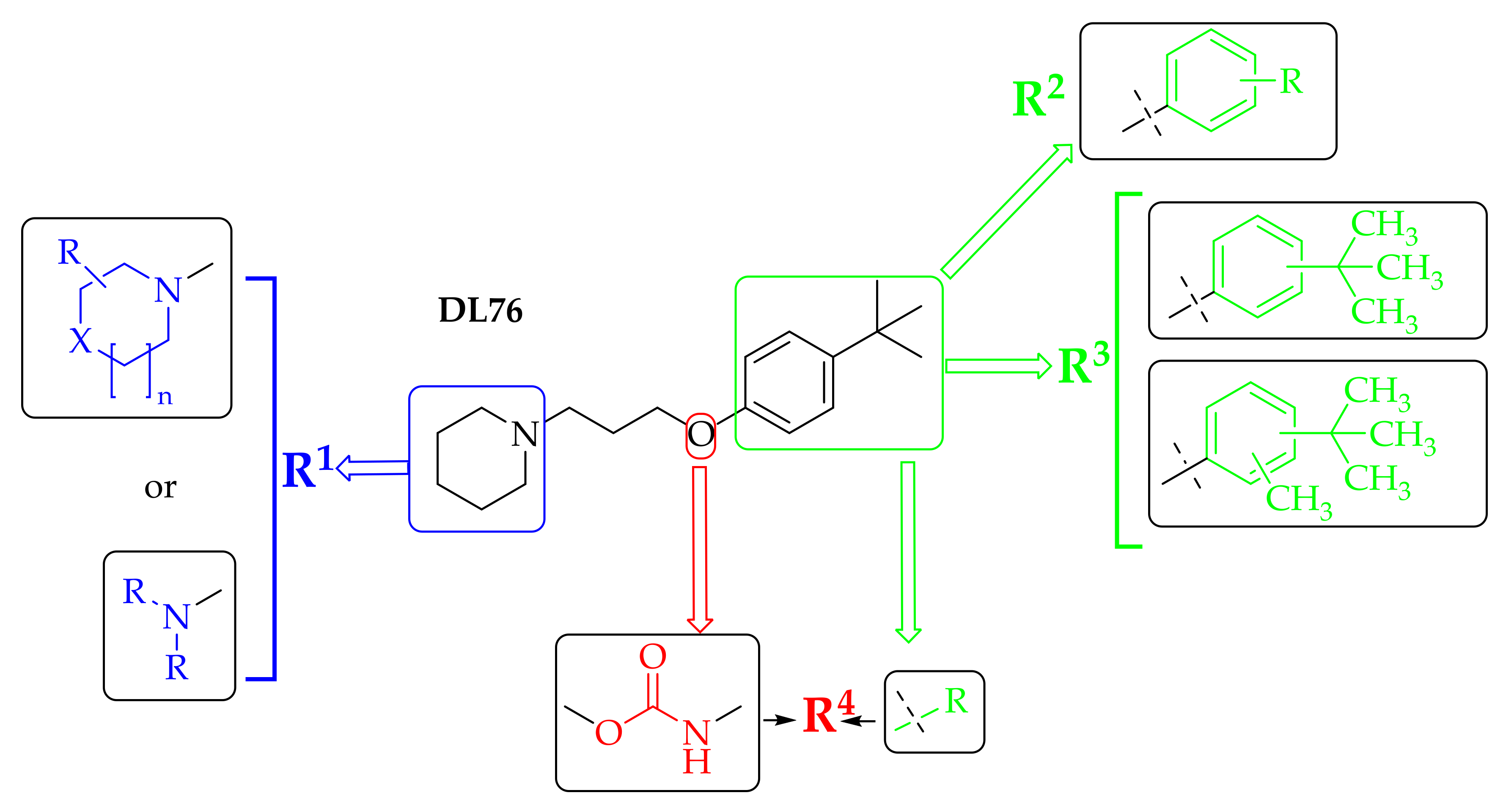
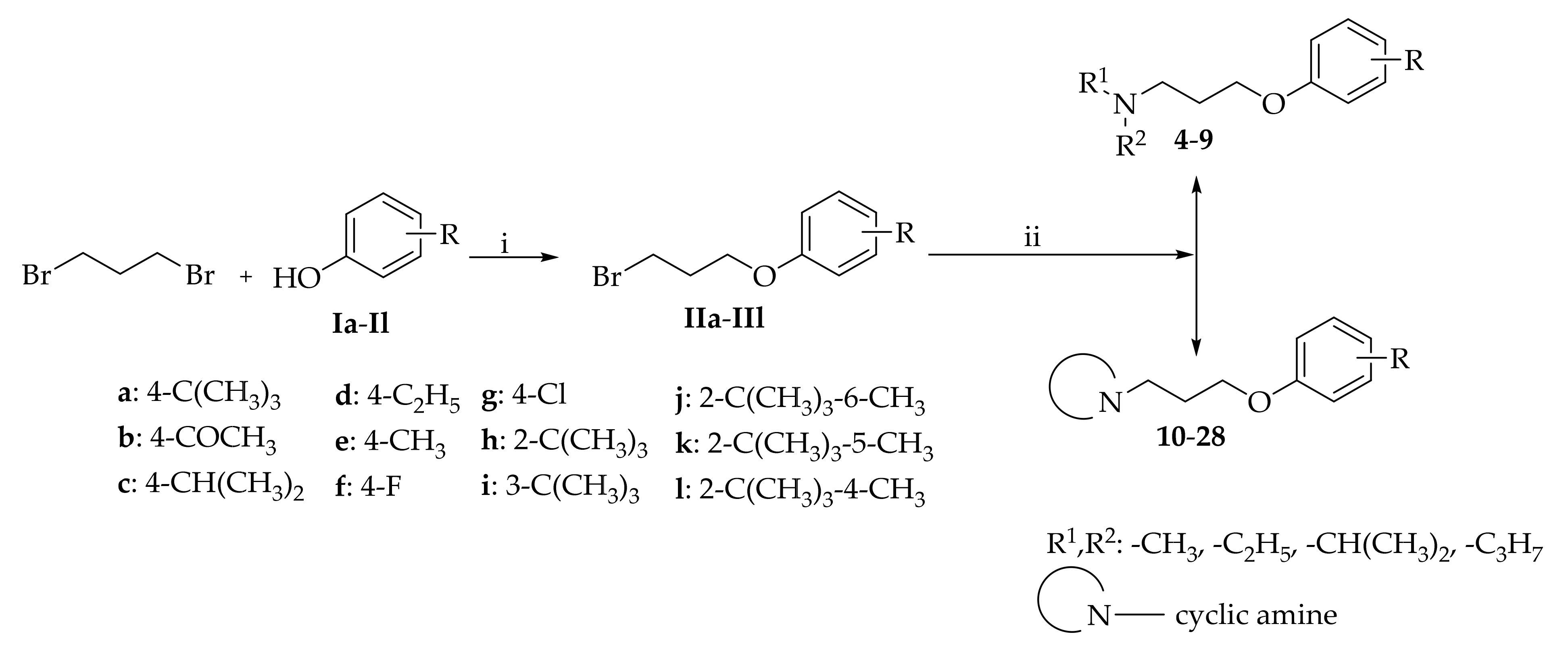
2.1. Synthesis of Compounds
- 3-(4-(tert-Butyl)phenoxy)-N,N-dimethylpropan-1-amine hydrogen oxalate (4)
- 3-(4-(tert-Butyl)phenoxy)-N-ethyl-N-methylpropan-1-amine hydrogen oxalate (5)
- 3-(4-(tert-Butyl)phenoxy)-N-isopropyl-N-methylpropan-1-amine hydrogen oxalate (6)
- 3-(4-(tert-Butyl)phenoxy)-N,N-diethylpropan-1-amine hydrogen oxalate (7)
- 3-(4-(tert-Butyl)phenoxy)-N,N-diisopropylpropan-1-amine hydrogen chloride (8)
- 3-(4-(tert-Butyl)phenoxy)-N,N-dipropylpropan-1-amine hydrogen oxalate (9)
- 1-(3-(4-(tert-Butyl)phenoxy)propyl)-2,6-dimethylpiperidine hydrogen chloride (11)
- 1-(3-(4-(tert-Butyl)phenoxy)propyl)-2-methylpyrrolidine hydrogen oxalate (13)
- 4-(3-(4-(tert-Butyl)phenoxy)propyl)morpholine hydrogen oxalate (14)
- 1-(3-(4-(tert-Butyl)phenoxy)propyl)-4-methylpiperazine hydrogen oxalate (15)
- 1-(3-(4-(tert-Butyl)phenoxy)propyl)-4-phenylpiperazine hydrogen oxalate (16)
- (Z)-1-(3-(4-(tert-Butyl)phenoxy)propyl)-4-(3-phenylallyl)piperazine hydrogen oxalate (17)
- 1-(4-(3-(Piperidin-1-yl)propoxy)phenyl)ethan-1-one hydrogen oxalate (18)
- 1-(3-(4-Isopropylphenoxy)propyl)piperidine hydrogen oxalate (19)
- 1-(3-(4-Ethylphenoxy)propyl)piperidine hydrogen oxalate (20)
- 1-(3-(p-Tolyloxy)propyl)piperidine hydrogen oxalate (21)
- 1-(3-(4-Fluorophenoxy)propyl)piperidine hydrogen oxalate (22)
- 1-(3-(4-chlorophenoxy)propyl)piperidine hydrogen oxalate (23)
- 1-(3-(2-(tert-Butyl)phenoxy)propyl)piperidine hydrogen oxalate (24)
- 1-(3-(3-(tert-Butyl)phenoxy)propyl)piperidine hydrogen chloride (25)
- 1-(3-(2-(tert-Butyl)-6-methylphenoxy)propyl)piperidine hydrogen oxalate (26)
- 1-(3-(2-(tert-Butyl)-5-methylphenoxy)propyl)piperidine hydrogen oxalate (27)
- 1-(3-(2-(tert-Butyl)-4-methylphenoxy)propyl)piperidine hydrogen oxalate (28)
- 3-(Piperidin-1-yl)propyl (4-(tert-butyl)phenyl)carbamate hydrogen oxalate (29)
2.2. Key Reagents (Cytotoxicity and In Vivo Pharmacology Studies)
2.3. In Vitro Biological Studies
2.3.1. Histamine H3 Receptor Affinity
2.3.2. Monoamine Oxidase B Inhibitory Activity
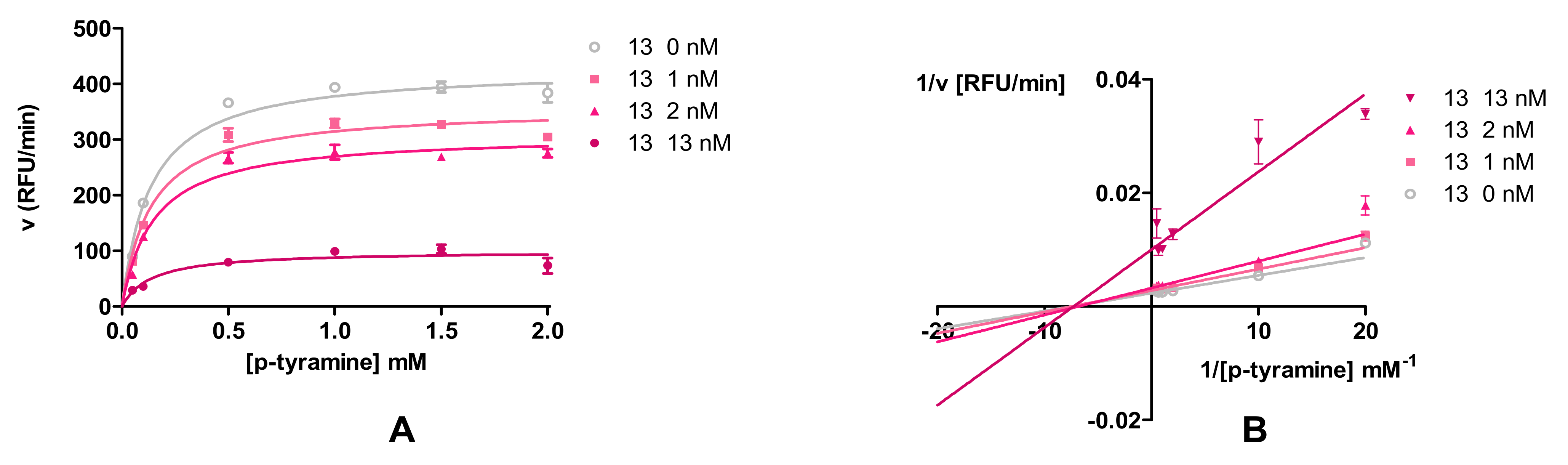
2.3.3. Modality of Monoamine Oxidase B Inhibition
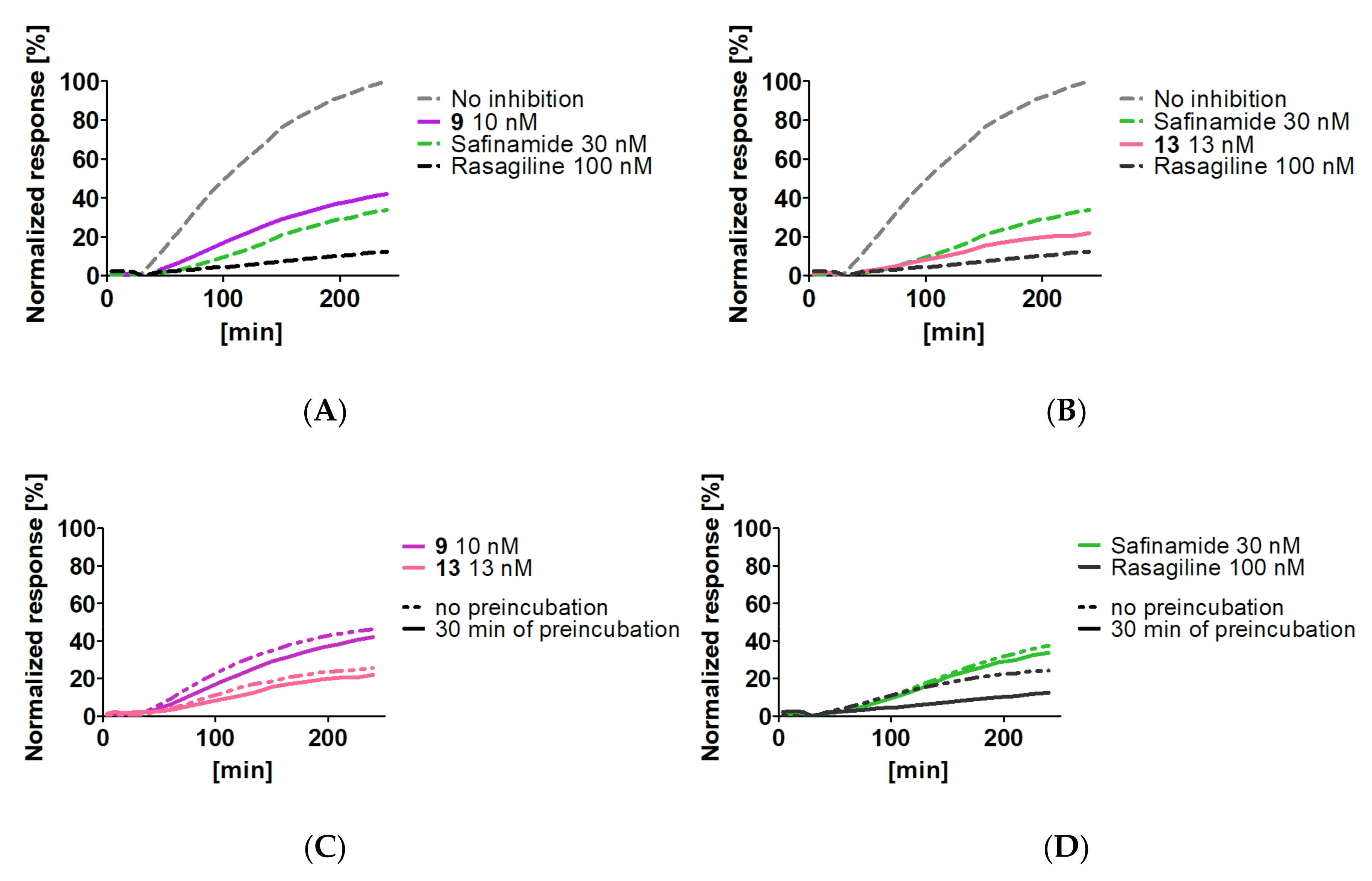
2.3.4. Reversibility of Monoamine Oxidase B Inhibition
2.3.5. Parallel Artificial Membrane Permeability
