

Novel Dimer Derivatives of PF-543 as Potential Antitumor Agents for the Treatment of Non-Small Cell Lung Cancer
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Cell Proliferation Assay
2.4. Colony Formation Assay
2.5. SK Activity Assay
2.6. Western Blotting
2.7. RNA Extraction and qRT-PCR
2.8. The Levels of Sphingosine, Ceramide, and S1P
2.9. Annexin V-FITC
2.10. Mitochondrial Membrane Potential Measurement
2.11. siRNA Transfection
2.12. In Vitro Metabolic Stability of PF-543 and Derivatives
2.13. Experimental Animals
2.14. Immunohistochemical Analysis
2.15. Serum Analysis
2.16. Statistical Analysis
2.17. Chemistry
2.17.1. Materials
2.17.2. Synthesis and Characterization of Compounds
(R)-(1-(4-(Bis(3-methyl-5-((phenylsulfonyl)methyl)benzyl)amino)phenethyl)pyrrolidin-2-yl)methanol (1)
1-(4-(Bis(3-methyl-5-((phenylsulfonyl)methyl)benzyl)amino)phenethyl)piperidin-4-ol (2)
(S)-1-(4-(Bis(3-methyl-5-((phenylsulfonyl)methyl)benzyl)amino)phenethyl)pyrrolidin-3-ol (3)
(1-(4-(Bis(3-methyl-5-((phenylsulfonyl)methyl)benzyl)amino)phenethyl)piperidin-4-yl)methanol (4)
