
BCR-ABL1 Tyrosine Kinase Complex Signaling Transduction: Challenges to Overcome Resistance in Chronic Myeloid Leukemia

 , and
, and
Abstract
:1. Introduction
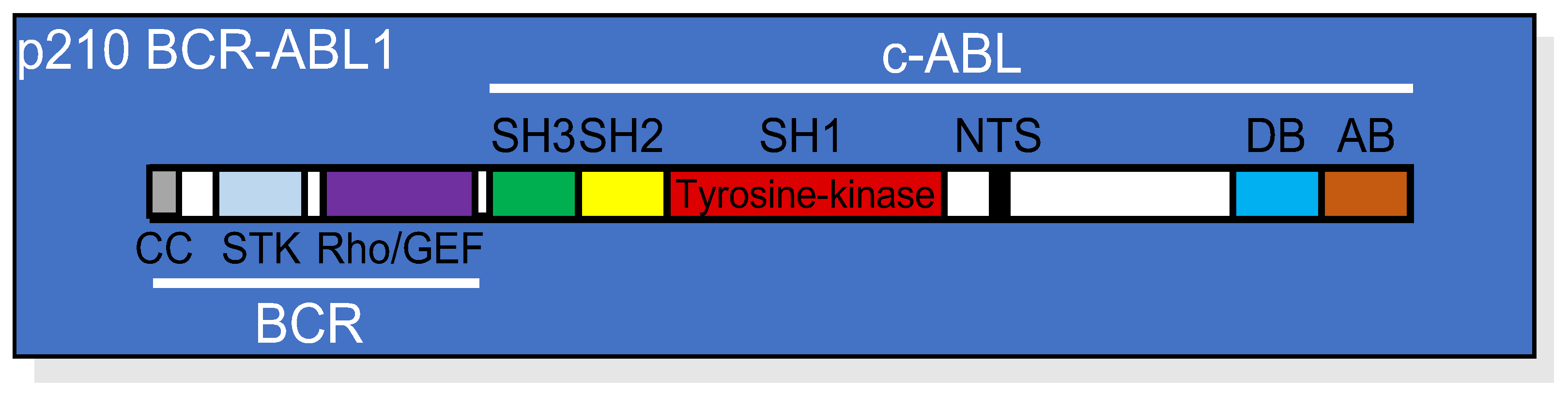
2. BCR-ABL1 Tyrosine Kinase-Dependent Signaling Cascade
2.1. BCR-ABL1 Activation of the RAS/RAF/MAPK Pathway
2.2. BCR-ABL1 Activation of the PI3K/AKT/mTOR Pathway
2.3. BCR-ABL1 Activation of the JAK/STAT Pathway
2.4. BCR-ABL1 Activation of the WNT/β-Catenin Pathway
2.5. BCR-ABL1 Activation of the PP2A Pathway
3. BCR-ABL1 Kinase-Independent Alternative Survival Signals
4. Resistance to Apoptosis in CML
5. Tyrosine Kinase Inhibitors and the Paradigm Shift of CML Treatment
5.1. Imatinib Mesylate
5.2. Nilotinib
5.3. Dasatinib
5.4. Bosutinib
5.5. Ponatinib
5.6. Asciminib
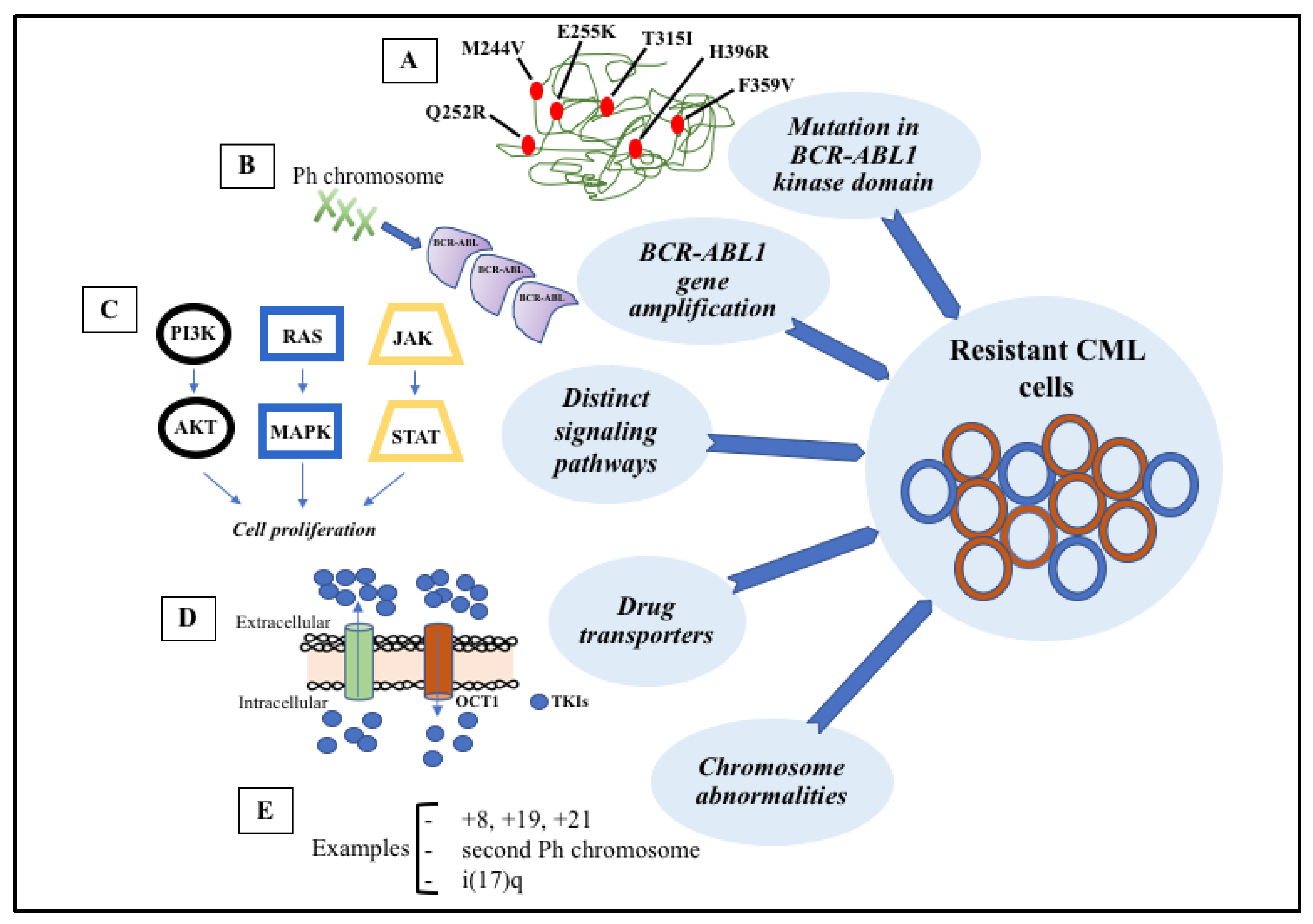
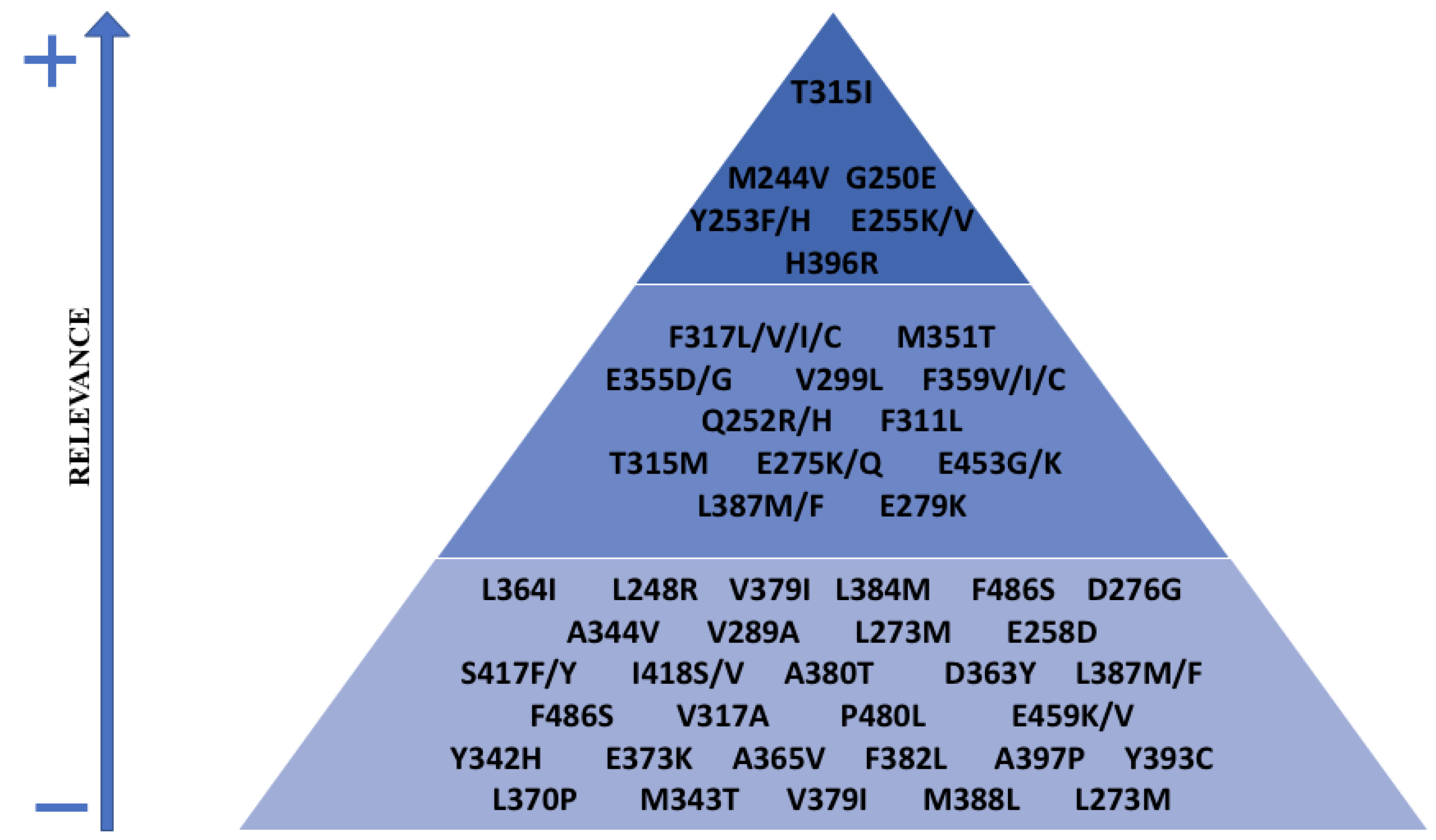
6. Mechanisms of Resistance to TKI
7. Challenges and Future Perspectives—Novel Agents and Combined Therapies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar]
- Pane, F.; Frigeri, F.; Sindona, M.; Luciano, L.; Ferrara, F.; Cimino, R.; Meloni, G.; Saglio, G.; Salvatore, F.; Rotoli, B. Neutrophilic-Chronic Myeloid Leukemia: A Distinct Disease with a Specific Molecular Marker (BCR/ABL With C3/A2 Junction). Blood 1996, 88, 2410–2414. [Google Scholar] [CrossRef]
- Walker, L.C.; Ganesan, T.S.; Dhut, S.; Gibbons, B.; Andrew Lister, T.; Rothbardt, J.; Young, B.D. Novel Chimaeric Protein Expressed in Philadelphia Positive Acute Lymphoblastic Leukaemia. Nature 1987, 329, 851–853. [Google Scholar] [CrossRef]
- Heisterkamp, N.; Stephenson·, J.R.; Groffen·, J.; Hansent, P.F.; de Klein, A.; Bartram, C.R.; Grosveld, G. Localization of the C-Abl Oncogene Adjacent to a Translocation Break Point in Chronic Myelocytic Leukaemia. Nature 1983, 306, 239–242. [Google Scholar] [CrossRef]
- Mcwhirter, J.R.; Wang, J.Y.J. An Actin-Binding Function Contributes to Transformation by the Bcr-Abl Oncoprotein of Philadelphia Chromosome-Positive Human Leukemias. EMBO J. 1993, 12, 1533–1546. [Google Scholar] [CrossRef]
- Peiris, M.N.; Li, F.; Donoghue, D.J. BCR: A promiscuous fusion partner in hematopoietic disorders. Oncotarget 2019, 10, 2738–2754. [Google Scholar] [CrossRef]
- Pendergast, A.M.; Quilliam, L.A.; Cripe, L.D.; Bassing, C.H.; Dai, Z.; Li, N.; Batzer, A.; Rabun, K.M.; Der, C.J.; Schlessinger, J.; et al. BCR-ABL-Induced Oncogenesis Is Mediated by Direct Interaction with the SH2 Domain of the GRB-2 Adaptor Protein. Cell 1993, 75, 175–785. [Google Scholar] [CrossRef]
- Van Etten, R.A.; Jackson, P.; Baltimore, D. The Mouse Type IV C-Abl Gene Product Is a Nuclear Protein, and Activation of Transforming Ability Is Associated with Cytoplasmic Localization. Cell 1989, 58, 669–678. [Google Scholar] [CrossRef] [Green Version]
- Druker, B.; Tamura, S.; Buchdungerz, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, O.; Lydon, N.B. Effects of a Selective Inhibitor of the Abl Tyrosine Kinase on the Growth of Bcr-Abl Positive Cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- Gambacorti-Passerini, C.; Antolini, L.; Mahon, F.X.; Guilhot, F.; Deininger, M.; Fava, C.; Nagler, A.; della Casa, C.M.; Morra, E.; Abruzzese, E.; et al. Multicenter Independent Assessment of Outcomes in Chronic Myeloid Leukemia Patients Treated with Imatinib. J. Natl. Cancer Inst. 2011, 103, 553–561. [Google Scholar] [CrossRef]
- Baccarani, M.; Cortes, J.; Pane, F.; Niederwieser, D.; Saglio, G.; Apperley, J.; Cervantes, F.; Deininger, M.; Gratwohl, A.; Guilhot, F.; et al. Chronic Myeloid Leukemia: An Update of Concepts and Management Recommendations of European LeukemiaNet. J. Clin. Oncol. 2009, 27, 6041–6051. [Google Scholar] [CrossRef] [Green Version]
- Bu, Q.; Cui, L.; Li, J.; Du, X.; Zou, W.; Ding, K.; Pan, J. SAHA and S116836, a Novel Tyrosine Kinase Inhibitor, Synergistically Induce Apoptosis in Imatinib-Resistant Chronic Myelogenous Leukemia Cells. Cancer Biol. Ther. 2014, 15, 951–962. [Google Scholar] [CrossRef]
- Chan, O.; Talati, C.; Isenalumhe, L.; Shams, S.; Nodzon, L.; Fradley, M.; Sweet, K.; Pinilla-Ibarz, J. Side-Effects Profile and Outcomes of Ponatinib in the Treatment of Chronic Myeloid Leukemia. Blood Adv. 2020, 4, 530–538. [Google Scholar] [CrossRef]
- Brehme, M.; Hantschel, O.; Colinge, J.; Kaupe, I.; Planyavsky, M.; Kö Cher, T.; Mechtler, K.; Bennett, K.L.; Superti-Furga, G. Charting the Molecular Network of the Drug Target Bcr-Abl. Proc. Natl. Acad. Sci. USA 2009, 106, 7414–7419. [Google Scholar] [CrossRef] [Green Version]
- Sattler, M.; Golam Mohi, M.; Pride, Y.B.; Quinnan, L.R.; Malouf, N.A.; Podar, K.; Gesbert, F.; Iwasaki, H.; Li, S.; van Etten, R.A.; et al. Critical Role for Gab2 in Transformation by BCR/ABL. Cancer Cell 2002, 1, 479–492. [Google Scholar] [CrossRef] [Green Version]
- Gregory, M.A.; Phang, T.L.; Neviani, P.; Alvarez-Calderon, F.; Eide, C.A.; O’Hare, T.; Zaberezhnyy, V.; Williams, R.T.; Druker, B.J.; Perrotti, D.; et al. Wnt/Ca2+/NFAT Signaling Maintains Survival of Ph+ Leukemia Cells upon Inhibition of Bcr-Abl. Cancer Cell 2010, 18, 74–87. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Chen, Y.; Douglas, L.; Li, S. β-Catenin Is Essential for Survival of Leukemic Stem Cells Insensitive to Kinase Inhibition in Mice with BCR-ABL-Induced Chronic Myeloid Leukemia. Leukemia 2009, 23, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Reins, J.; Mossner, M.; Neumann, M.; Platzbecker, U.; Schumann, C.; Thiel, E.; Hofmann, W.K. Transcriptional Down-Regulation of the Wnt Antagonist SFRP1 in Haematopoietic Cells of Patients with Different Risk Types of MDS. Leuk. Res. 2010, 34, 1610–1616. [Google Scholar] [CrossRef]
- Cortez, D.; Kadlec, L.; Pendergast, A.M. Structural and Signaling Requirements for BCR-ABL-Mediated Transformation and Inhibition of Apoptosis. Mol. Cell. Biol. 1995, 15, 5531–5541. [Google Scholar] [CrossRef] [Green Version]
- Castellano, E.; Downward, J. Ras Interaction with PI3K: More than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Danisz, K.; Blasiak, J. Role of Anti-Apoptotic Pathways Activated by BCR/ABL in the Resistance of Chronic Myeloid Leukemia Cells to Tyrosine Kinase Inhibitors. Acta Biochim. Pol. 2013, 4, 503–514. [Google Scholar] [CrossRef]