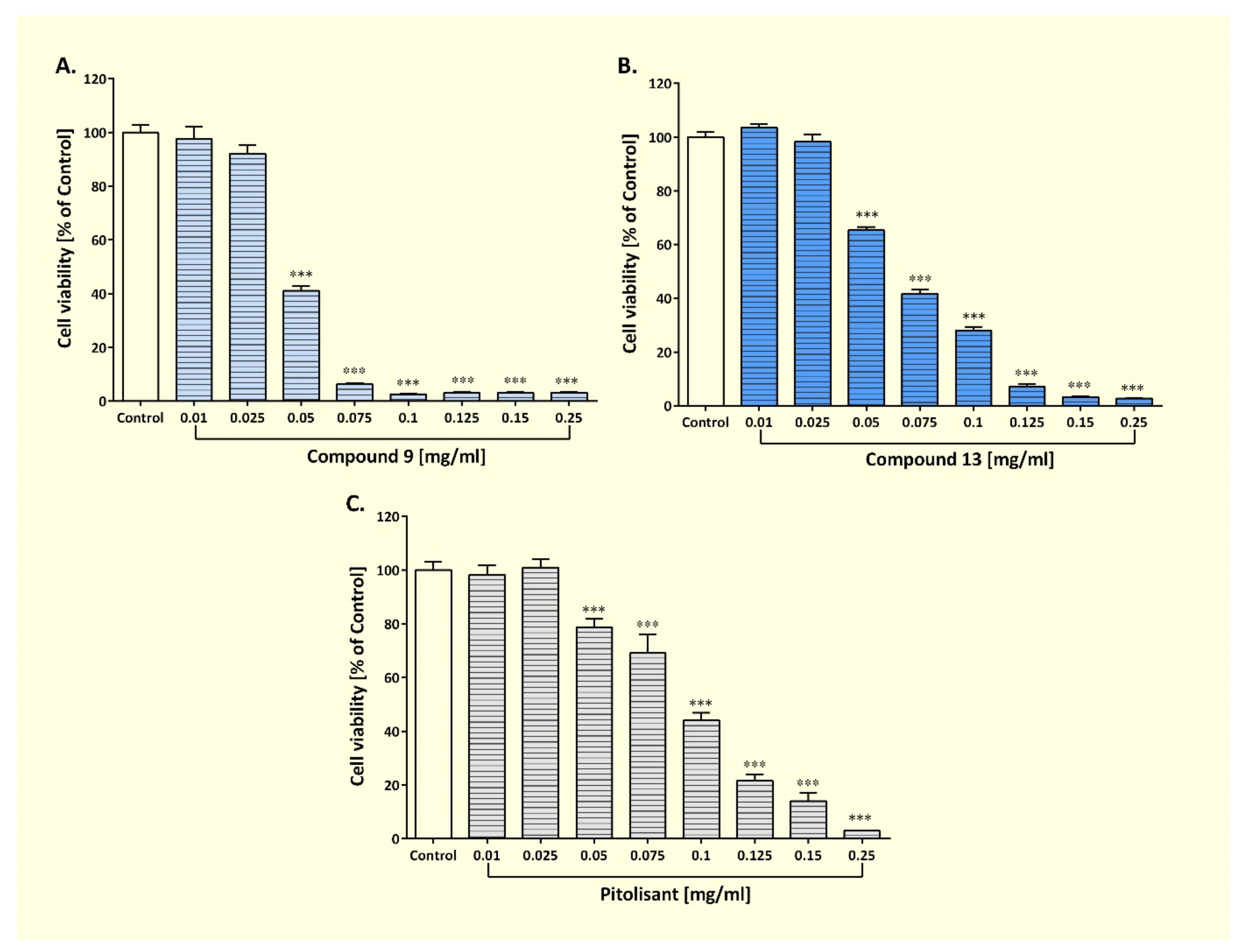
2.3.6. Evaluation of the Cytotoxicity of Compounds 9 and 13
Cell Cultures
MTT Cell Viability Test
2.4. Animals and Pharmacological Treatment
2.5. Sample Preparation and Biochemical Analyzes
2.5.1. HNMT and MAOs Activities
2.5.2. HPLC Detection of Monoamines and Their Metabolites in Rat Brain Tissue Samples
2.6. Statistical Analysis
3. Results and Discussion
3.1. Chemistry
3.2. In Vitro Pharmacological Studies
3.2.1. Histamine H3 Receptor Affinity of Tested Compounds
3.2.2. Human MAO B Inhibitory Activity of Tested Compounds
3.2.3. Modality of Human MAO B Reversible Inhibition of Compounds 9 and 13
3.2.4. Reversibility of Monoamine Oxidase B Inhibition of Compounds 9 and 13
3.2.5. Permeability of Compounds 9 and 13
3.2.6. Effect of Compounds 9 and 13 on the Viability of Human Astrocyte Cell Lines
3.3. Preliminary Verification of In Vivo Activity of the Compound 13
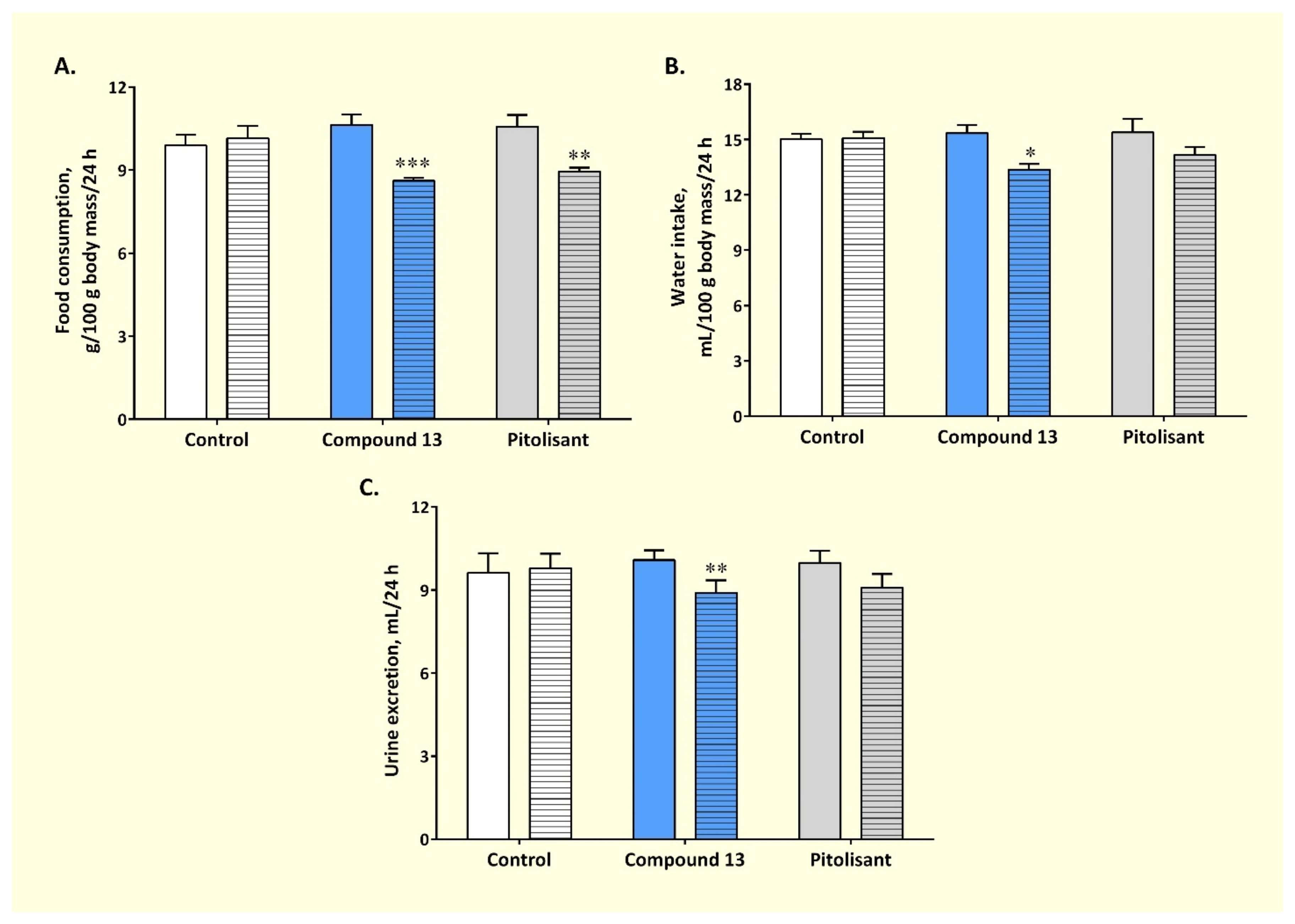
3.3.1. Effect on Sub-Chronic Administration of Compound 13 on Feeding Behavior
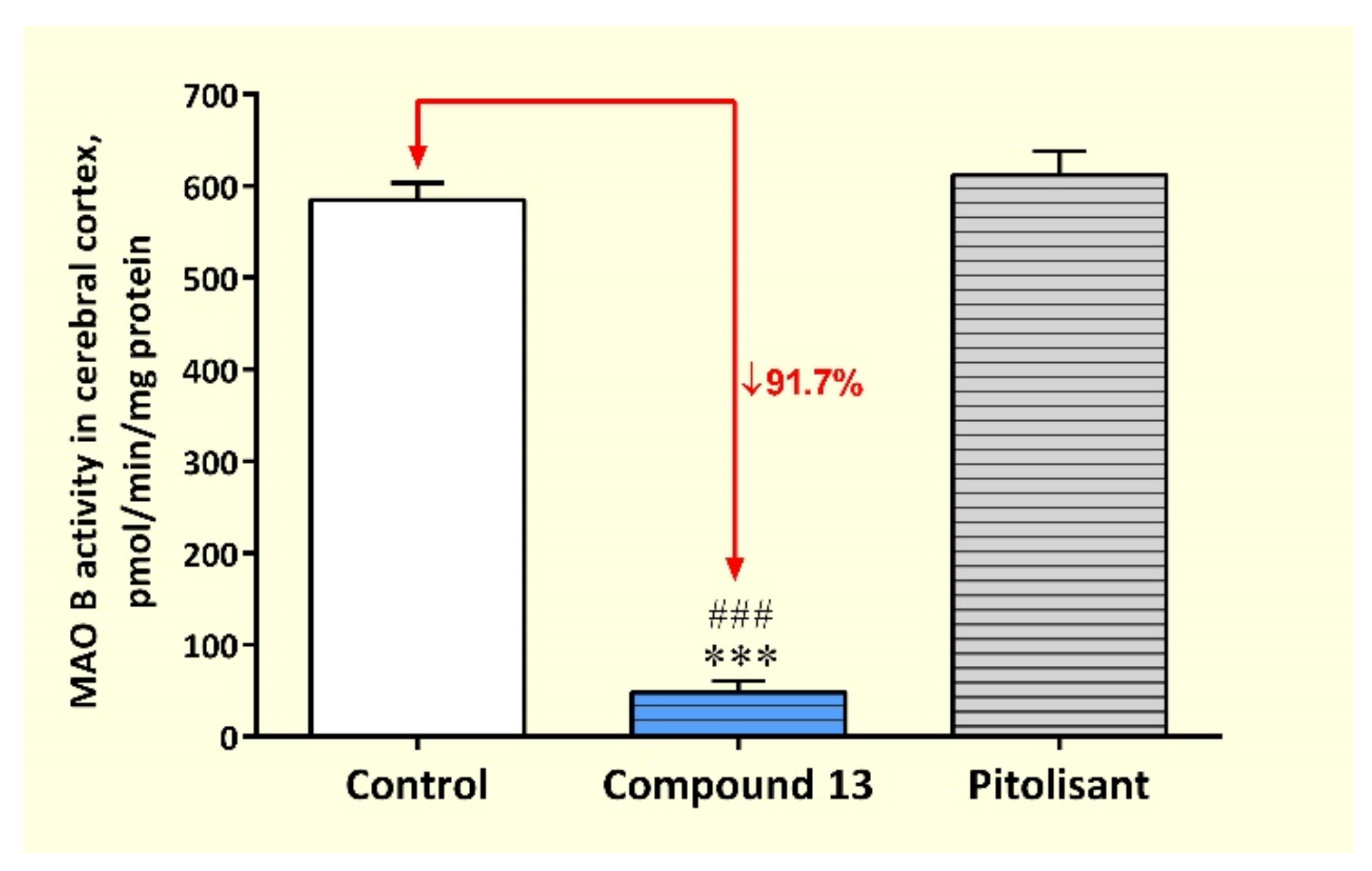
3.3.2. Activity of MAOs and HNMT in Rat Cerebral Cortex after Sub-Chronic Administration of Compound 13
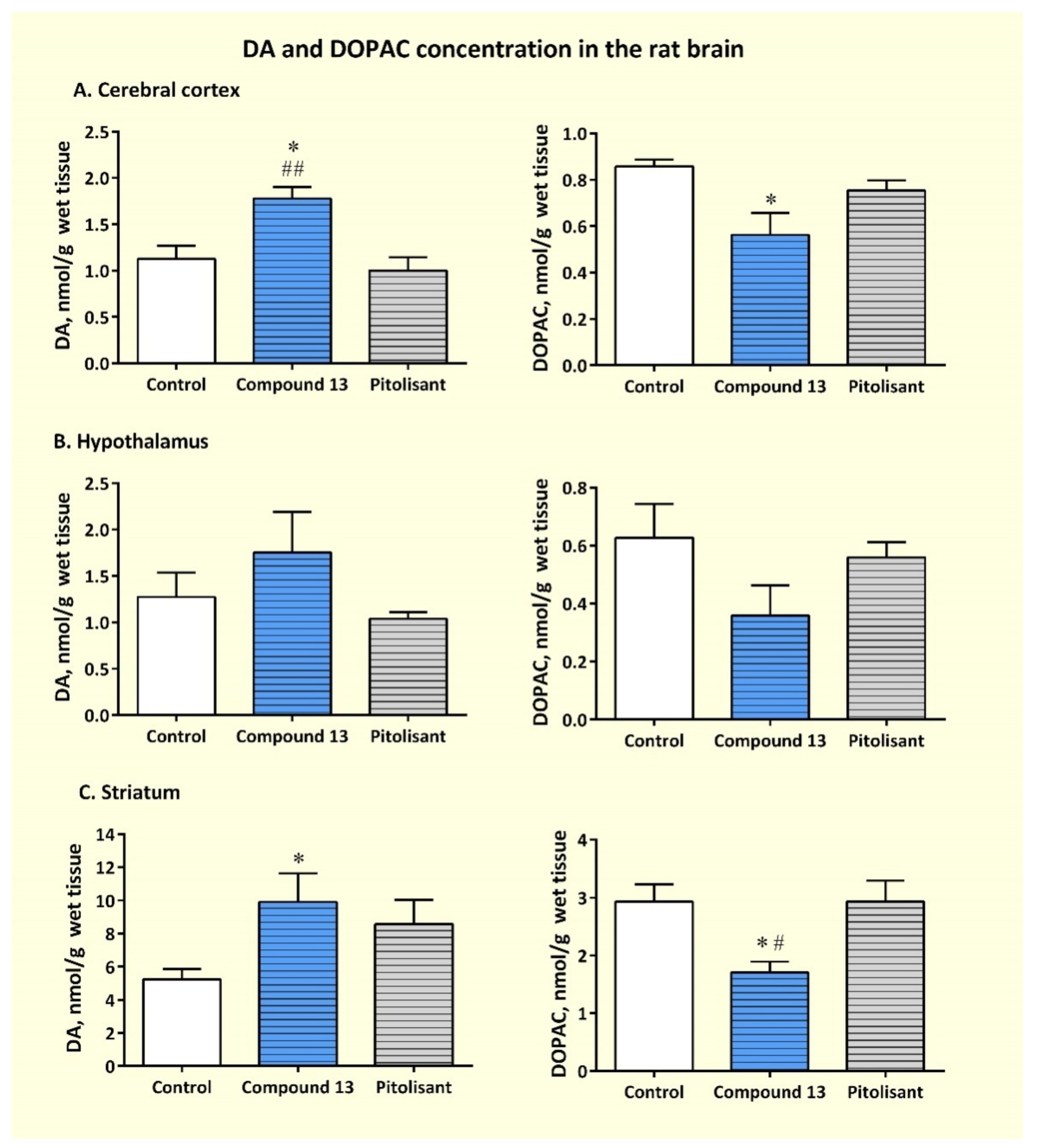
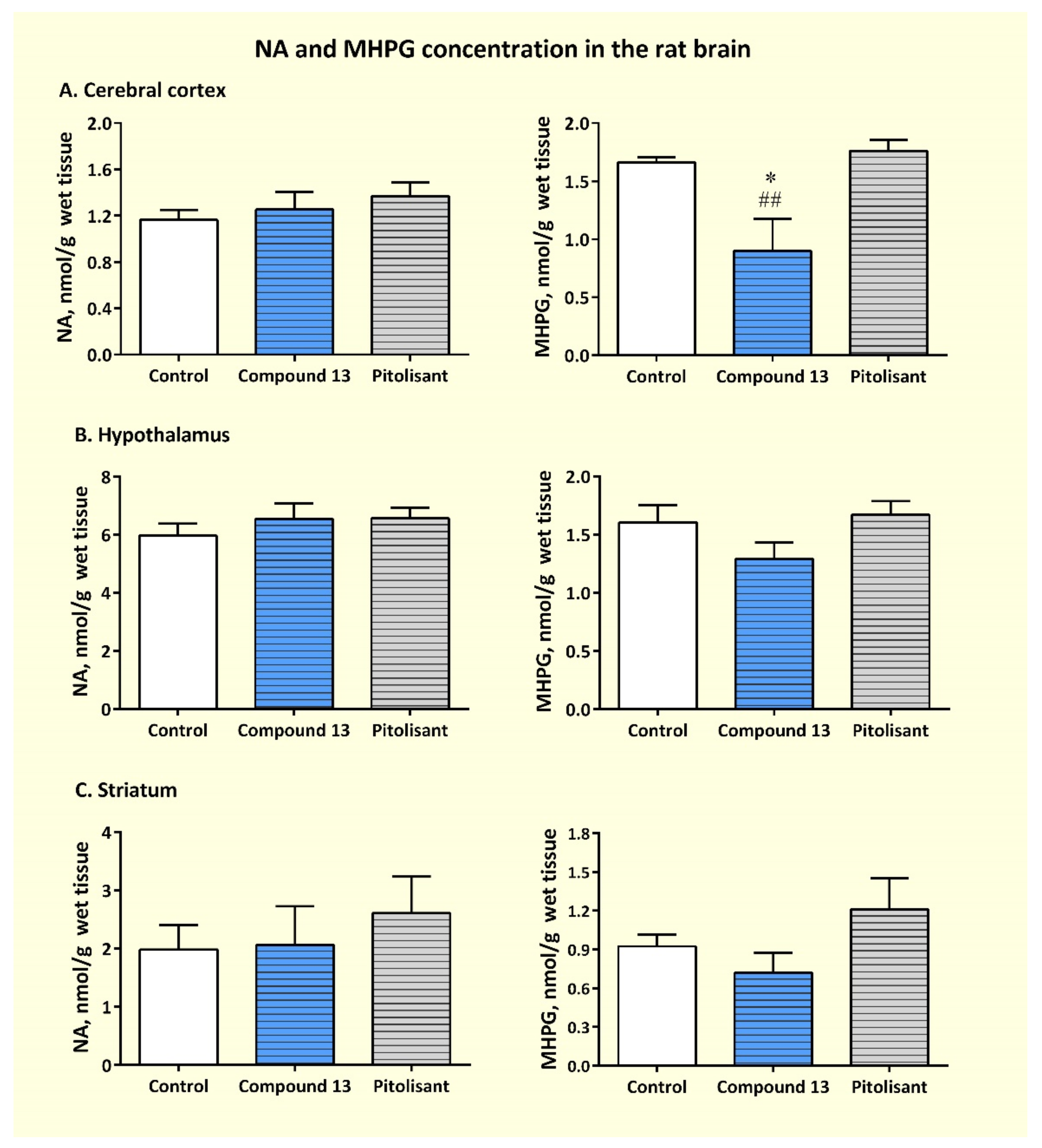
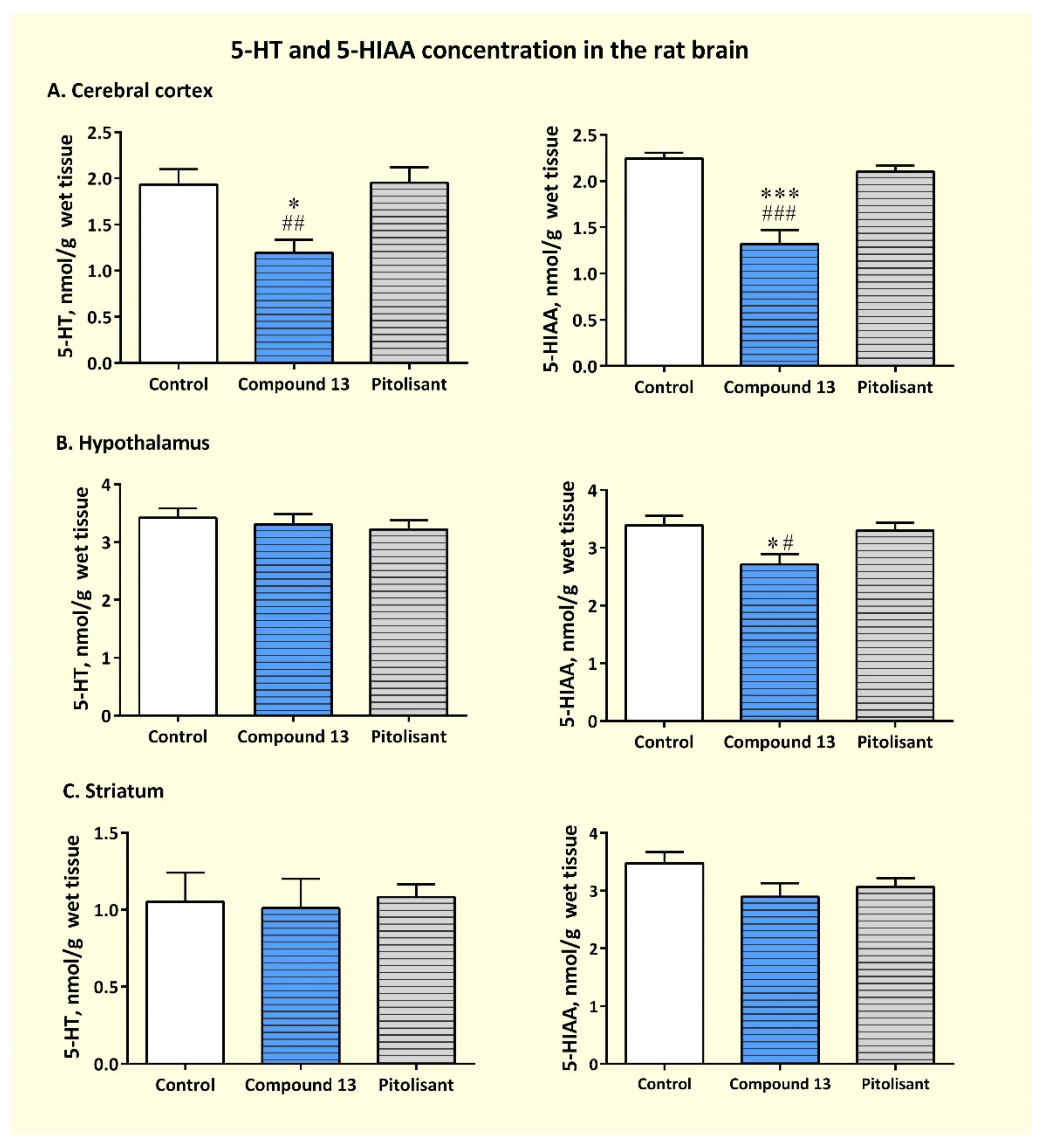
3.3.3. Effects of Sub-Chronic Administration of Compound 13 on Cerebral Concentration of Monoamines and Their Metabolites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef]
- Ma, H.; Huang, B.; Zhang, Y. Recent advances in multitarget-directed ligands targeting G-protein-coupled receptors. Drug. Discov. Today 2020, 25, 1682–1692. [Google Scholar] [CrossRef]
- Kuder, K.J.; Załuski, M.; Schabikowski, J.; Latacz, G.; Olejarz-Maciej, A.; Jaśko, P.; Doroz-Płonka, A.; Brockmann, A.; Müller, C.E.; Kieć-Kononowicz, K. Novel, Dual Target-Directed Annelated Xanthine Derivatives Acting on Adenosine Receptors and Monoamine Oxidase B. ChemMedChem. 2020, 15, 772–786. [Google Scholar] [CrossRef] [PubMed]
- Affini, A.; Hagenow, S.; Zivkovic, A.; Marco-Contelles, J.; Stark, H. Novel indanone derivatives as MAO B/H3R dual-targeting ligands for treatment of Parkinson’s disease. Eur. J. Med. Chem. 2018, 148, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, K.; Hagenow, S.; Affini, A.; Reiner, D.; Stark, H. Rasagiline derivatives combined with histamine H3 receptor properties. Bioorg. Med. Chem. Lett. 2019, 29, 126612. [Google Scholar] [CrossRef]
