
In Silico Screening of Available Drugs Targeting Non-Small Cell Lung Cancer Targets: A Drug Repurposing Approach

 ,
,  ,
,
Abstract
:1. Introduction
2. Methodology
2.1. Dataset
2.2. Protein and Ligand Preparation
2.3. Binding Site Analysis and Grid Generation
2.4. Glide Docking and MM/GBSA Analysis
2.5. Scoring Functions
2.5.1. RF-Score Analysis
2.5.2. Tanimoto Coefficient Calculation
2.6. Molecular Dynamics (MD) Simulations
2.7. End-Point Binding Free Energy Calculations
3. Result and Discussion
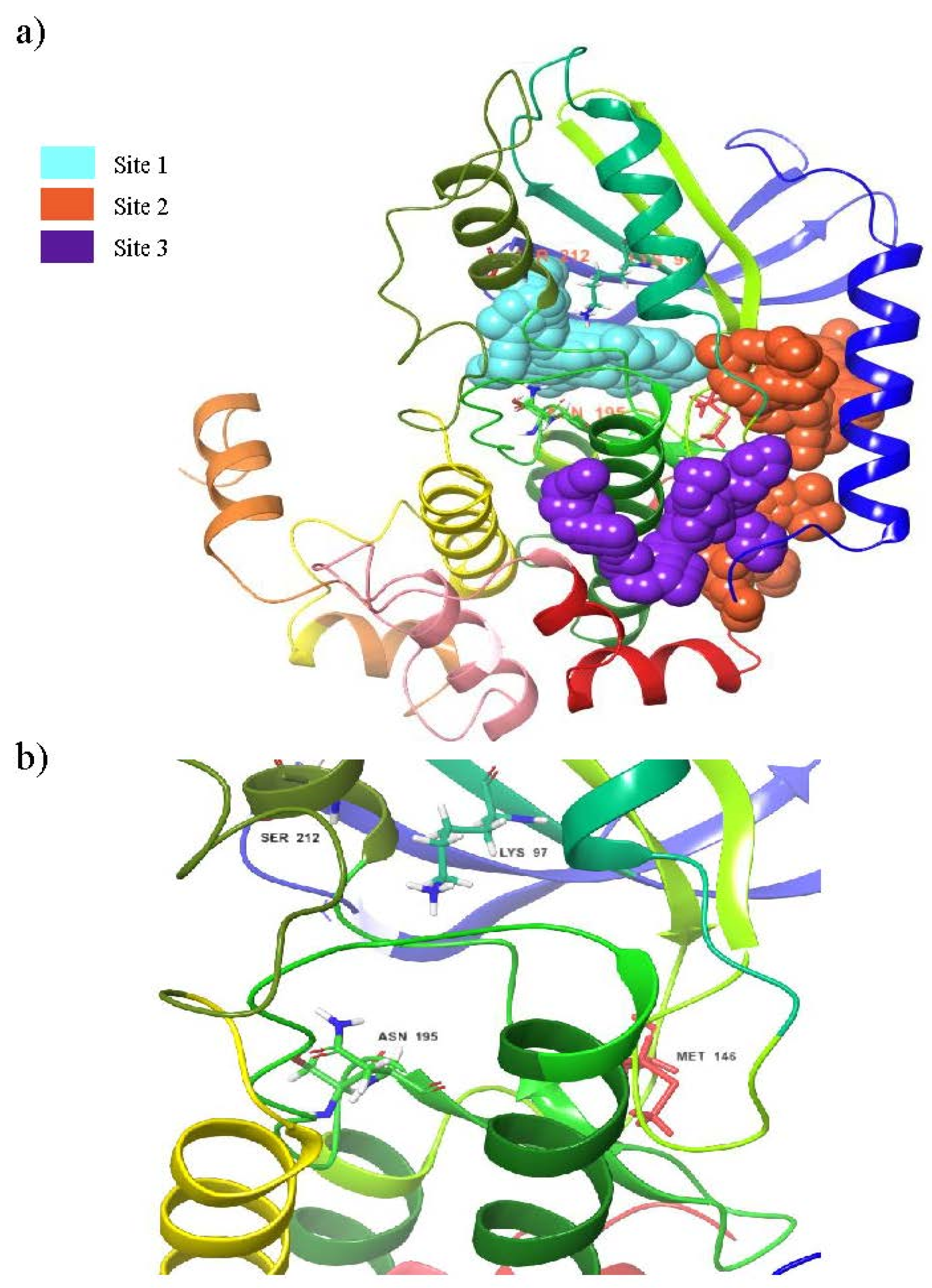
3.1. Binding Site Prediction
3.2. Validation of Molecular Docking
3.3. Virtual Screening
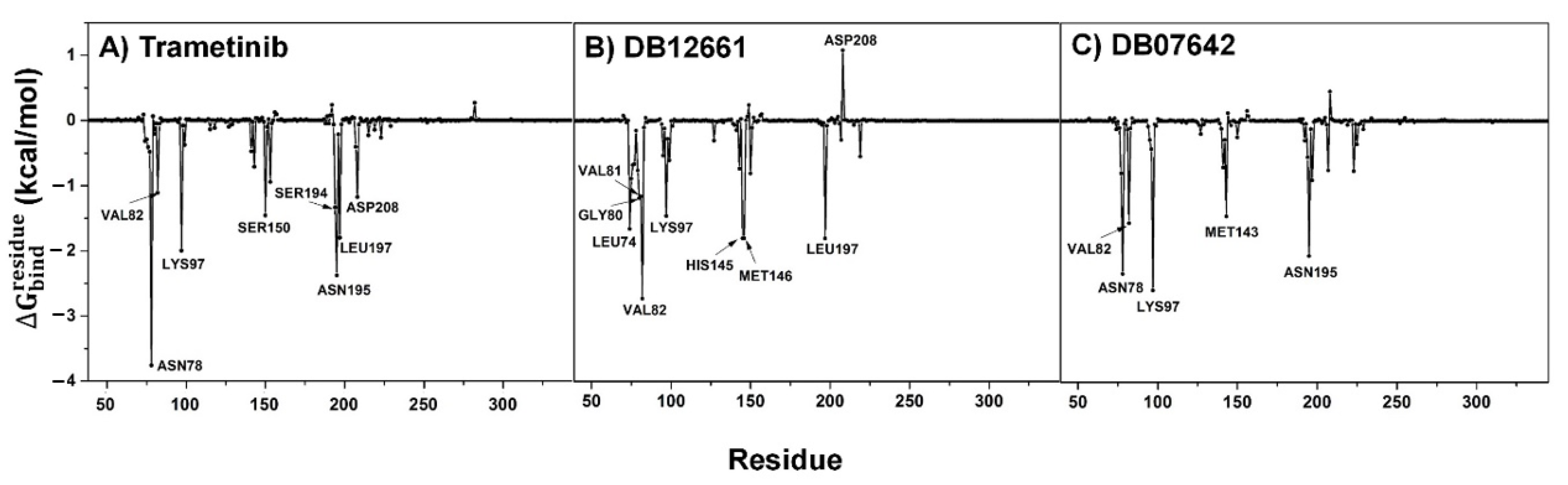
3.4. MM/GBSA Analysis
3.5. Structural Properties of Hit Compounds
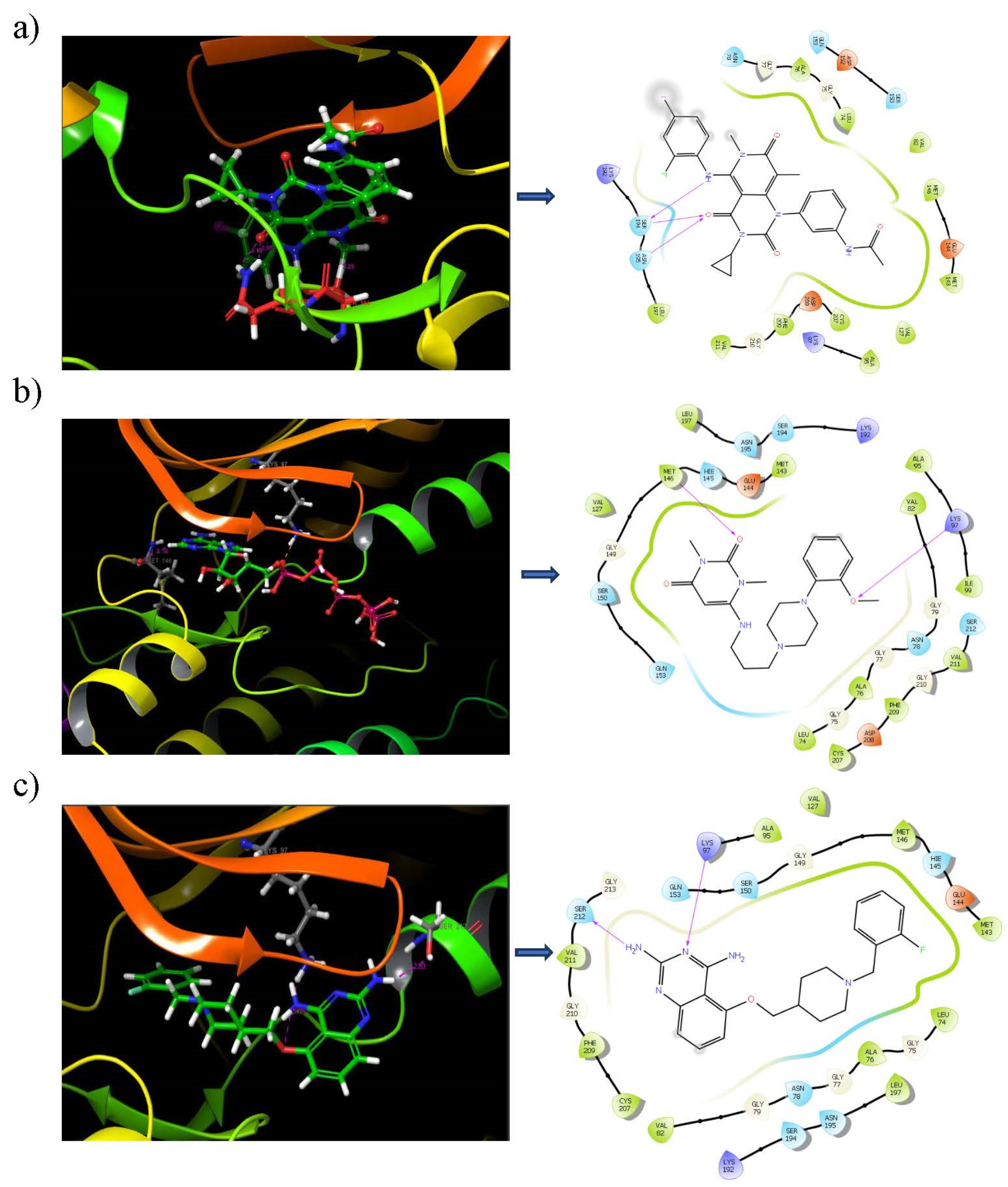
3.6. Binding Mode Analysis
3.7. Binding Analysis of Lead Compounds with PIM1
3.8. SIE-Based Free Energy of Binding
3.9. Key Binding Residues
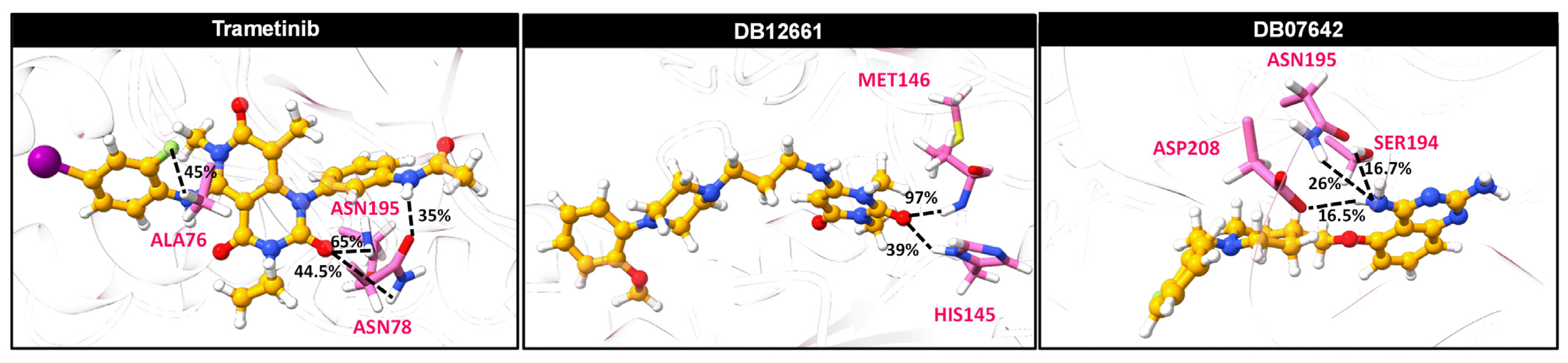
3.10. Ligand–Protein Hydrogen Bonding
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bradley, S.H.; Kennedy, M.P.T.; Neal, R.D. Recognising lung cancer in primary care. Adv. Ther. 2019, 36, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, P.S.; Raina, V. Lung cancer: Prevalent trends & emerging concepts. Indian J. Med. Res. 2015, 141, 5–7. [Google Scholar] [PubMed]
- Li, S.; Xu, S.; Liang, X.; Xue, Y.; Mei, J.; Ma, Y.; Liu, Y.; Liu, Y. Nanotechnology: Breaking the current treatment limits of lung cancer. Adv. Healthc. Mater. 2021, 10, 2100078. [Google Scholar] [CrossRef]
- Yuan, M.; Huang, L.-L.; Chen, J.-H.; Wu, J.; Xu, Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct. Target. 2019, 4, 61. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Liu, Y.; Yang, S.; Wu, X.; Li, H.; Wang, Q. Mek inhibitors for the treatment of non-small cell lung cancer. J. Hematol. Oncol. 2021, 14, 1. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-RAS (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Yaeger, R.; Corcoran, R.B. Targeting alterations in the RAF–MEK pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegedüs, L.; Okumus, Ö.; Livingstone, E.; Baranyi, M.; Kovács, I.; Döme, B.; Tóvári, J.; Bánkfalvi, Á.; Schadendorf, D.; Aigner, C. Allosteric and ATP-competitive MEK-inhibition in a novel spitzoid melanoma model with a RAF-and phosphorylation-independent mutation. Cancers 2021, 13, 829. [Google Scholar] [CrossRef]