- Ren, R. Mechanisms of BCR-ABL in the Pathogenesis of Chronic Myelogenous Leukaemia. Nat. Rev. Cancer 2005, 5, 172–183. [Google Scholar] [CrossRef]
- Hochhaus, A.; Kreil, S.; Corbin, A.S.; la Rosée, P.; Müller, M.C.; Lahaye, T.; Hanfstein, B.; Schoch, C.; Cross, N.C.P.; Berger, U.; et al. Molecular and Chromosomal Mechanisms of Resistance to Imatinib (STI571) Therapy. Leukemia 2002, 16, 2190–2196. [Google Scholar] [CrossRef] [Green Version]
- Bertacchini, J.; Heidari, N.; Mediani, L.; Capitani, S.; Shahjahani, M.; Ahmadzadeh, A.; Saki, N. Targeting PI3K/AKT/MTOR Network for Treatment of Leukemia. Cell. Mol. Life Sci. 2015, 72, 2337–2347. [Google Scholar] [CrossRef]
- Toker, A.; Marmiroli, S. Signaling Specificity in the Akt Pathway in Biology and Disease. Adv. Biol. Regul. 2014, 55, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Neshat, M.S.; Raitano, A.B.; Wang, H.-G.; Reed, J.C.; Sawyers, C.L. The Survival Function of the Bcr-Abl Oncogene Is Mediated by Bad-Dependent and-Independent Pathways: Roles for Phosphatidylinositol 3-Kinase and Raf. Mol. Cell Biol. 2000, 20, 1179–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minshall, C.; Arkins, S.; Freund, G.C.; Kelley, K.W. Requirement for Phosphatidylinositol 3′-Kinase to Protect Hemopoietic Progenitors Against Apoptosis Depends Upon the Extracellular Survival Factor. J. Immunol. 1996, 156, 939–947. [Google Scholar]
- Yao, R.; Cooper, G.M. Requirement for Phosphatidylinositol-3 Kinase in the Prevention of Apoptosis by Nerve Growth Factor. Science 1995, 267, 2003–2006. [Google Scholar] [CrossRef] [PubMed]
- Kulik, G.; Klippel, A.; Weber, M.J. Antiapoptotic Signalling by the Insulin-Like Growth Factor I Receptor, Phosphatidylinositol 3-Kinase, and Akt. Mol. Cell Biol. 1997, 17, 1595–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarante-Mendes, G.P.; Jascur, T.; Nishioka, W.K.; Mustelin, T.; Green, D.R. Bcr-Abl-Mediated Resistance to Apoptosis Is Independent of PI 3-Kinase Activity. Cell Death Differ. 1997, 4, 548–554. [Google Scholar] [CrossRef] [Green Version]
- Dinner, S.; Platanias, L.C. Targeting the MTOR Pathway in Leukemia. J. Cell. Biochem. 2016, 117, 1745–1752. [Google Scholar] [CrossRef] [PubMed]
- Alves, R.; Gonçalves, A.C.; Jorge, J.; Alves, J.; Alves da Silva, A.; Freitas-Tavares, P.; Nascimento Costa, J.M.; Almeida, A.M.; Sarmento-Ribeiro, A.B. Everolimus in Combination with Imatinib Overcomes Resistance in Chronic Myeloid Leukaemia. Med. Oncol. 2019, 36, 30. [Google Scholar] [CrossRef] [PubMed]
- Burgering, B.M.T.; Medema, R.H. Decisions on Life and Death: FOXO Forkhead Transcription Factors Are in Command When PKB/Akt Is off Duty. J. Leukoc. Biol. 2003, 73, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Jacquel, A.; Luciano, F.; Robert, G.; Auberger, P. Implication and Regulation of AMPK during Physiological and Pathological Myeloid Differentiation. Int. J. Mol. Sci. 2018, 19, 2991. [Google Scholar] [CrossRef] [Green Version]
- Vakana, E.; Platanias, L.C. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2011, 2, 49. [Google Scholar]
- Burchert, A.; Wang, Y.; Cai, D.; von Bubnoff, N.; Paschka, P.; Müller-Brüsselbach, S.; Ottmann, O.G.; Duyster, J.; Hochhaus, A.; Neubauer, A. Compensatory PI3-Kinase/Akt/MTor Activation Regulates Im’atinib Resistance Development. Leukemia 2005, 19, 1774–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Romani, J.; Zaborski, M.; MacLeod, R.A.F.; Nagel, S.; Drexler, H.G.; Quentmeier, H. Inhibition of PI3K/MTOR Overcomes Nilotinib Resistance in BCR-ABL1 Positive Leukemia Cells through Translational down-Regulation of MDM2. PLoS ONE 2013, 8, e83510. [Google Scholar]
- Quentmeier, H.; Eberth, S.; Romani, J.; Zaborski, M.; Drexler, H.G. BCR-ABL1-Independent PI3Kinase Activation Causing Imatinib-Resistance. J. Hematol. Oncol. 2011, 4, 6. [Google Scholar] [CrossRef] [Green Version]
- Joha, S.; Nugues, A.L.; Hétuin, D.; Berthon, C.; Dezitter, X.; Dauphin, V.; Mahon, F.X.; Roche-Lestienne, C.; Preudhomme, C.; Quesnel, B.; et al. GILZ Inhibits the MTORC2/AKT Pathway in BCR-ABL + Cells. Oncogene 2012, 31, 1419–1430. [Google Scholar] [CrossRef] [Green Version]
- Carayol, N.; Vakana, E.; Sassano, A.; Kaur, S.; Goussetis, D.J.; Glaser, H.; Druker, B.J.; Donato, N.J.; Altman, J.K.; Barr, S.; et al. Critical Roles for MTORC2-and Rapamycin-Insensitive MTORC1-Complexes in Growth and Survival of BCR-ABL-Expressing Leukemic Cells. Proc. Natl. Acad. Sci. USA 2010, 107, 12469–12474. [Google Scholar] [CrossRef] [Green Version]
- Vakana, E.; Sassano, A.; Platanias, L.C. Induction of Autophagy by Dual MTORC1-MTORC2 Inhibition in BCR-ABL-Expressing Leukemic Cells. Autophagy 2010, 6, 966–967. [Google Scholar] [CrossRef]
- Airiau, K.; Mahon, F.X.; Josselin, M.; Jeanneteau, M.; Belloc, F. PI3K/MTOR Pathway Inhibitors Sensitize Chronic Myeloid Leukemia Stem Cells to Nilotinib and Restore the Response of Progenitors to Nilotinib in the Presence of Stem Cell Factor. Cell Death Dis. 2013, 4, e827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilaria, R.L.; van Etten, R.A. P210 and P190 BCR/ABL Induce the Tyrosine Phosphorylation and DNA Binding Activity of Multiple Specific STAT Family Members. J. Biol. Biochem. 1996, 271, 6188–6195. [Google Scholar]
- Xie, S.; Wang, Y.; Liu, J.; Sun, T.; Wilson, M.B.; Smithgall, T.E.; Arlinghaus, R.B. Involvement of Jak2 Tyrosine Phosphorylation in Bcr ± Abl Transformation. Oncogene 2001, 20, 6188–6195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoelbl, A.; Schuster, C.; Kovacic, B.; Zhu, B.; Wickre, M.; Hoelzl, M.A.; Fajmann, S.; Grebien, F.; Warsch, W.; Stengl, G.; et al. Stat5 Is Indispensable for the Maintenance of Bcr/Abl-Positive Leukaemia. EMBO Mol. Med. 2010, 2, 98–110. [Google Scholar] [CrossRef]
- Gallipoli, P.; Cook, A.; Rhodes, S.; Hopcroft, L.; Wheadon, H.; Whetton, A.D.; Jørgensen, H.G.; Bhatia, R.; Holyoake, T.L. JAK2/STAT5 Inhibition by Nilotinib with Ruxolitinib Contributes to the Elimination of CML CD34 + Cells in Vitro and in Vivo. Blood 2014, 124, 1492–1501. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.K.; Nichols, G.L.; Rothman, P. Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J. Immunol. 2021, 159, 4720–4728. [Google Scholar]