- Łażewska, D.; Olejarz-Maciej, A.; Kaleta, M.; Bajda, M.; Siwek, A.; Karcz, T.; Doroz-Płonka, A.; Cichoń, U.; Kuder, K.; Kieć-Kononowicz, K. 4-tert-Pentylphenoxyalkyl derivatives—Histamine H3 receptor ligands and monoamine oxidase B inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 3596–3600. [Google Scholar] [CrossRef] [PubMed]
- Łażewska, D.; Olejarz-Maciej, A.; Reiner, D.; Kaleta, M.; Latacz, G.; Zygmunt, M.; Doroz-Płonka, A.; Karcz, T.; Frank, A.; Stark, H.; et al. Dual Target Ligands with 4-tert-Butylphenoxy Scaffold as Histamine H3 Receptor Antagonists and Monoamine Oxidase B Inhibitors. Int. J. Mol. Sci. 2020, 21, 3411. [Google Scholar] [CrossRef]
- Hagenow, S.; Stasiak, A.; Ramsay, R.R.; Stark, H. Ciproxifan, a histamine H3 receptor antagonist, reversibly inhibits monoamine oxidase A and B. Sci. Rep. 2017, 7, 40541. [Google Scholar] [CrossRef] [Green Version]
- Ligneau, X.; Morisset, S.; Tardivel-Lacombe, J.; Gbahou, F.; Ganellin, C.R.; Stark, H.; Schunack, W.; Schwartz, J.C.; Arrang, J.M. Distinct pharmacology of rat and human histamine H(3) receptors: Role of two amino acids in the third transmembrane domain. Br. J. Pharmacol. 2000, 131, 1247–1250. [Google Scholar] [CrossRef] [Green Version]
- Łazewska, D.; Kieć-Kononowicz, K.; Elz, S.; Pertz, H.H.; Stark, H.; Schunack, W. Piperidine-containing histamine H3 receptor antagonists of the carbamate series: The alkyl derivatives. Pharmazie 2005, 60, 403–410. [Google Scholar]
- Łażewska, D.; Kaleta, M.; Schwed, J.S.; Karcz, T.; Mogilski, S.; Latacz, G.; Olejarz, A.; Siwek, A.; Kubacka, M.; Lubelska, A.; et al. Biphenyloxy-alkyl-piperidine and azepane derivatives as histamine H3 receptor ligands. Bioorg. Med. Chem. 2017, 25, 5341–5354. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Łażewska, D.; Bajda, M.; Kaleta, M.; Zaręba, P.; Doroz-Płonka, A.; Siwek, A.; Alachkar, A.; Mogilski, S.; Saad, A.; Kuder, K.; et al. Rational design of new multitarget histamine H3 receptor ligands as potential candidates for treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2020, 207, 112743. [Google Scholar] [CrossRef] [PubMed]