1,3-Bis(bromomethyl)-5-methylbenzene (5)
1-(Bromomethyl)-3-methyl-5-((phenylsulfonyl)methyl)benzene (6)
2-(4-(Bis(3-methyl-5-((phenylsulfonyl)methyl)benzyl)amino)phenyl)ethanol (7)
4-(2-Bromoethyl)-N,N-bis(3-methyl-5-((phenylsulfonyl)methyl)benzyl)aniline (8)
3. Results
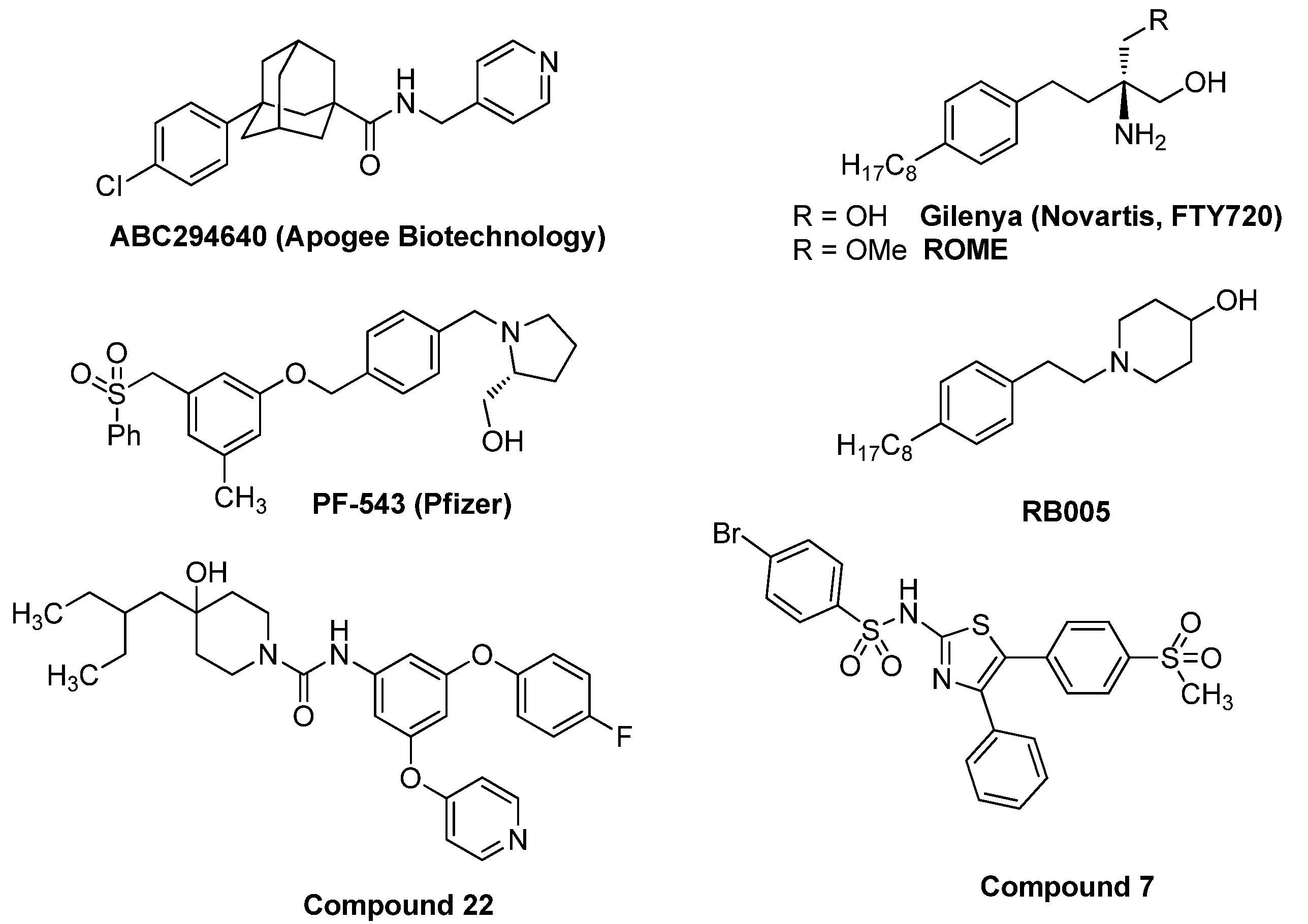
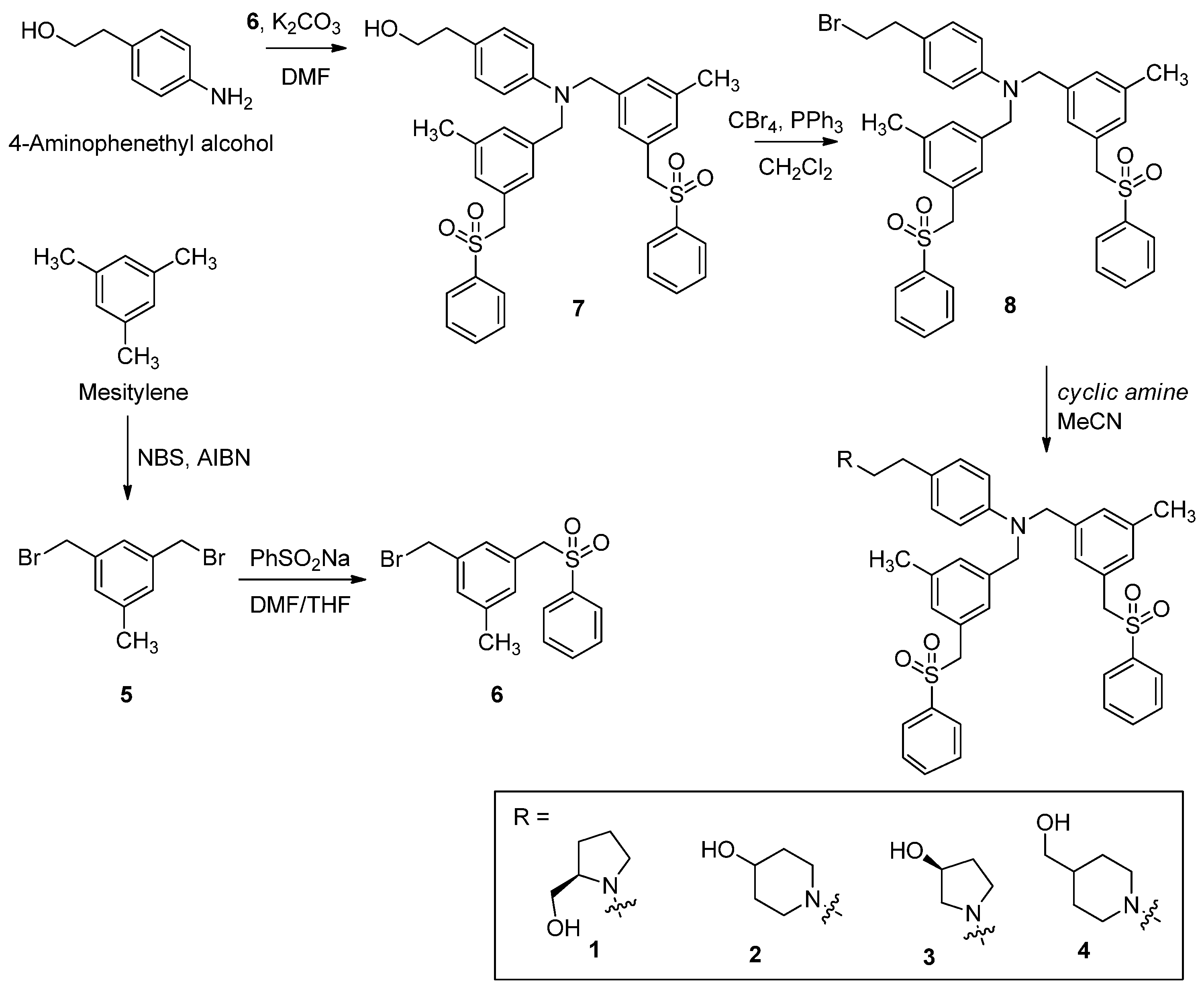
3.1. Synthesis of PF-543 Derivatives
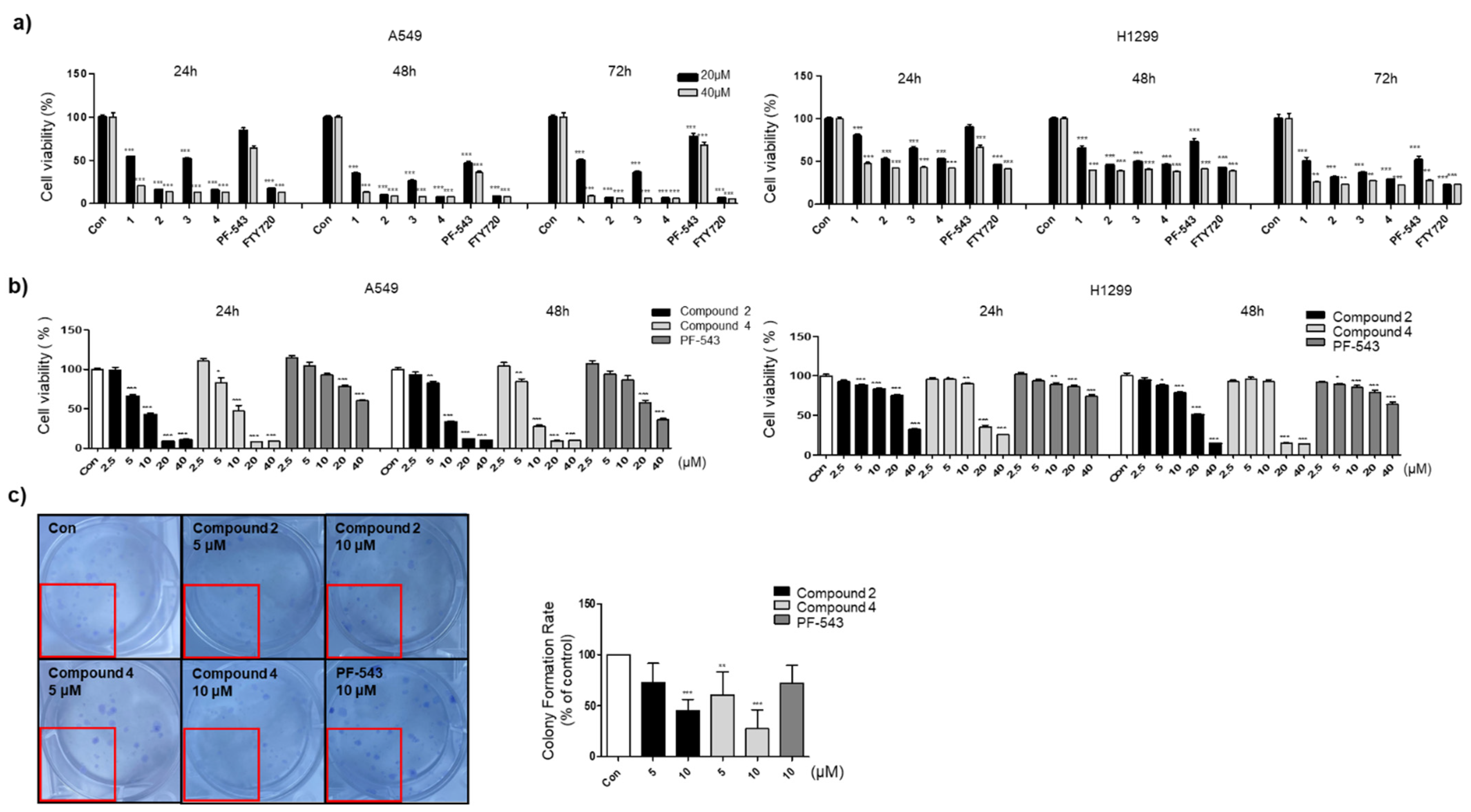
3.2. Compounds 2 and 4 Reduce the Viability of Lung Cancer Cell Lines Compared to PF-543
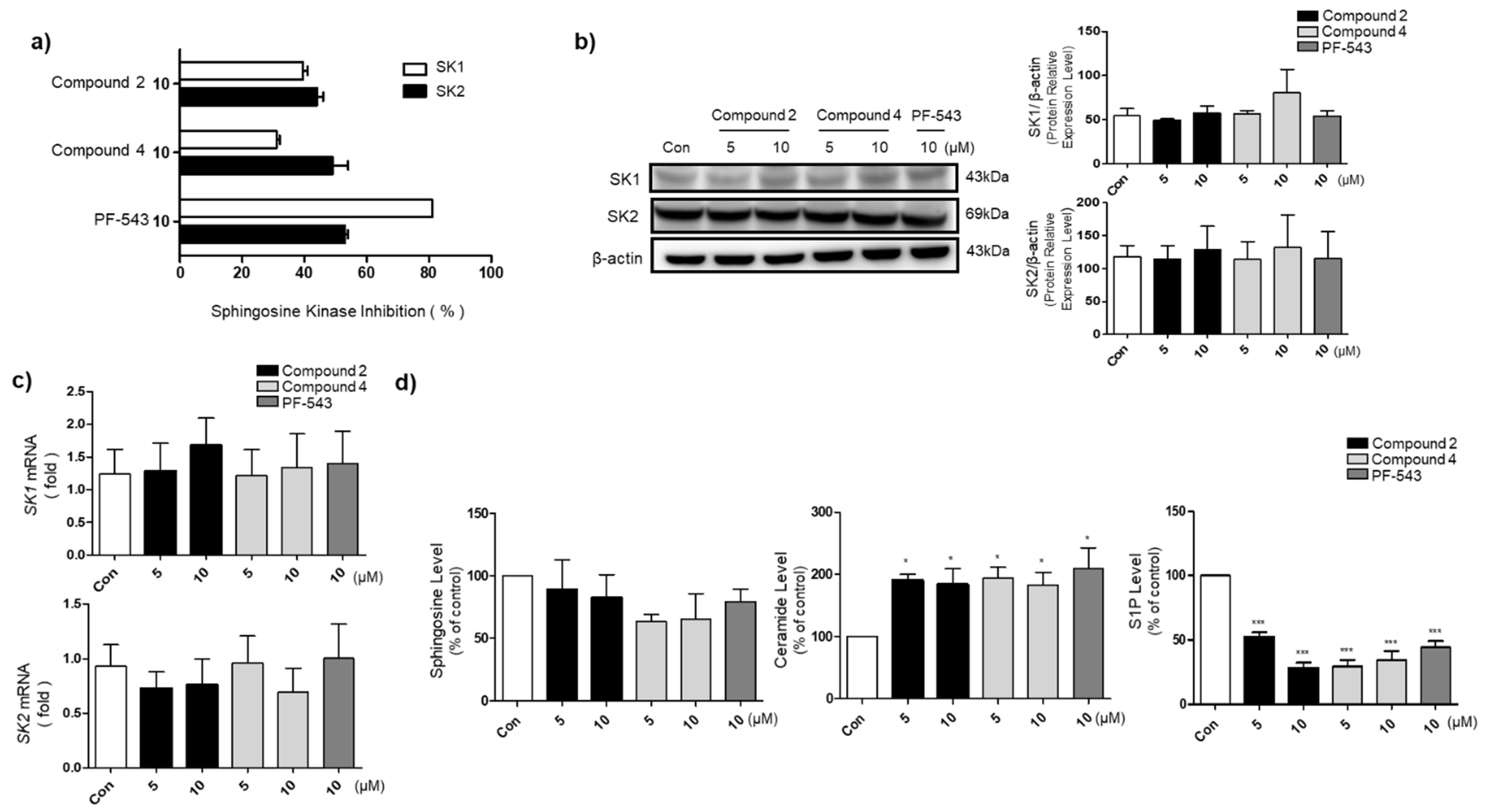
3.3. Compounds 2 and 4 Reduce S1P Formation and Increase Ceramide Levels in A549 Cells
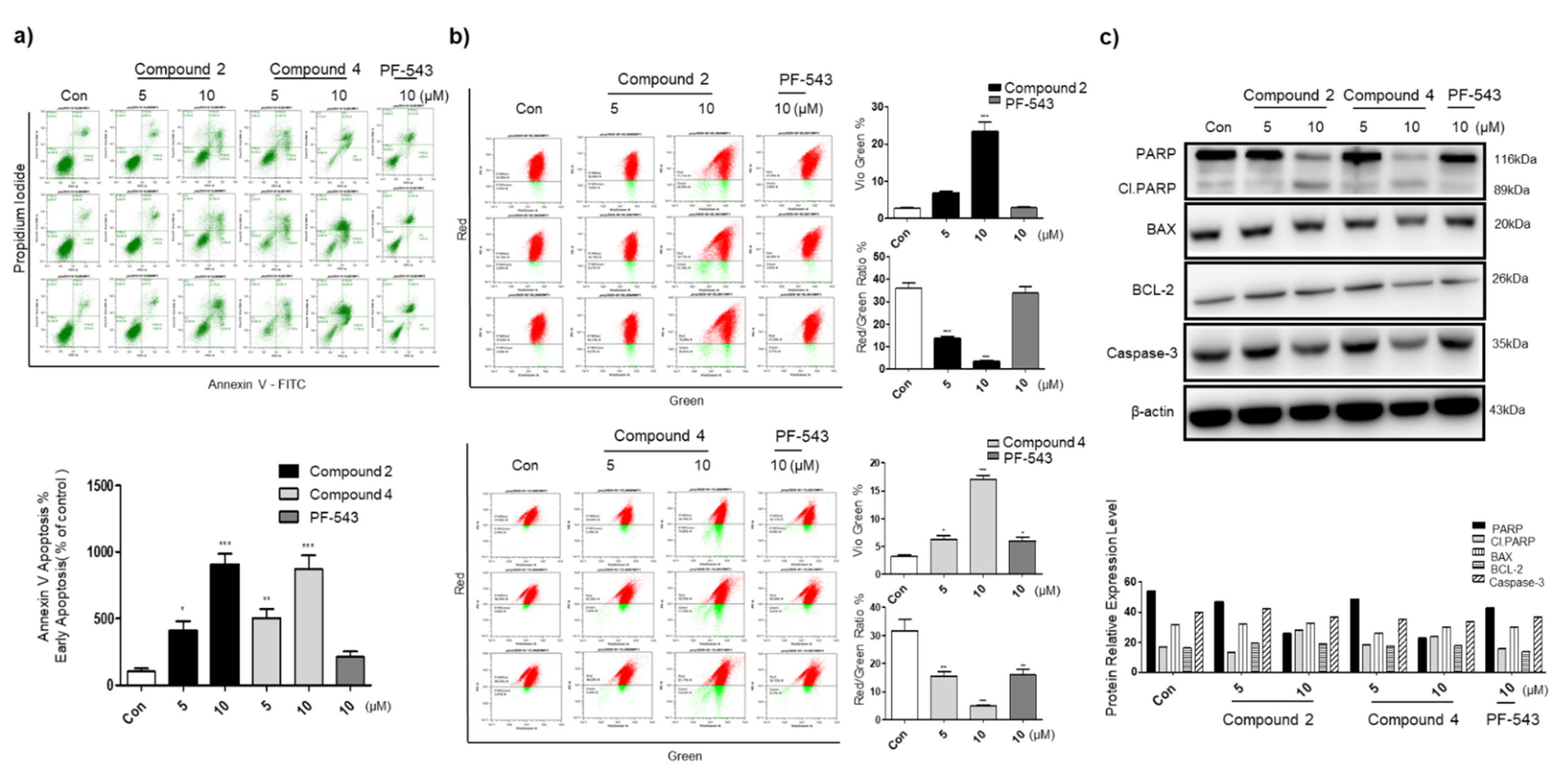
3.4. Compounds 2 and 4 Induce Apoptosis through Mitochondrial Membrane Potential Depolarization and Active Intrinsic Apoptosis Pathway
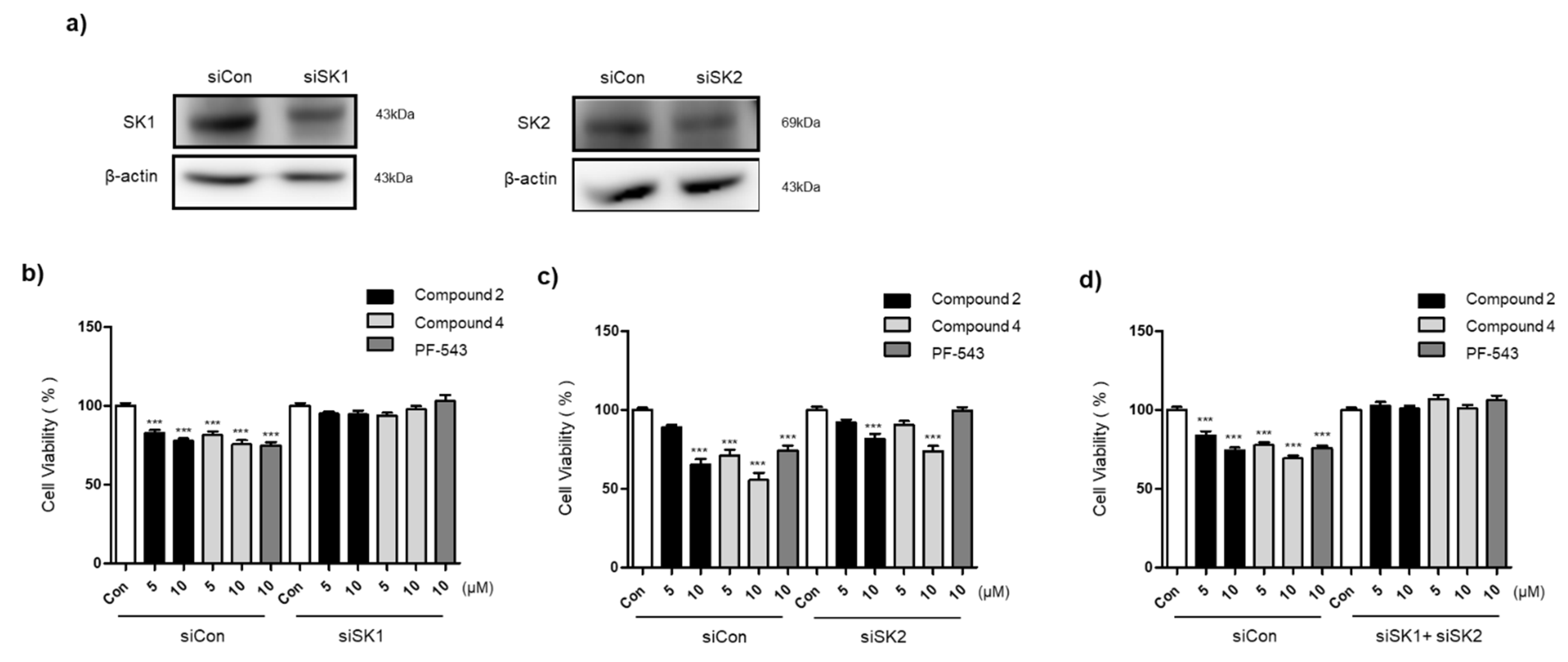
3.5. Compounds 2 and 4 Selectively Act on SK1 to Induce Cytotoxicity
3.6. The Bulky Tail Structure of Compounds 2 and 4 Increases MS
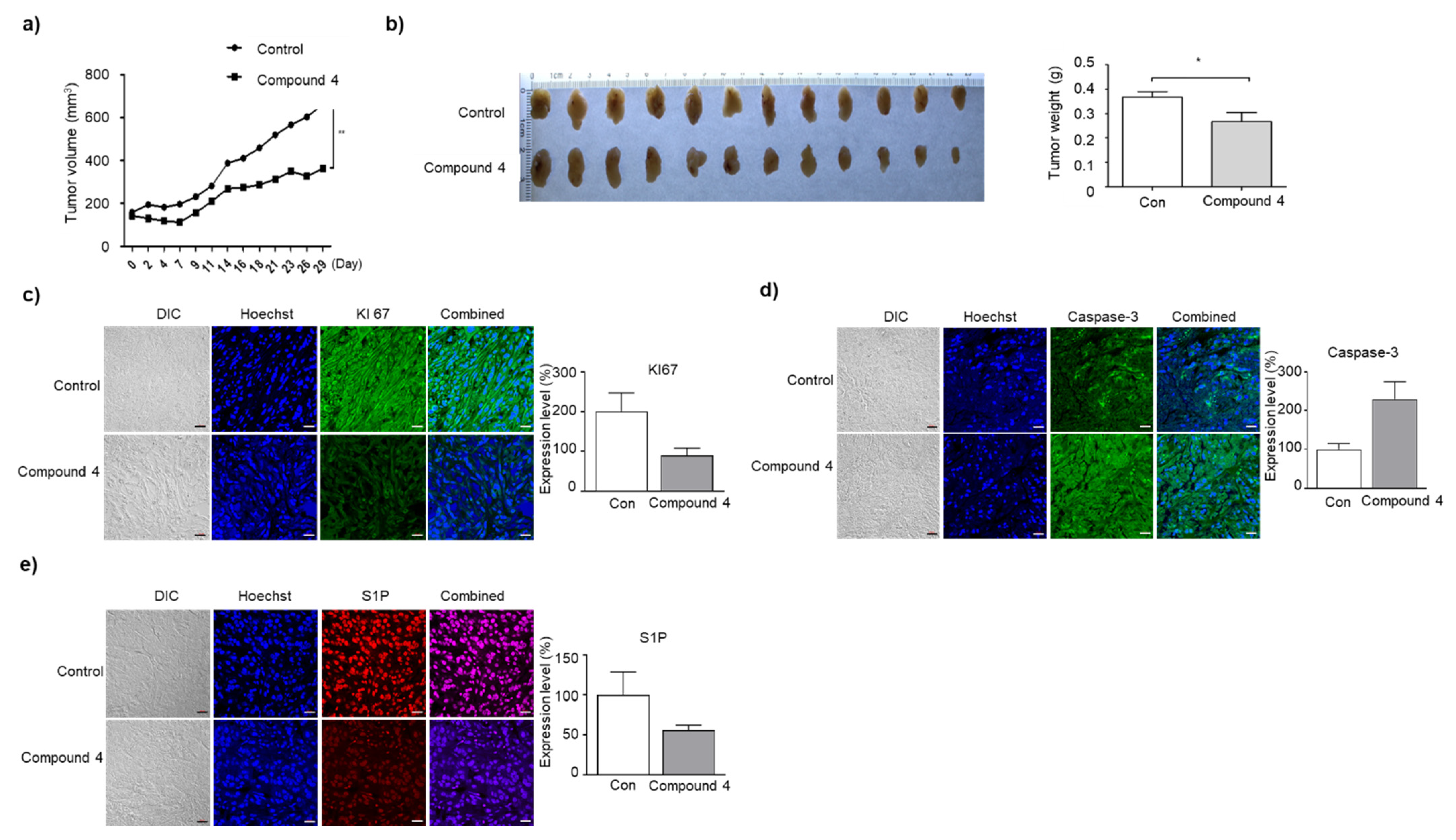
3.7. Compound 4 Induces Growth Inhibition and Apoptosis of A549 Cells in BALB/c Nude Mice Tumor Xenograft