- Horita, M.; Andreu, E.J.; Benito, A.; Arbona, C.; Sanz, C.; Benet, I.; Prosper, F.; Luis Fernandez-Luna, J. Blockade of the Bcr-Abl Kinase Activity Induces Apoptosis of Chronic Myelogenous Leukemia Cells by Suppressing Signal Transducer and Activator of Transcription 5-Dependent Expression of Bcl-XL. J. Exp. Med. 2000, 191, 977–984. [Google Scholar] [CrossRef]
- Gesbert, F.; Griffin, J.D. Bcr/Abl Activates Transcription of the Bcl-X Gene through STAT5. Blood 2000, 96, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Nieborowska-Skorska, M.; Hoser, G.; Kossev, P.; Wasik, M.A.; Skorski, T. Complementary Functions of the Antiapoptotic Protein A1 and Serine/Threonine Kinase Pim-1 in the BCR/ABL-Mediated Leukemogenesis. Blood 2002, 99, 4531–4539. [Google Scholar] [CrossRef] [Green Version]
- Aichberger, K.J.; Mayerhofer, M.; Krauth, M.-T.; Skvara, H.; Florian, S.; Sonneck, K.; Akgul, C.; Derdak, S.; Pickl, W.F.; Wacheck, V.; et al. Identification of Mcl-1 as a BCR/ABL-Dependent Target in Chronic Myeloid Leukemia (CML): Evidence for Cooperative Antileukemic Effects of Imatinib and Mcl-1 Antisense Oligonucleotides. Blood 2005, 105, 3303–3311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, A.K.; Chakraborty, S.N.; Wang, Y.; Kantarjian, H.; Sun, X.; Hood, J.; Perrotti, D.; Arlinghaus, R.B. Jak2 Inhibition Deactivates Lyn Kinase through the SET-PP2A-SHP1 Pathway, Causing Apoptosis in Drug-Resistant Cells from Chronic Myelogenous Leukemia Patients. Oncogene 2009, 28, 1669–1681. [Google Scholar] [CrossRef] [Green Version]
- Fetisov, T.I.; Lesovaya, E.A.; Yakubovskaya, M.G.; Kirsanov, K.I.; Belitsky, G.A. Alterations in WNT Signaling in Leukemias. Biochemistry 2018, 83, 1448–1458. [Google Scholar] [CrossRef]
- Soares-Lima, S.C.; Pombo-de-Oliveira, M.S.; Carneiro, F.R.G. The Multiple Ways Wnt Signaling Contributes to Acute Leukemia Pathogenesis. J. Leukoc. Biol. 2020, 108, 1081–1099. [Google Scholar] [CrossRef]
- Coluccia, A.M.L.; Vacca, A.; Dũach, M.; Mologni, L.; Redaelli, S.; Bustos, V.H.; Benati, D.; Pinna, L.A.; Gambacorti-Passerini, C. Bcr-Abl Stabilizes β-Catenin in Chronic Myeloid Leukemia through Its Tyrosine Phosphorylation. EMBO J. 2007, 26, 1456–1466. [Google Scholar] [CrossRef] [Green Version]
- Luu, H.H.; Zhang, R.; Haydon, R.C.; Rayburn, E.; Kang, Q.; Si, W.; Park, J.K.; Wang, H.; Peng, Y.; Jiang, W.; et al. Wnt/beta-catenin signaling pathway as a novel cancer drug target. Curr. Cancer Drug Targets 2004, 4, 653–671. [Google Scholar] [CrossRef]
- Arrigoni, E.; del Re, M.; Galimberti, S.; Restante, G.; Rofi, E.; Crucitta, S.; Baratè, C.; Petrini, M.; Danesi, R.; di Paolo, A. Concise Review: Chronic Myeloid Leukemia: Stem Cell Niche and Response to Pharmacologic Treatment. Stem Cells Transl. Med. 2018, 7, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Nagao, R.; Ashihara, E.; Kimura, S.; Strovel, J.W.; Yao, H.; Takeuchi, M.; Tanaka, R.; Hayashi, Y.; Hirai, H.; Padia, J.; et al. Growth Inhibition of Imatinib-Resistant CML Cells with the T315I Mutation and Hypoxia-Adaptation by AV65—A Novel Wnt/β-Catenin Signaling Inhibitor. Cancer Lett. 2011, 312, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef]
- Neviani, P.; Santhanam, R.; Trotta, R.; Notari, M.; Blaser, B.W.; Liu, S.; Mao, H.; Chang, J.S.; Galietta, A.; Uttam, A.; et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell 2005, 8, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Neviani, P.; Santhanam, R.; Oaks, J.J.; Eiring, A.M.; Notari, M.; Blaser, B.W.; Liu, S.; Trotta, R.; Muthusamy, N.; Gambacorti-Passerini, C.; et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J. Clin. Investig. 2007, 117, 2408–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neviani, P.; Harb, J.G.; Oaks, J.J.; Santhanam, R.; Walker, C.J.; Ellis, J.J.; Ferenchak, G.; Dorrance, A.M.; Paisie, C.A.; Eiring, A.M.; et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J Clin. Investig. 2013, 123, 4144–4157. [Google Scholar] [CrossRef] [PubMed]
- Gregor, T.; Bosakova, M.K.; Nita, A.; Abraham, S.P.; Fafilek, B.; Cernohorsky, N.H.; Rynes, J.; Foldynova-Trantirkova, S.; Zackova, D.; Mayer, J.; et al. Elucidation of Protein Interactions Necessary for the Maintenance of the BCR–ABL Signaling Complex. Cell. Mol. Life Sci. 2020, 77, 3885–3903. [Google Scholar] [CrossRef]
- Bueno-da-Silva, A.E.B.; Brumatti, G.; Russo, F.O.; Green, D.R.; Amarante-Mendes, G.P. Bcr-Abl-Mediated Resistance to Apoptosis Is Independent of Constant Tyrosine-Kinase Activity. Cell Death Differ. 2003, 10, 592–598. [Google Scholar] [CrossRef]
- Ma, L.; Shan, Y.; Bai, R.; Xue, L.; Eide, C.A.; Ou, J.; Zhu, L.J.; Hutchinson, L.; Cerny, J.; Khoury, H.J.; et al. A Therapeutically Targetable Mechanism of BCR-ABL-Independent Imatinib Resistance in Chronic Myeloid Leukemia. Sci. Transl. Med. 2014, 6, 252ra121. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Liu, P.; Wang, P.; Zhou, Z.; Fang, Q.; Wang, J. PKC-β/Alox5 Axis Activation Promotes Bcr-Abl-Independent TKI-Resistance in Chronic Myeloid Leukemia. J. Cell. Physiol. 2021, 236, 6312–6327. [Google Scholar] [CrossRef]
- Mitchell, R.; Hopcroft, L.E.M.; Baquero, P.; Allan, E.K.; Hewit, K.; James, D.; Hamilton, G.; Mukhopadhyay, A.; O’Prey, J.; Hair, A.; et al. Targeting BCR-ABL-Independent TKI Resistance in Chronic Myeloid Leukemia by MTOR and Autophagy Inhibition. J. Natl. Cancer Inst. 2018, 110, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Giustacchini, A.; Thongjuea, S.; Barkas, N.; Woll, P.S.; Povinelli, B.J.; Booth, C.A.G.; Sopp, P.; Norfo, R.; Rodriguez-Meira, A.; Ashley, N.; et al. Single-Cell Transcriptomics Uncovers Distinct Molecular Signatures of Stem Cells in Chronic Myeloid Leukemia. Nat. Med. 2017, 23, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Bhatia, R. Preservation of quiescent chronic myelogenous leukemia stem cells by the bone marrow microenvironment. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2018; Volume 1100, pp. 97–110. [Google Scholar]
- Loscocco, F.; Visani, G.; Galimberti, S.; Curti, A.; Isidori, A. BCR-ABL Independent Mechanisms of Resistance in Chronic Myeloid Leukemia. Front. Oncol. 2019, 9, 939. [Google Scholar] [CrossRef] [Green Version]
- Raitano, A.B.; Whang, Y.E.; Sawyers, C.L. Signal Transduction by Wild-Type and Leukemogenic Abl Proteins. Biochim. Biophys. Acta 1997, 1333, F201–F216. [Google Scholar] [CrossRef]
