
Drug Targeting and Nanomedicine: Lessons Learned from Liver Targeting and Opportunities for Drug Innovation


Abstract
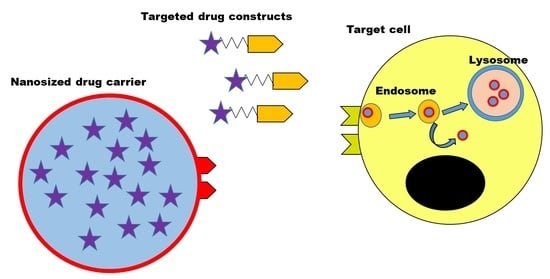
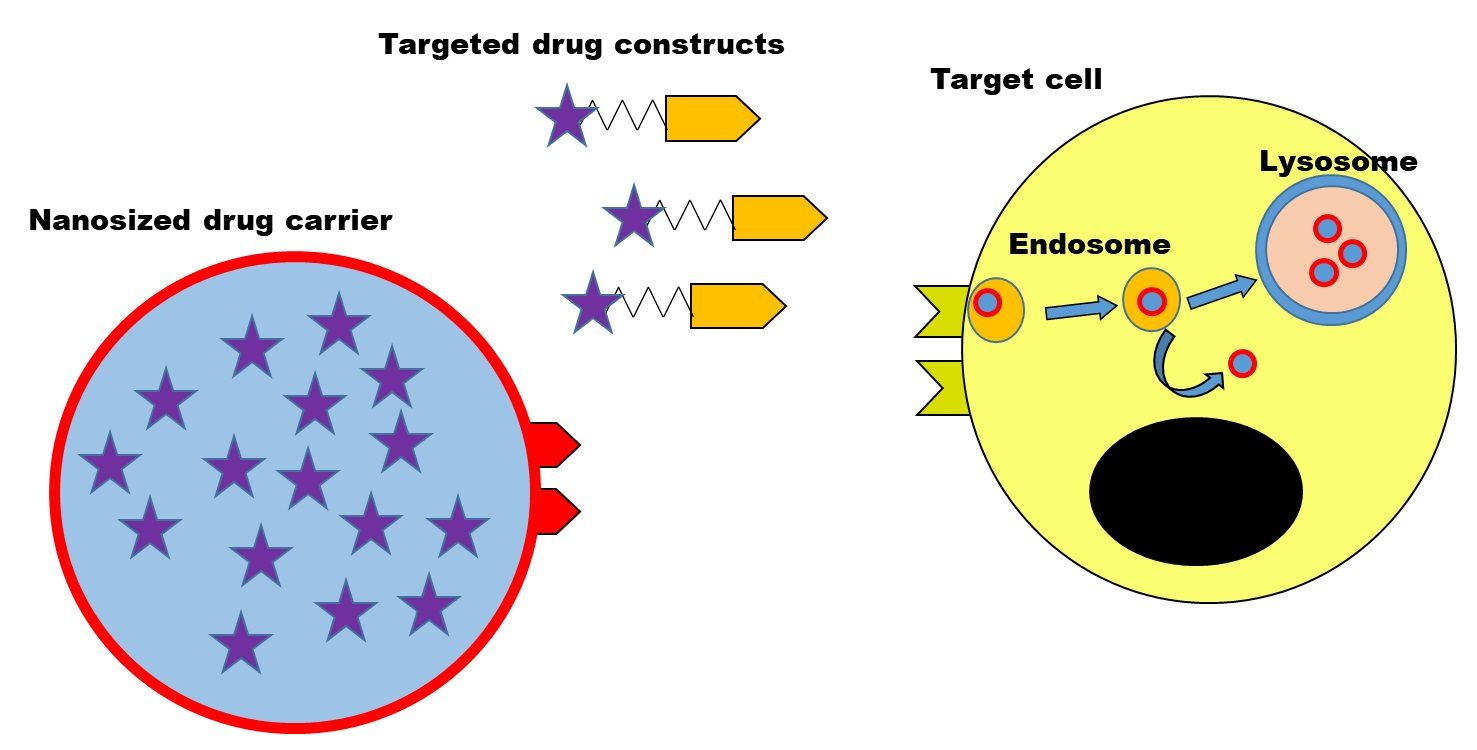
:
1. Introduction
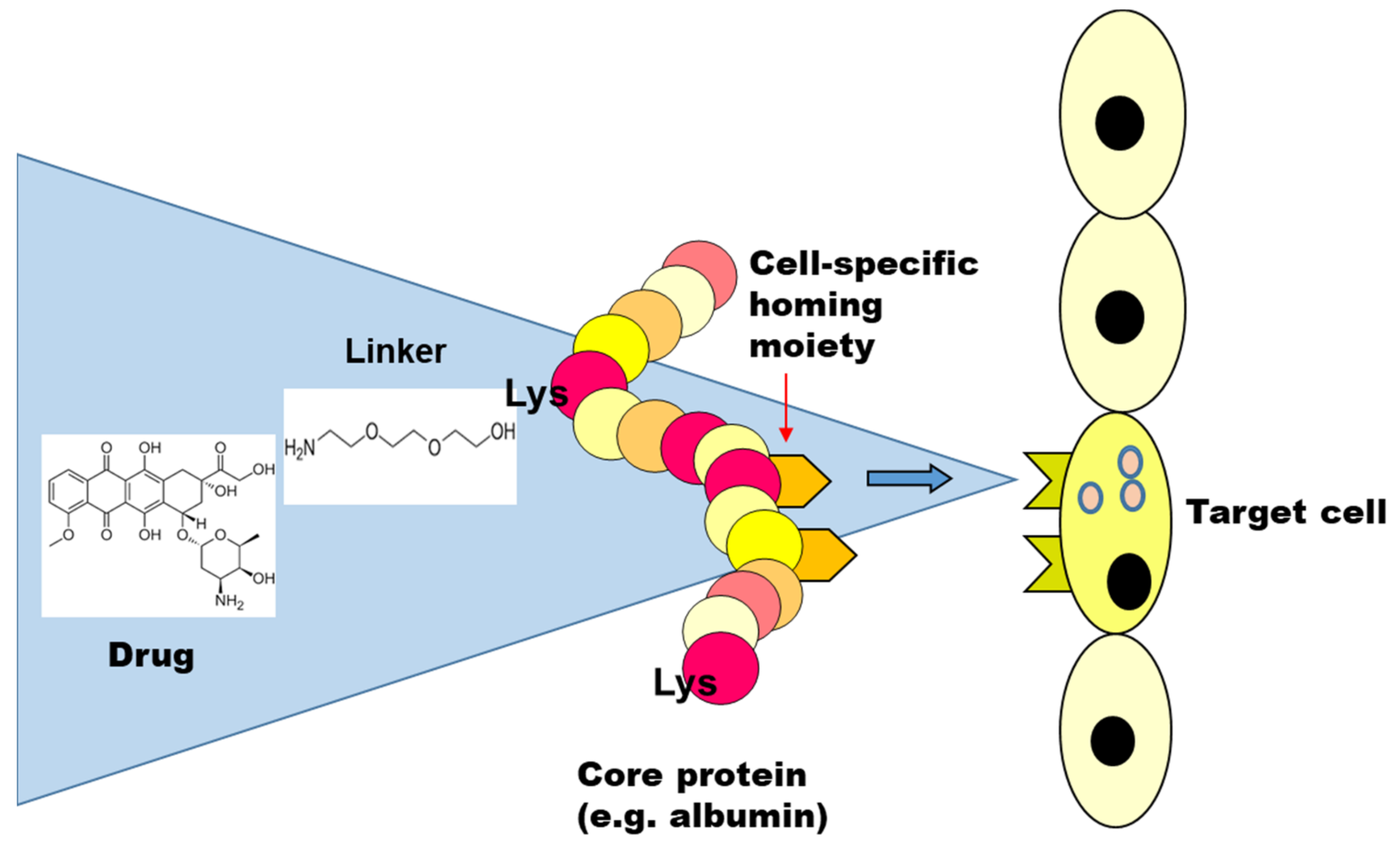
2. Drug Targeting Constructs
3. Drug Targeting Approaches in Fibrotic Livers
4. Other Monomeric Carriers
5. Nanomedicines to Delivery Drugs into Target Cells
6. Controlling the Interactions of Targeted Drugs and Nanomedicines with Cells
6.1. Interactions with Biological Fluids
6.2. Interactions with Cells and Intracellular Fate
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Ravi Kiran, A.V.V.V.; Kusuma Kumari, G.; Krishnamurthy, P.T.; Khaydarov, R.R. Tumor microenvironment and nanotherapeutics: Intruding the tumor fort. Biomater. Sci. 2021, 9, 7667–7704. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Feron, O.; Preat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, E.A.; Ferguson, E.L.; Thomas, D.W. The enhanced permeability retention effect: A new paradigm for drug targeting in infection. J. Antimicrob. Chemother. 2013, 68, 257–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera, E. Liposomal anthracyclines in metastatic breast cancer: Clinical update. Oncologist 2003, 8, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Sindhwani, S.; Syed, A.M.; Ngai, J.; Kingston, B.R.; Maiorino, L.; Rothschild, J.; MacMillan, P.; Zhang, Y.; Rajesh, N.U.; Hoang, T.; et al. The entry of nanoparticles into solid tumours. Nat. Mater. 2020, 19, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil(R)—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Hrkach, J.; Langer, R. From micro to nano: Evolution and impact of drug delivery in treating disease. Drug Deliv. Transl. Res. 2020, 10, 567–570. [Google Scholar] [CrossRef]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Darjuan, M.M.; Mercer, J.E.; Chen, S.; van der Meel, R.; Thewalt, J.L.; Tam, Y.Y.C.; Cullis, P.R. On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and siRNA. ACS Nano 2018, 12, 4787–4795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, L.; Maiseyeu, A. Low-density lipoprotein nanomedicines: Mechanisms of targeting, biology, and theranostic potential. Drug Deliv. 2021, 28, 408–421. [Google Scholar] [CrossRef]
- Sebak, A.A.; Gomaa, I.E.O.; ElMeshad, A.N.; Farag, M.H.; Breitinger, U.; Breitinger, H.G.; AbdelKader, M.H. Distinct Proteins in Protein Corona of Nanoparticles Represent a Promising Venue for Endogenous Targeting—Part II: In vitro and in vivo Kinetics Study. Int. J. Nanomed. 2020, 15, 9539–9556. [Google Scholar] [CrossRef]
- Lammers, T.; Kiessling, F.; Ashford, M.; Hennink, W.; Crommelin, D.; Storm, G. Cancer nanomedicine: Is targeting our target? Nat. Rev. Mater. 2016, 1, 16069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germain, M.; Caputo, F.; Metcalfe, S.; Tosi, G.; Spring, K.; Aslund, A.K.O.; Pottier, A.; Schiffelers, R.; Ceccaldi, A.; Schmid, R. Delivering the power of nanomedicine to patients today. J. Control. Release 2020, 326, 164–171. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Dvorak, H.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, J.; Yuan, Z. Strategies and challenges to improve the performance of tumor-associated active targeting. J. Mater. Chem. B 2020, 8, 3959–3971. [Google Scholar] [CrossRef]
- Yang, M.; Wu, E.; Tang, W.; Qian, J.; Zhan, C. Interplay between nanomedicine and protein corona. J. Mater. Chem. B 2021, 9, 6713–6727. [Google Scholar] [CrossRef]
- Monopoli, M.P.; Åberg, C.; Salvati, A.; Dawson, K.A. Biomolecular coronas provide the biological identity of nanosized materials. Nat. Nanotechnol 2012, 7, 779–786. [Google Scholar] [CrossRef]
- Nel, A.E.; Madler, L.; Velegol, D.; Xia, T.; Hoek, E.M.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Poelstra, K.; Beljaars, L.; Melgert, B.N. Cell-specific delivery of biologicals: Problems, pitfalls and possibilities of antifibrotic compounds in the liver. Drug Discov. Today 2013, 18, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Baah, S.; Laws, M.; Rahman, K.M. Antibody-Drug Conjugates—A Tutorial Review. Molecules 2021, 26, 2943. [Google Scholar] [CrossRef] [PubMed]
- Kostova, V.; Desos, P.; Starck, J.B.; Kotschy, A. The Chemistry Behind ADCs. Pharmaceuticals 2021, 14, 422. [Google Scholar] [CrossRef]
- Poelstra, K.; Prakash, J.; Beljaars, L. Drug targeting to the diseased liver. J. Control. Release 2012, 161, 188–197. [Google Scholar] [CrossRef]
- Caligiuri, A.; Gentilini, A.; Pastore, M.; Gitto, S.; Marra, F. Cellular and Molecular Mechanisms Underlying Liver Fibrosis Regression. Cells 2021, 10, 2759. [Google Scholar] [CrossRef]
- Wang, X.; Ikejima, K.; Kon, K.; Arai, K.; Aoyama, T.; Okumura, K.; Abe, W.; Sato, N.; Watanabe, S. Ursolic acid ameliorates hepatic fibrosis in the rat by specific induction of apoptosis in hepatic stellate cells. J. Hepatol. 2011, 55, 379–387. [Google Scholar] [CrossRef]
- Javary, J.; Allain, N.; Ezzoukhry, Z.; Di Tommaso, S.; Dupuy, J.W.; Costet, P.; Dugot-Senant, N.; Saltel, F.; Moreau, V.; Dubus, P; et al. Reptin/RUVBL2 is required for hepatocyte proliferation in vivo, liver regeneration and homeostasis. Liver Int. 2021, 41, 1423–1429. [Google Scholar] [CrossRef]
- Nelson, D.R.; Tu, Z.; Soldevila-Pico, C.; Abdelmalek, M.; Zhu, H.; Xu, Y.L.; Cabrera, R.; Liu, C.; Davis, G.L. Long-term interleukin 10 therapy in chronic hepatitis C patients has a proviral and anti-inflammatory effect. Hepatology 2003, 38, 859–868. [Google Scholar] [CrossRef]