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Li, W.; Ren, L.; Liu, J.; Pang, X.; Chen, X.; Kang, D.; Wang, J.; Du, G. The sphingosine kinase-1/sphingosine-1-phosphate axis in cancer: Potential target for anticancer therapy. Pharmacol. Ther. 2019, 195, 85–99. [Google Scholar] [CrossRef]
- Pyne, N.J.; El Buri, A.; Adams, D.R.; Pyne, S. Sphingosine 1-phosphate and cancer. Adv. Biol. Regul. 2018, 68, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Pitman, M.R.; Costabile, M.; Pitson, S.M. Recent advances in the development of sphingosine kinase inhibitors. Cell Signal. 2016, 28, 1349–1363. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Brinkmann, V. A mechanistically novel, first oral therapy for multiple sclerosis: The development of fingolimod (FTY720, Gilenya). Discov. Med. 2011, 12, 213–228. [Google Scholar]
- Lim, K.G.; Sun, C.; Bittman, R.; Pyne, N.J.; Pyne, S. (R)-FTY720 methyl ether is a specific sphingosine kinase 2 inhibitor: Effect on sphingosine kinase 2 expression in HEK 293 cells and actin rearrangement and survival of MCF-7 breast cancer cells. Cell Signal. 2011, 23, 1590–1595. [Google Scholar] [CrossRef]