- Sattler, M.; Salgia, R. Activation of Hematopoietic Growth Factor Signal Transduction Pathways by the Human Oncogene BCR/ABL. Cytokine Growth Factor Rev. 1997, 8, 63–79. [Google Scholar] [CrossRef]
- Zou, X.; Calame, K. Signaling Pathways Activated by Oncogenic Forms of Abl Tyrosine Kinase. J. Biol. Chem. 1999, 274, 18141–18144. [Google Scholar] [CrossRef] [Green Version]
- Dormer, P.; Lau, B.; Wilmanns, W. Kinetics of bone marrow cell production in human acute and chronic myeloid leukemias. Leuk. Res. 1980, 4, 231–237. [Google Scholar] [CrossRef]
- Bedi, A.; Zehnbauer, B.A.; Barber, J.P.; Sharkis, S.J.; Jones, R.J. Inhibition of apoptosis by BCR-ABL in chronic myeloid leukemia. Blood 1994, 83, 2038–2044. [Google Scholar] [CrossRef] [Green Version]
- Mcgahon, A.J.; Brown, D.G.; Martin, S.J.; Amarante-Mendes, G.P.; Cotter, T.G.; Cohen, G.M.; Green, D.R. Downregulation of Bcr-Abl in K562 Cells Restores Susceptibility to Apoptosis: Characterization of the Apoptotic Death. Cell Death Differ. 1997, 4, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Amarante-Mendes, G.P.; Finucane, D.M.; Martin, S.J.; Cotter, T.G.; Salvesen, G.S.; Green, D.R. Anti-Apoptotic Oncogenes Prevent Caspase-Dependent and Independent Commitment for Cell Death. Cell Death Differ. 1998, 5, 298–306. [Google Scholar] [CrossRef]
- Fernandez-Luna, J.L. Bcr-Abl and Inhibition of Apoptosis in Chronic Myelogenous Leukemia Cells. Apoptosis 2000, 5, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Brumatti, G.; Weinlich, R.; Chehab, C.F.; Yon, M.; Amarante-Mendes, G.P. Comparison of the Anti-Apoptotic Effects of Bcr-Abl, Bcl-2 and Bcl-XL Following Diverse Apoptogenic Stimuli. FEBS Lett. 2003, 541, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Ghaffari, S.; Jagani, Z.; Kitidis, C.; Lodish, H.F.; Khosravi-Far, R. Cytokines and BCR-ABL Mediate Suppression of TRAIL-Induced Apoptosis through Inhibition of Forkhead FOXO3a Transcription Factor. Proc. Natl. Acad. Sci. USA 2003, 100, 6523–6528. [Google Scholar] [CrossRef] [Green Version]
- Pereira, W.O.; Amarante-Mendes, G.P. Apoptosis: A Programme of Cell Death or Cell Disposal? Scand. J. Immunol. 2011, 73, 401–407. [Google Scholar] [CrossRef] [Green Version]
- Diepstraten, S.T.; Anderson, M.A.; Czabotar, P.E.; Lessene, G.; Strasser, A.; Kelly, G.L. The Manipulation of Apoptosis for Cancer Therapy Using BH3-Mimetic Drugs. Nat. Rev. Cancer 2021, 22, 45–64. [Google Scholar] [CrossRef]
- Benito, A.; Silva, M.; Grillot, D.; Nuiiez, G.; Luis Fernandez-Luna, J. Apoptosis Induced by Erythroid Differentiation of Human Leukemia Cell Lines Is Inhibited by Bcl-X. Blood 1996, 87, 3837–3843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.F.; Huang, W.R.; Duan, H.F.; Wang, H.; Wu, C.T.; Wang, L.S. Sphingosine Kinase-1 Mediates BCR/ABL-Induced Upregulation of Mcl-1 in Chronic Myeloid Leukemia Cells. Oncogene 2007, 26, 7904–7908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez-Castellanos, S.; Cruz, M.; Rabelo, L.; Godínez, R.; Reyes-Maldonado, E.; Riebeling-Navarro, C. Differences in BCL-XL expression and STAT5 phosphorylation in chronic myeloid leukaemia patients. Eur. J. Haematol. 2004, 72, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Puthalakath, H.; Cragg, M.S.; Kelly, P.N.; Bouillet, P.; Huang, D.C.; Kimura, S.; Ottmann, O.G.; Druker, B.J.; Villunger, A.; et al. Bim and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc. Natl. Acad. Sci. USA 2006, 103, 14907–14912. [Google Scholar] [CrossRef] [Green Version]
- Ng, K.P.; Hillmer, A.M.; Chuah, C.T.H.; Juan, W.C.; Ko, T.K.; Teo, A.S.M.; Ariyaratne, P.N.; Takahashi, N.; Sawada, K.; Fei, Y.; et al. A Common BIM Deletion Polymorphism Mediates Intrinsic Resistance and Inferior Responses to Tyrosine Kinase Inhibitors in Cancer. Nat. Med. 2012, 18, 521–528. [Google Scholar] [CrossRef] [PubMed]
- McGahon, A.J.; Nishioka, W.K.; Martin, S.J.; Mahboubi, A.; Cotter, T.G.; Green, D.R. Regulation of the Fas Apoptotic Cell Death Pathway by Abl. J. Biol. Chem. 1995, 270, 22625–22631. [Google Scholar] [CrossRef] [Green Version]
- Selleri, C.; Maciejewski, J.P. The Role of FAS-Mediated Apoptosis in Chronic Myelogenous Leukemia. Leuk. Lymphoma 2000, 37, 283–297. [Google Scholar] [CrossRef]
- De Carvalho, D.D.; Binato, R.; Pereira, W.O.; Leroy, J.M.G.; Colassanti, M.D.; Proto-Siqueira, R.; Bueno-Da-Silva, A.E.B.; Zago, M.A.; Zanichelli, M.A.; Abdelhay, E.; et al. BCR-ABL-Mediated Upregulation of PRAME Is Responsible for Knocking down TRAIL in CML Patients. Oncogene 2011, 30, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor–related apoptosis–inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef]
- Wang, S.; El-Deiry, W.S. TRAIL and Apoptosis Induction by TNF-Family Death Receptors. Oncogene 2003, 22, 8628–8633. [Google Scholar] [CrossRef] [Green Version]
- Amarante-Mendes, G.P.; Griffith, T.S. Therapeutic Applications of TRAIL Receptor Agonists in Cancer and Beyond. Pharmacol. Ther. 2015, 155, 117–131. [Google Scholar] [CrossRef] [Green Version]
- De Carvalho, D.; Mello, B.; Pereira, W.; Amarante-Mendes, G. PRAME/EZH2-mediated regulation of TRAIL: A new target for cancer therapy. Curr. Mol. Med. 2013, 13, 296–304. [Google Scholar] [CrossRef]
- Deming, P.B.; Schafer, Z.T.; Tashker, J.S.; Potts, M.B.; Deshmukh, M.; Kornbluth, S. Bcr-Abl-Mediated Protection from Apoptosis Downstream of Mitochondrial Cytochrome c Release. Mol. Cell. Biol. 2004, 24, 10289–10299. [Google Scholar] [CrossRef] [Green Version]
- Lugo, T.G.; Pendergast, A.M.; Muller, A.J.; Witte, O.N. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 1990, 247, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.W.N.; Goldman, J.M.; Melo, J.V. The molecular biology of chronic myeloid leukemia. Blood 2000, 96, 3343–3356. [Google Scholar] [CrossRef]
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, L.; Guilhot, J.; Krahnke, T.; Guerci-Bresler, A.; Druker, B.J.; Larson, R.A.; O’Brien, S.; So, C.; Massimini, G.; Guilhot, F. Survival Advantage from Imatinib Compared with the Combination Interferon-α plus Cytarabine in Chronic-Phase Chronic Myelogenous Leukemia: Historical Comparison between Two Phase 3 Trials. Blood 2006, 108, 1478–1484. [Google Scholar] [CrossRef]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.N.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef]