- Heigener, D.F.; Gandara, D.R.; Reck, M. Targeting of MEK in lung cancer therapeutics. Lancet Respir. Med. 2015, 3, 319–327. [Google Scholar] [CrossRef]
- Zhao, Y.; Adjei, A.A. The clinical development of MEK inhibitors. Nat. Rev. Clin. Oncol. 2014, 11, 385–400. [Google Scholar] [CrossRef]
- Menzies, A.M.; Long, G.V. Dabrafenib and trametinib, alone and in combination for BRAF-mutant metastatic melanoma. Clin. Cancer Res. 2014, 20, 2035–2043. [Google Scholar] [CrossRef] [Green Version]
- Odogwu, L.; Mathieu, L.; Blumenthal, G.; Larkins, E.; Goldberg, K.B.; Griffin, N.; Bijwaard, K.; Lee, E.Y.; Philip, R.; Jiang, X. Fda approval summary: Dabrafenib and trametinib for the treatment of metastatic non-small cell lung cancers harboring BRAF V600E mutations. Oncologist 2018, 23, 740. [Google Scholar] [CrossRef] [Green Version]
- Renouf, D.J.; Velazquez-Martin, J.P.; Simpson, R.; Siu, L.L.; Bedard, P.L. Ocular toxicity of targeted therapies. J. Clin. Oncol. 2012, 30, 3277–3286. [Google Scholar] [CrossRef]
- Jin, J.; Guo, Q.; Xie, J.; Jin, D.; Zhu, Y. Combination of MEK inhibitor and the JAK2-STAT3 pathway inhibition for the therapy of colon cancer. Pathol. Oncol. Res. 2019, 25, 769–775. [Google Scholar] [CrossRef]
- Sato, H.; Yamamoto, H.; Sakaguchi, M.; Shien, K.; Tomida, S.; Shien, T.; Ikeda, H.; Hatono, M.; Torigoe, H.; Namba, K.; et al. Combined inhibition of MEK and PI3K pathways overcomes acquired resistance to EGFR-TKIs in non-small cell lung cancer. Cancer Sci. 2018, 109, 3183–3196. [Google Scholar] [CrossRef]
