
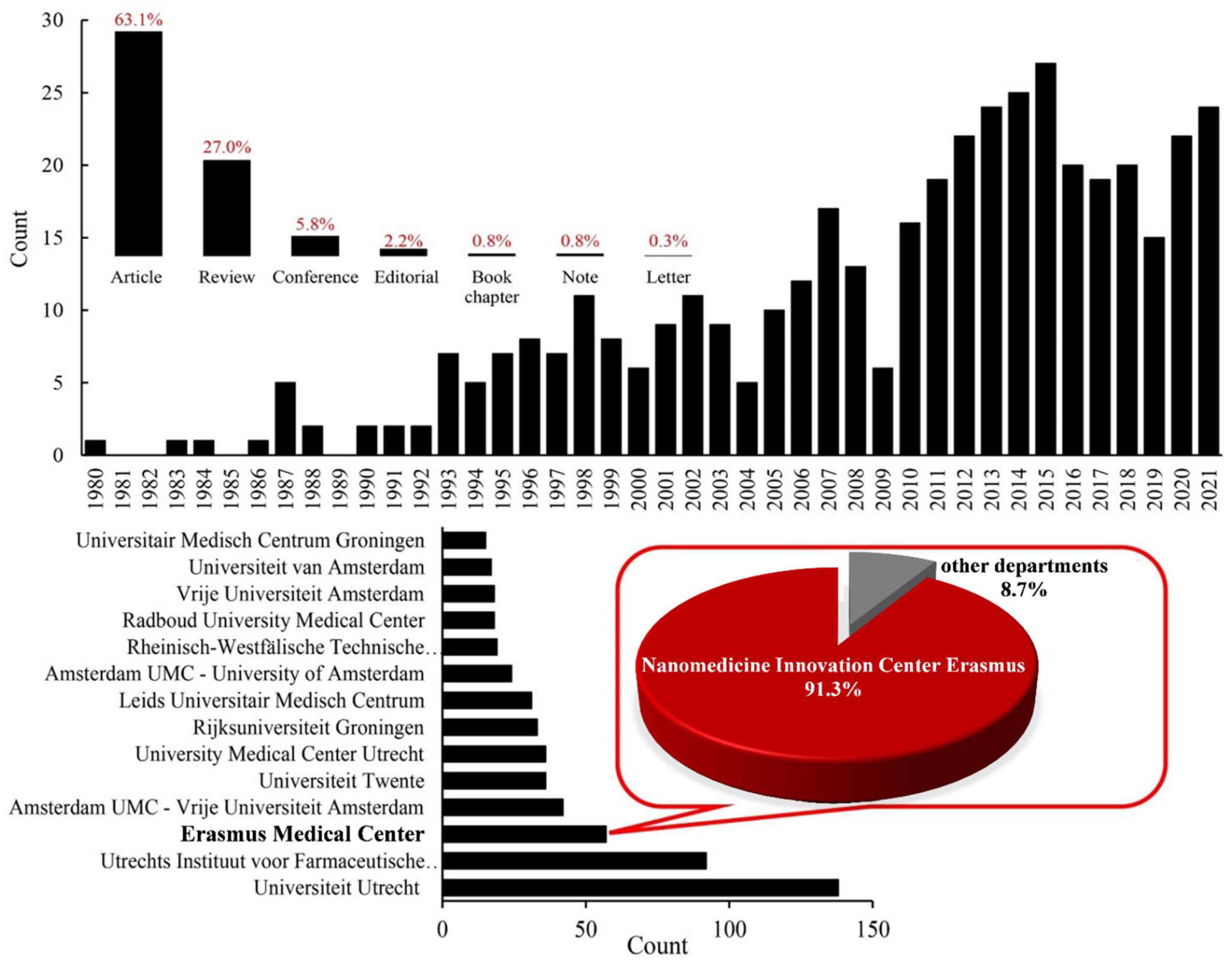
Liposomal Drug Delivery Systems for Cancer Therapy: The Rotterdam Experience

 ,
,
Abstract
:1. Introduction
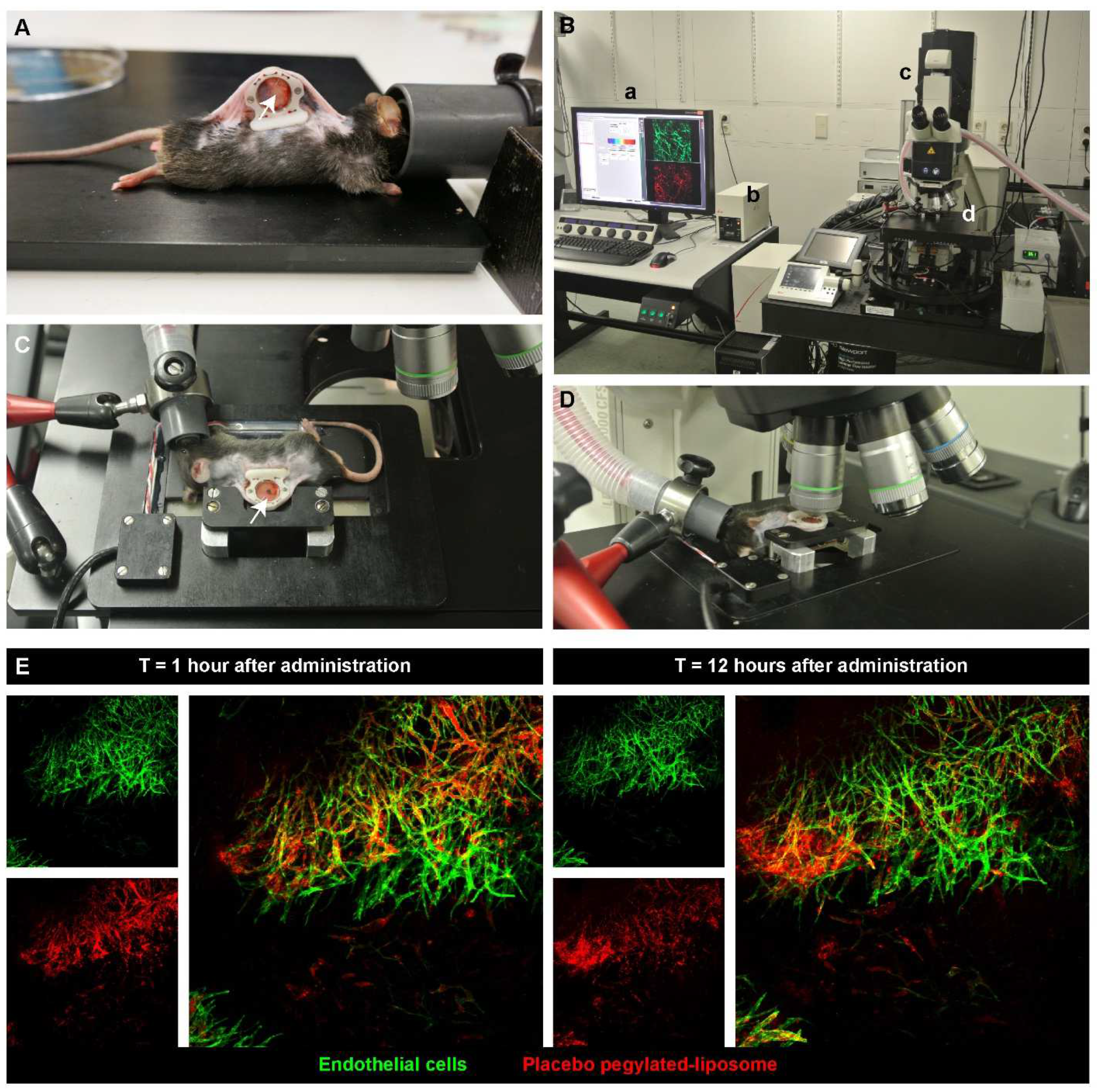
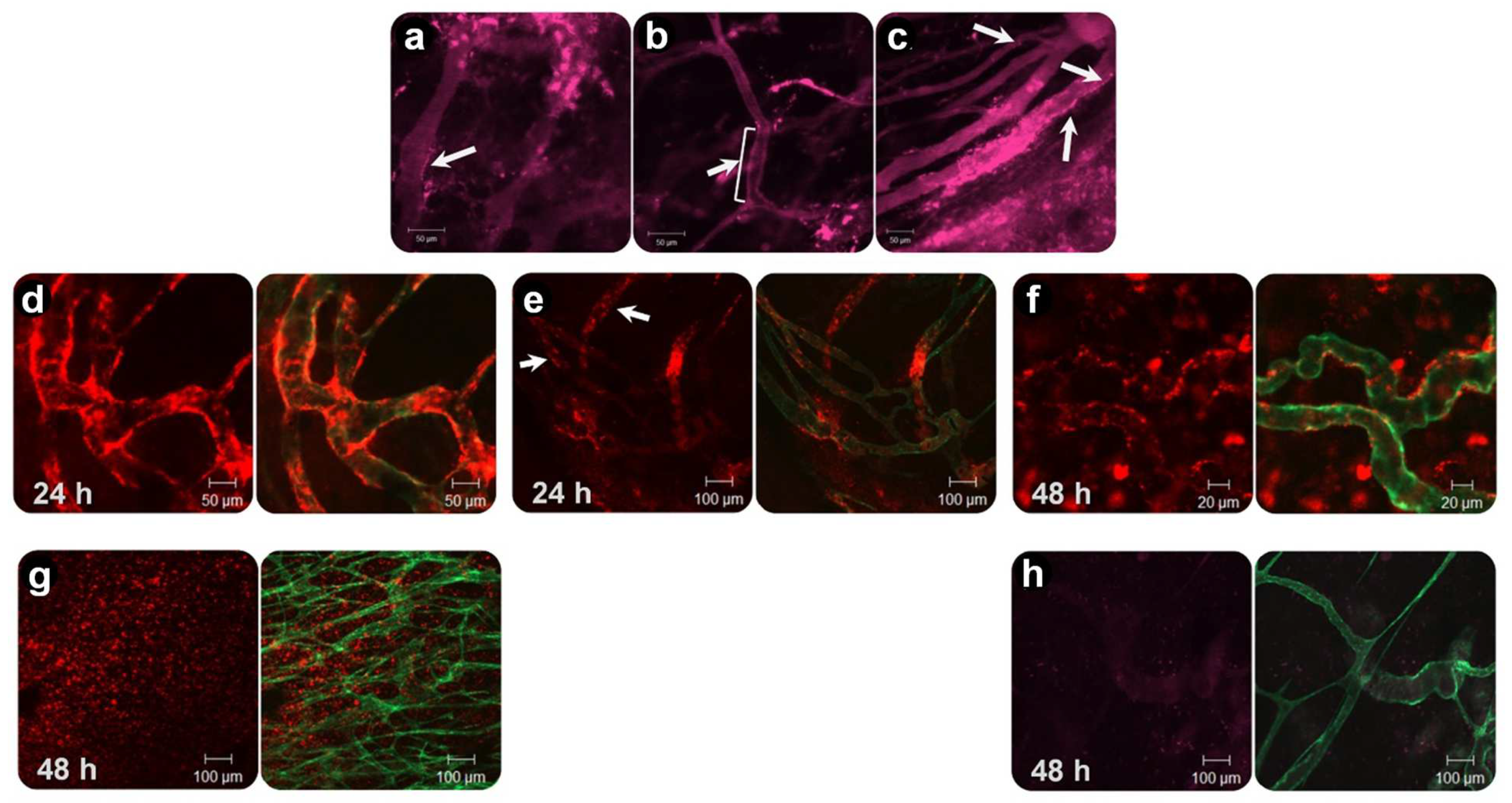
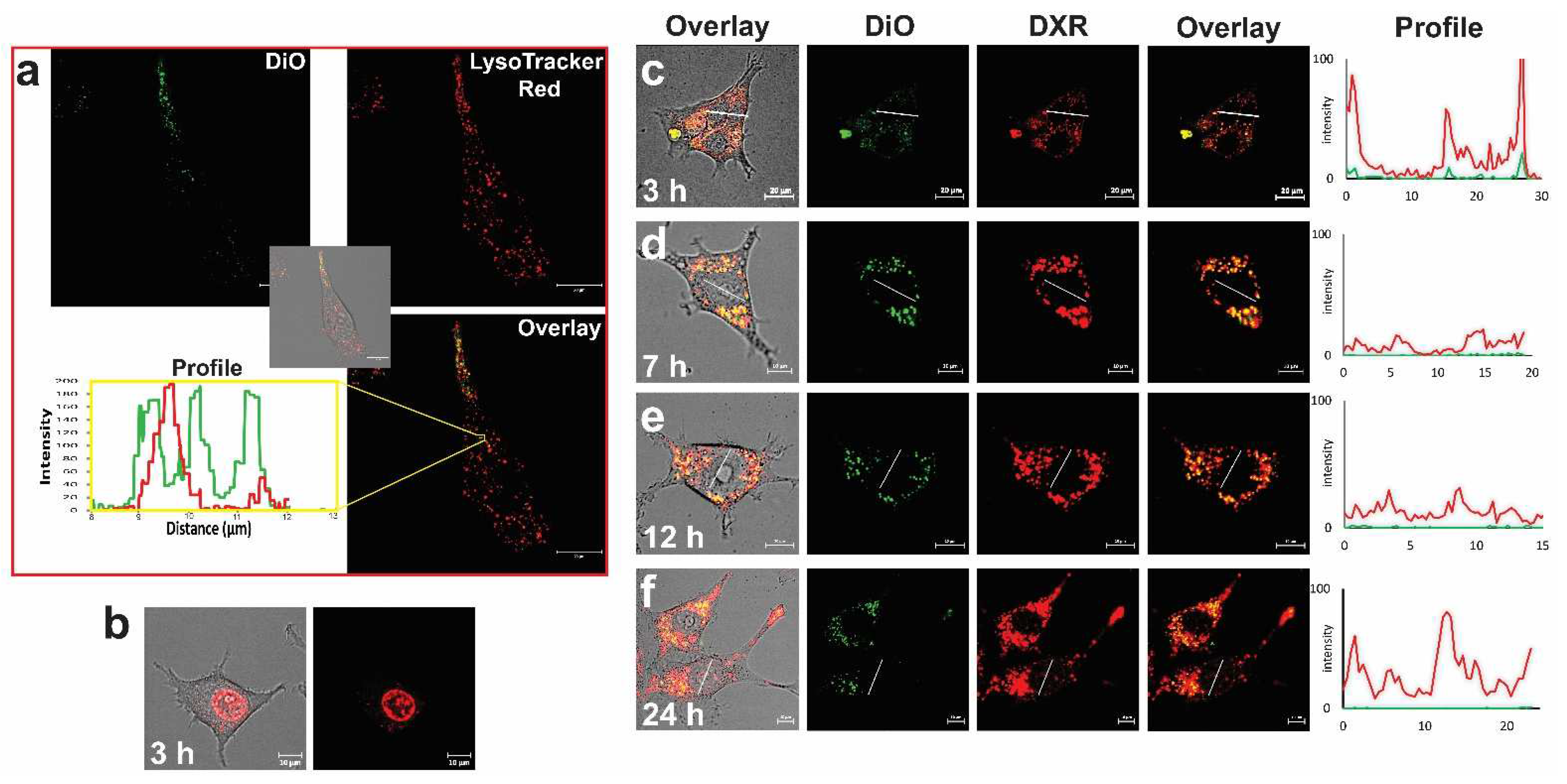