- Nelson, D.A.; Petty, C.C.; Bost, K.L. Infection with murine gammaherpesvirus 68 exacerbates inflammatory bowel disease in IL-10-deficient mice. Inflamm. Res. 2009, 58, 881–889. [Google Scholar] [CrossRef]
- Kang, D.J.; Betrapally, N.S.; Ghosh, S.A.; Sartor, R.B.; Hylemon, P.B.; Gillevet, P.M.; Sanyal, A.J.; Heuman, D.M.; Carl, D.; Zhou, H.; et al. Gut microbiota drive the development of neuroinflammatory response in cirrhosis in mice. Hepatology 2016, 64, 1232–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2019, 72, 558–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, F.; Grander, C.; Effenberger, M.; Adolph, T.E.; Tilg, H. Gut Dysfunction and Non-alcoholic Fatty Liver Disease. Front. Endocrinol. 2019, 10, 611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacke, F.; Weiskirchen, R. An update on the recent advances in antifibrotic therapy. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 1143–1152. [Google Scholar] [CrossRef]
- Brigstock, D.R. Extracellular Vesicles in Organ Fibrosis: Mechanisms, Therapies, and Diagnostics. Cells 2021, 10, 1596. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef]
- Jensen, C.; Nissen, N.I.; Von Arenstorff, C.S.; Karsdal, M.A.; Willumsen, N. Serological assessment of collagen fragments and tumor fibrosis may guide immune checkpoint inhibitor therapy. J. Exp. Clin. Cancer Res. 2021, 40, 326. [Google Scholar] [CrossRef] [PubMed]
- Hurkmans, D.P.; Jensen, C.; Koolen, S.L.W.; Aerts, J.; Karsdal, M.A.; Mathijssen, R.H.J.; Willumsen, N. Blood-based extracellular matrix biomarkers are correlated with clinical outcome after PD-1 inhibition in patients with metastatic melanoma. J. Immunother. Cancer 2020, 8, e001193. [Google Scholar] [CrossRef]
- Poelstra, K.; Schuppan, D. Targeted therapy of liver fibrosis/cirrhosis and its complications. J. Hepatol. 2011, 55, 726–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.A.; Friedman, S.L. Inflammatory and fibrotic mechanisms in NAFLD-Implications for new treatment strategies. J. Intern. Med. 2021, 291, 11–31. [Google Scholar] [CrossRef]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Huisman, T.M.; Dieterich, D.T.; Friedman, S.L. Experimental and Investigational Targeted Therapies for the Management of Fibrosis in NASH: An Update. J. Exp. Pharm. 2021, 13, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Park, E.I.; Mi, Y.; Unverzagt, C.; Gabius, H.J.; Baenziger, J.U. The asialoglycoprotein receptor clears glycoconjugates terminating with sialic acid alpha 2,6GalNAc. Proc. Natl. Acad. Sci. USA 2005, 102, 17125–17129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, A.A.; Devarajan, P.V. Asialoglycoprotein receptor mediated hepatocyte targeting—Strategies and applications. J. Control. Release 2015, 203, 126–139. [Google Scholar] [CrossRef]
- Ivanenkov, Y.A.; Majouga, A.G.; Petrov, R.A.; Petrov, S.A.; Kovalev, S.V.; Maklakova, S.Y.; Yamansarov, E.Y.; Saltykova, I.V.; Deyneka, E.V.; Filkov, G.I.; et al. Synthesis and biological evaluation of novel doxorubicin-containing ASGP-R-targeted drug-conjugates. Bioorg. Med. Chem. Lett. 2018, 28, 503–508. [Google Scholar] [CrossRef]
- Ahmed, M.; Narain, R. Carbohydrate-based materials for targeted delivery of drugs and genes to the liver. Nanomedicine 2015, 10, 2263–2288. [Google Scholar] [CrossRef]
- Teran-Saavedra, N.G.; Sarabia-Sainz, J.A.; Velázquez-Contreras, E.F.; Montfort, G.R.-C.; Pedroza-Montero, M.; Vazquez-Moreno, L. Albumin-Albumin/Lactosylated Core-Shell Nanoparticles: Therapy to Treat Hepatocellular Carcinoma for Controlled Delivery of Doxorubicin. Molecules 2020, 25, 5432. [Google Scholar] [CrossRef]
- Singh, L.; Indermun, S.; Govender, M.; Kumar, P.; du Toit, L.C.; Choonara, Y.E.; Pillay, V. Drug Delivery Strategies for Antivirals against Hepatitis B Virus. Viruses 2018, 10, 267. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.C.; Gantzel, R.H.; Clària, J.; Trebicka, J.; Møller, H.J.; Grønbæk, H. Macrophage Activation Markers, CD163 and CD206, in Acute-on-Chronic Liver Failure. Cells 2020, 9, 1175. [Google Scholar] [CrossRef]
- Janeiro, A.L.; Padilla-Ansala, C.; de Andrea, C.E.; Hardisson, D.; Melero, I. Prognostic value of macrophage polarization markers in epithelial neoplasms and melanoma. A systematic review and meta-analysis. Mod. Pathol. 2020, 33, 1458–1465. [Google Scholar] [CrossRef]
- Cheng, P.; Li, S.; Chen, H. Macrophages in Lung Injury, Repair, and Fibrosis. Cells 2021, 10, 436. [Google Scholar] [CrossRef]
- Melgert, B.N.; Olinga, P.; Van Der Laan, J.J.; Weert, B.; Cho, J.; Schuppan, D.; Groothuis, G.M.; Meijer, D.K.; Poelstra, K. Targeting dexamethasone to Kupffer cells: Effects on liver inflammation and fibrosis in rats. Hepatology 2001, 34, 719–728. [Google Scholar] [CrossRef]
- Melgert, B.N.; Olinga, P.; Weert, B.; Slooff, M.J.; Meijer, D.K.; Poelstra, K.; Groothuis, G.M. Cellular distribution and handling of liver-targeting preparations in human livers studied by a liver lobe perfusion. Drug Metab. Dispos. 2001, 29, 361–367. [Google Scholar]
- Melgert, B.N.; Olinga, P.; Jack, V.K.; Molema, G.; Meijer, D.K.; Poelstra, K. Dexamethasone coupled to albumin is selectively taken up by rat nonparenchymal liver cells and attenuates LPS-induced activation of hepatic cells. J. Hepatol. 2000, 32, 603–611. [Google Scholar] [CrossRef]
- Melgert, B.N.; Weert, B.; Schellekens, H.; Meijer, D.K.; Poelstra, K. The pharmacokinetic and biological activity profile of dexamethasone targeted to sinusoidal endothelial and Kupffer cells. J. Drug Target. 2003, 11, 1–10. [Google Scholar] [CrossRef]