- Milojkovic, D.; Apperley, J.F. Mechanisms of Resistance to Imatinib and Second-Generation Tyrosine Inhibitors in Chronic Myeloid Leukemia. Clin. Cancer Res. 2009, 15, 7519–7527. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, E.; Manley, P.W.; Breitenstein, W.; Brüggen, J.; Cowan-Jacob, S.W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; et al. Characterization of AMN107, a Selective Inhibitor of Native and Mutant Bcr-Abl. Cancer Cell 2005, 7, 129–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, A.; Cowan-Jacob, S.W.; Manley, P.W.; Rgen Mestan, J.; Griffin, J.D. Identification of BCR-ABL Point Mutations Conferring Resistance to the Abl Kinase Inhibitor AMN107 (Nilotinib) by a Random Mutagenesis Study. Blood 2007, 109, 5011–5015. [Google Scholar] [CrossRef] [Green Version]
- Saglio, G.; Kim, D.W.; Issaragrisil, S.; le Coutre, P.; Etienne, G.; Lobo, C.; Pasquini, R.; Clark, R.E.; Hochhaus, A.; Hughes, T.P.; et al. Nilotinib versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia. N. Engl. J. Med. 2010, 362, 2251–2259. [Google Scholar] [CrossRef] [Green Version]
- Hughes, L.L.; Wang, M.; Page, D.L.; Gray, R.; Solin, L.J.; Davidson, N.E.; Lowen, M.A.; Ingle, J.N.; Recht, A.; Wood, W.C. Local Excision Alone without Irradiation for Ductal Carcinoma in Situ of the Breast: A Trial of the Eastern Cooperative Oncology Group. J. Clin. Oncol. 2009, 27, 5319–5324. [Google Scholar] [CrossRef]
- Hochhaus, A.; Saglio, G.; Larson, R.A.; Kim, D.-W.; Etienne, G.; Rosti, G.; de Souza, C.; Kurokawa, M.; Kalaycio, M.E.; Hoenekopp, A.; et al. Nilotinib Is Associated with a Reduced Incidence of BCR-ABL Mutations vs Imatinib in Patients with Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase. Blood 2013, 121, 3703–3708. [Google Scholar] [CrossRef]
- Mahon, F.X.; Hayette, S.; Lagarde, V.; Belloc, F.; Turcq, B.; Nicolini, F.; Belanger, C.; Manley, P.W.; Leroy, C.; Etienne, G.; et al. Evidence That Resistance to Nilotinib May Be Due to BCR-ABL, Pgp, or Src Kinase Overexpression. Cancer Res. 2008, 68, 9809–9816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, L.J.; Lee, F.Y.; Chen, P.; Norris, D.; Barrish, J.C.; Behnia, K.; Castaneda, S.; Cornelius, L.A.M.; Das, J.; Doweyko, A.M.; et al. Discovery of N-(2-Chloro-6-Methylphenyl)-2-(6-(4-(2-Hydroxyethyl)- Piperazin-1-Yl)-2-Methylpyrimidin-4-Ylamino)Thiazole-5-Carboxamide (BMS-354825), a Dual Src/Abl Kinase Inhibitor with Potent Antitumor Activity in Preclinical Assays. J. Med. Chem. 2004, 47, 6658–6661. [Google Scholar] [CrossRef]
- Tokarski, J.S.; Newitt, J.A.; Chang, C.Y.J.; Cheng, J.D.; Wittekind, M.; Kiefer, S.E.; Kish, K.; Lee, F.Y.F.; Borzillerri, R.; Lombardo, L.J.; et al. The Structure of Dasatinib (BMS-354825) Bound to Activated ABL Kinase Domain Elucidates Its Inhibitory Activity against Imatinib-Resistant ABL Mutants. Cancer Res. 2006, 66, 5790–5797. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.P.; Tran, C.; Lee, F.Y.; Chen, P.; Norris, D.; Sawyers, C.L. Overriding Imatinib Resistance with a Novel ABL Kinase Inhibitor. Science 2004, 305, 399–401. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.P.; Guilhot, F.; Cortes, J.E.; Schiffer, C.A.; le Coutre, P.; Brümmendorf, T.H.; Brümmendorf, B.; Kantarjian, H.M.; Hochhaus, A.; Rousselot, P.; et al. Long-Term Outcome with Dasatinib after Imatinib Failure in Chronic-Phase Chronic Myeloid Leukemia: Follow-up of a Phase 3 Study. Blood 2014, 123, 2317–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, J.E.; Jiang, Q.; Wang, J.; Weng, J.; Zhu, H.; Liu, X.; Hochhaus, A.; Kim, D.W.; Radich, J.; Savona, M.; et al. Dasatinib vs. Imatinib in Patients with Chronic Myeloid Leukemia in Chronic Phase (CML-CP) Who Have Not Achieved an Optimal Response to 3 Months of Imatinib Therapy: The DASCERN Randomized Study. Leukemia 2020, 34, 2064–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puttini, M.; Coluccia, A.M.L.; Boschelli, F.; Cleris, L.; Marchesi, E.; Donella-Deana, A.; Ahmed, S.; Redaelli, S.; Piazza, R.; Magistroni, V.; et al. In Vitro and in Vivo Activity of SKI-606, a Novel Src-Abl Inhibitor, against Imatinib-Resistant Bcr-Abl+ Neoplastic Cells. Cancer Res. 2006, 66, 11314–11322. [Google Scholar] [CrossRef] [Green Version]
- Remsing Rix, L.L.; Rix, U.; Colinge, J.; Hantschel, O.; Bennett, K.L.; Stranzl, T.; Müller, A.; Baumgartner, C.; Valent, P.; Augustin, M.; et al. Global Target Profile of the Kinase Inhibitor Bosutinib in Primary Chronic Myeloid Leukemia Cells. Leukemia 2009, 23, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.E.; Kantarjian, H.M.; Brümmendorf, T.H.; Kim, D.W.; Turkina, A.G.; Shen, Z.X.; Pasquini, R.; Khoury, H.J.; Arkin, S.; Volkert, A.; et al. Safety and Efficacy of Bosutinib (SKI-606) in Chronic Phase Philadelphia Chromosome-Positive Chronic Myeloid Leukemia Patients with Resistance or Intolerance to Imatinib. Blood 2011, 118, 4567–4576. [Google Scholar] [CrossRef] [Green Version]
- Redaelli, S.; Piazza, R.; Rostagno, R.; Magistroni, V.; Perini, P.; Marega, M.; Gambacorti-Passerini, C.; Boschelli, F. Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J. Clin. Oncol. 2008, 27, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Bartolovic, K.; Balabanov, S.; Hartmann, U.; Komor, M.; Boehmler, A.M.; Bühring, H.J.; Möhle, R.; Hoelzer, D.; Kanz, L.; Hofmann, W.K.; et al. Inhibitory Effect of Imatinib on Normal Progenitor Cells in Vitro. Blood 2004, 103, 523–529. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Cortes, J. Molecular biology of bcr-abl1–positive chronic myeloid leukemia. Blood 2009, 113, 1619–1630. [Google Scholar] [CrossRef] [Green Version]
- O’Hare, T.; Shakespeare, W.C.; Zhu, X.; Eide, C.A.; Rivera, V.M.; Wang, F.; Adrian, L.T.; Zhou, T.; Huang, W.S.; Xu, Q.; et al. AP24534, a Pan-BCR-ABL Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315I Mutant and Overcomes Mutation-Based Resistance. Cancer Cell 2009, 16, 401–412. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.E.; Kim, D.-W.; Pinilla-Ibarz, J.; le Coutre, P.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. A Phase 2 Trial of Ponatinib in Philadelphia Chromosome–Positive Leukemias. N. Engl. J. Med. 2013, 369, 1783–1796. [Google Scholar] [CrossRef] [Green Version]