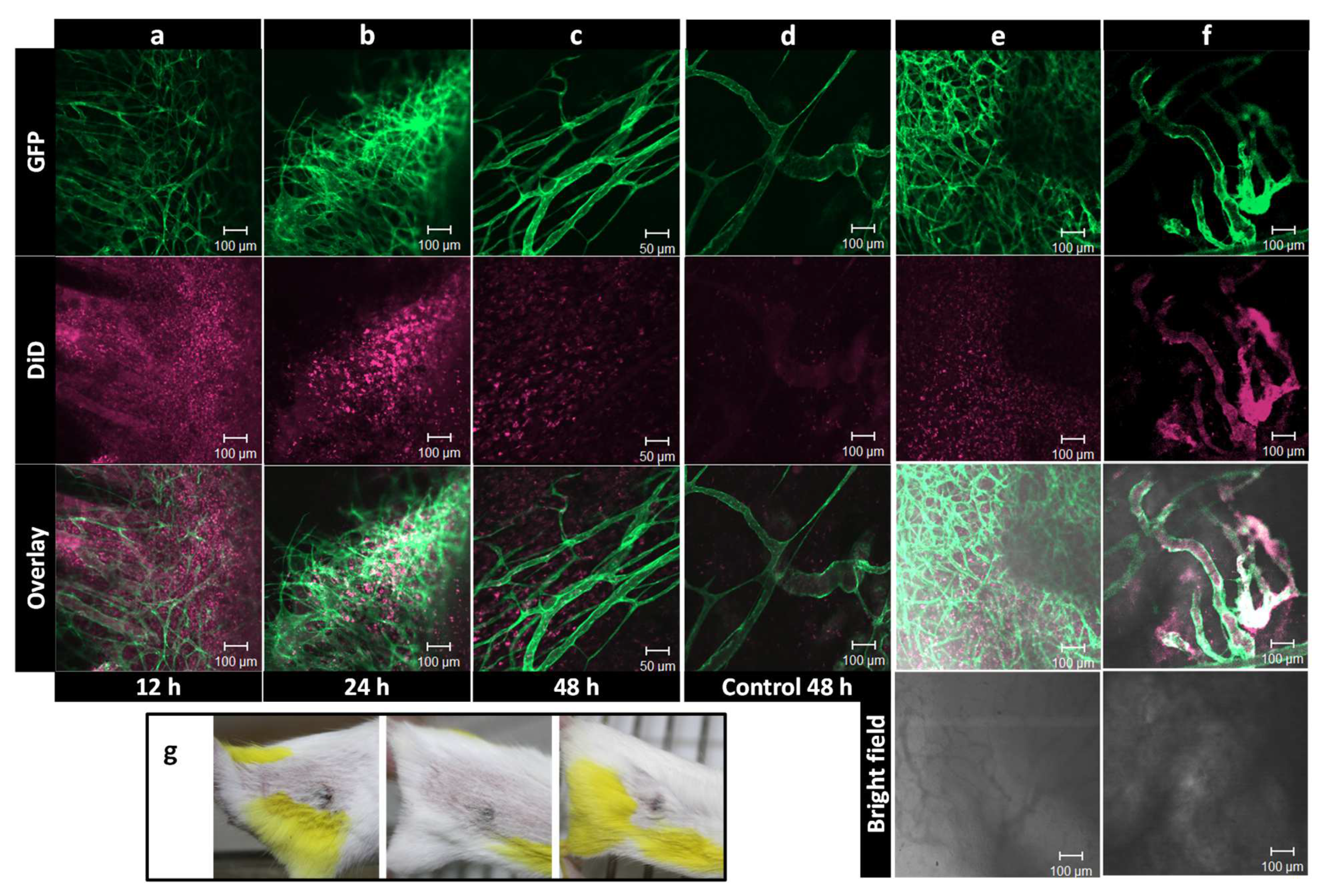
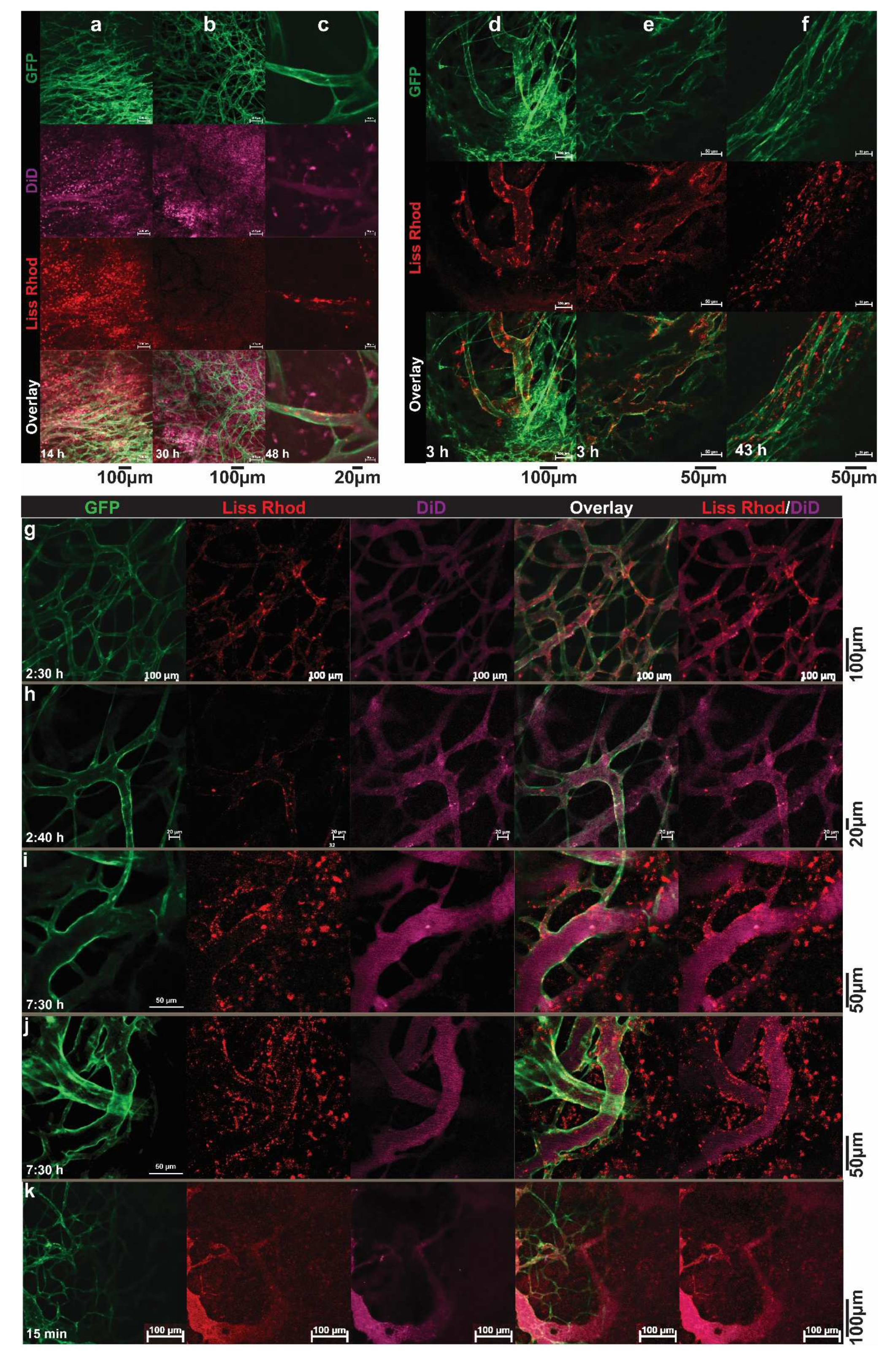
2. Visualizing the Intratumoral Fate of Nanomedicines by Intravital Microscopy
3. Augmentation of Vascular Permeability by Tumor Necrosis Factor α