- Bruneau, A.; Hundertmark, J.; Guillot, A.; Tacke, F. Molecular and Cellular Mediators of the Gut-Liver Axis in the Progression of Liver Diseases. Front. Med. 2021, 8, 725390. [Google Scholar] [CrossRef]
- Schippers, M.; Beljaars, L.; Post, E.; Lotersztajn, S.; Reker-Smit, C.; Han, B.; Munoz-Llancao, P.; Schmidt, M.; Poelstra, K. Upregulation of Epac-1 in Hepatic Stellate Cells by Prostaglandin E2 in Liver Fibrosis Is Associated with Reduced Fibrogenesis. J. Pharm. Exp. 2017, 363, 126–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Do, D.C.; Ishmael, F.T.; Squadrito, M.L.; Tang, H.M.; Tang, H.L.; Hsu, M.H.; Qiu, L.; Li, C.; Zhang, Y.; et al. Mannose receptor modulates macrophage polarization and allergic inflammation through miR-511-3p. J. Allergy Clin. Immunol. 2018, 141, 350–364.e8. [Google Scholar] [CrossRef] [Green Version]
- Peled, E.; Sosnik, A. Amphiphilic galactomannan nanoparticles trigger the alternative activation of murine macrophages. J. Control. Release 2021, 339, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Beljaars, L.; Molema, G.; Weert, B.; Bonnema, H.; Olinga, P.; Groothuis, G.M.; Meijer, D.K.; Poelstra, K. Albumin modified with mannose 6-phosphate: A potential carrier for selective delivery of antifibrotic drugs to rat and human hepatic stellate cells. Hepatology 1999, 29, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Beljaars, L.; Weert, B.; Geerts, A.; Meijer, D.K.; Poelstra, K. The preferential homing of a platelet derived growth factor receptor-recognizing macromolecule to fibroblast-like cells in fibrotic tissue. Biochem. Pharm. 2003, 66, 1307–1317. [Google Scholar] [CrossRef]
- Beljaars, L.; Molema, G.; Schuppan, D.; Geerts, A.; De Bleser, P.J.; Weert, B.; Meijer, D.K.; Poelstra, K. Successful targeting to rat hepatic stellate cells using albumin modified with cyclic peptides that recognize the collagen type VI receptor. J. Biol. Chem. 2000, 275, 12743–12751. [Google Scholar] [CrossRef] [Green Version]
- Greupink, R.; Bakker, H.I.; Bouma, W.; Reker-Smit, C.; Meijer, D.K.; Beljaars, L.; Poelstra, K. The antiproliferative drug doxorubicin inhibits liver fibrosis in bile duct-ligated rats and can be selectively delivered to hepatic stellate cells in vivo. J. Pharm. Exp. 2006, 317, 514–521. [Google Scholar] [CrossRef]
- Greupink, R.; Bakker, H.I.; Reker-Smit, C.; van Loenen-Weemaes, A.M.; Kok, R.J.; Meijer, D.K.; Beljaars, L.; Poelstra, K. Studies on the targeted delivery of the antifibrogenic compound mycophenolic acid to the hepatic stellate cell. J. Hepatol. 2005, 43, 884–892. [Google Scholar] [CrossRef]
- Hagens, W.I.; Beljaars, L.; Mann, D.A.; Wright, M.C.; Julien, B.; Lotersztajn, S.; Reker-Smit, C.; Poelstra, K. Cellular targeting of the apoptosis-inducing compound gliotoxin to fibrotic rat livers. J. Pharm. Exp. 2008, 324, 902–910. [Google Scholar] [CrossRef] [Green Version]
- Hagens, W.I.; Mattos, A.; Greupink, R.; de Jager-Krikken, A.; Reker-Smit, C.; van Loenen-Weemaes, A.; Gouw, I.A.; Poelstra, K.; Beljaars, L. Targeting 15d-prostaglandin J2 to hepatic stellate cells: Two options evaluated. Pharm. Res. 2007, 24, 566–574. [Google Scholar] [CrossRef] [Green Version]
- Gonzalo, T.; Talman, E.G.; van de Ven, A.; Temming, K.; Greupink, R.; Beljaars, L.; Reker-Smit, C.; Meijer, D.K.; Molema, G.; Poelstra, K.; et al. Selective targeting of pentoxifylline to hepatic stellate cells using a novel platinum-based linker technology. J. Control. Release 2006, 111, 193–203. [Google Scholar] [CrossRef]
- Rachmawati, H.; Reker-Smit, C.; Lub-de Hooge, M.N.; van Loenen-Weemaes, A.; Poelstra, K.; Beljaars, L. Chemical modification of interleukin-10 with mannose 6-phosphate groups yields a liver-selective cytokine. Drug Metab. Disposition. 2007, 35, 814–821. [Google Scholar] [CrossRef] [Green Version]
- Van Beuge, M.M.; Prakash, J.; Lacombe, M.; Post, E.; Reker-Smit, C.; Beljaars, L.; Poelstra, K. Enhanced effectivity of an ALK5-inhibitor after cell-specific delivery to hepatic stellate cells in mice with liver injury. PLoS ONE 2013, 8, e56442. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, F.; Teekamp, N.; Post, E.; Schuppan, D.; Kim, Y.O.; Zuidema, J.; Steendam, R.; Klose, M.H.M.; Meier-Menches, S.M.; Casini, A.; et al. The antifibrotic potential of a sustained release formulation of a PDGFbeta-receptor targeted rho kinase inhibitor. J. Control. Release 2019, 296, 250–257. [Google Scholar] [CrossRef]
- Van Beuge, M.M.; Prakash, J.; Lacombe, M.; Post, E.; Reker-Smit, C.; Beljaars, L.; Poelstra, K. Increased liver uptake and reduced hepatic stellate cell activation with a cell-specific conjugate of the Rho-kinase inhibitor Y27632. Pharm. Res. 2011, 28, 2045–2054. [Google Scholar] [CrossRef] [Green Version]
- Van Beuge, M.M.; Prakash, J.; Lacombe, M.; Gosens, R.; Post, E.; Reker-Smit, C.; Beljaars, L.; Poelstra, K. Reduction of fibrogenesis by selective delivery of a Rho kinase inhibitor to hepatic stellate cells in mice. J. Pharm. Exp. 2011, 337, 628–635. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.; Frohn, F.; Magdaleno, F.; Reker-Smit, C.; Schierwagen, R.; Schierwagen, I.; Uschner, F.E.; van Dijk, F.; Fürst, D.O.; Djudjaj, S.; et al. Rho-kinase inhibitor coupled to peptide-modified albumin carrier reduces portal pressure and increases renal perfusion in cirrhotic rats. Sci. Rep. 2019, 9, 2256. [Google Scholar] [CrossRef] [Green Version]