- Baek, D.J.; MacRitchie, N.; Pyne, N.J.; Pyne, S.; Bittman, R. Synthesis of selective inhibitors of sphingosine kinase 1. Chem. Commun. 2013, 49, 2136–2138. [Google Scholar] [CrossRef] [PubMed]
- Britten, C.D.; Garrett-Mayer, E.; Chin, S.H.; Shirai, K.; Ogretmen, B.; Bentz, T.A.; Brisendine, A.; Anderton, K.; Cusack, S.L.; Maines, L.W.; et al. A Phase I Study of ABC294640, a First-in-Class Sphingosine Kinase-2 Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 4642–4650. [Google Scholar] [CrossRef] [PubMed]
- Schnute, M.E.; McReynolds, M.D.; Kasten, T.; Yates, M.; Jerome, G.; Rains, J.W.; Hall, T.; Chrencik, J.; Kraus, M.; Cronin, C.N.; et al. Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 2012, 444, 79–88. [Google Scholar] [CrossRef]
- Ju, T.; Gao, D.; Fang, Z.Y. Targeting colorectal cancer cells by a novel sphingosine kinase 1 inhibitor PF-543. Biochem. Biophys. Res. Commun. 2016, 470, 728–734. [Google Scholar] [CrossRef]
- Kim, S.B.; Lee, T.; Moon, H.S.; Ki, S.H.; Oh, Y.S.; Lee, J.Y.; Kim, S.B.; Park, J.E.; Kwon, Y.; Kim, S.; et al. Verification of the Necessity of the Tolyl Group of PF-543 for Sphingosine Kinase 1 Inhibitory Activity. Molecules 2020, 25, 2484. [Google Scholar] [CrossRef]
- Adams, D.R.; Tawati, S.; Berretta, G.; Rivas, P.L.; Baiget, J.; Jiang, Z.; Alsfouk, A.; Mackay, S.P.; Pyne, N.J.; Pyne, S. Topographical Mapping of Isoform-Selectivity Determinants for J-Channel-Binding Inhibitors of Sphingosine Kinases 1 and 2. J. Med. Chem. 2019, 62, 3658–3676. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, K.; Shinozaki, K.; Kanaji, T.; Kurata, H.; Naganawa, A.; Otsuki, K.; Matsushita, T.; Sekiguchi, T.; Kakuuchi, A.; Seko, T. Discovery of novel S1P2 antagonists. Part 1: Discovery of 1, 3-bis(aryloxy)benzene derivatives. Bioorg. Med. Chem. Lett. 2015, 25, 1479–1482. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Mahapatra, A.D.; Mohammad, T.; Gupta, P.; Alajmi, M.F.; Hussain, A.; Rehman, M.T.; Datta, B.; Hassan, M.I. Design and Development of Novel Urea, Sulfonyltriurea, and Sulfonamide