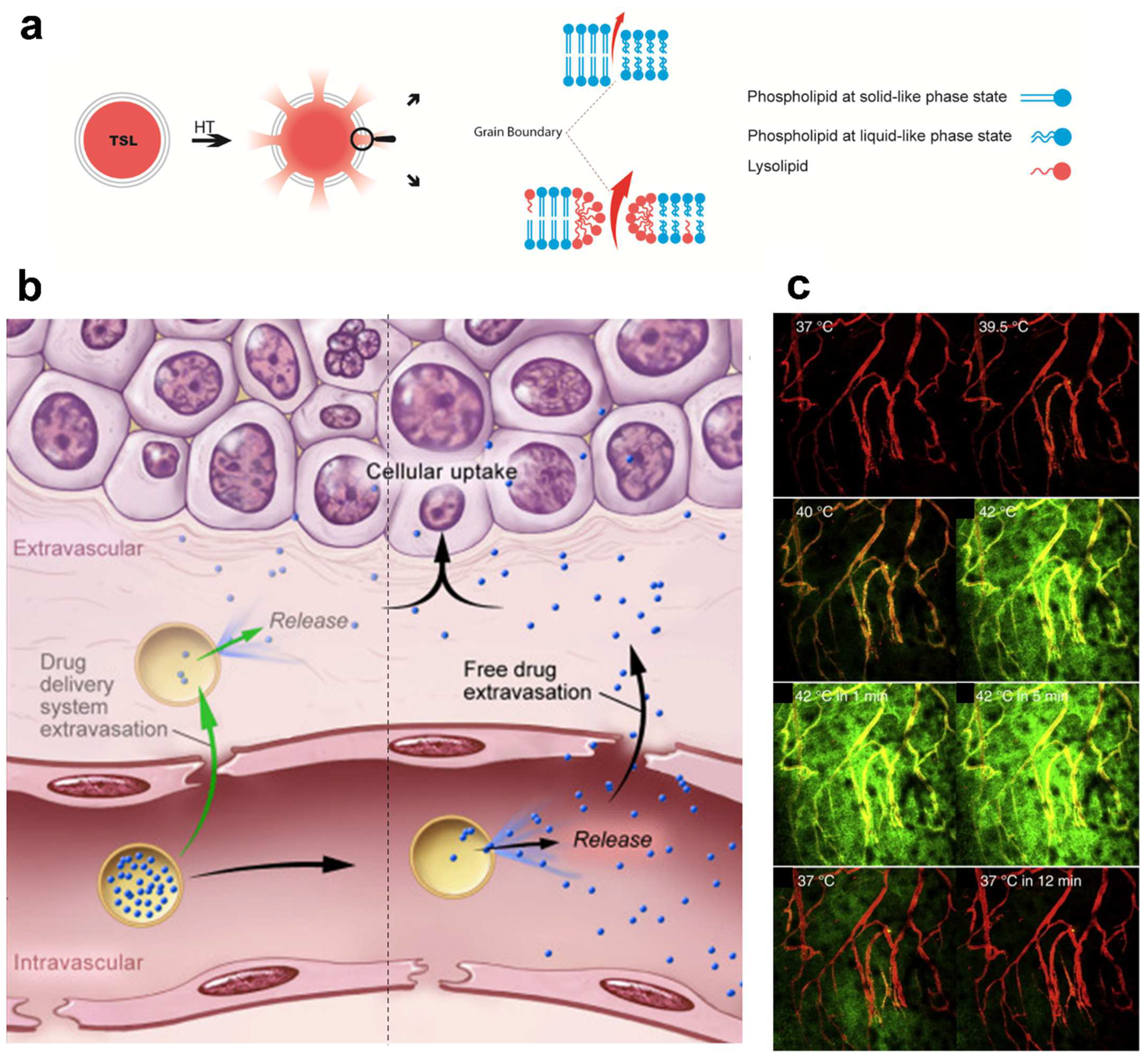
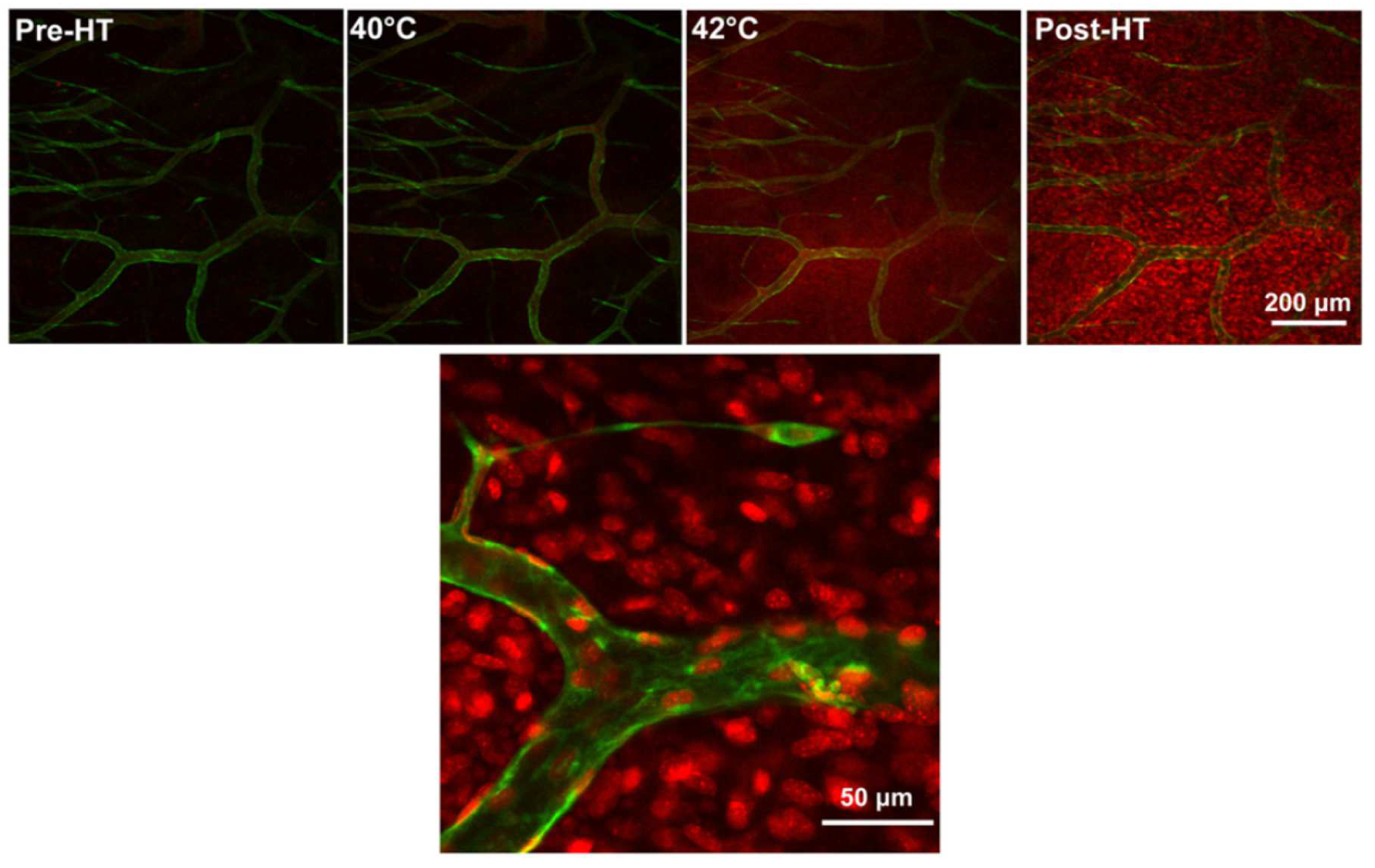
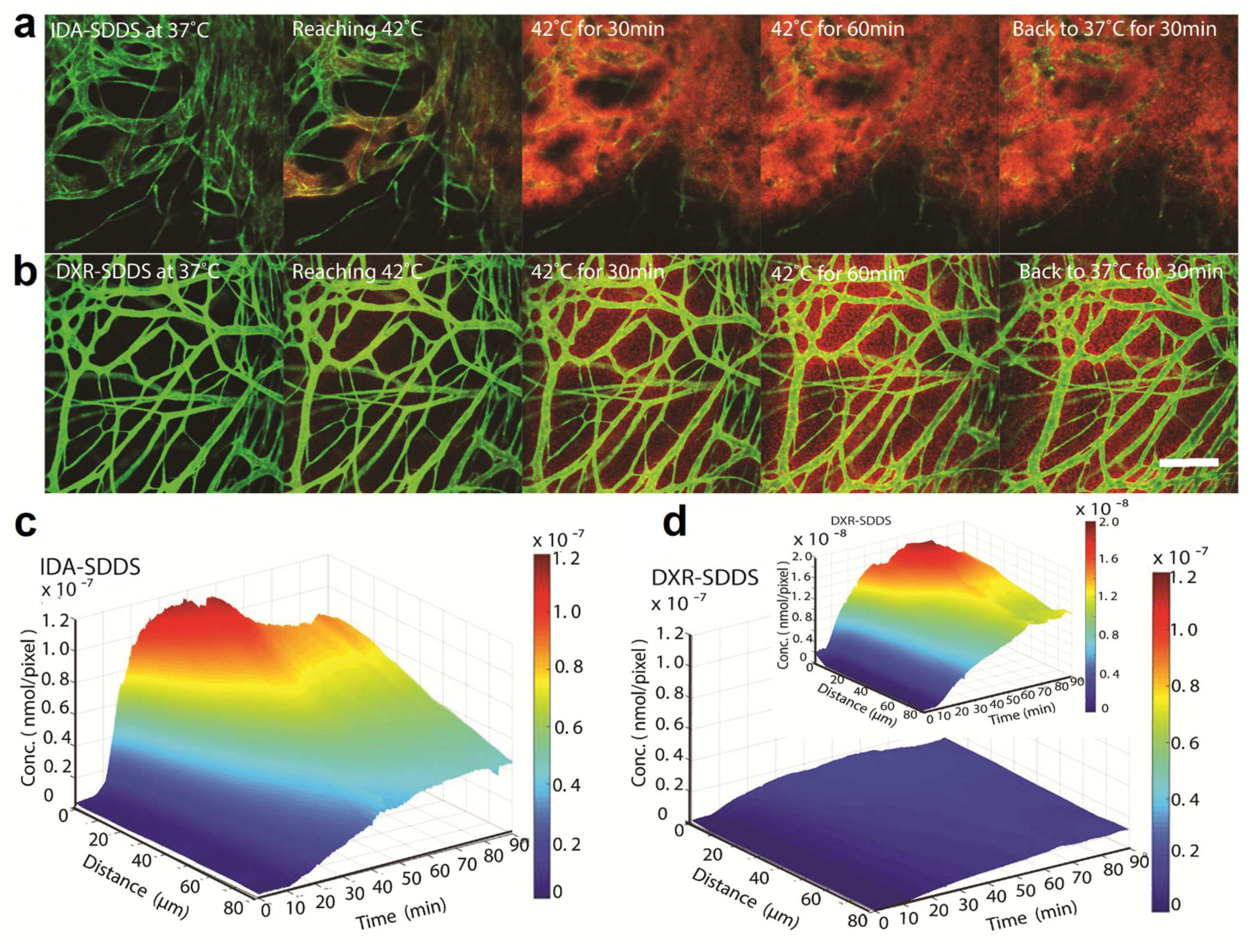
4. Application of Heat- and Temperature-Sensitive Liposomes
- (a)
- (b)
- (c)
- Impairing the cellular mechanisms that repair DNA damages [97].