- Gonzalo, T.; Beljaars, L.; van de Bovenkamp, M.; Temming, K.; van Loenen, A.M.; Reker-Smit, C.; Meijer, D.K.; Lacombe, M.; Opdam, F.; Kéri, G.; et al. Local inhibition of liver fibrosis by specific delivery of a platelet-derived growth factor kinase inhibitor to hepatic stellate cells. J. Pharm. Exp. Ther. 2007, 321, 856–865. [Google Scholar] [CrossRef] [Green Version]
- Moreno, M.; Gonzalo, T.; Kok, R.J.; Sancho-Bru, P.; van Beuge, M.; Swart, J.; Prakash, J.; Temming, K.; Fondevila, C.; Beljaars, L.; et al. Reduction of advanced liver fibrosis by short-term targeted delivery of an angiotensin receptor blocker to hepatic stellate cells in rats. Hepatology 2010, 51, 942–952. [Google Scholar] [CrossRef]
- Poosti, F.; Bansal, R.; Yazdani, S.; Prakash, J.; Post, E.; Klok, P.; van den Born, J.; de Borst, M.H.; van Goor, H.; Poelstra, K.; et al. Selective delivery of IFN-gamma to renal interstitial myofibroblasts: A novel strategy for the treatment of renal fibrosis. FASEB J. 2015, 29, 1029–1042. [Google Scholar] [CrossRef]
- Bansal, R.; Prakash, J.; De Ruiter, M.; Poelstra, K. Interferon gamma peptidomimetic targeted to hepatic stellate cells ameliorates acute and chronic liver fibrosis in vivo. J. Control. Release 2014, 179, 18–24. [Google Scholar] [CrossRef]
- Iwakiri, Y.; Trebicka, J. Portal hypertension in cirrhosis: Pathophysiological mechanisms and therapy. JHEP Rep. 2021, 3, 100316. [Google Scholar] [CrossRef]
- Klein, S.; Van Beuge, M.M.; Granzow, M.; Beljaars, L.; Schierwagen, R.; Kilic, S.; Heidari, I.; Huss, S.; Sauerbruch, T.; Poelstra, K.; et al. HSC-specific inhibition of Rho-kinase reduces portal pressure in cirrhotic rats without major systemic effects. J. Hepatol. 2012, 57, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Qu, K.; Liu, T.; Lin, T.; Zhang, X.; Cui, R.; Liu, S.; Meng, F.; Zhang, J.; Tai, M.; Wan, Y.; et al. Tyrosine kinase inhibitors: Friends or foe in treatment of hepatic fibrosis? Oncotarget 2016, 7, 67650–67660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Alqahtani, S.; Hu, X. Aromatic Rings as Molecular Determinants for the Molecular Recognition of Protein Kinase Inhibitors. Molecules 2021, 26, 1176. [Google Scholar] [CrossRef] [PubMed]
- Turdo, A.; D’Accardo, C.; Glaviano, A.; Porcelli, G.; Colarossi, C.; Colarossi, L.; Mare, M.; Faldetta, N.; Modica, C.; Pistone, G.; et al. Targeting Phosphatases and Kinases: How to Checkmate Cancer. Front. Cell Dev. Biol. 2021, 9, 690306. [Google Scholar] [CrossRef]
- Matucci, A.; Vivarelli, E.; Nencini, F.; Maggi, E.; Vultaggio, A. Strategies Targeting Type 2 Inflammation: From Monoclonal Antibodies to JAK-Inhibitors. Biomedicines 2021, 9, 1497. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.J.; Sharma, B.; Chawla, P.A. Recent developments in mitogen activated protein kinase inhibitors as potential anticancer agents. Bioorg. Chem. 2021, 114, 105161. [Google Scholar] [CrossRef]
- Rockey, D.C.; Maher, J.J.; Jarnagin, W.R.; Gabbiani, G.; Friedman, S.L. Inhibition of rat hepatic lipocyte activation in culture by interferon-gamma. Hepatology 1992, 16, 776–784. [Google Scholar] [CrossRef]
- Rockey, D.C.; Chung, J.J. Interferon gamma inhibits lipocyte activation and extracellular matrix mRNA expression during experimental liver injury: Implications for treatment of hepatic fibrosis. J. Investig. Med. 1994, 42, 660–670. [Google Scholar] [PubMed]
- Muir, A.J.; Sylvestre, P.B.; Rockey, D.C. Interferon gamma-1b for the treatment of fibrosis in chronic hepatitis C infection. J. Viral Hepat. 2006, 13, 322–328. [Google Scholar] [CrossRef]
- Ivashkiv, L.B. IFNgamma: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef]
- Bansal, R.; Prakash, J.; de Ruijter, M.; Beljaars, L.; Poelstra, K. Peptide-modified albumin carrier explored as a novel strategy for a cell-specific delivery of interferon gamma to treat liver fibrosis. Mol. Pharm. 2011, 8, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Prakash, J.; Post, E.; Beljaars, L.; Schuppan, D.; Poelstra, K. Novel engineered targeted interferon-gamma blocks hepatic fibrogenesis in mice. Hepatology 2011, 54, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Poosti, F.; Pham, B.T.; Oosterhuis, D.; Poelstra, K.; van Goor, H.; Olinga, P.; Hillebrands, J.L. Precision-cut kidney slices (PCKS) to study development of renal fibrosis and efficacy of drug targeting ex vivo. Dis. Model Mech. 2015, 8, 1227–1236. [Google Scholar] [CrossRef]
- Bansal, R.; Tomar, T.; Ostman, A.; Poelstra, K.; Prakash, J. Selective targeting of interferon gamma to stromal fibroblasts and pericytes as a novel therapeutic approach to inhibit angiogenesis and tumor growth. Mol. Cancer 2012, 11, 2419–2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poosti, F.; Bansal, R.; Yazdani, S.; Prakash, J.; Beljaars, L.; van den Born, J.; de Borst, M.H.; van Goor, H.; Hillebrands, J.L.; Poelstra, K. Interferon gamma peptidomimetic targeted to interstitial myofibroblasts attenuates renal fibrosis after unilateral ureteral obstruction in mice. Oncotarget 2016, 7, 54240–54252. [Google Scholar] [CrossRef] [Green Version]
- Bansal, R.; Prakash, J.; De Ruiter, M.; Poelstra, K. Targeted recombinant fusion proteins of IFNγ and mimetic IFNγ with PDGFβR bicyclic peptide inhibits liver fibrogenesis in vivo. PLoS One 2014, 9, e89878. [Google Scholar] [CrossRef]
- Tanaka, M.; Hasegawa, M.; Yoshimoto, N.; Hoshikawa, K.; Mukai, T. Preparation of Lipid Nanodisks Containing Apolipoprotein E-Derived Synthetic Peptides for Biocompatible Delivery Vehicles Targeting Low-Density Lipoprotein Receptor. Biol. Pharm. Bull. 2019, 42, 1376–1383. [Google Scholar] [CrossRef]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef]