- Cortes, J.; Tamura, K.; DeAngelo, D.J.; De Bono, J.; Lorente, D.; Minden, M.; Uy, G.L.; Kantarjian, H.; Chen, L.S.; Gandhi, V. Phase I studies of azd1208, a proviral integration moloney virus kinase inhibitor in solid and haematological cancers. Br. J. Cancer 2018, 118, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Le, X.; Antony, R.; Razavi, P.; Treacy, D.J.; Luo, F.; Ghandi, M.; Castel, P.; Scaltriti, M.; Baselga, J.; Garraway, L.A. Systematic functional characterization of resistance to PI3K inhibition in breast cancer. Cancer Discov. 2016, 6, 1134–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef]
- Sohraby, F.; Bagheri, M.; Aryapour, H. Performing an in silico repurposing of existing drugs by combining virtual screening and molecular dynamics simulation. In Computational Methods for Drug Repurposing; Springer: Cham, Switzerland, 2019; pp. 23–43. [Google Scholar]
- Wójcikowski, M.; Ballester, P.J.; Siedlecki, P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci. Rep. 2017, 7, 46710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurz, R.P.; Sastri, C.; D’Amico, D.C.; Herberich, B.; Jackson, C.L.; Pettus, L.H.; Tasker, A.S.; Wu, B.; Guerrero, N.; Lipford, J.R. Discovery of imidazopyridazines as potent PIM-1/2 kinase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 5580–5590. [Google Scholar] [CrossRef]
- Protein Data Bank; Nakae, S.; Kitamura, M.; Shirai, T.; Tada, T. Structure of the Human Mitogen-Activated Protein Kinase Kinase 1 (MEK1). 2014. Available online: https://datamed.org/display-item.php?repository=0002&id=5952ebec5152c64c3b126f08&query=MAP2K1 (accessed on 3 May 2021).
- Rohini, K.; Ramanathan, K.; Shanthi, V. Multi-dimensional screening strategy for drug repurposing with statistical framework—A new road to influenza drug discovery. Cell Biochem. Biophys. 2019, 77, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Patschull, A.O.; Gooptu, B.; Ashford, P.; Daviter, T.; Nobeli, I. In silico assessment of potential druggable pockets on the surface of α1-antitrypsin conformers. PLoS ONE 2012, 7, e36612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger. Sitemap, Schrödinger Release-2020; Schrödinger: New York, NY, USA, 2020. [Google Scholar]
- Zhou, H.; Wang, C.; Deng, T.; Tao, R.; Li, W. Novel urushiol derivatives as HDAC8 inhibitors: Rational design, virtual screening, molecular docking and molecular dynamics studies. J. Biomol. Struct. Dyn. 2018, 36, 1966–1978. [Google Scholar] [CrossRef]