Derivatives as Potential Inhibitors of Sphingosine Kinase 1. Pharmaceuticals 2020, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, J.; Li, G.; Li, Y.; Xu, C.; Li, M.; Xu, G.; Fu, S. Prognostic significance of sphingosine kinase 2 expression in non-small cell lung cancer. Tumour Biol. 2014, 35, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Gachechiladze, M.; Tichý, T.; Kolek, V.; Grygárková, I.; Klein, J.; Mgebrishvili, G.; Kharaishvili, G.; Janíková, M.; Smičková, P.; Cierna, L.; et al. Sphingosine kinase-1 predicts overall survival outcomes in non-small cell lung cancer patients treated with carboplatin and navelbine. Oncol. Lett. 2019, 18, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Miyake, N.; Chikumi, H.; Yamaguchi, K.; Takata, M.; Takata, M.; Okada, K.; Kitaura, T.; Nakamoto, M.; Yamasaki, A. Effect of Cetuximab and EGFR Small Interfering RNA Combination Treatment in NSCLC Cell Lines with Wild Type EGFR and Use of KRAS as a Possible Biomarker for Treatment Responsiveness. Yonago Acta Med. 2019, 62, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Minakata, K.; Takahashi, F.; Nara, T.; Hashimoto, M.; Tajima, K.; Murakami, A.; Nurwidya, F.; Yae, S.; Koizumi, F.; Moriyama, H.; et al. Hypoxia induces gefitinib resistance in non-small-cell lung cancer with both mutant and wild-type epidermal growth factor receptors. Cancer Sci. 2012, 103, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Oh, Y.S.; Kim, K.J.; Cho, S.W.; Park, S.K.; Baek, D.J.; Park, E.-Y. Synthesis of PP2A-Activating PF-543 Derivatives and Investigation of Their Inhibitory Effects on Pancreatic Cancer Cells. Molecules 2022, 27, 3346. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequences |
|---|---|
| SK1 | Forward: 5′-GGCGTCATGCATCTGTTCTA-3′ |
| Reverse: 5′-ACACACCTTTCCCATCCTTG-3′ | |
| SK2 | Forward: 5′-ATCTCTGAAGCTGGGCTGTCC-3′ |
| Reverse: 5′-CTCCCAGTCAGGGCGATCTA-3′ |
| MS | HLM | DLM | RLM | MLM |
|---|---|---|---|---|
| Compound 2 | 18.1 | 18.6 | 29.1 | 24.6 |
| Compound 4 | 30.2 | 26.1 | 22.9 | 38.5 |
| PF-543 | 6.4 | 7.3 | 7.8 | 9.4 |
| Verapamil | 13.4 |
| Con | Compound 4 | |
|---|---|---|
| ALT | 101.61 ± 5.27 | 93.64 ± 11.28 |