- Sato, Y.; Kinami, Y.; Hashiba, K.; Harashima, H. Different kinetics for the hepatic uptake of lipid nanoparticles between the apolipoprotein E/low density lipoprotein receptor and the N-acetyl-d-galactosamine/asialoglycoprotein receptor pathway. J. Control. Release 2020, 322, 217–226. [Google Scholar] [CrossRef]
- Sun, G.; Hsueh, P.Y.; Janib, S.M.; Hamm-Alvarez, S.; MacKay, J.A. Design and cellular internalization of genetically engineered polypeptide nanoparticles displaying adenovirus knob domain. J. Control. Release 2011, 155, 218–226. [Google Scholar] [CrossRef] [Green Version]
- Ruan, G.X.; Chen, Y.Z.; Yao, X.L.; Du, A.; Tang, G.P.; Shen, Y.Q.; Tabata, Y.; Gao, J.Q. Macrophage mannose receptor-specific gene delivery vehicle for macrophage engineering. Acta Biomater. 2014, 10, 1847–1855. [Google Scholar] [CrossRef] [PubMed]
- Schoemaker, M.H.; Rots, M.G.; Beljaars, L.; Ypma, A.Y.; Jansen, P.L.; Poelstra, K.; Moshage, H.; Haisma, H.J. PDGF-receptor beta-targeted adenovirus redirects gene transfer from hepatocytes to activated stellate cells. Mol. Pharm. 2008, 5, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Meijer, D.K.; Beljaars, L.; Molema, G.; Poelstra, K. Disease-induced drug targeting using novel peptide-ligand albumins. J. Control. Release 2001, 72, 157–164. [Google Scholar] [CrossRef]
- Beljaars, L.; Olinga, P.; Molema, G.; de Bleser, P.; Geerts, A.; Groothuis, G.M.; Meijer, D.K.; Poelstra, K. Characteristics of the hepatic stellate cell-selective carrier mannose 6-phosphate modified albumin (M6P28-HSA). Liver 2001, 21, 320–328. [Google Scholar] [CrossRef]
- Beljaars, L.; Meijer, D.K.; Poelstra, K. Targeting hepatic stellate cells for cell-specific treatment of liver fibrosis. Front. Biosci. 2002, 7, 214. [Google Scholar] [CrossRef]
- Hagens, W.I.; Olinga, P.; Meijer, D.K.; Groothuis, G.M.; Beljaars, L.; Poelstra, K. Gliotoxin non-selectively induces apoptosis in fibrotic and normal livers. Liver Int. 2006, 26, 232–239. [Google Scholar] [CrossRef]
- Greupink, R.; Bakker, H.I.; van Goor, H.; de Borst, M.H.; Beljaars, L.; Poelstra, K. Mannose-6-phosphate/insulin-Like growth factor-II receptors may represent a target for the selective delivery of mycophenolic acid to fibrogenic cells. Pharm. Res. 2006, 23, 1827–1834. [Google Scholar] [CrossRef]
- Yang, N.; Singh, S.; Mahato, R.I. Targeted TFO delivery to hepatic stellate cells. J. Control. Release 2011, 155, 326–330. [Google Scholar] [CrossRef] [Green Version]
- Okimoto, S.; Kuroda, S.; Tashiro, H.; Kobayashi, T.; Taogoshi, T.; Matsuo, H.; Ohdan, H. Vitamin A-coupled liposomal Rho-kinase inhibitor ameliorates liver fibrosis without systemic adverse effects. Hepatol. Res. 2019, 49, 663–675. [Google Scholar] [CrossRef]
- Qiao, J.B.; Fan, Q.Q.; Xing, L.; Cui, P.F.; He, Y.J.; Zhu, J.C.; Wang, L.; Pang, T.; Oh, Y.K.; Zhang, C.; et al. Vitamin A-decorated biocompatible micelles for chemogene therapy of liver fibrosis. J. Control. Release 2018, 283, 113–125. [Google Scholar] [CrossRef]
- El-Mezayen, N.S.; El-Hadidy, W.F.; El-Refaie, W.M.; Shalaby, T.I.; Khattab, M.M.; El-Khatib, A.S. Hepatic stellate cell-targeted imatinib nanomedicine versus conventional imatinib: A novel strategy with potent efficacy in experimental liver fibrosis. J. Control. Release 2017, 266, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.W.; Ikenaga, N.; Liu, S.B.; Sverdlov, D.Y.; Vaid, K.A.; Dixit, R.; Weinreb, P.H.; Violette, S.; Sheppard, D.; Schuppan, D.; et al. Integrin alphavbeta6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology 2016, 63, 217–232. [Google Scholar] [CrossRef] [Green Version]
- Patsenker, E.; Popov, Y.; Stickel, F.; Jonczyk, A.; Goodman, S.L.; Schuppan, D. Inhibition of integrin alphavbeta6 on cholangiocytes blocks transforming growth factor-beta activation and retards biliary fibrosis progression. Gastroenterology 2008, 135, 660–670. [Google Scholar] [CrossRef] [Green Version]
- Popov, Y.; Patsenker, E.; Stickel, F.; Zaks, J.; Bhaskar, K.R.; Niedobitek, G.; Kolb, A.; Friess, H.; Schuppan, D. Integrin alphavbeta6 is a marker of the progression of biliary and portal liver fibrosis and a novel target for antifibrotic therapies. J. Hepatol. 2008, 48, 453–464. [Google Scholar] [CrossRef]
- Tarrus, M.; van der Sloot, A.A.; Temming, K.; Lacombe, M.; Opdam, F.; Quax, W.J.; Molema, G.; Poelstra, K.; Kok, R.J. RGD-avidin-biotin pretargeting to alpha v beta 3 integrin enhances the proapoptotic activity of TNF alpha related apoptosis inducing ligand (TRAIL). Apoptosis 2008, 13, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Temming, K.; Lacombe, M.; Moorlag, H.E.; Asgeirsdottir, S.A.; Orfi, L.; Keri, G.; Poelstra, K.; Molema, G.; Kok, R.J. Targeting of the VEGF-kinase inhibitor PTK787 to angiogenic vasculature using RGD-equipped albumin carrier molecules. J. Control. Release 2006, 116, e57. [Google Scholar] [CrossRef]
- Temming, K.; Lacombe, M.; Schaapveld, R.Q.; Orfi, L.; Keri, G.; Poelstra, K.; Molema, G.; Kok, R.J. Rational design of RGD-albumin conjugates for targeted delivery of the VEGF-R kinase inhibitor PTK787 to angiogenic endothelium. Chem. Med. Chem. 2006, 1, 1200–1203. [Google Scholar] [CrossRef]