- Borkotoky, S.; Meena, C.K.; Murali, A. Interaction analysis of T7 RNA polymerase with heparin and its low molecular weight derivatives—An in silico approach. Bioinform. Biol. Insights 2016, 10, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Ballester, P.J.; Mitchell, J.B. A machine learning approach to predicting protein–ligand binding affinity with applications to molecular docking. Bioinformatics 2010, 26, 1169–1175. [Google Scholar] [CrossRef] [Green Version]
- Williams, C. Reverse fingerprinting, similarity searching by group fusion and fingerprint bit importance. Mol. Divers. 2006, 10, 311–332. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Kammarabutr, J.; Mahalapbutr, P.; Nutho, B.; Kungwan, N.; Rungrotmongkol, T. Low susceptibility of asunaprevir towards R155K and D168A point mutations in HCV NS3/4A protease: A molecular dynamics simulation. J. Mol. Graph. 2019, 89, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Mahalapbutr, P.; Wonganan, P.; Chavasiri, W.; Rungrotmongkol, T. Butoxy mansonone G inhibits STAT3 and AKT signaling pathways in non-small cell lung cancers: Combined experimental and theoretical investigations. Cancers 2019, 11, 437. [Google Scholar] [CrossRef] [Green Version]
- Meeprasert, A.; Hannongbua, S.; Rungrotmongkol, T. Key binding and susceptibility of NS3/4A serine protease inhibitors against hepatitis C virus. J. Chem. Inf. Model. 2014, 54, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Nutho, B.; Mahalapbutr, P.; Hengphasatporn, K.; Pattaranggoon, N.C.; Simanon, N.; Shigeta, Y.; Hannongbua, S.; Rungrotmongkol, T. Why are lopinavir and ritonavir effective against the newly emerged coronavirus 2019? Atomistic insights into the inhibitory mechanisms. Biochemistry 2020, 59, 1769–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nutho, B.; Rungrotmongkol, T. Binding recognition of substrates in NS2B/NS3 serine protease of zika virus revealed by molecular dynamics simulations. J. Mol. Graph. Model. 2019, 92, 227–235. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald: An N⋅log(N) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Uberuaga, B.P.; Anghel, M.; Voter, A.F. Synchronization of trajectories in canonical molecular-dynamics simulations: Observation, explanation, and exploitation. J. Chem. Phys. 2004, 120, 6363–6374. [Google Scholar] [CrossRef]