- Khorashad, J.S.; Kelley, T.W.; Szankasi, P.; Mason, C.C.; Soverini, S.; Adrian, L.T.; Eide, C.A.; Zabriskie, M.S.; Lange, T.; Estrada, J.C.; et al. BCR-ABL1 Compound Mutations in Tyrosine Kinase Inhibitor-Resistant CML: Frequency and Clonal Relationships. Blood 2013, 121, 489–498. [Google Scholar] [CrossRef]
- Lipton, J.H.; Chuah, C.; Guerci-Bresler, A.; Rosti, G.; Simpson, D.; Assouline, S.; Etienne, G.; Nicolini, F.E.; le Coutre, P.; Clark, R.E.; et al. Ponatinib versus Imatinib for Newly Diagnosed Chronic Myeloid Leukaemia: An International, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2016, 17, 612–621. [Google Scholar] [CrossRef]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 Recommendations for Treating Chronic Myeloid Leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wylie, A.A.; Schoepfer, J.; Jahnke, W.; Cowan-Jacob, S.W.; Loo, A.; Furet, P.; Marzinzik, A.L.; Pelle, X.; Donovan, J.; Zhu, W.; et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 2017, 543, 733–737. [Google Scholar] [CrossRef]
- Schoepfer, J.; Jahnke, W.; Berellini, G.; Buonamici, S.; Cotesta, S.; Cowan-Jacob, S.W.; Dodd, S.; Drueckes, P.; Fabbro, D.; Gabriel, T.; et al. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J. Med. Chem. 2018, 61, 8120–8135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hehlmann, R. The New Eln Recommendations for Treating Cml. J. Clin. Med. 2020, 9, 3671. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; Branford, S.; Nicolini, F.E.; Talpaz, M.; Deininger, M.W.N.; Martinelli, G.; Müller, M.C.; Radich, J.P.; Shah, N.P. Implications of BCR-ABL1 Kinase Domain-Mediated Resistance in Chronic Myeloid Leukemia. Leuk. Res. 2014, 38, 10–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eck, M.J.; Manley, P.W. The Interplay of Structural Information and Functional Studies in Kinase Drug Design: Insights from BCR-Abl. Curr. Opin. Cell Biol. 2009, 21, 288–295. [Google Scholar] [CrossRef]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, J.M.; Melo, J.V. Targeting the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1084–1086. [Google Scholar] [CrossRef]
- Griswold, I.J.; MacPartlin, M.; Bumm, T.; Goss, V.L.; O’Hare, T.; Lee, K.A.; Corbin, A.S.; Stoffregen, E.P.; Smith, C.; Johnson, K.; et al. Kinase Domain Mutants of Bcr-Abl Exhibit Altered Transformation Potency, Kinase Activity, and Substrate Utilization, Irrespective of Sensitivity to Imatinib. Mol. Cell. Biol. 2006, 26, 6082–6093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, E.; Tumian, N.R.; Azma, R.Z.; Sharifah, N.A.; Salwati, S.; Hamidah, N.H.; Elias, M.H.; Wong, C.L. Primary imatinib resistance in chronic myeloid leukemia patients in a developing country: BCR-ABL kinase domain mutations or BCR-ABL independent mechanisms? Malays. J. Pathol. 2017, 39, 107–113. [Google Scholar]
- Jabbour, E.; Kantarjian, H.; Jones, D.; Talpaz, M.; Bekele, N.; O’Brien, S.; Zhou, X.; Luthra, R.; Garcia-Manero, G.; Giles, F.; et al. Frequency and Clinical Significance of BCR-ABL Mutations in Patients with Chronic Myeloid Leukemia Treated with Imatinib Mesylate. Leukemia 2006, 20, 1767–1773. [Google Scholar] [CrossRef]
- Barnes, D.J.; Palaiologou, D.; Panousopoulou, E.; Schultheis, B.; Yong, A.S.M.; Wong, A.; Pattacini, L.; Goldman, J.M.; Melo, J.V. Bcr-Abl Expression Levels Determine the Rate of Development of Resistance to Imatinib Mesylate in Chronic Myeloid Leukemia. Cancer Res. 2005, 65, 8912–8919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, B.; Fioretos, T.; Mitelman, F. Cytogenetic and Molecular Genetic Evolution of Chronic Myeloid Leukemia. Acta Haematol. 2002, 107, 76–94. [Google Scholar] [CrossRef]
- Alhuraiji, A.; Kantarjian, H.; Boddu, P.; Ravandi, F.; Borthakur, G.; DiNardo, C.; Daver, N.; Kadia, T.; Pemmaraju, N.; Pierce, S.; et al. Prognostic significance of additional chromosomal abnormalities at the time of diagnosis in patients with chronic myeloid leukemia treated with frontline tyrosine kinase inhibitors. Am. J. Hematol. 2018, 93, 84–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Cortes, J.E.; Tang, G.; Khoury, J.D.; Wang, S.; Bueso-Ramos, C.E.; DiGiuseppe, J.A.; Chen, Z.; Kantarjian, H.M.; Medeiros, L.J.; et al. Risk stratification of chromosomal abnormalities in chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Blood 2016, 127, 2742–2750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baccarani, M.; Deininger, M.W.; Rosti, G.; Hochhaus, A.; Soverini, S.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Guilhot, F.; et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013, 122, 872–874. [Google Scholar] [CrossRef]
- Lahaye, T.; Riehm, B.; Berger, U.; Paschka, P.; Müller, M.C.; Kreil, S.; Merx, K.; Schwindel, U.; Schoch, C.; Hehlmann, R.; et al. Response and Resistance in 300 Patients with BCR-ABL-Positive Leukemias Treated with Imatinib in a Single Center: A 4.5-Year Follow-Up. Cancer 2005, 103, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Shoukier, M.; Kubiak, M.; Cortes, J. Review of New-Generation Tyrosine Kinase Inhibitors for Chronic Myeloid Leukemia. Curr. Oncol. Rep. 2021, 23, 91. [Google Scholar] [CrossRef]
- Abdulmawjood, B.; Costa, B.; Roma-Rodrigues, C.; Baptista, P.V.; Fernandes, A.R. Genetic Biomarkers in Chronic Myeloid Leukemia: What Have We Learned So Far? Int. J. Mol. Sci. 2021, 22, 12516. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, S.; Grassi, S.; Baratè, C.; Guerrini, F.; Ciabatti, E.; Perutelli, F.; Ricci, F.; Del Genio, G.; Montali, M.; Barachini, S.; et al. The Polycomb BMI1 Protein Is Co-expressed With CD26+ in Leukemic Stem Cells of Chronic Myeloid Leukemia. Front. Oncol. 2018, 8, 555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelinek, J.; Gharibyan, V.; Estecio, M.R.; Kondo, K.; He, R.; Chung, W.; Lu, Y.; Zhang, N.; Liang, S.; Kantarjian, H.M.; et al. Aberrant DNA methylation is associated with disease progression, resistance to imatinib and shortened survival in chronic myelogenous leukemia. PLoS ONE 2011, 6, e22110. [Google Scholar] [CrossRef]
- Branford, S.; Wang, P.; Yeung, D.T.; Thomson, D.; Purins, A.; Wadham, C.; Shahrin, N.H.; Marum, J.E.; Nataren, N.; Parker, W.T.; et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood 2018, 13, 948–961. [Google Scholar] [CrossRef] [PubMed]