- (d)
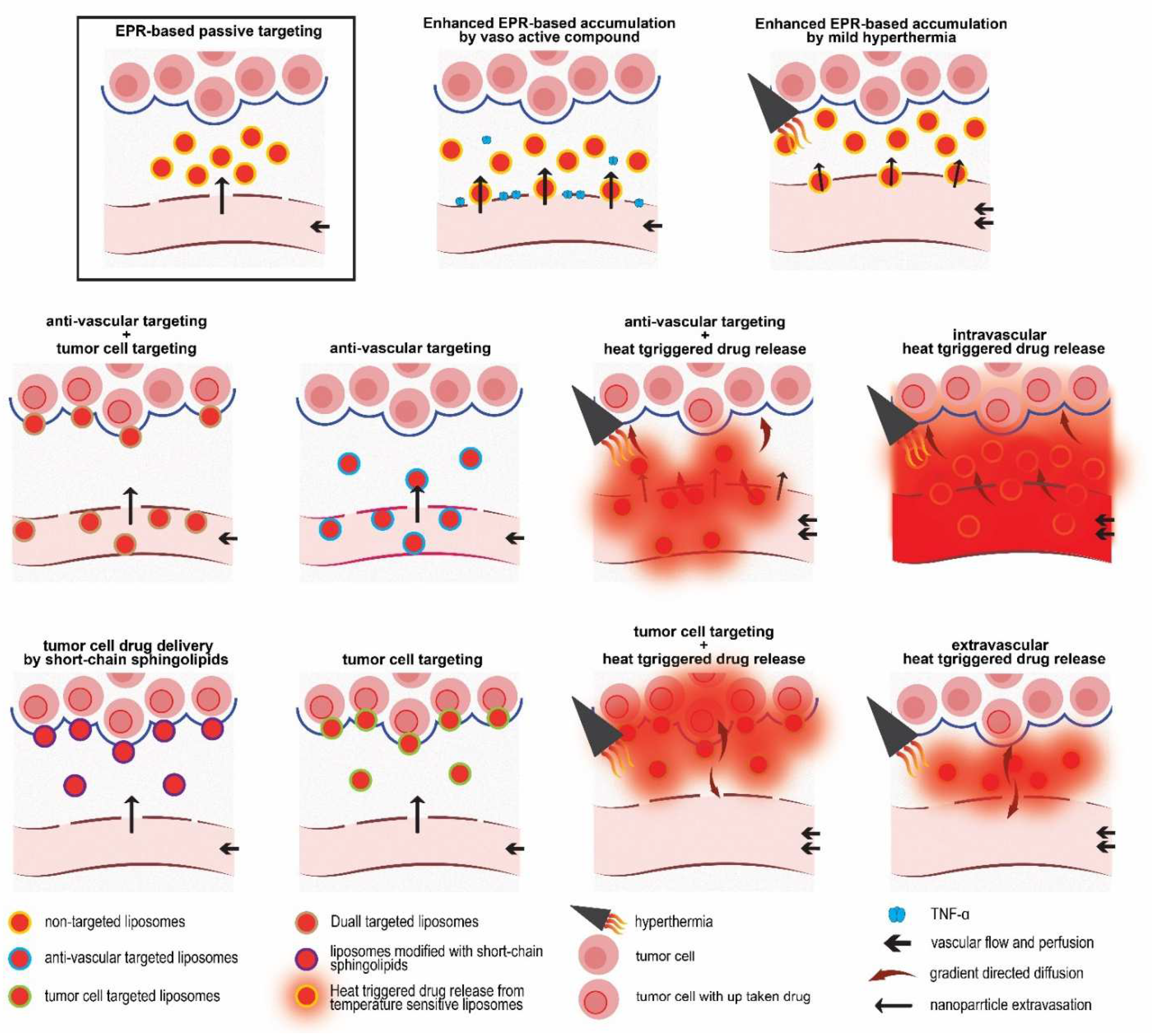
5. Active-Targeted Drug Delivery in Cancer Therapy
5.1. Anti-Vascular Targeting
Combination of Anti-Vascular Targeting and Hyperthermia
- Cationic charged TSL:
- RGD-modified TSL
5.2. Tumor Cell Targeting
5.3. Combination of Vascular Targeting and Tumor Cell Targeting
6. Enhancing Cellular Drug Delivery by Short Chain Sphingolipids
7. Liposome-Based Cancer Immunotherapy
8. A NICE Perspective
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Dickens, E.; Ahmed, S. Principles of cancer treatment by chemotherapy. Surgery 2018, 36, 134–138. [Google Scholar]
- Sharifi, M.; Cho, W.C.; Ansariesfahani, A.; Tarharoudi, R.; Malekisarvar, H.; Sari, S.; Bloukh, S.H.; Edis, Z.; Amin, M.; Gleghorn, J.P. An Updated Review on EPR-Based Solid Tumor Targeting Nanocarriers for Cancer Treatment. Cancers 2022, 14, 2868. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Papahadjopoulos, D.; Allen, T.M.; Gabizon, A.; Mayhew, E.; Matthay, K.; Huang, S.K.; Lee, K.D.; Woodle, M.C.; Lasic, D.D.; Redemann, C.; et al. Sterically stabilized liposomes: Improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc. Natl. Acad. Sci. USA 1991, 88, 11460–11464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakor, A.S.; Gambhir, S.S. Nanooncology: The future of cancer diagnosis and therapy. CA Cancer J. Clin. 2013, 63, 395–418. [Google Scholar] [CrossRef]
- Wang, A.Z.; Langer, R.; Farokhzad, O.C. Nanoparticle delivery of cancer drugs. Annu. Rev. Med. 2012, 63, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Douer, D. Efficacy and Safety of Vincristine Sulfate Liposome Injection in the Treatment of Adult Acute Lymphocytic Leukemia. Oncologist 2016, 21, 840–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deitcher, O.R.; Glaspy, J.; Gonzalez, R.; Sato, T.; Bedikian, A.Y.; Segarini, K.; Silverman, J.; Deitcher, S.R. High-dose vincristine sulfate liposome injection (Marqibo) Is not associated with clinically meaningful hematologic toxicity. Clin. Lymphoma Myeloma Leuk. 2014, 14, 197–202. [Google Scholar] [CrossRef]
- Silverman, J.A.; Deitcher, S.R. Marqibo(R) (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother. Pharmacol. 2013, 71, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Passero, F.C., Jr.; Grapsa, D.; Syrigos, K.N.; Saif, M.W. The safety and efficacy of Onivyde (irinotecan liposome injection) for the treatment of metastatic pancreatic cancer following gemcitabine-based therapy. Expert Rev. Anticancer Ther. 2016, 16, 697–703. [Google Scholar] [CrossRef]
- Zhang, H. Onivyde for the therapy of multiple solid tumors. Onco Targets Ther. 2016, 9, 3001–3007. [Google Scholar] [CrossRef] [Green Version]
- Lamb, Y.N.; Scott, L.J. Liposomal Irinotecan: A Review in Metastatic Pancreatic Adenocarcinoma. Drugs 2017, 77, 785–792. [Google Scholar] [CrossRef]
- Hare, J.I.; Lammers, T.; Ashford, M.B.; Puri, S.; Storm, G.; Barry, S.T. Challenges and strategies in anti-cancer nanomedicine development: An industry perspective. Adv. Drug Deliv. Rev. 2017, 108, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Lammers, T.; Kiessling, F.; Ashford, M.; Hennink, W.; Crommelin, D.; Storm, G. Cancer nanomedicine: Is targeting our target? Nat. Rev. Mater. 2016, 1, 16069. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Eberhard, A.; Kahlert, S.; Goede, V.; Hemmerlein, B.; Plate, K.H.; Augustin, H.G. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: Implications for antiangiogenic tumor therapies. Cancer Res. 2000, 60, 1388–1393. [Google Scholar] [PubMed]
- El Emir, E.; Qureshi, U.; Dearling, J.L.; Boxer, G.M.; Clatworthy, I.; Folarin, A.A.; Robson, M.P.; Nagl, S.; Konerding, M.A.; Pedley, R.B. Predicting response to radioimmunotherapy from the tumor microenvironment of colorectal carcinomas. Cancer Res. 2007, 67, 11896–11905. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Shields, A.F.; Siegel, B.A.; Miller, K.D.; Krop, I.; Ma, C.X.; LoRusso, P.M.; Munster, P.N.; Campbell, K.; Gaddy, D.F.; et al. (64)Cu-MM-302 Positron Emission Tomography Quantifies Variability of Enhanced Permeability and Retention of Nanoparticles in Relation to Treatment Response in Patients with Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 4190–4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Islam, W.; Maeda, H. Exploiting the dynamics of the EPR effect and strategies to improve the therapeutic effects of nanomedicines by using EPR effect enhancers. Adv. Drug Deliv. Rev. 2020, 157, 142–160. [Google Scholar] [CrossRef]
- Shi, Y.; van der Meel, R.; Chen, X.; Lammers, T. The EPR effect and beyond: Strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics 2020, 10, 7921–7924. [Google Scholar] [CrossRef]
- Golombek, S.K.; May, J.N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef]
- Gregoriadis, G. Liposomology: Delivering the message. J. Liposome Res. 2018, 28, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; ten Hagen, T.L.; Schipper, D.; Wijnberg, T.M.; van Rhoon, G.C.; Eggermont, A.M.; Lindner, L.H.; Koning, G.A. Triggered content release from optimized stealth thermosensitive liposomes using mild hyperthermia. J. Control. Release 2010, 143, 274–279. [Google Scholar] [CrossRef]
- Li, L.; ten Hagen, T.L.; Haeri, A.; Soullie, T.; Scholten, C.; Seynhaeve, A.L.; Eggermont, A.M.; Koning, G.A. A novel two-step mild hyperthermia for advanced liposomal chemotherapy. J. Control. Release 2014, 174, 202–208. [Google Scholar] [CrossRef]
- Lokerse, W.J.; Kneepkens, E.C.; ten Hagen, T.L.; Eggermont, A.M.; Grull, H.; Koning, G.A. In depth study on thermosensitive liposomes: Optimizing formulations for tumor specific therapy and in vitro to in vivo relations. Biomaterials 2016, 82, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Ten Hagen, T.L.M. Inhomogeneous crystal grain formation in DPPC-DSPC based thermosensitive liposomes determines content release kinetics. J. Control. Release 2017, 247, 64–72. [Google Scholar] [CrossRef]
- Lu, T.; Haemmerich, D.; Liu, H.; Seynhaeve, A.L.B.; van Rhoon, G.C.; Houtsmuller, A.B.; Ten Hagen, T.L.M. Externally triggered smart drug delivery system encapsulating idarubicin shows superior kinetics and enhances tumoral drug uptake and response. Theranostics 2021, 11, 5700–5712. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Mansourian, M.; Koning, G.A.; Badiee, A.; Jaafari, M.R.; Ten Hagen, T.L. Development of a novel cyclic RGD peptide for multiple targeting approaches of liposomes to tumor region. J. Control. Release 2015, 220, 308–315. [Google Scholar] [CrossRef]
- Dicheva, B.M.; ten Hagen, T.L.M.; Seynhaeve, A.L.B.; Amin, M.; Eggermont, A.M.M.; Koning, G.A. Enhanced Specificity and Drug Delivery in Tumors by cRGD—Anchoring Thermosensitive Liposomes. Pharm. Res.-Dordr 2015, 32, 3862–3876. [Google Scholar] [CrossRef] [Green Version]
- Dicheva, B.M.; ten Hagen, T.L.; Li, L.; Schipper, D.; Seynhaeve, A.L.; van Rhoon, G.C.; Eggermont, A.M.; Lindner, L.H.; Koning, G.A. Cationic thermosensitive liposomes: A novel dual targeted heat-triggered drug delivery approach for endothelial and tumor cells. Nano Lett. 2013, 13, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Dicheva, B.M.; Seynhaeve, A.L.; Soulie, T.; Eggermont, A.M.; Ten Hagen, T.L.; Koning, G.A. Pharmacokinetics, Tissue Distribution and Therapeutic Effect of Cationic Thermosensitive Liposomal Doxorubicin Upon Mild Hyperthermia. Pharm. Res. 2016, 33, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Bagheri, M.; Mansourian, M.; Jaafari, M.R.; Ten Hagen, T.L. Regulation of in vivo behavior of TAT-modified liposome by associated protein corona and avidity to tumor cells. Int. J. Nanomed. 2018, 13, 7441–7455. [Google Scholar] [CrossRef] [Green Version]
- Zalba, S.; Contreras, A.M.; Haeri, A.; Ten Hagen, T.L.; Navarro, I.; Koning, G.; Garrido, M.J. Cetuximab-oxaliplatin-liposomes for epidermal growth factor receptor targeted chemotherapy of colorectal cancer. J. Control. Release 2015, 210, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Mansourian, M.; Burgers, P.C.; Amin, B.; Jaafari, M.R.; ten Hagen, T.L.M. Increased Targeting Area in Tumors by Dual-Ligand Modification of Liposomes with RGD and TAT Peptides. Pharmaceutics 2022, 14, 458. [Google Scholar] [CrossRef] [PubMed]
- van Lummel, M.; van Blitterswijk, W.J.; Vink, S.R.; Veldman, R.J.; van der Valk, M.A.; Schipper, D.; Dicheva, B.M.; Eggermont, A.M.; ten Hagen, T.L.; Verheij, M.; et al. Enriching lipid nanovesicles with short-chain glucosylceramide improves doxorubicin delivery and efficacy in solid tumors. FASEB J. 2011, 25, 280–289. [Google Scholar] [CrossRef] [Green Version]
- Pedrosa, L.R.; van Hell, A.; Suss, R.; van Blitterswijk, W.J.; Seynhaeve, A.L.; van Cappellen, W.A.; Eggermont, A.M.; ten Hagen, T.L.; Verheij, M.; Koning, G.A. Improving intracellular doxorubicin delivery through nanoliposomes equipped with selective tumor cell membrane permeabilizing short-chain sphingolipids. Pharm. Res. 2013, 30, 1883–1895. [Google Scholar] [CrossRef]
- Cordeiro Pedrosa, L.R.; van Cappellen, W.A.; Steurer, B.; Ciceri, D.; ten Hagen, T.L.; Eggermont, A.M.; Verheij, M.; Goni, F.M.; Koning, G.A.; Contreras, F.X. C8-glycosphingolipids preferentially insert into tumor cell membranes and promote chemotherapeutic drug uptake. Biochim. Biophys. Acta 2015, 1848, 1656–1670. [Google Scholar] [CrossRef] [Green Version]