- Berendsen, H.J.; Postma, J.V.; van Gunsteren, W.F.; DiNola, A.R.H.J.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Naïm, M.; Bhat, S.; Rankin, K.N.; Dennis, S.; Chowdhury, S.F.; Siddiqi, I.; Drabik, P.; Sulea, T.; Bayly, C.I.; Jakalian, A.; et al. Solvated interaction energy (SIE) for scoring protein−ligand binding affinities. 1. Exploring the parameter space. J. Chem. Inf. Model. 2007, 47, 122–133. [Google Scholar] [CrossRef]
- Ghattas, M.A.; Raslan, N.; Sadeq, A.; Al Sorkhy, M.; Atwater, N. Druggability analysis and classification of protein tyrosine phosphatase active sites. Drug Des. Dev. Ther. 2016, 10, 3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.-K.; Park, J.-I. Mek1/2 inhibitors: Molecular activity and resistance mechanisms. Semin. Oncol. 2015, 42, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.-T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep docking: A deep learning platform for augmentation of structure based drug discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef]
- Backman, T.W.; Cao, Y.; Girke, T. Chemmine tools: An online service for analyzing and clustering small molecules. Nucleic Acids Res. 2011, 39, W486–W491. [Google Scholar] [CrossRef]
- Tripathi, S.K.; Muttineni, R.; Singh, S.K. Extra precision docking, free energy calculation and molecular dynamics simulation studies of CDK2 inhibitors. J. Theor. Biol. 2013, 334, 87–100. [Google Scholar] [CrossRef]
- Abdel-Hamid, M.K.; McCluskey, A. In silico docking, molecular dynamics and binding energy insights into the bolinaquinone-clathrin terminal domain binding site. Molecules 2014, 19, 6609–6622. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.D.; Muthusamy, K. Molecular modeling, quantum polarized ligand docking and structure-based 3D-QSAR analysis of the imidazole series as dual AT1 and ETa receptor antagonists. Acta Pharmacol. Sin. 2013, 34, 1592–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, R.; Mishra, R.; Kumar, R.; Mazumder, A.; Kumar, A. Pyridine moiety: Recent advances in cancer treatment. Indian J. Pharm. Sci. 2021, 83, 162–185. [Google Scholar] [CrossRef]