- Jóźwiak-Bębenista, M.; Sokołowska, P.; Siatkowska, M.; Panek, C.A.; Komorowski, P.; Kowalczyk, E.; Wiktorowska-Owczarek, A. The Importance of Endoplasmic Reticulum Stress as a Novel Antidepressant Drug Target and Its Potential Impact on CNS Disorders. Pharmaceutics 2022, 14, 846. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Tan, A.S. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): Subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys. 1993, 303, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Stockert, J.C.; Horobin, R.W.; Colombo, L.L.; Blázquez-Castro, A. Tetrazolium salts and formazan products in Cell Biology: Viability assessment, fluorescence imaging, and labeling perspectives. Acta Histochem. 2018, 120, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, J.C. The histamine H3 receptor: From discovery to clinical trials with pitolisant. Br. J. Pharmacol. 2011, 163, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Gorain, B.; Sengupta, P.; Dutta, S.; Pandey, M.; Choudhury, H. Pharmacology of Histamine, Its Receptors and Antagonists in the Modulation of Physiological Functions. In Frontiers in Pharmacology of Neurotransmitters; Kumar, P., Deb, P.K., Eds.; Springer: Singapore, 2020; pp. 213–240. [Google Scholar] [CrossRef]
- Stasiak, A.; Mussur, M.; Unzeta, M.; Łażewska, D.; Kieć-Kononowicz, K.; Fogel, W.A. The central histamine level in rat model of vascular dementia. J. Physiol. Pharmacol. 2011, 62, 549–558. [Google Scholar]
- Stasiak, A.; Mussur, M.; Unzeta, M.; Samadi, A.; Marco-Contelles, J.L.; Fogel, W.A. Effects of novel monoamine oxidases and cholinesterases targeting compounds on brain neurotransmitters and behavior in rat model of vascular dementia. Curr. Pharm. Des. 2014, 20, 161–171. [Google Scholar] [CrossRef] [Green Version]
- Glowinski, J.; Iversen, L.L. Regional studies of catecholamines in the rat brain. I. The disposition of [3H]norepinephrine, [3H]dopamine and [3H]dopa in various regions of the brain. J. Neurochem. 1966, 13, 655–669. [Google Scholar] [CrossRef]
- Fowler, C.J.; Tipton, K.F. Deamination of 5-hydroxytryptamine by both forms of monoamine oxidase in the rat brain. J. Neurochem. 1982, 38, 733–736. [Google Scholar] [CrossRef]
- Gómez, N.; Balsa, D.; Unzeta, M. A comparative study of some kinetic and molecular properties of microsomal and mitochondrial monoamine oxidase. Biochem. Pharmacol. 1988, 37, 3407–3413. [Google Scholar] [CrossRef]