- Temming, K.; Lacombe, M.; van der Hoeven, P.; Prakash, J.; Gonzalo, T.; Dijkers, E.C.; Orfi, L.; Kéri, G.; Poelstra, K.; Molema, G.; et al. Delivery of the p38 MAPkinase inhibitor SB202190 to angiogenic endothelial cells: Development of novel RGD-equipped and PEGylated drug-albumin conjugates using platinum(II)-based drug linker technology. Bioconjugate Chem. 2006, 17, 1246–1255. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Park, K.; Kim, J.; Kim, K.S.; Hahn, S.K. Target specific intracellular delivery of siRNA/PEI-HA complex by receptor mediated endocytosis. Mol. Pharm. 2009, 6, 727–737. [Google Scholar] [CrossRef]
- Yamada, Y.; Hashida, M.; Hayashi, Y.; Tabata, M.; Hyodo, M.; Ara, M.N.; Ohga, N.; Hida, K.; Harashima, H. An approach to transgene expression in liver endothelial cells using a liposome-based gene vector coated with hyaluronic acid. J. Pharm. Sci 2013, 102, 3119–3127. [Google Scholar] [CrossRef]
- Lin, L.; Cai, M.; Deng, S.; Huang, W.; Huang, J.; Huang, X.; Huang, M.; Wang, Y.; Shuai, X.; Zhu, K. Amelioration of cirrhotic portal hypertension by targeted cyclooxygenase-1 siRNA delivery to liver sinusoidal endothelium with polyethylenimine grafted hyaluronic acid. Nanomedicine 2017, 13, 2329–2339. [Google Scholar] [CrossRef] [PubMed]
- Beljaars, L.; Poelstra, K.; Molema, G.; Meijer, D.K. Targeting of sugar- and charge-modified albumins to fibrotic rat livers: The accessibility of hepatic cells after chronic bile duct ligation. J. Hepatol. 1998, 29, 579–588. [Google Scholar] [CrossRef]
- Van Dijk, F.; Teekamp, N.; Beljaars, L.; Post, E.; Zuidema, J.; Steendam, R.; Kim, Y.O.; Frijlink, H.W.; Schuppan, D.; Poelstra, K.; et al. Pharmacokinetics of a sustained release formulation of PDGFbeta-receptor directed carrier proteins to target the fibrotic liver. J. Control. Release 2018, 269, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Adrian, J.E.; Poelstra, K.; Kamps, J.A. Addressing liver fibrosis with liposomes targeted to hepatic stellate cells. J. Liposome Res. 2007, 17, 205–218. [Google Scholar] [CrossRef]
- Adrian, J.E.; Kamps, J.A.; Scherphof, G.L.; Meijer, D.K.; van Loenen-Weemaes, A.M.; Reker-Smit, C.; Terpstra, P.; Poelstra, K. A novel lipid-based drug carrier targeted to the non-parenchymal cells, including hepatic stellate cells, in the fibrotic livers of bile duct ligated rats. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1430–1439. [Google Scholar] [CrossRef] [Green Version]
- Adrian, J.E.; Poelstra, K.; Scherphof, G.L.; Molema, G.; Meijer, D.K.; Reker-Smit, C.; Morselt, H.W.; Kamps, J.A. Interaction of targeted liposomes with primary cultured hepatic stellate cells: Involvement of multiple receptor systems. J. Hepatol. 2006, 44, 560–567. [Google Scholar] [CrossRef]
- Adrian, J.E.; Poelstra, K.; Scherphof, G.L.; Meijer, D.K.; van Loenen-Weemaes, A.M.; Reker-Smit, C.; Morselt, H.W.; Zwiers, P.; Kamps, J.A. Effects of a new bioactive lipid-based drug carrier on cultured hepatic stellate cells and liver fibrosis in bile duct-ligated rats. J. Pharm. Exp. 2007, 321, 536–543. [Google Scholar] [CrossRef] [Green Version]
- Adrian, J.E.; Kamps, J.A.; Poelstra, K.; Scherphof, G.L.; Meijer, D.K.; Kaneda, Y. Delivery of viral vectors to hepatic stellate cells in fibrotic livers using HVJ envelopes fused with targeted liposomes. J. Drug Target. 2007, 15, 75–82. [Google Scholar] [CrossRef]
- Tee, J.K.; Peng, F.; Ho, H.K. Effects of inorganic nanoparticles on liver fibrosis: Optimizing a double-edged sword for therapeutics. Biochem. Pharmacol. 2019, 160, 24–33. [Google Scholar] [CrossRef]
- Poilil Surendran, S.; George Thomas, R.; Moon, M.J.; Jeong, Y.Y. Nanoparticles for the treatment of liver fibrosis. Int. J. Nanomed. 2017, 12, 6997–7006. [Google Scholar] [CrossRef] [Green Version]
- Ergen, C.; Niemietz, P.M.; Heymann, F.; Baues, M.; Gremse, F.; Pola, R.; van Bloois, L.; Storm, G.; Kiessling, F.; Trautwein, C.; et al. Liver fibrosis affects the targeting properties of drug delivery systems to macrophage subsets in vivo. Biomaterials 2019, 206, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef] [PubMed]
- Salvati, A.; Åberg, C.; dos Santos, T.; Varela, J.; Pinto, P.; Lynch, I.; Dawson, K.A. Experimental and theoretical comparison of intracellular import of polymeric nanoparticles and small molecules: Toward models of uptake kinetics. Nanomedicine 2011, 7, 818–826. [Google Scholar] [CrossRef]
- Rennick, J.J.; Johnston, A.P.R.; Parton, R.G. Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nat. Nanotechnol. 2021, 16, 266–276. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Owens, D.E., III; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, T.; Lynch, I.; Lindman, S.; Berggard, T.; Thulin, E.; Nilsson, H.; Dawson, K.A.; Linse, S. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvati, A.; Pitek, A.S.; Monopoli, M.P.; Prapainop, K.; Bombelli, F.B.; Hristov, D.R.; Kelly, P.M.; Åberg, C.; Mahon, E.; Dawson, K.A. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat. Nanotechnol. 2013, 8, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Dai, Q.; Walkey, C.; Chan, W.C. Polyethylene glycol backfilling mitigates the negative impact of the protein corona on nanoparticle cell targeting. Angew. Chem. Int. Ed. 2014, 53, 5093–5096. [Google Scholar] [CrossRef]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214. [Google Scholar] [CrossRef] [PubMed]
- Parodi, A.; Quattrocchi, N.; van de Ven, A.A.; Chiappini, C.; Evangelopoulos, M.; Martinez, J.O.; Brown, B.S.; Khaled, S.Z.; Yazdi, I.K.; Enzo, M.V.; et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat. Nanotechnol. 2013, 8, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, P.L.; Harada, T.; Christian, D.A.; Pantano, D.A.; Tsai, R.K.; Discher, D.E. Minimal “Self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science 2013, 339, 971–975. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.M.; Zhang, L.; Aryal, S.; Cheung, C.; Fang, R.H.; Zhang, L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. USA 2011, 108, 10980–10985. [Google Scholar] [CrossRef] [Green Version]