- Kuwazuru, Y.; Yoshimura, A.; Hanada, S.; Ichikawa, M.; Saito, T.; Uozumi, K.; Utsunomiya, A.; Arima, T.; Akiyama, S.I. Expression of the multidrug transporter, P-glycoprotein, in chronic myelogenous leukaemia cells in blast crisis. Br. J. Haematol. 1990, 74, 24–29. [Google Scholar] [CrossRef]
- Thomas, J.; Wang, L.; Clark, R.E.; Pirmohamed, M. Active Transport of Imatinib into and out of Cells: Implications for Drug Resistance. Blood 2004, 104, 3739–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrish, E.; Copeland, A.; Moujalled, D.M.; Powell, J.A.; Silke, N.; Lin, A.; Jarman, K.E.; Sandow, J.J.; Ebert, G.; Mackiewicz, L.; et al. Clinical MDR1 inhibitors enhance Smac-mimetic bioavailability to kill murine LSCs and improve survival in AML models. Blood Adv. 2020, 4, 5062–5077. [Google Scholar] [CrossRef]
- Vasconcelos, F.C.; Silva, K.L.; Souza, P.S.; Silva, L.F.; Moellmann-Coelho, A.; Klumb, C.E.; Maia, R.C. Variation of MDR proteins expression and activity levels according to clinical status and evolution of CML patients. Cytom. B Clin. Cytom. 2011, 80, 158–166. [Google Scholar] [CrossRef]
- Mahon, F.X.; Deininger, M.W.N.; Schultheis, B.; Rome Chabrol, J.; Reiffers, J.; Goldman, J.M.; Melo, J.V. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the tyrosine kinase inhibitor STI571: Diverse mechanisms of resistance. Blood 2000, 96, 1070–1079. [Google Scholar] [CrossRef]
- Donato, N.J.; Wu, J.Y.; Stapley, J.; Gallick, G.; Lin, H.; Arlinghaus, R.; Talpaz, M. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood 2003, 101, 690–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiring, A.M.; Kraft, I.L.; Page, B.D.; O’Hare, T.; Gunning, P.T.; Deininger, M.W. STAT3 as a mediator of BCR-ABL1-independent resistance in chronic myeloid leukemia. Leuk. Suppl. 2014, 3, S5–S6. [Google Scholar] [CrossRef] [PubMed]
- Aggoune, D.; Sorel, N.; Bonnet, M.L.; Goujon, J.M.; Tarte, K.; Hérault, O.; Domenech, J.; Réa, D.; Legros, L.; Johnson-Ansa, H.; et al. Bone marrow mesenchymal stromal cell (MSC) gene profiling in chronic myeloid leukemia (CML) patients at diagnosis and in deep molecular response induced by tyrosine kinase inhibitors (TKIs). Leuk. Res. 2017, 60, 94–102. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, X.; Liu, Y.; Yang, L.; Huang, S.; Lu, L.; Wang, S.; Guo, Q.; Zhao, L. BM microenvironmental protection of CML cells from imatinib through Stat5/NF-κB signaling and reversal by Wogonin. Oncotarget 2016, 7, 24436–24454. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.; Isringhausen, S.; Li, H.; Paterson, A.J.; He, J.; Gomariz, Á.; Nagasawa, T.; Nombela-Arrieta, C.; Bhatia, R. Mesenchymal Niche-Specific Expression of Cxcl12 Controls Quiescence of Treatment-Resistant Leukemia Stem Cells. Cell Stem Cell. 2019, 24, 769–784.e6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cai, D.; Brendel, C.; Barett, C.; Erben, P.; Manley, P.W.; Hochhaus, A.; Neubauer, A.; Burchert, A. Adaptive secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF) mediates imatinib and nilotinib resistance in BCR/ABL+ progenitors via JAK-2/STAT-5 pathway activation. Blood 2007, 109, 2147–2155. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, E.; Wright, R.D.; McMillin, D.W.; Mitsiades, C.; Ray, A.; Barrett, R.; Adamia, S.; Stone, R.; Galinsky, I.; Kung, A.L.; et al. Stromal-mediated protection of tyrosine kinase inhibitor-treated BCR-ABL-expressing leukemia cells. Mol. Cancer Ther. 2008, 7, 1121–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.; Kharabi Masouleh, B.; Loges, S.; Cauwenberghs, S.; Fraisl, P.; Maes, C.; Jonckx, B.; De Keersmaecker, K.; Kleppe, M.; Tjwa, M.; et al. Loss or inhibition of stromal-derived PlGF prolongs survival of mice with imatinib-resistant Bcr-Abl1(+) leukemia. Cancer Cell 2011, 19, 740–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floyd, M.D.; Gervasini, G.; Masica, A.L.; Mayo, G.; George, A.L.; Bhat, K.; Kim, R.B.; Wilkinson, G.R. Genotype–phenotype associations for common CYP3A4 and CYP3A5 variants in the basal and induced metabolism of midazolam in European-and African-American men and women. Pharm. Genom. 2003, 13, 595–606. [Google Scholar] [CrossRef]
- Pellicano, F.; Scott, M.T.; Helgason, G.V.; Hopcroft, L.E.M.; Allan, E.K.; Aspinall-O’Dea, M.; Copland, M.; Pierce, A.; Huntly, B.J.P.; Whetton, A.D.; et al. The Antiproliferative Activity of Kinase Inhibitors in Chronic Myeloid Leukemia Cells Is Mediated by FOXO Transcription Factors. Stem Cells 2014, 32, 2324–2337. [Google Scholar] [CrossRef] [Green Version]
- Wagle, M.; Eiring, A.M.; Wongchenko, M.; Lu, S.; Guan, Y.; Wang, Y.; Lackner, M.; Amler, L.; Hampton, G.; Deininger, M.W.; et al. A Role for FOXO1 in BCR-ABL1-Independent Tyrosine Kinase Inhibitor Resistance in Chronic Myeloid Leukemia. Leukemia 2016, 30, 1493–1501. [Google Scholar] [CrossRef] [Green Version]
- Moon, R.T.; Kohn, A.D.; de Ferrari, G.V.; Kaykas, A. WNT and β-Catenin Signalling: Diseases and Therapies. Nat. Rev. Genet. 2004, 5, 691–701. [Google Scholar] [CrossRef]
- Eiring, A.M.; Khorashad, J.S.; Anderson, D.J.; Yu, F.; Redwine, H.M.; Mason, C.C.; Reynolds, K.R.; Clair, P.M.; Gantz, K.C.; Zhang, T.Y.; et al. β-Catenin Is Required for Intrinsic but Not Extrinsic BCR-ABL1 Kinase-Independent Resistance to Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Leukemia 2015, 29, 2328–2337. [Google Scholar] [CrossRef]
- Ben-Batalla, I.; Erdmann, R.; Jorgensen, H.; Mitchell, R.; Ernst, T.; Von Amsberg, G.; Schafhausen, P.; Velthaus, J.L.; Rankin, S.; Clark, R.E.; et al. Axl Blockade by BGB324 Inhibits BCR-ABL Tyrosine Kinase Inhibitor-Sensitive and -Resistant Chronic Myeloid Leukemia. Clin. Cancer Res. 2017, 23, 2289–2300. [Google Scholar] [CrossRef] [Green Version]
- Gioia, R.; Tregoat, C.; Dumas, P.Y.; Lagarde, V.; Prouzet-Mauleon, V.; Desplat, V.; Sirvent, A.; Praloran, V.; Lippert, E.; Villacreces, A.; et al. CBL controls a tyrosine kinase network involving AXL, SYK and LYN in nilotinib-resistant chronic myeloid leukaemia. J. Pathol. 2015, 237, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.L.; Nie, D.N.; Li, J.; Du, X.; Lu, Y.H.; Li, Y.Q.; Liu, C.; Zhou, J.F.; Pan, J.X. Gas6/AXL Signaling Regulates Self-Renewal of Chronic Myelogenous Leukemia Stem Cells by Stabilizing beta-Catenin. Clin. Cancer Res. 2017, 23, 2842–2855. [Google Scholar] [CrossRef] [Green Version]