- Taylor, K.M.; Snyder, S.H. Isotopic microassay of histamine, histidine, histidine decarboxylase and histamine methyltransferase in brain tissue. J. Neurochem. 1972, 19, 1343–1358. [Google Scholar] [CrossRef] [PubMed]
- Fogel, W.A.; Andrzejewski, W.; Maslinski, C. Brain histamine in rats with hepatic encephalopathy. J. Neurochem. 1991, 56, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein Measurement with Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Kobrzycka, A.T.; Stankiewicz, A.M.; Goscik, J.; Gora, M.; Burzynska, B.; Iwanicka-Nowicka, R.; Pierzchala-Koziec, K.; Wieczorek, M. Hypothalamic NeurochemicalChanges in Long-Term RecoveredBilateral SubdiaphragmaticVagotomized Rats. Front. Behav. Neurosci. 2022, 16, 869526. [Google Scholar] [CrossRef]
- Sander, K.; Kottke, T.; Weizel, L.; Stark, H. Kojic acid derivatives as histamine H(3) receptor ligands. Chem. Pharm. Bull. (Tokyo) 2010, 58, 1353–1361. [Google Scholar] [CrossRef] [Green Version]
- Copeland, R.A. Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods Biochem. Anal. 2005, 46, 1–265. [Google Scholar]
- Ramsay, R.R.; Albreht, A. Kinetics, mechanism, and inhibition of monoamine oxidase. J. Neural Transm. 2018, 125, 1659–1683. [Google Scholar] [CrossRef] [Green Version]
- Esbenshade, T.A.; Fox, G.B.; Cowart, M.D. Histamine H3 receptor antagonists: Preclinical promise for treating obesity and cognitive disorders. Mol. Interv. 2006, 6, 77–88. [Google Scholar] [CrossRef]
- Guryn, R.; Staszewski, M.; Stasiak, A.; McNaught Flores, D.; Fogel, W.A.; Leurs, R.; Walczyński, K. Non-Imidazole Histamine H3 Ligands. Part VII. Synthesis, In Vitro and In Vivo Characterization of 5-Substituted-2-thiazol-4-n-propylpiperazines. Molecules 2018, 23, 326. [Google Scholar] [CrossRef] [Green Version]
- Staszewski, M.; Stasiak, A.; Karcz, T.; McNaught Flores, D.; Fogel, W.A.; Kieć-Kononowicz, K.; Leurs, R.; Walczyński, K. Design, synthesis, and in vitro and in vivo characterization of 1-{4-[4-(substituted)piperazin-1-yl]butyl}guanidines and their piperidine analogues as histamine H3 receptor antagonists. MedChemComm 2019, 10, 234–251. [Google Scholar] [CrossRef] [PubMed]
- Provensi, G.; Blandina, P.; Passani, M.B. The histaminergic system as a target for the prevention of obesity and metabolic syndrome. Neuropharmacology 2016, 106, 3–12. [Google Scholar] [CrossRef]