- Zalba, S.; Seynhaeve, A.L.B.; Brouwers, J.F.; Suss, R.; Verheij, M.; Ten Hagen, T.L.M. Sensitization of drug resistant sarcoma tumors by membrane modulation via short chain sphingolipid-containing nanoparticles. Nanoscale 2020, 12, 16967–16979. [Google Scholar] [CrossRef]
- Cordeiro Pedrosa, L.R.; van Tellingen, O.; Soullie, T.; Seynhaeve, A.L.; Eggermont, A.M.; Ten Hagen, T.L.; Verheij, M.; Koning, G.A. Plasma membrane targeting by short chain sphingolipids inserted in liposomes improves anti-tumor activity of mitoxantrone in an orthotopic breast carcinoma xenograft model. Eur. J. Pharm. Biopharm 2015, 94, 207–219. [Google Scholar] [CrossRef]
- Saeed, M.; van Brakel, M.; Zalba, S.; Schooten, E.; Rens, J.A.; Koning, G.A.; Debets, R.; Ten Hagen, T.L. Targeting melanoma with immunoliposomes coupled to anti-MAGE A1 TCR-like single-chain antibody. Int. J. Nanomed. 2016, 11, 955–975. [Google Scholar] [CrossRef] [Green Version]
- Saeed, M.; Zalba, S.; Seynhaeve, A.L.B.; Debets, R.; Ten Hagen, T.L.M. Liposomes targeted to MHC-restricted antigen improve drug delivery and antimelanoma response. Int. J. Nanomed. 2019, 14, 2069–2089. [Google Scholar] [CrossRef] [PubMed]
- Merino, M.; Lozano, T.; Casares, N.; Lana, H.; Troconiz, I.F.; Ten Hagen, T.L.M.; Kochan, G.; Berraondo, P.; Zalba, S.; Garrido, M.J. Dual activity of PD-L1 targeted Doxorubicin immunoliposomes promoted an enhanced efficacy of the antitumor immune response in melanoma murine model. J. Nanobiotechnol. 2021, 19, 102. [Google Scholar] [CrossRef]
- Seynhaeve, A.L.B.; Dicheva, B.M.; Hoving, S.; Koning, G.A.; Ten Hagen, T.L.M. Intact Doxil is taken up intracellularly and released doxorubicin sequesters in the lysosome: Evaluated by in vitro/in vivo live cell imaging. J. Control. Release 2013, 172, 330–340. [Google Scholar] [CrossRef]
- Seynhaeve, A.L.B.; Ten Hagen, T.L.M. An adapted dorsal skinfold model used for 4D intravital followed by whole-mount imaging to reveal endothelial cell-pericyte association. Sci. Rep. 2021, 11, 20389. [Google Scholar] [CrossRef] [PubMed]
- Seynhaeve, A.L.B.; Ten Hagen, T.L.M. Intravital Microscopy of Tumor-associated Vasculature Using Advanced Dorsal Skinfold Window Chambers on Transgenic Fluorescent Mice. J. Vis. Exp. 2018, 131, e55115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seynhaeve, A.L.B.; Oostinga, D.; van Haperen, R.; Eilken, H.M.; Adams, S.; Adams, R.H.; Ten Hagen, T.L.M. Spatiotemporal endothelial cell—Pericyte association in tumors as shown by high resolution 4D intravital imaging. Sci. Rep. 2018, 8, 9596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreher, M.R.; Liu, W.; Michelich, C.R.; Dewhirst, M.W.; Yuan, F.; Chilkoti, A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J. Natl. Cancer Inst. 2006, 98, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Manzoor, A.A.; Lindner, L.H.; Landon, C.D.; Park, J.Y.; Simnick, A.J.; Dreher, M.R.; Das, S.; Hanna, G.; Park, W.; Chilkoti, A.; et al. Overcoming limitations in nanoparticle drug delivery: Triggered, intravascular release to improve drug penetration into tumors. Cancer Res. 2012, 72, 5566–5575. [Google Scholar] [CrossRef] [Green Version]
- Seynhaeve, A.L.; Hoving, S.; Schipper, D.; Vermeulen, C.E.; de Wiel-Ambagtsheer, G.; van Tiel, S.T.; Eggermont, A.M.; Ten Hagen, T.L. Tumor necrosis factor alpha mediates homogeneous distribution of liposomes in murine melanoma that contributes to a better tumor response. Cancer Res. 2007, 67, 9455–9462. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Lokerse, W.J.M.; Seynhaeve, A.L.B.; Koning, G.A.; Ten Hagen, T.L.M. Formulation and optimization of idarubicin thermosensitive liposomes provides ultrafast triggered release at mild hyperthermia and improves tumor response. J. Control. Release 2015, 220, 425–437. [Google Scholar] [CrossRef]
- Pedrosa, L.R.; Ten Hagen, T.L.; Suss, R.; van Hell, A.; Eggermont, A.M.; Verheij, M.; Koning, G.A. Short-chain glycoceramides promote intracellular mitoxantrone delivery from novel nanoliposomes into breast cancer cells. Pharm. Res. 2015, 32, 1354–1367. [Google Scholar] [CrossRef] [PubMed]
- Djanashvili, K.; ten Hagen, T.L.; Blange, R.; Schipper, D.; Peters, J.A.; Koning, G.A. Development of a liposomal delivery system for temperature-triggered release of a tumor targeting agent, Ln(III)-DOTA-phenylboronate. Bioorg. Med. Chem. 2011, 19, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Dicheva, B.M.; ten Hagen, T.L.; Schipper, D.; Seynhaeve, A.L.; van Rhoon, G.C.; Eggermont, A.M.; Koning, G.A. Targeted and heat-triggered doxorubicin delivery to tumors by dual targeted cationic thermosensitive liposomes. J. Control. Release 2014, 195, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, S.K.; Monsky, W.L.; Yuan, F.; Roberts, W.G.; Griffith, L.; Torchilin, V.P.; Jain, R.K. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proc. Natl. Acad. Sci. USA 1998, 95, 4607–4612. [Google Scholar] [CrossRef] [Green Version]
- Hashizume, H.; Baluk, P.; Morikawa, S.; McLean, J.W.; Thurston, G.; Roberge, S.; Jain, R.K.; McDonald, D.M. Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 2000, 156, 1363–1380. [Google Scholar] [CrossRef] [Green Version]
- Schiffelers, R.M.; Koning, G.A.; ten Hagen, T.L.; Fens, M.H.; Schraa, A.J.; Janssen, A.P.; Kok, R.J.; Molema, G.; Storm, G. Anti-tumor efficacy of tumor vasculature-targeted liposomal doxorubicin. J. Control. Release 2003, 91, 115–122. [Google Scholar] [CrossRef]
- Priester, M.I.; Curto, S.; Seynhaeve, A.L.B.; Perdomo, A.C.; Amin, M.; Agnass, P.; Salimibani, M.; Faridi, P.; Prakash, P.; van Rhoon, G.C.; et al. Preclinical Studies in Small Animals for Advanced Drug Delivery Using Hyperthermia and Intravital Microscopy. Cancers 2021, 13, 5146. [Google Scholar] [CrossRef]
- Seynhaeve, A.L.B.; Amin, M.; Haemmerich, D.; van Rhoon, G.C.; Ten Hagen, T.L.M. Hyperthermia and smart drug delivery systems for solid tumor therapy. Adv. Drug Deliv. Rev. 2020, 163–164, 125–144. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Stylianopoulos, T.; Boucher, Y.; Jain, R.K. Delivery of molecular and nanoscale medicine to tumors: Transport barriers and strategies. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 281–298. [Google Scholar] [CrossRef]
- Grunhagen, D.J.; de Wilt, J.H.; ten Hagen, T.L.; Eggermont, A.M. Technology insight: Utility of TNF-alpha-based isolated limb perfusion to avoid amputation of irresectable tumors of the extremities. Nat. Clin. Pract. Oncol. 2006, 3, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Lejeune, F.; Lienard, D.; Eggermont, A. Regional administration of recombinant tumour necrosis factor-alpha in cancer, with special reference to melanoma. BioDrugs 1998, 9, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Cherix, S.; Speiser, M.; Matter, M.; Raffoul, W.; Lienard, D.; Theumann, N.; Mouhsine, E.; Mirimanoff, R.O.; Leyvraz, S.; Lejeune, F.J.; et al. Isolated limb perfusion with tumor necrosis factor and melphalan for non-resectable soft tissue sarcomas: Long-term results on efficacy and limb salvage in a selected group of patients. J. Surg. Oncol. 2008, 98, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunstein, F.; Santos, I.D.; Ferreira, L.M.; van Tiel, S.T.; Eggermont, A.M.; Ten Hagen, T.L. Histamine combined with melphalan in isolated limb perfusion for the treatment of locally advanced soft tissue sarcomas: Preclinical studies in rats. Acta Cir. Bras. 2005, 20, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Hoving, S.; Brunstein, F.; aan de Wiel-Ambagtsheer, G.; van Tiel, S.T.; de Boeck, G.; de Bruijn, E.A.; Eggermont, A.M.; ten Hagen, T.L. Synergistic antitumor response of interleukin 2 with melphalan in isolated limb perfusion in soft tissue sarcoma-bearing rats. Cancer Res. 2005, 65, 4300–4308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoving, S.; Seynhaeve, A.L.; van Tiel, S.T.; aan de Wiel-Ambagtsheer, G.; de Bruijn, E.A.; Eggermont, A.M.; ten Hagen, T.L. Early destruction of tumor vasculature in tumor necrosis factor-alpha-based isolated limb perfusion is responsible for tumor response. Anticancer Drugs 2006, 17, 949–959. [Google Scholar] [CrossRef]
- van der Veen, A.H.; de Wilt, J.H.; Eggermont, A.M.; van Tiel, S.T.; Seynhaeve, A.L.; ten Hagen, T.L. TNF-alpha augments intratumoural concentrations of doxorubicin in TNF-alpha-based isolated limb perfusion in rat sarcoma models and enhances anti-tumour effects. Br. J. Cancer 2000, 82, 973–980. [Google Scholar] [CrossRef]
- de Wilt, J.H.; ten Hagen, T.L.; de Boeck, G.; van Tiel, S.T.; de Bruijn, E.A.; Eggermont, A.M. Tumour necrosis factor alpha increases melphalan concentration in tumour tissue after isolated limb perfusion. Br. J. Cancer 2000, 82, 1000–1003. [Google Scholar] [CrossRef]
- Verhoef, C.; de Wilt, J.H.; Grunhagen, D.J.; van Geel, A.N.; ten Hagen, T.L.; Eggermont, A.M. Isolated limb perfusion with melphalan and TNF-alpha in the treatment of extremity sarcoma. Curr. Treat. Options Oncol. 2007, 8, 417–427. [Google Scholar] [CrossRef] [Green Version]
- van Rhoon, G.C.; Franckena, M.; ten Hagen, T.L.M. A moderate thermal dose is sufficient for effective free and TSL based thermochemotherapy. Adv. Drug Deliv. Rev. 2020, 163–164, 145–156. [Google Scholar] [CrossRef]