- Caracciolo, G.; Cardarelli, F.; Pozzi, D.; Salomone, F.; Maccari, G.; Bardi, G.; Capriotti, A.L.; Cavaliere, C.; Papi, M.; Laganà, A. Selective targeting capability acquired with a protein corona adsorbed on the surface of 1,2-dioleoyl-3-trimethylammonium propane/DNA nanoparticles. ACS Appl. Mater. Interfaces 2013, 5, 13171–13179. [Google Scholar] [CrossRef]
- Chen, D.; Ganesh, S.; Wang, W.; Lupieri, A.; Amiji, M. Role of vitronectin-rich protein corona on tumor-specific siRNA delivery and transfection with lipid nanoparticles. Nanomedicine 2021, 16, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J.; Shamenkov, D.; Petrov, V.; Ramge, P.; Cychutek, K.; Koch-Brandt, C.; Alyautdin, R. Apolipoprotein-mediated transport of nanoparticle-bound drugs across the blood-brain barrier. J. Drug Target. 2002, 10, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Francia, V.; Yang, K.; Deville, S.; Reker-Smit, C.; Nelissen, I.; Salvati, A. Corona Composition Can Affect the Mechanisms Cells Use to Internalize Nanoparticles. ACS Nano 2019, 13, 11107–11121. [Google Scholar] [CrossRef]
- Lara, S.; Alnasser, F.; Polo, E.; Garry, D.; Lo Giudice, M.C.; Hristov, D.R.; Rocks, L.; Salvati, A.; Yan, Y.; Dawson, K.A. Identification of Receptor Binding to the Biomolecular Corona of Nanoparticles. ACS Nano 2017, 11, 1884–1893. [Google Scholar] [CrossRef]
- Bros, M.; Nuhn, L.; Simon, J.; Moll, L.; Mailander, V.; Landfester, K.; Grabbe, S. The Protein Corona as a Confounding Variable of Nanoparticle-Mediated Targeted Vaccine Delivery. Front. Immunol. 2018, 9, 1760. [Google Scholar] [CrossRef]
- Takeuchi, T.; Kitayama, Y.; Sasao, R.; Yamada, T.; Toh, K.; Matsumoto, Y.; Kataoka, K. Molecularly Imprinted Nanogels Acquire Stealth In Situ by Cloaking Themselves with Native Dysopsonic Proteins. Angew. Chem. Int. Ed. 2017, 56, 7088–7092. [Google Scholar] [CrossRef]
- Schottler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailänder, V.; Wurm, F.R. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016, 11, 372–377. [Google Scholar] [CrossRef]
- Aliyandi, A.; Reker-Smit, C.; Bron, R.; Zuhorn, I.S.; Salvati, A. Correlating Corona Composition and Cell Uptake to Identify Proteins Affecting Nanoparticle Entry into Endothelial Cells. ACS Biomater. Sci. Eng. 2021, 7, 5573–5584. [Google Scholar] [CrossRef]
- Fedeli, C.; Segat, D.; Tavano, R.; Bubacco, L.; De Franceschi, G.; de Laureto, P.P.; Lubian, E.; Selvestrel, F.; Mancin, F.; Papini, E. The functional dissection of the plasma corona of SiO[2]-NPs spots histidine rich glycoprotein as a major player able to hamper nanoparticle capture by macrophages. Nanoscale 2015, 7, 17710–17728. [Google Scholar] [CrossRef]
- Ritz, S.; Schottler, S.; Kotman, N.; Baier, G.; Musyanovych, A.; Kuharev, J.; Landfester, K.; Schild, H.; Jahn, O.; Tenzer, S.; et al. Protein corona of nanoparticles: Distinct proteins regulate the cellular uptake. Biomacromolecules 2015, 16, 1311–1321. [Google Scholar] [CrossRef]
- Palchetti, S.; Digiacomo, L.; Pozzi, D.; Peruzzi, G.; Micarelli, E.; Mahmoudi, M.; Caracciolo, G. Nanoparticles-cell association predicted by protein corona fingerprints. Nanoscale 2016, 8, 12755–12763. [Google Scholar] [CrossRef] [Green Version]
- Walkey, C.D.; Olsen, J.B.; Song, F.; Liu, R.; Guo, H.; Olsen, D.W.; Cohen, Y.; Emili, A.; Chan, W.C. Protein corona fingerprinting predicts the cellular interaction of gold and silver nanoparticles. ACS Nano 2014, 8, 2439–2455. [Google Scholar] [CrossRef]
- Liu, R.; Jiang, W.; Walkey, C.D.; Chan, W.C.; Cohen, Y. Prediction of nanoparticles-cell association based on corona proteins and physicochemical properties. Nanoscale 2015, 7, 9664–9675. [Google Scholar] [CrossRef]
- Yang, K.; Mesquita, B.; Horvatovich, P.; Salvati, A. Tuning liposome composition to modulate corona formation in human serum and cellular uptake. Acta Biomater. 2020, 106, 314–327. [Google Scholar] [CrossRef]
- Aliyandi, A.; Zuhorn, I.S.; Salvati, A. Disentangling Biomolecular Corona Interactions With Cell Receptors and Implications for Targeting of Nanomedicines. Front. Bioeng. Biotechnol. 2020, 8, 599454. [Google Scholar] [CrossRef]
- Francia, V.; Montizaan, D.; Salvati, A. Interactions at the cell membrane and pathways of internalization of nano-sized materials for nanomedicine. Beilstein. J. Nanotechnol. 2020, 11, 338–353. [Google Scholar] [CrossRef] [PubMed]
- Iversen, T.G.; Skotland, T.; Sandvig, K. Endocytosis and intracellular transport of nanoparticles: Present knowledge and need for future studies. Nano Today 2011, 6, 176. [Google Scholar] [CrossRef]
- Francia, V.; Reker-Smit, C.; Boel, G.; Salvati, A. Limits and challenges in using transport inhibitors to characterize how nano-sized drug carriers enter cells. Nanomedicine 2019, 14, 1533–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rejman, J.; Oberle, V.; Zuhorn, I.S.; Hoekstra, D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem. J. 2004, 377, 159–169. [Google Scholar] [CrossRef]
- Degors, I.M.S.; Wang, C.; Rehman, Z.U.; Zuhorn, I.S. Carriers Break Barriers in Drug Delivery: Endocytosis and Endosomal Escape of Gene Delivery Vectors. Acc. Chem. Res. 2019, 52, 1750–1760. [Google Scholar] [CrossRef] [Green Version]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef]