- Schapira, A.H.V. Monoamine Oxidase B Inhibitors for the Treatment of Parkinson’s Disease. CNS Drugs 2011, 25, 1061–1071. [Google Scholar] [CrossRef]
- Fowler, J.S.; Volkow, N.D.; Wang, G.J.; Logan, J.; Pappas, N.; Shea, C.; MacGregor, R. Age-related increases in brain monoamine oxidase B in living healthy human subjects. Neurobiol. Aging 1997, 18, 431–435. [Google Scholar] [CrossRef]
- Tan, Y.Y.; Jenner, P.; Chen, S.D. Monoamine Oxidase-B Inhibitors for the Treatment of Parkinson’s Disease: Past, Present, and Future. J. Parkinsons Dis. 2022, 12, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Bakhle, Y.S. Monoamine oxidase: Isoforms and inhibitors in Parkinson’s disease and depressive illness. Br. J. Pharmacol. 2006, 147, S287–S296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pytliak, M.; Vargová, V.; Mechírová, V.; Felšöci, M. Serotonin receptors—From molecular biology to clinical applications. Physiol. Res. 2011, 60, 15–25. [Google Scholar] [CrossRef]
- McCorvy, J.D.; Roth, B.L. Structure and function of serotonin G protein-coupled receptors. Pharmacol. Ther. 2015, 150, 129–142. [Google Scholar] [CrossRef] [Green Version]
- You, I.J.; Wright, S.R.; Garcia-Garcia, A.L.; Tapper, A.R.; Gardner, P.D.; Koob, G.F.; Leonardo, E.D.; Bohn, L.M.; Wee, S. 5-HT1A Autoreceptors in the Dorsal Raphe Nucleus Convey Vulnerability to Compulsive Cocaine Seeking. Neuropsychopharmacology 2016, 41, 1210–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, R.; Huereca, D.; Lyons, J.G.; Andrade, E.M.; McGregor, K.M. 5-HT1A Receptor-Mediated Autoinhibition and the Control of Serotonergic Cell Firing. ACS Chem. Neurosci. 2015, 6, 1110–1115. [Google Scholar] [CrossRef] [Green Version]
- Sharp, T.; Boothman, L.; Raley, J.; Queree, P. Important messages in the ‘post’: Recent discoveries in 5-HT 707 neurone feedback control. Trends Pharmacol. Sci. 2007, 28, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Altieri, S.C.; Garcia-Garcia, A.L.; Leonardo, E.D.; Andrews, A.M. Rethinking 5-HT1A receptors: Emerging modes of inhibitory feedback of relevance to emotion-related behavior. ACS Chem. Neurosci. 2013, 4, 72–83. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
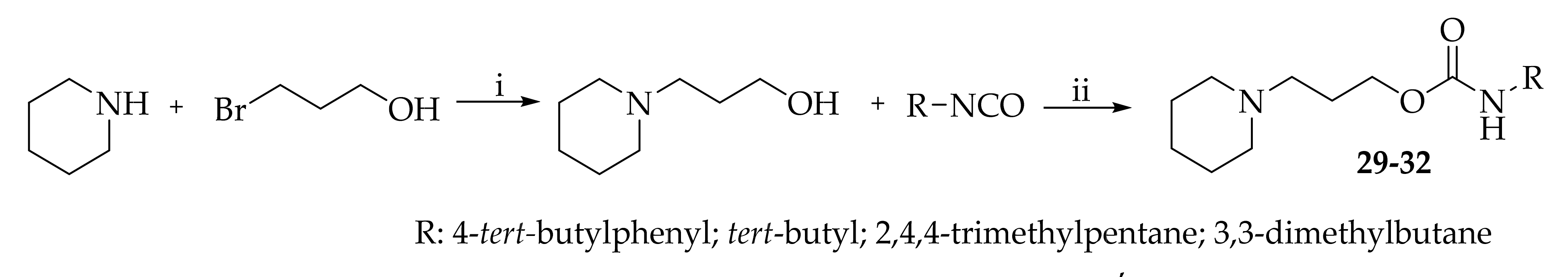{kind=link}
| Compound | Structure | hH3R a,b Ki [nM] ± SEM a or Ki [nM] [95%CI] b (% Inh.) c | hMAO B d IC50 [nM] (% of Inh.) e |
|---|---|---|---|