- Kong, L.; Chen, Q.; Campbell, F.; Snaar-Jagalska, E.; Kros, A. Light-Triggered Cancer Cell Specific Targeting and Liposomal Drug Delivery in a Zebrafish Xenograft Model. Adv. Healthc. Mater. 2020, 9, 1901489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoving, S.; Seynhaeve, A.L.; van Tiel, S.T.; Eggermont, A.M.; ten Hagen, T.L. Addition of low-dose tumor necrosis factor-alpha to systemic treatment with STEALTH liposomal doxorubicin (Doxil) improved anti-tumor activity in osteosarcoma-bearing rats. Anticancer Drugs 2005, 16, 667–674. [Google Scholar] [CrossRef]
- Ten Hagen, T.L.; Van Der Veen, A.H.; Nooijen, P.T.; Van Tiel, S.T.; Seynhaeve, A.L.; Eggermont, A.M. Low-dose tumor necrosis factor-alpha augments antitumor activity of stealth liposomal doxorubicin (DOXIL) in soft tissue sarcoma-bearing rats. Int. J. Cancer 2000, 87, 829–837. [Google Scholar] [CrossRef]
- Brouckaert, P.; Takahashi, N.; van Tiel, S.T.; Hostens, J.; Eggermont, A.M.M.; Seynhaeve, A.L.B.; Fiers, W.; ten Hagen, T.L.M. Tumor necrosis factor-α augmented tumor response in B16BL6 melanoma-bearing mice treated with stealth liposomal doxorubicin (Doxil®) correlates with altered Doxil® pharmacokinetics. Int. J. Cancer 2004, 109, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Seetharamu, N.; Kim, E.; Hochster, H.; Martin, F.; Muggia, F. Phase II study of liposomal cisplatin (SPI-77) in platinum-sensitive recurrences of ovarian cancer. Anticancer Res. 2010, 30, 541–545. [Google Scholar]
- Lee, E.S.; Gao, Z.; Bae, Y.H. Recent progress in tumor pH targeting nanotechnology. J. Control. Release 2008, 132, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Yatvin, M.B.; Kreutz, W.; Horwitz, B.A.; Shinitzky, M. pH-sensitive liposomes: Possible clinical implications. Science 1980, 210, 1253–1255. [Google Scholar] [CrossRef]
- Feng, L.Z.; Dong, Z.L.; Tao, D.L.; Zhang, Y.C.; Liu, Z. The acidic tumor microenvironment: A target for smart cancer nano-theranostics. Natl. Sci. Rev. 2018, 5, 269–286. [Google Scholar] [CrossRef] [Green Version]
- Andresen, T.L.; Thompson, D.H.; Kaasgaard, T. Enzyme-triggered nanomedicine: Drug release strategies in cancer therapy. Mol. Membr. Biol. 2010, 27, 353–363. [Google Scholar] [CrossRef]
- Ong, W.; Yang, Y.; Cruciano, A.C.; McCarley, R.L. Redox-triggered contents release from liposomes. J. Am. Chem. Soc. 2008, 130, 14739–14744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Mo, S.; Coussios, C.C.; Seymour, L.; Carlisle, R. Ultrasound-enhanced drug delivery for cancer. Expert Opin. Drug Deliv. 2012, 9, 1525–1538. [Google Scholar] [CrossRef]
- Eggen, S.; Fagerland, S.M.; Morch, Y.; Hansen, R.; Sovik, K.; Berg, S.; Furu, H.; Bohn, A.D.; Lilledahl, M.B.; Angelsen, A.; et al. Ultrasound-enhanced drug delivery in prostate cancer xenografts by nanoparticles stabilizing microbubbles. J. Control. Release 2014, 187, 39–49. [Google Scholar] [CrossRef]
- Huang, S.L. Liposomes in ultrasonic drug and gene delivery. Adv. Drug Deliv. Rev. 2008, 60, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.J.; Romanowski, M. Light-activated content release from liposomes. Theranostics 2012, 2, 1020–1036. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Zhang, Y. Photocontrolled nanoparticle delivery systems for biomedical applications. Acc. Chem. Res. 2014, 47, 3052–3060. [Google Scholar] [CrossRef]
- Li, L.; Ten Hagen, T.L.; Bolkestein, M.; Gasselhuber, A.; Yatvin, J.; van Rhoon, G.C.; Eggermont, A.M.; Haemmerich, D.; Koning, G.A. Improved intratumoral nanoparticle extravasation and penetration by mild hyperthermia. J. Control. Release 2013, 167, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Gaber, M.H.; Wu, N.Z.; Hong, K.; Huang, S.K.; Dewhirst, M.W.; Papahadjopoulos, D. Thermosensitive liposomes: Extravasation and release of contents in tumor microvascular networks. Int. J. Radiat. Oncol. Biol. Phys. 1996, 36, 1177–1187. [Google Scholar] [CrossRef]
- Nittayacharn, P.; Yuan, H.X.; Hernandez, C.; Bielecki, P.; Zhou, H.; Exner, A.A. Enhancing Tumor Drug Distribution With Ultrasound-Triggered Nanobubbles. J. Pharm. Sci. 2019, 108, 3091–3098. [Google Scholar] [CrossRef] [PubMed]
- Rizzitelli, S.; Giustetto, P.; Cutrin, J.C.; Delli Castelli, D.; Boffa, C.; Ruzza, M.; Menchise, V.; Molinari, F.; Aime, S.; Terreno, E. Sonosensitive theranostic liposomes for preclinical in vivo MRI-guided visualization of doxorubicin release stimulated by pulsed low intensity non-focused ultrasound. J. Control. Release 2015, 202, 21–30. [Google Scholar] [CrossRef]
- Griffin, R.J.; Ogawa, A.; Williams, B.W.; Song, C.W. Hyperthermic enhancement of tumor radiosensitization strategies. Immunol. Investig. 2005, 34, 343–359. [Google Scholar] [CrossRef]
- Griffin, R.J.; Dings, R.P.; Jamshidi-Parsian, A.; Song, C.W. Mild temperature hyperthermia and radiation therapy: Role of tumour vascular thermotolerance and relevant physiological factors. Int. J. Hyperth. 2010, 26, 256–263. [Google Scholar] [CrossRef]
- Lepock, J.R. Cellular effects of hyperthermia: Relevance to the minimum dose for thermal damage. Int. J. Hyperth. 2003, 19, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Roti, J.L.R. Cellular responses to hyperthermia (40-46 degrees C): Cell killing and molecular events. Int. J. Hyperth. 2008, 24, 3–15. [Google Scholar] [CrossRef]
- Overgaard, J.; Grau, C.; Lindegaard, J.C.; Horsman, M.R. The Potential of Using Hyperthermia to Eliminate Radioresistant Hypoxic Cells. Radiother. Oncol. 1991, 20, 113–116. [Google Scholar] [CrossRef]
- Oei, A.L.; Vriend, L.E.M.; Crezee, J.; Franken, N.A.P.; Krawczyk, P.M. Effects of hyperthermia on DNA repair pathways: One treatment to inhibit them all. Radiat Oncol. 2015, 10, 165. [Google Scholar] [CrossRef] [Green Version]
- Skitzki, J.J.; Repasky, E.A.; Evans, S.S. Hyperthermia as an immunotherapy strategy for cancer. Curr. Opin Investig. Dr. 2009, 10, 550–558. [Google Scholar]
- Lee, S.; Son, B.; Park, G.; Kim, H.; Kang, H.; Jeon, J.; Youn, H.; Youn, B. Immunogenic Effect of Hyperthermia on Enhancing Radiotherapeutic Efficacy. Int. J. Mol. Sci. 2018, 19, 2795. [Google Scholar] [CrossRef] [Green Version]
- Toraya-Brown, S.; Sheen, M.R.; Zhang, P.; Chen, L.; Baird, J.R.; Demidenko, E.; Turk, M.J.; Hoopes, P.J.; Conejo-Garcia, J.R.; Fiering, S. Local hyperthermia treatment of tumors induces CD8(+) T cell-mediated resistance against distal and secondary tumors. Nanomedicine 2014, 10, 1273–1285. [Google Scholar] [CrossRef] [Green Version]
- Tsang, Y.W.; Huang, C.C.; Yang, K.L.; Chi, M.S.; Chiang, H.C.; Wang, Y.S.; Andocs, G.; Szasz, A.; Li, W.T.; Chi, K.H. Improving immunological tumor microenvironment using electro-hyperthermia followed by dendritic cell immunotherapy. BMC Cancer 2015, 15, 708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issels, R.D.; Lindner, L.H.; Verweij, J.; Wust, P.; Reichardt, P.; Schem, B.C.; Abdel-Rahman, S.; Daugaard, S.; Salat, C.; Wendtner, C.M.; et al. Neo-adjuvant chemotherapy alone or with regional hyperthermia for localised high-risk soft-tissue sarcoma: A randomised phase 3 multicentre study. Lancet Oncol. 2010, 11, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Issels, R.D.; Lindner, L.H.; Verweij, J.; Wessalowski, R.; Reichardt, P.; Wust, P.; Ghadjar, P.; Hohenberger, P.; Angele, M.; Salat, C.; et al. Effect of Neoadjuvant Chemotherapy Plus Regional Hyperthermia on Long-term Outcomes Among Patients With Localized High-Risk Soft Tissue Sarcoma: The EORTC 62961-ESHO 95 Randomized Clinical Trial. JAMA Oncol. 2018, 4, 483–492. [Google Scholar] [CrossRef]
- Killock, D. Sarcoma: Local hyperthermia improves survival. Nat. Rev. Clin. Oncol. 2018, 15, 266. [Google Scholar] [CrossRef]
- Issels, R.; Kampmann, E.; Kanaar, R.; Lindner, L.H. Hallmarks of hyperthermia in driving the future of clinical hyperthermia as targeted therapy: Translation into clinical application. Int. J. Hyperth. 2016, 32, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.K.; Stauffer, P.R.; Hong, K.; Guo, J.W.; Phillips, T.L.; Huang, A.; Papahadjopoulos, D. Liposomes and hyperthermia in mice: Increased tumor uptake and therapeutic efficacy of doxorubicin in sterically stabilized liposomes. Cancer Res. 1994, 54, 2186–2191. [Google Scholar] [PubMed]
- Kong, G.; Braun, R.D.; Dewhirst, M.W. Hyperthermia enables tumor-specific nanoparticle delivery: Effect of particle size. Cancer Res. 2000, 60, 4440–4445. [Google Scholar]
- Kong, G.; Braun, R.D.; Dewhirst, M.W. Characterization of the effect of hyperthermia on nanoparticle extravasation from tumor vasculature. Cancer Res. 2001, 61, 3027–3032. [Google Scholar]
- Amin, M.; Huang, W.; Seynhaeve, A.L.B.; Ten Hagen, T.L.M. Hyperthermia and Temperature-Sensitive Nanomaterials for Spatiotemporal Drug Delivery to Solid Tumors. Pharmaceutics 2020, 12, 1007. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Lammers, T.; Ten Hagen, T.L.M. Temperature-sensitive polymers to promote heat-triggered drug release from liposomes: Towards bypassing EPR. Adv. Drug Deliv. Rev. 2022, 189, 114503. [Google Scholar] [CrossRef]
- Papahadjopoulos, D.; Jacobson, K.; Nir, S.; Isac, T. Phase transitions in phospholipid vesicles. Fluorescence polarization and permeability measurements concerning the effect of temperature and cholesterol. Biochim. Biophys. Acta 1973, 311, 330–348. [Google Scholar] [CrossRef]