- Miles, J.A.; Ng, J.H.; Sreenivas, B.Y.; Courageux, C.; Igert, A.; Dias, J.; McGeary, R.P.; Brazzolotto, X.; Ross, B.P. Discovery of drug-like acetylcholinesterase inhibitors by rapid virtual screening of a 6.9 million compound database. Chem. Biol. Drug Des. 2021, 97, 1048–1058. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. MEK1/2 dual-specificity protein kinases: Structure and regulation. Biochem. Biophys. Res. Commun. 2012, 417, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Xie, L.; Bourne, P.E. Insights into the binding mode of MEK Type-III inhibitors. A step towards discovering and designing allosteric kinase inhibitors across the human kinome. PLoS ONE 2017, 12, e0179936. [Google Scholar] [CrossRef] [Green Version]
- Varalda, M.; Antona, A.; Bettio, V.; Roy, K.; Vachamaram, A.; Yellenki, V.; Massarotti, A.; Baldanzi, G.; Capello, D. Psychotropic drugs show anticancer activity by disrupting mitochondrial and lysosomal function. Front. Oncol. 2020, 10, 2148. [Google Scholar] [CrossRef]
- Jin, F.; Gao, D.; Wu, Q.; Liu, F.; Chen, Y.; Tan, C.; Jiang, Y. Exploration of N-(2-aminoethyl) piperidine-4-carboxamide as a potential scaffold for development of VEGFR-2, ERK-2 and ABL-1 multikinase inhibitor. Bioorg. Med. Chem. 2013, 21, 5694–5706. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I. An insight into the therapeutic potential of quinazoline derivatives as anticancer agents. MedChemComm 2017, 8, 871–885. [Google Scholar]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into protein-ligand interactions: Mechanisms, models, and methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sites | Site Score | Dscore | Binding Pocket Region |
|---|---|---|---|