| DL76 |  | 57.5 ± 6.4 a 38 [8; 181] b | 48 ± 15 |
 | |||
| R1 | |||
| 4 |  | (24%) c | 19 ± 2 |
| 5 |  | 484 ± 15 a | 9 ± 0 |
| 6 |  | 900 ± 75 a | 6 ± 1 |
| 7 |  | (32%) c | 15 ± 4 |
| 8 |  | (8%) c | 37 ± 6 |
| 9 |  | 323 ± 73 a | 2 ± 0 |
| 10 f |  | 69 b,f [49; 96] | 11 ± 1f |
| 11 |  | 97 ± 3 a | 9 ± 1 |
| 12f |  | 371 b,f [136; 1009] | 2.7 ± 0.4 f |
| 13 |  | 25 ± 9 a | 4.0 ± 0.3 |
| 14 |  | (11%) c | 192 ± 14 |
| 15 |  | (0%) c | 665 ± 118 |
| 16 |  | 535 ± 41 a | (14%) e |
| 17 |  | (0%) c | (7%) e |
| Compound | Structure | hH3R a Ki [nM] [95%CI] | hMAO B b IC50 [nM] (% of Inh.) c |
|---|---|---|---|
 | |||
| R2 | |||
| 18 |  | 15 [5; 45] | (19%) |
| 19 |  | 52 [24; 113] | 21 ± 3 |
| 20 |  | 61 [21; 178] | 70 ± 2 |
| 21 |  | 43 [10; 178] | 755 ± 106 |
| 22 |  | 93 [16; 536] | (29%) |
| 23 |  | 83 [12; 561] | 1058 ± 37 |
| Compound | Structure | hH3R a Ki [nM] ± SEM (% Inh.) b | hMAO B c IC50 [nM] (% of Inh.) d |
|---|---|---|---|
 | |||
| R3 | |||
| 24 | 2-tert-Butyl | (13%) | (4%) |
| 25 | 3-tert-Butyl | 340 ± 23 | 1021 ± 69 |
| 26 | 2-tert-Butyl-6-methyl | (0%) | (0%) |
| 27 | 2-tert-Butyl-5-methyl | (0%) | (4%) |
| 28 | 2-tert-Butyl-4-methyl | 448 ± 59 | (7%) |
| Compound | Structure | hH3R a (% Inh.) b | hMAO B c IC50 ± SEM [nM] (% of Inh.) d |
|---|---|---|---|
 | |||
| R4 | |||
| 29 |  | (32%) | 2325 ± 436 |
| 30 |  | (15%) | (47%) |
| 31 |  | (19%) | (41%) |
| 32 |  | (20%) | 925 ± 29 |
| Parameter | Compound | ||
|---|---|---|---|
| Safinamide | 9 | 13 | |
| app. KM | ↑ curvilinearly with ↑ [I] | No effect a | No effect a |
| app. Vmax | ↓ curvilinearly with ↑ [I] | ↓ curvilinearly with ↑ [I] | ↓ curvilinearly with ↑ [I] |
| app. Vmax/app. KM | ↓ curvilinearly with ↑ [I] | ↓ curvilinearly with ↑ [I] | ↓ curvilinearly with ↑ [I] |
| Lines on LB plot | Lines intersect to the left of y-axis and above x-axis | Lines intersect to the left of y-axis directly on x-axis | Lines intersect to the left of y-axis directly on x-axis |
| Mode of inhibition from kinetic values and LB plot | Mixed mode | Noncompetitive | Noncompetitive |
| Affinity | Free enzyme > enzyme-substrate complex | Free enzyme ≈ enzyme-substrate complex | Free enzyme ≈ enzyme-substrate complex |
| Compound | a,bPe (×10−6cm/s) ± SD |
|---|---|
| 9 | ---- --c |
| 13 | 16.72 ± 0.06 |
| caffeine | 15.1 ± 0.4 |
| Compound 9 | Compound 13 | Pitolisant | |