- Phillips, M.C.; Hauser, H.; Paltauf, F. The inter- and intra-molecular mixing of hydrocarbon chains in lecithin-water systems. Chem. Phys. Lipids 1972, 8, 127–133. [Google Scholar] [CrossRef]
- Ten Hagen, T.L.M.; Dreher, M.R.; Zalba, S.; Seynhaeve, A.L.B.; Amin, M.; Li, L.; Haemmerich, D. Drug transport kinetics of intravascular triggered drug delivery systems. Commun. Biol. 2021, 4, 920. [Google Scholar] [CrossRef]
- Yatvin, M.B.; Weinstein, J.N.; Dennis, W.H.; Blumenthal, R. Design of liposomes for enhanced local release of drugs by hyperthermia. Science 1978, 202, 1290–1293. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Magin, R.L.; Yatvin, M.B.; Zaharko, D.S. Liposomes and local hyperthermia: Selective delivery of methotrexate to heated tumors. Science 1979, 204, 188–191. [Google Scholar] [CrossRef]
- Yatvin, M.B.; Muhlensiepen, H.; Porschen, W.; Weinstein, J.N.; Feinendegen, L.E. Selective delivery of liposome-associated cis-dichlorodiammineplatinum(II) by heat and its influence on tumor drug uptake and growth. Cancer Res. 1981, 41, 1602–1607. [Google Scholar]
- Iga, K.; Hamaguchi, N.; Igari, Y.; Ogawa, Y.; Gotoh, K.; Ootsu, K.; Toguchi, H.; Shimamoto, T. Enhanced antitumor activity in mice after administration of thermosensitive liposome encapsulating cisplatin with hyperthermia. J. Pharmacol. Exp. Ther. 1991, 257, 1203–1207. [Google Scholar]
- Maruyama, K.; Unezaki, S.; Takahashi, N.; Iwatsuru, M. Enhanced delivery of doxorubicin to tumor by long-circulating thermosensitive liposomes and local hyperthermia. Biochim. Biophys. Acta 1993, 1149, 209–216. [Google Scholar] [CrossRef]
- Gaber, M.H.; Hong, K.; Huang, S.K.; Papahadjopoulos, D. Thermosensitive sterically stabilized liposomes: Formulation and in vitro studies on mechanism of doxorubicin release by bovine serum and human plasma. Pharm. Res. 1995, 12, 1407–1416. [Google Scholar] [CrossRef]
- Sadeghi, N.; Deckers, R.; Ozbakir, B.; Akthar, S.; Kok, R.J.; Lammers, T.; Storm, G. Influence of cholesterol inclusion on the doxorubicin release characteristics of lysolipid-based thermosensitive liposomes. Int. J. Pharm. 2018, 548, 778–782. [Google Scholar] [CrossRef]
- Landon, C.D.; Park, J.Y.; Needham, D.; Dewhirst, M.W. Nanoscale Drug Delivery and Hyperthermia: The Materials Design and Preclinical and Clinical Testing of Low Temperature-Sensitive Liposomes Used in Combination with Mild Hyperthermia in the Treatment of Local Cancer. Open Nanomed. J. 2011, 3, 38–64. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; ten Hagen, T.L.; Hossann, M.; Suss, R.; van Rhoon, G.C.; Eggermont, A.M.; Haemmerich, D.; Koning, G.A. Mild hyperthermia triggered doxorubicin release from optimized stealth thermosensitive liposomes improves intratumoral drug delivery and efficacy. J. Control. Release 2013, 168, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Anyarambhatla, G.R.; Needham, D. Enhancement of the phase transition permeability of DPPC liposomes by incorporation of MPPC: A new temperature-sensitive liposome for use with mild hyperthermia. J. Liposome Res. 1999, 9, 491–506. [Google Scholar] [CrossRef]
- Mills, J.K.; Needham, D. Lysolipid incorporation in dipalmitoylphosphatidylcholine bilayer membranes enhances the ion permeability and drug release rates at the membrane phase transition. Biochim. Biophys. Acta 2005, 1716, 77–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindner, L.H.; Eichhorn, M.E.; Eibl, H.; Teichert, N.; Schmitt-Sody, M.; Issels, R.D.; Dellian, M. Novel temperature-sensitive liposomes with prolonged circulation time. Clin. Cancer Res. 2004, 10, 2168–2178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossann, M.; Wiggenhorn, M.; Schwerdt, A.; Wachholz, K.; Teichert, N.; Eibl, H.; Issels, R.D.; Lindner, L.H. In vitro stability and content release properties of phosphatidylglyceroglycerol containing thermosensitive liposomes. Biochim. Biophys. Acta 2007, 1768, 2491–2499. [Google Scholar] [CrossRef] [Green Version]
- Brummelhuis, I.S.G.; Simons, M.; Lindner, L.H.; Kort, S.; de Jong, S.; Hossann, M.; Witjes, J.A.; Oosterwijk, E. DPPG2-based thermosensitive liposomes as drug delivery system for effective muscle-invasive bladder cancer treatment in vivo. Int. J. Hyperth. 2021, 38, 1415–1424. [Google Scholar] [CrossRef]
- Gasselhuber, A.; Dreher, M.R.; Rattay, F.; Wood, B.J.; Haemmerich, D. Comparison of conventional chemotherapy, stealth liposomes and temperature-sensitive liposomes in a mathematical model. PLoS ONE 2012, 7, e47453. [Google Scholar] [CrossRef]
- Chen, Q.; Krol, A.; Wright, A.; Needham, D.; Dewhirst, M.W.; Yuan, F. Tumor microvascular permeability is a key determinant for antivascular effects of doxorubicin encapsulated in a temperature sensitive liposome. Int. J. Hyperth. 2008, 24, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Tong, S.; Dewhirst, M.W.; Yuan, F. Targeting tumor microvessels using doxorubicin encapsulated in a novel thermosensitive liposome. Mol. Cancer Ther. 2004, 3, 1311–1317. [Google Scholar] [CrossRef]
- Al-Jamal, W.T.; Kostarelos, K. Mild hyperthermia accelerates doxorubicin clearance from tumour-extravasated temperature-sensitive liposomes. Nanotheranostics 2022, 6, 230–242. [Google Scholar] [CrossRef]
- Poon, R.T.; Borys, N. Lyso-thermosensitive liposomal doxorubicin: A novel approach to enhance efficacy of thermal ablation of liver cancer. Expert Opin. Pharmacother. 2009, 10, 333–343. [Google Scholar] [CrossRef]
- Dou, Y.; Hynynen, K.; Allen, C. To heat or not to heat: Challenges with clinical translation of thermosensitive liposomes. J. Control. Release 2017, 249, 63–73. [Google Scholar] [CrossRef]
- Lokerse, W.J.M.; Lazarian, A.; Kleinhempel, A.; Petrini, M.; Schwarz, P.; Hossann, M.; Holdt, L.M.; Mailander, V.; Lindner, L.H. Mechanistic investigation of thermosensitive liposome immunogenicity and understanding the drivers for circulation half-life: A polyethylene glycol versus 1,2-dipalmitoyl-sn-glycero-3-phosphodiglycerol study. J. Control. Release 2021, 333, 1–15. [Google Scholar] [CrossRef]
- van Valenberg, F.J.P.; Brummelhuis, I.S.G.; Lindner, L.H.; Kuhnle, F.; Wedmann, B.; Schweizer, P.; Hossann, M.; Witjes, J.A.; Oosterwijk, E. DPPG2-Based Thermosensitive Liposomes with Encapsulated Doxorubicin Combined with Hyperthermia Lead to Higher Doxorubicin Concentrations in the Bladder Compared to Conventional Application in Pigs: A Rationale for the Treatment of Muscle-Invasive Bladder Cancer. Int. J. Nanomed. 2021, 16, 75–88. [Google Scholar] [CrossRef]
- Harrington, K.J.; Mohammadtaghi, S.; Uster, P.S.; Glass, D.; Peters, A.M.; Vile, R.G.; Stewart, J.S. Effective targeting of solid tumors in patients with locally advanced cancers by radiolabeled pegylated liposomes. Clin. Cancer Res. 2001, 7, 243–254. [Google Scholar]
- Uziely, B.; Jeffers, S.; Isacson, R.; Kutsch, K.; Wei-Tsao, D.; Yehoshua, Z.; Libson, E.; Muggia, F.M.; Gabizon, A. Liposomal doxorubicin: Antitumor activity and unique toxicities during two complementary phase I studies. J. Clin. Oncol. 1995, 13, 1777–1785. [Google Scholar] [CrossRef]
- Gabizon, A.A.; Patil, Y.; La-Beck, N.M. New insights and evolving role of pegylated liposomal doxorubicin in cancer therapy. Drug Resist. Updat. 2016, 29, 90–106. [Google Scholar] [CrossRef]
- Anchordoquy, T.J.; Barenholz, Y.; Boraschi, D.; Chorny, M.; Decuzzi, P.; Dobrovolskaia, M.A.; Farhangrazi, Z.S.; Farrell, D.; Gabizon, A.; Ghandehari, H.; et al. Mechanisms and Barriers in Cancer Nanomedicine: Addressing Challenges, Looking for Solutions. ACS Nano 2017, 11, 12–18. [Google Scholar] [CrossRef]
- Denekamp, J. Vascular endothelium as the vulnerable element in tumours. Acta Radiol. Oncol. 1984, 23, 217–225. [Google Scholar] [CrossRef]
- Corti, A.; Pastorino, F.; Curnis, F.; Arap, W.; Ponzoni, M.; Pasqualini, R. Targeted Drug Delivery and Penetration Into Solid Tumors. Med. Res. Rev. 2012, 32, 1078–1091. [Google Scholar] [CrossRef]
- Maeda, N.; Takeuchi, Y.; Takada, M.; Sadzuka, Y.; Namba, Y.; Oku, N. Anti-neovascular therapy by use of tumor neovasculature-targeted long-circulating liposome. J. Control. Release 2004, 100, 41–52. [Google Scholar] [CrossRef]
- Oku, N.; Asai, T.; Watanabe, K.; Kuromi, K.; Nagatsuka, M.; Kurohane, K.; Kikkawa, H.; Ogino, K.; Tanaka, M.; Ishikawa, D.; et al. Anti-neovascular therapy using novel peptides homing to angiogenic vessels. Oncogene 2002, 21, 2662–2669. [Google Scholar] [CrossRef] [Green Version]
- Alessi, P.; Ebbinghaus, C.; Neri, D. Molecular targeting of angiogenesis. Biochim. Biophys. Acta-Rev. Cancer 2004, 1654, 39–49. [Google Scholar] [CrossRef]
- Bikfalvi, A.; Bicknell, R. Recent advances in angiogenesis, anti-angiogenesis and vascular targeting. Trends Pharmacol. Sci. 2002, 23, 576–582. [Google Scholar] [CrossRef]
- Eichhorn, M.E.; Strieth, S.; Dellian, M. Anti-vascular tumor therapy: Recent advances, pitfalls and clinical perspectives. Drug Resist. Updates 2004, 7, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. The RGD story: A personal account. Matrix Biol. 2003, 22, 459–465. [Google Scholar] [CrossRef]