| AST | 98.21 ± 6.53 | 93.44 ± 14.57 |
| ALP | 100.52 ± 4.34 | 79.54 ± 7.08 *** |
| Sphingosine | 4.37 ± 5.39 | 7.15 ± 6.61 |
| Ceramide | 6.87 ± 3.26 | 6.2 ± 3.24 |
| S1P | 10.2 ± 5.49 | 11.43 ± 5.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.B.; Limbu, K.R.; Oh, Y.S.; Kim, S.L.; Park, S.K.; Baek, D.J.; Park, E.-Y. Novel Dimer Derivatives of PF-543 as Potential Antitumor Agents for the Treatment of Non-Small Cell Lung Cancer. Pharmaceutics 2022, 14, 2035. https://doi.org/10.3390/pharmaceutics14102035
Kim SB, Limbu KR, Oh YS, Kim SL, Park SK, Baek DJ, Park E-Y. Novel Dimer Derivatives of PF-543 as Potential Antitumor Agents for the Treatment of Non-Small Cell Lung Cancer. Pharmaceutics. 2022; 14(10):2035. https://doi.org/10.3390/pharmaceutics14102035
Chicago/Turabian StyleKim, Su Bin, Khem Raj Limbu, Yoon Sin Oh, Soo Lim Kim, Seung Ki Park, Dong Jae Baek, and Eun-Young Park. 2022. "Novel Dimer Derivatives of PF-543 as Potential Antitumor Agents for the Treatment of Non-Small Cell Lung Cancer" Pharmaceutics 14, no. 10: 2035. https://doi.org/10.3390/pharmaceutics14102035