| 1 | 1.067 | 0.995 | LEU74, GLY75, ALA76, GLY77, ASN78, GLY79, GLY80, VAL82, ALA95, LYS97, ILE99, VAL127, MET143, GLU144, HIS145, MET146, GLY149, SER150, ASP152, GLN153, LYS192, SER194, ASN195, LEU197, CYS207, ASP208, PHE209, GLY210, VAL211, SER212 |
| 2 | 1.028 | 1.05 | GLU39, GLN45, GLN46, ARG49, LEU50, ALA52, PHE53, LEU54, GLN56, LYS57, LEU92, VAL93, HIS119, GLU120, CYS121, ASN122, SER123, PRO124, TYR125, ILE126, VAL127, GLY128, PHE129, TYR130, GLU144, HIS145, MET146, ASP147, LYS168, ILE171, ALA172, LYS175, ASN199, ARG201, GLY202, GLU203, ILE204, LYS205, ASP365, VAL369, ASP370, PHE371, ALA372 |
| 3 | 0.974 | 1.005 | GLU39, LEU40, GLU41, LEU42, GLN46, ASN122, SER123, PRO124, TYR125, ILE174, LYS175, THR178, TYR179, ARG181, GLU182, LYS183, VAL242, LEU352, LYS353, MET356 |
| 4 | 0.819 | 0.782 | LEU118, HIS119, ILE126, LEU180, HIS184, LYS185, ILE186, MET187, HIS188, ARG189, ASP208, PHE209, GLY210, GLY213, GLN214, ASP217 |
| 5 | 0.702 | 0.673 | VAL254, VAL258, PRO262, PRO265, PRO266, LEU271, PRO321, PRO322, PRO323, LYS324, LEU325, PRO326, SER327, GLN335, ASN339 |
| Compound ID | Docking Score (kcal/mol) | ΔGbind (kcal/mol) | ΔGbind Coulomb | ΔGbind Lipophilic | ΔGbind Solv GB | ΔGbind vdW | Ligand Strain Energy |
|---|---|---|---|---|---|---|---|
| Reference | −3.423 | −46.137 | −13.639 | −32.888 | 32.888 | −43.528 | 24.43 |
| DB12661 | −7.051 | −87.013 | −16.84 | −46.647 | 31.692 | −57.476 | 5.29 |
| DB07642 | −6.174 | −83.845 | −20.352 | −42.431 | 28.151 | −55.062 | 8.453 |
| DB02366 | −7.427 | −76.925 | −34.282 | −36.488 | 39.865 | −47.657 | 7.09 |
| DB08251 | −11.98 | −75.956 | −34.186 | −24.74 | 27.909 | −44.926 | 3.995 |
| DB01771 | −7.775 | −75.093 | −28.532 | −45.543 | 38.739 | −46.271 | 10.615 |
| DB12847 | −6.716 | −66.948 | −29.293 | −28.799 | 31.254 | −41.632 | 4.669 |
| DB07177 | −6.989 | −65.876 | −14.264 | −51.153 | 31.763 | −39.082 | 18.693 |
| DB13174 | −9.287 | −64.939 | −22.947 | −21.409 | 20.618 | −42.359 | 2.315 |
| DB07125 | −8.416 | −63.963 | −20.194 | −26.628 | 25.206 | −42.305 | 8.554 |
| DB07773 | −9.256 | −61.255 | −31.925 | −29.541 | 32.325 | −36.44 | 7.628 |
| DB07546 | −6.456 | −61.064 | −24.4 | −37.67 | 35.031 | −36.04 | 9.162 |