- Heckmann, D.; Meyer, A.; Marinelli, L.; Zahn, G.; Stragies, R.; Kessler, H. Probing integrin selectivity: Rational design of highly active and selective ligands for the alpha 5 beta 1 and alpha v beta 3 integrin receptor. Angew. Chem. Int. Ed. 2007, 46, 3571–3574. [Google Scholar] [CrossRef] [PubMed]
- Temming, K.; Schiffelers, R.M.; Molema, G.; Kok, R.J. RGD-based strategies for selective delivery of therapeutics and imaging agents to the tumour vasculature. Drug Resist. Updat 2005, 8, 381–402. [Google Scholar] [CrossRef]
- Salvati, M.; Cordero, F.M.; Pisaneschi, F.; Melani, F.; Gratteri, P.; Cini, N.; Bottoncetti, A.; Brandi, A. Synthesis, SAR and in vitro evaluation of new cyclic Arg-Gly-Asp pseudopentapeptides containing a s-cis peptide bond as integrin alphavbeta3 and alphavbeta5 ligands. Bioorg. Med. Chem. 2008, 16, 4262–4271. [Google Scholar] [CrossRef]
- Heckmann, D.; Kesster, H. Design and chemical synthesis of integrin ligands. Integrins 2007, 426, 463. [Google Scholar] [CrossRef]
- Amin, M.; Badiee, A.; Jaafari, M.R. Improvement of pharmacokinetic and antitumor activity of PEGylated liposomal doxorubicin by targeting with N-methylated cyclic RGD peptide in mice bearing C-26 colon carcinomas. Int. J. Pharm. 2013, 458, 324–333. [Google Scholar] [CrossRef]
- Vincent, S.; DePace, D.; Finkelstein, S. Distribution of anionic sites on the capillary endothelium in an experimental brain tumor model. Microcirc Endothel. Lymphat. 1988, 4, 45–67. [Google Scholar]
- Ran, S.; Downes, A.; Thorpe, P.E. Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res. 2002, 62, 6132–6140. [Google Scholar]
- Ho, E.A.; Ramsay, E.; Ginj, M.; Anantha, M.; Bregman, I.; Sy, J.; Woo, J.; Osooly-Talesh, M.; Yapp, D.T.; Bally, M.B. Characterization of cationic liposome formulations designed to exhibit extended plasma residence times and tumor vasculature targeting properties. J. Pharm. Sci. 2010, 99, 2839–2853. [Google Scholar] [CrossRef]
- Torchilin, V.P.; Rammohan, R.; Weissig, V.; Levchenko, T.S. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 8786–8791. [Google Scholar] [CrossRef] [Green Version]
- Fretz, M.M.; Koning, G.A.; Mastrobattista, E.; Jiskoot, W.; Storm, G. OVCAR-3 cells internalize TAT-peptide modified liposomes by endocytosis. Biochim. Biophys. Acta 2004, 1665, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seguin, J.; Nicolazzi, C.; Mignet, N.; Scherman, D.; Chabot, G.G. Vascular density and endothelial cell expression of integrin alpha v beta 3 and E-selectin in murine tumours. Tumour Biol. 2012, 33, 1709–1717. [Google Scholar] [CrossRef] [Green Version]
- Contreras, F.X.; Villar, A.V.; Alonso, A.; Kolesnick, R.N.; Goni, F.M. Sphingomyelinase activity causes transbilayer lipid translocation in model and cell membranes. J. Biol. Chem. 2003, 278, 37169–37174. [Google Scholar] [CrossRef] [Green Version]
- Contreras, F.X.; Basanez, G.; Alonso, A.; Herrmann, A.; Goni, F.M. Asymmetric addition of ceramides but not dihydroceramides promotes transbilayer (flip-flop) lipid motion in membranes. Biophys. J. 2005, 88, 348–359. [Google Scholar] [CrossRef] [Green Version]
- Veldman, R.J.; Zerp, S.; van Blitterswijk, W.J.; Verheij, M. N-hexanoyl-sphingomyelin potentiates in vitro doxorubicin cytotoxicity by enhancing its cellular influx. Br. J. Cancer 2004, 90, 917–925. [Google Scholar] [CrossRef]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.W.; Kim, H.; Cho, H.; Kim, M.S.; Paek, S.H.; Park, H.J.; Griffin, R.J.; Terezakis, S.; Cho, L.C. HIF-1 alpha Inhibition Improves Anti-Tumor Immunity and Promotes the Efficacy of Stereotactic Ablative Radiotherapy (SABR). Cancers 2022, 14, 3273. [Google Scholar] [CrossRef] [PubMed]
- Rastakhiz, S.; Yazdani, M.; Shariat, S.; Arab, A.; Momtazi-Borojeni, A.A.; Barati, N.; Mansourian, M.; Amin, M.; Abbasi, A.; Saberi, Z.; et al. Preparation of nanoliposomes linked to HER2/neu-derived (P5) peptide containing MPL adjuvant as vaccine against breast cancer. J. Cell Biochem. 2019, 120, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Wang, L.; Li, H.; Miao, F.; Zhang, Z.; Hu, C.; Yu, W.; Tang, Q.; Shao, G. Application of lipid nanovesicle drug delivery system in cancer immunotherapy. J. Nanobiotechnol. 2022, 20, 214. [Google Scholar] [CrossRef]
- Gu, Z.; Da Silva, C.G.; Van der Maaden, K.; Ossendorp, F.; Cruz, L.J. Liposome-Based Drug Delivery Systems in Cancer Immunotherapy. Pharmaceutics 2020, 12, 1054. [Google Scholar] [CrossRef]
- Fobian, S.F.; Cheng, Z.; Ten Hagen, T.L.M. Smart Lipid-Based Nanosystems for Therapeutic Immune Induction against Cancers: Perspectives and Outlooks. Pharmaceutics 2021, 14, 26. [Google Scholar] [CrossRef]
- Saeed, M.; Schooten, E.; van Brakel, M.; Cole, D.K.; Ten Hagen, T.L.M.; Debets, R. T Cells Expressing a TCR-Like Antibody Selected Against the Heteroclitic Variant of a Shared MAGE-A Epitope Do Not Recognise the Cognate Epitope. Cancers 2020, 12, 1255. [Google Scholar] [CrossRef] [PubMed]
- Merino, M.; Contreras, A.; Casares, N.; Troconiz, I.F.; Ten Hagen, T.L.; Berraondo, P.; Zalba, S.; Garrido, M.J. A new immune-nanoplatform for promoting adaptive antitumor immune response. Nanomedicine 2019, 17, 13–25. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Composition mol:mol | Encapsulated Compound | Study Type | Cell Line | Tumor Model | Function | Ref |
|---|---|---|---|---|---|---|---|
| DPPC:DSPC: mPEG2000-DSPE | 80: (20 − x): x (x = 1, 3, 4, 5, 6, 10) | CF | In vitro in vivo | BLM | BLM melanoma | TSL | [24] |
| DPPC:DSPC: mPEG2000-DSPE | 55:40:5 80:15:5 | DXR | In vitro in vivo | Different cell lines | BLM melanoma | TSL | [25] |
| DPPC:DSPC: mPEG2000-DSPE | x: (100 − x): 5 (x = 80, 70, 60, 50) | DXR | In vivo | - | BFS-1 sarcoma | TSL | [26] |
| DPPC:DSPC: mPEG2000-DSPE | x: (100 − x): 5 (x = 100, 80, 60, 40, 20, 0) | CF | In vitro | - | - | TSL | [27] |
| DPPC:DSPC: mPEG2000-DSPE | 60:35:5 for IDA -TSL 70:25:5 for DXR -TSL | IDA DXR | In vitro in vivo | B16BL6, BLM, BFS-1 | BLM melanoma | TSL | [28] |
| HSPC:Chol: mPEG2000-DSPE | 56.1:38.2:5.5 | DXR | In vitro in vivo | Different cell lines | C26 colon carcinoma B16F10 and BLM melanoma | Vascular targeting by RGD peptide | [29] |
| DPPC:DSPC: mPEG2000-DSPE | 70:25:5 | DXR | In vitro in vivo | B16BL6 and B16F10 | B16BL6 melanoma | TSL + Vascular targeting by RGD peptide | [30] |
| DPPC:DSPC: DPTAP mPEG2000-DSPE | 62.5:25:10:5 | CF | In vitro in vivo | BLM and HUVEC | B16BL6 melanoma | TSL + Vascular targeting by positive charge | [31] |
| DPPC:DSPC: DPTAP: mPEG2000-DSPE | 62.5:25:7.5:5 | DXR | In vitro in vivo | B16 and LLC | B16BL6 melanoma | TSL + Vascular targeting by positive charge | [32] |
| HSPC:Chol: mPEG2000-DSPE | 56.1:38.2:5.5 | DXR | In vitro in vivo | Different cell lines | C26 colon carcinoma | Targeting tumor cells with TAT | [33] |
| HSPC:Chol: mPEG2000-DSPE: Mal-PEG2000 DSPE | 1.85:1:0.12:0.03 | Oxaliplatin | In vitro in vivo | Different cell lines | SW-480 colorectal cancer | Targeting tumor cells with Cetuximab | [34] |
| HSPC:Chol: mPEG2000-DSPE | 56.1:38.2:5.5 | DXR | In vitro in vivo | Different cell lines | C26 colon carcinoma B16F10 melanoma | Dual anti-vascular and tumor cell targeting with RGD and TAT peptides | [35] |
| HSPC:Chol: mPEG2000-DSPE: C8-glucosylceramide | 1.85:1:0.15:15 | DXR | In vitro in vivo | B16 cells A431 | A431 epidermoid carcinoma | Short-chain sphingolipids | [36] |
| HSPC:Chol: mPEG2000-DSPE: C8-GluCer, C8-GalCer, C8-LacCer | 1.85:1:0.15:15 | DXR | In vitro | Different cell lines | - | Short-chain sphingolipids | [37] |
| HSPC:Chol: mPEG DSPE: C8-GlcCer, C8-GalCer | 1.85:1:0.15:10 | DXR | In vitro | Different cell lines | - | Short-chain sphingolipids | [38] |
| HSPC:Chol: mPEG2000-DSPE: C8-GluCer | 1.85:1:0.15:10 | DXR | In vitro in vivo | Different cell lines | MES-SA or MES-SA/MX2 uterine sarcoma | Short-chain sphingolipids | [39] |
| HSPC:Chol: mPEG2000-DSPE: C8-GlcCer, C8-GalCer | 1.85:1:0.15:10 | Mitoxantrone | In vivo | - | MDA-MB-231 and MCF-7 breast cancer | Short-chain sphingolipids | [40] |
| HSPC:Chol: mPEG2000-DSPE: maleimide mPEG2000-DSPE | 55:40:4:1 | In vitro | Different cell lines | Immunotherapy with Anti-MAGE A1 T-cell receptor (TCR)-like single-chain antibody | [41] | ||
| HSPC: Chol: mPEG2000-DSPE: maleimide mPEG2000-DSPE | 55:40:4:1 | DXR | In vitro in vivo | Different cell lines | G43 and Mel78 melanoma | Immunotherapy Anti-MAGE A1 TCR-like single-chain antibody | [42] |
| HSPC: Chol: mPEG2000-DSPE | 1.85:1:0.12 | DXR | In vitro in vivo | B16OVA murine melanoma | B16OVA B16OVA | Immunotherapy Monovalent-variable fragments (Fab’) of α-PD-L1 | [43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amin, M.; Seynhaeve, A.L.B.; Sharifi, M.; Falahati, M.; ten Hagen, T.L.M. Liposomal Drug Delivery Systems for Cancer Therapy: The Rotterdam Experience. Pharmaceutics 2022, 14, 2165. https://doi.org/10.3390/pharmaceutics14102165
Amin M, Seynhaeve ALB, Sharifi M, Falahati M, ten Hagen TLM. Liposomal Drug Delivery Systems for Cancer Therapy: The Rotterdam Experience. Pharmaceutics. 2022; 14(10):2165. https://doi.org/10.3390/pharmaceutics14102165
Chicago/Turabian StyleAmin, Mohamadreza, Ann L. B. Seynhaeve, Majid Sharifi, Mojtaba Falahati, and Timo L. M. ten Hagen. 2022. "Liposomal Drug Delivery Systems for Cancer Therapy: The Rotterdam Experience" Pharmaceutics 14, no. 10: 2165. https://doi.org/10.3390/pharmaceutics14102165