- Neubauer, A.; Fiebeler, A.; Graham, D.K.; Obryan, J.P.; Schmidt, C.A.; Barckow, P.; Serke, S.; Siegert, W.; Snodgrass, H.R.; Huhn, D.; et al. Expression of Axl, a Transforming Receptor Tyrosine Kinase, in Normal and Malignant Hematopoiesis. Blood 1994, 84, 1931–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koptyra, M.; Falinski, R.; Nowicki, M.O.; Stoklosa, T.; Majsterek, I.; Nieborowska-Skorska, M.; Blasiak, J.; Skorski, T. BCR/ABL Kinase Induces Self-Mutagenesis via Reactive Oxygen Species to Encode Imatinib Resistance. Blood 2006, 108, 319–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrgazov, K.; Lucini, C.B.; Valent, P.; Hantschel, O.; Lion, T. BCR-ABL1 Compound Mutants Display Differential and Dose-Dependent Responses to Ponatinib. Haematologica 2018, 103, e10–e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pemovska, T.; Johnson, E.; Kontro, M.; Repasky, G.A.; Chen, J.; Wells, P.; Cronin, C.N.; Mctigue, M.; Kallioniemi, O.; Porkka, K.; et al. Axitinib Effectively Inhibits BCR-ABL1(T315I) with a Distinct Binding Conformation. Nature 2015, 519, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Leverson, J.D.; Sampath, D.; Souers, A.J.; Rosenberg, S.H.; Fairbrother, W.J.; Amiot, M.; Konopleva, M.; Letai, A. Found in Translation: How Preclinical Research Is Guiding the Clinical Development of the BCL2-Selective Inhibitor Venetoclax. Cancer Discov. 2017, 7, 1376–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.W. Therapeutic development and current uses of BCL-2 inhibition. Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 1–9. [Google Scholar] [CrossRef]
- Parry, N.; Wheadon, H.; Copland, M. The application of BH3 mimetics in myeloid leukemias. Cell Death Dis. 2021, 12, 222. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Ko, T.K.; Chuah, C.T.; Huang, J.W.; Ng, K.P.; Ong, S.T. The BCL2 inhibitor ABT-199 significantly enhances imatinib-induced cell death in chronic myeloid leukemia progenitors. Oncotarget 2014, 5, 9033–9038. [Google Scholar] [CrossRef] [Green Version]
- Carter, B.Z.; Mak, P.Y.; Mu, H.; Zhou, H.; Mak, D.H.; Schober, W.; Leverson, J.D.; Zhang, B.; Bhatia, R.; Huang, X.; et al. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci. Transl. Med. 2016, 8, 355ra117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burslem, G.M.; Crews, C.M. Small-molecule modulation of protein homeostasis. Chem. Rev. 2017, 117, 11269–11301. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; He, Y.; Zhang, X.; Yuan, Y.; Pu, S.; Kong, Q.; Zheng, G.; Zhou, D. PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene 2020, 39, 4909–4924. [Google Scholar] [CrossRef]
- Liu, J.; Ma, J.; Liu, Y.; Xia, J.; Li, Y.; Wang, Z.P.; Wei, W. PROTACs: A novel strategy for cancer therapy. Semin. Cancer Biol. 2020, 67, 171–179. [Google Scholar] [CrossRef]
- He, Y.; Khan, S.; Huo, Z.; Lv, D.; Zhang, X.; Liu, X.; Yuan, Y.; Hromas, R.; Xu, M.; Zheng, G.; et al. Proteolysis targeting chimeras (PROTACs) are emerging therapeutics for hematologic malignancies. J. Hematol. Oncol. 2020, 13, 103. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Schultz, A.R.; Bondeson, D.P.; Eide, C.A.; Stevens, S.L.S.; Druker, B.J.; Crews, C.M. Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation. Cancer Res. 2019, 79, 4744–4753. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Ren, C.; Liu, L.; Chen, J.; Shao, Y.; Sun, N.; Sun, R.; Kong, Y.; Ding, X.; Zhang, X.; et al. Discovery of SIAIS178 as an Effective BCR-ABL Degrader by Recruiting Von Hippel-Lindau (VHL) E3 Ubiquitin Ligase. J. Med. Chem. 2019, 62, 9281–9298. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gao, H.; Sun, X.; Sun, Y.; Qiu, Y.; Weng, Q.; Rao, Y. Global PROTAC Toolbox for Degrading BCR-ABL Overcomes Drug-Resistant Mutants and Adverse Effects. J. Med. Chem. 2020, 63, 8567–8583. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, A.; Li, X.; Chen, C.; Qi, Z.; Hu, C.; Wang, W.; Wu, H.; Huang, T.; Zhao, M.; et al. Discovery and characterization of a novel highly potent and selective type II native and drug-resistant V299L mutant BCR-ABL inhibitor (CHMFL-ABL-039) for Chronic Myeloid Leukemia (CML). Cancer Biol. Ther. 2019, 20, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, P.; Gao, Y.; Song, Y.; Zhu, Y.; Su, H.; Yang, B.; Li, L.; Li, G.; Gong, N.; et al. Discovery of a Candidate Containing an (S)-3,3-Difluoro-1-(4-methylpiperazin-1-yl)-2,3-dihydro-1H-inden Scaffold as a Highly Potent Pan-Inhibitor of the BCR-ABL Kinase Including the T315I-Resistant Mutant for the Treatment of Chronic Myeloid Leukemia. J. Med. Chem. 2021, 6, 7434–7452. [Google Scholar] [CrossRef]
- Lu, T.; Cao, J.; Zou, F.; Li, X.; Wang, A.; Wang, W.; Liang, H.; Liu, Q.; Hu, C.; Chen, C.; et al. Discovery of a highly potent kinase inhibitor capable of overcoming multiple imatinib-resistant ABL mutants for chronic myeloid leukemia (CML). Eur. J. Pharmacol. 2021, 897, 173944. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time | Optimal | Warning | Failure |
|---|---|---|---|
| Baseline | NA | High risk ACA, high risk ELTS score | NA |
| 3 months | BCR-ABL1 ≤10% | BCR-ABL1 >10% | BCR-ABL1 >10% if confirmed within 1–3 months |
| 6 months | BCR-ABL1 ≤1% | BCR-ABL1 >1–10% | BCR-ABL1 >10% |
| 12 months | BCR-ABL1 ≤0.1% | BCR-ABL1 >0.1–1% | BCR-ABL1 >1% |
| Any time | BCR-ABL1 ≤0.1% | >0.1%; loss of ≤0.1% (MMR) * | BCR-ABL1 >1%, resistance mutations high risk ACA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amarante-Mendes, G.P.; Rana, A.; Datoguia, T.S.; Hamerschlak, N.; Brumatti, G. BCR-ABL1 Tyrosine Kinase Complex Signaling Transduction: Challenges to Overcome Resistance in Chronic Myeloid Leukemia. Pharmaceutics 2022, 14, 215. https://doi.org/10.3390/pharmaceutics14010215
Amarante-Mendes GP, Rana A, Datoguia TS, Hamerschlak N, Brumatti G. BCR-ABL1 Tyrosine Kinase Complex Signaling Transduction: Challenges to Overcome Resistance in Chronic Myeloid Leukemia. Pharmaceutics. 2022; 14(1):215. https://doi.org/10.3390/pharmaceutics14010215
Chicago/Turabian StyleAmarante-Mendes, Gustavo P., Aamir Rana, Tarcila Santos Datoguia, Nelson Hamerschlak, and Gabriela Brumatti. 2022. "BCR-ABL1 Tyrosine Kinase Complex Signaling Transduction: Challenges to Overcome Resistance in Chronic Myeloid Leukemia" Pharmaceutics 14, no. 1: 215. https://doi.org/10.3390/pharmaceutics14010215