| DB02849 | −8.72 | −59.793 | −49.808 | −16.914 | 42.084 | −35.493 | 5.028 |
| DB02709 | −7.091 | −59.576 | −21.878 | −29.309 | 21.114 | −32.041 | 3.817 |
| DB04241 | −8.469 | −57.965 | −46.177 | −23.207 | 30.706 | −27.2 | 10.366 |
| DrugBank ID | 2D Strucure | Stars a | CNS b | QPlogS c | HOA d |
|---|---|---|---|---|---|
| Reference |  | 1 | −2 | −8.042 | 1 |
| DB08251 |  | 1 | −2 | −3.274 | 1 |
| DB13174 |  | 0 | −2 | −2.449 | 2 |
| DB07773 |  | 0 | −2 | −1.457 | 1 |
| DB02849 |  | 1 | −2 | −2.647 | 2 |
| DB04241 |  | 0 | −2 | −3.902 | 2 |
| DB07125 |  | 0 | −2 | −1.666 | 1 |
| DB01771 |  | 0 | −2 | −2.794 | 3 |
| DB02366 |  | 0 | −2 | −5.171 | 3 |
| DB02709 |  | 0 | −2 | −0.905 | 2 |
| DB12661 |  | 0 | 0 | −5.177 | 3 |
| DB07177 |  | 0 | −2 | −4.861 | 3 |
| DB12847 |  | 0 | −2 | −3.52 | 2 |
| DB07546 |  | 0 | −2 | −5.71 | 3 |
| DB07642 |  | 0 | −1 | −3.874 | 3 |
| Compounds | Energy Components | ||||
|---|---|---|---|---|---|
| EvdW | Eele | Reaction Field | Cavity | ΔGbind | |
| Trametinib | −51.05 ± 0.34 | −9.58 ± 0.20 | 19.25 ± 0.26 | −9.05 ± 0.07 | −8.17 ± 0.04 |
| DB12661 | −52.08 ± 0.32 | −4.29 ± 0.17 | 12.18 ± 0.24 | −8.52 ± 0.05 | −8.41 ± 0.04 |
| DB07642 | −43.91 ± 0.37 | −6.90 ± 0.21 | 14.62 ± 0.36 | −8.02 ± 0.06 | −7.52 ± 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thirunavukkarasu, M.K.; Suriya, U.; Rungrotmongkol, T.; Karuppasamy, R. In Silico Screening of Available Drugs Targeting Non-Small Cell Lung Cancer Targets: A Drug Repurposing Approach. Pharmaceutics 2022, 14, 59. https://doi.org/10.3390/pharmaceutics14010059
Thirunavukkarasu MK, Suriya U, Rungrotmongkol T, Karuppasamy R. In Silico Screening of Available Drugs Targeting Non-Small Cell Lung Cancer Targets: A Drug Repurposing Approach. Pharmaceutics. 2022; 14(1):59. https://doi.org/10.3390/pharmaceutics14010059
Chicago/Turabian StyleThirunavukkarasu, Muthu Kumar, Utid Suriya, Thanyada Rungrotmongkol, and Ramanathan Karuppasamy. 2022. "In Silico Screening of Available Drugs Targeting Non-Small Cell Lung Cancer Targets: A Drug Repurposing Approach" Pharmaceutics 14, no. 1: 59. https://doi.org/10.3390/pharmaceutics14010059