|---|---|---|---|
| IC50 24 h | 144.16 μM (0.055 mg/mL) | 229.84 μM (0.084 mg/mL) | 346.06 μM (0.115 mg/mL) |
| IC50 72 h | 123.19 μM (0.047 mg/mL) | 142.28 μM (0.052 mg/mL) | 240.74 μM (0.08 mg/mL) |
| Brain Region | Group | MHPG/NA | DOPAC/DA | HVA/DA | 5-HIAA/5-HT |
|---|---|---|---|---|---|
| CTX | Control | 1.466 ± 0.131 | 0.886 ± 0.107 | 0.391 ± 0.042 | 1.229 ± 0.185 |
| Compound 13 | 0.790 ± 0.275 * | 0.364 ±0.080 **,## | 0.254 ± 0.015 *,## | 1.211 ± 0.202 | |
| Pitolisant | 1.344 ± 0.118 | 0.923 ±0.132 | 0.449 ± 0.035 | 1.105 ±0.087 | |
| HPT | Control | 0.268 ±0.018 | 0.563 ± 0.075 | 0.328 ± 0.118 | 0.820 ±0.139 |
| Compound 13 | 0.205 ±0.030 | 0.359 ±0.117 | 0.329 ± 0.091 | 0.810 ±0.061 | |
| Pitolisant | 0.257 ±0.021 | 0.538 ± 0.037 | 0.276 ± 0.030 | 0.981 ±0.051 | |
| STR | Control | 0.470 ± 0.032 | 0.513± 0.036 | 0.472 ±0.384 | 3.029 ± 0.672 |
| Compound 13 | 0.329 ± 0.057 | 0.207 ± 0.067 * | 0.486 ± 0.388 | 2.456 ± 0.908 | |
| Pitolisant | 0.455 ± 0.046 | 0.376 ± 0.074 | ND | 2.879 ± 0.222 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łażewska, D.; Siwek, A.; Olejarz-Maciej, A.; Doroz-Płonka, A.; Wiktorowska-Owczarek, A.; Jóźwiak-Bębenista, M.; Reiner-Link, D.; Frank, A.; Sromek-Trzaskowska, W.; Honkisz-Orzechowska, E.; et al. Dual Targeting Ligands—Histamine H3 Receptor Ligands with Monoamine Oxidase B Inhibitory Activity—In Vitro and In Vivo Evaluation. Pharmaceutics 2022, 14, 2187. https://doi.org/10.3390/pharmaceutics14102187
Łażewska D, Siwek A, Olejarz-Maciej A, Doroz-Płonka A, Wiktorowska-Owczarek A, Jóźwiak-Bębenista M, Reiner-Link D, Frank A, Sromek-Trzaskowska W, Honkisz-Orzechowska E, et al. Dual Targeting Ligands—Histamine H3 Receptor Ligands with Monoamine Oxidase B Inhibitory Activity—In Vitro and In Vivo Evaluation. Pharmaceutics. 2022; 14(10):2187. https://doi.org/10.3390/pharmaceutics14102187
Chicago/Turabian StyleŁażewska, Dorota, Agata Siwek, Agnieszka Olejarz-Maciej, Agata Doroz-Płonka, Anna Wiktorowska-Owczarek, Marta Jóźwiak-Bębenista, David Reiner-Link, Annika Frank, Wioletta Sromek-Trzaskowska, Ewelina Honkisz-Orzechowska, and et al. 2022. "Dual Targeting Ligands—Histamine H3 Receptor Ligands with Monoamine Oxidase B Inhibitory Activity—In Vitro and In Vivo Evaluation" Pharmaceutics 14, no. 10: 2187. https://doi.org/10.3390/pharmaceutics14102187