- Vtyurina, N.; Åberg, C.; Salvati, A. Imaging of nanoparticle uptake and kinetics of intracellular trafficking in individual cells. Nanoscale 2021, 13, 10436–10446. [Google Scholar] [CrossRef] [PubMed]
- Åberg, C.; Piattelli, V.; Montizaan, D.; Salvati, A. Sources of variability in nanoparticle uptake by cells. Nanoscale 2021, 13, 17530–17546. [Google Scholar] [CrossRef]
- Garcia Romeu, H.; Deville, S.; Salvati, A. Time- and Space-Resolved Flow-Cytometry of Cell Organelles to Quantify Nanoparticle Uptake and Intracellular Trafficking by Cells. Small 2021, 17, e2100887. [Google Scholar] [CrossRef]
- Dowden, H.; Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov. 2019, 18, 495–496. [Google Scholar] [CrossRef]
- Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2020. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2021, 26, 627. [Google Scholar] [CrossRef] [PubMed]
- Al Musaimi, O.; Al Shaer, D.; Albericio, F.; de la Torre, B.G. 2020 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2021, 14, 145. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, S.; Bowman, D.M. COVID-19 may become nanomedicine’s finest hour yet. Nat. Nanotechnol. 2021, 16, 362–364. [Google Scholar] [CrossRef]
- Chung, Y.H.; Beiss, V.; Fiering, S.N.; Steinmetz, N.F. COVID-19 Vaccine Frontrunners and Their Nanotechnology Design. ACS Nano 2020, 14, 12522–12537. [Google Scholar] [CrossRef]
- Madamsetty, V.S.; Mukherjee, A.; Mukherjee, S. Recent Trends of the Bio-Inspired Nanoparticles in Cancer Theranostics. Front. Pharmacol. 2019, 10, 1264. [Google Scholar] [CrossRef] [PubMed]
- Torok, N.J.; Dranoff, J.A.; Schuppan, D.; Friedman, S.L. Strategies and endpoints of antifibrotic drug trials: Summary and recommendations from the AASLD Emerging Trends Conference, Chicago, June 2014. Hepatology 2015, 62, 627–634. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Target Cell | Target Receptor | Homing Ligand | Delivered Drug | Reference |
|---|---|---|---|---|
| Hepatocytes | Asialoglycoprotein-receptor (ASGP-R) | Galactose or Lactose | Antiviral Antitumor Hepatoprotective | [44,45,46] |
| Low-density lipoprotein Receptor (LDL-R) | APO-E | RNA-based drugs | [97,98,99] | |
| Coxsackie and adenovirus cell adhesion receptor (CAR) | Adenoviruses Adenoviral-derived ligands | Genes | [100] | |
| Macrophages | CD206 | Mannose | Anti-inflammatory | [52,53,57,101] |
| Hepatic Stellate Cells and Myofibroblasts | Platelet Derived Growth Factor β-receptor (PDGF-β-R) | pPB peptide | Antifibrotic Anti-proliferative Anti-inflammatory Pro-apototic Rho-Kinase inhibitors Collagen synthesis inhibitors Tyrosin-kinase inhibitor Angiotensin inhibitor | [57,63,68,72,102] |
| Insulin-like-Growth Factor II receptor (IGFII-R) | Mannose-6-phosphate | Anti-fibrotic | [65,68,71,73,103,104,105,106,107,108] | |
| Vitamin A-receptor | Retinoic acid | Anti- collagen chaparone glycoprotein (gp46)- siRNA | [99,109,110,111] | |
| Progenitor cells/Cholangiocytes | Integrin Avβ6-receptor | αvβ6 ligand/antibody | [112,113,114] | |
| Endothelial Cells | Scavenger receptor | Succinylated molecules | Anti-inflammatory | [54] |
| Integrin receptor | RGD-peptides | Antiangiogenic Anti-inflammator Kinase inhibitors | [115,116,117,118] | |
| Hyaluronic Acid-recptor | Hyaluronic acid | [119,120,121] |
| Nanoparticle Modification or Corona Component | Effect Reported | Selected Examples |
|---|---|---|
| Corona formation | Can mask targeting ligands in vitro | [139] |
| Opsonin proteins in the corona | Activation of immune cells, nanoparticle removal from circulation | [135,136,137] |
| PEGylation | Reduced protein adsorption and/or binding of dysopsonin proteins in the corona such as clusterin | [135,141,152] |
| Dysopsonin proteins in the corona | Prolonged circulation time | [150] |
| Albumin in the corona | Prolonged circulation time | [151] |
| Clusterin (apolipoprotein J) in the corona | Prolonged circulation time | [152] |
| Histidine rich glycoprotein in the corona | Prolonged circulation time | [153,154] |
| CD47 functionalization | Marker of self, “don’t eat me” signal for immune cells, prolonged circulation | [143] |
| Leukocytes cell membrane coating | Nanoparticle camouflage, prolonged circulation and increased accumulation in inflamed areas | [142] |
| Red-cell membrane coating | Nanoparticle camouflage, prolonged circulation | [144] |
| Apolipoprotein B in the corona | Uptake mediated by LDLR | [148,149] |
| Apolipoprotein E in the corona | In vivo targeting of liver hepatocytes via LDLR | [11] |
| Apolipoprotein E in the corona | Promotes nanoparticle transcytosis across the blood brain barrier | [147] |
| Vitronectin in the corona | Increased uptake via ανβ3 integrin receptor in vitro and in vivo | [145,146] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvati, A.; Poelstra, K. Drug Targeting and Nanomedicine: Lessons Learned from Liver Targeting and Opportunities for Drug Innovation. Pharmaceutics 2022, 14, 217. https://doi.org/10.3390/pharmaceutics14010217
Salvati A, Poelstra K. Drug Targeting and Nanomedicine: Lessons Learned from Liver Targeting and Opportunities for Drug Innovation. Pharmaceutics. 2022; 14(1):217. https://doi.org/10.3390/pharmaceutics14010217
Chicago/Turabian StyleSalvati, Anna, and Klaas Poelstra. 2022. "Drug Targeting and Nanomedicine: Lessons Learned from Liver Targeting and Opportunities for Drug Innovation" Pharmaceutics 14, no. 1: 217. https://doi.org/10.3390/pharmaceutics14010217