
Impact of Formulation Conditions on Lipid Nanoparticle Characteristics and Functional Delivery of CRISPR RNP for Gene Knock-Out and Correction

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Reagents
2.1.1. SpCas9 Protein Production and Purification
2.1.2. SpCas9 Characterization and Stability Study
2.2. Lipid Nanoparticle Formulation
2.3. Physical Characterization of Lipid Nanoparticles
2.4. Quantification of RNP Complexed with LNP
2.5. Stability of Lipid Nanoparticles in Human Plasma
2.6. Cell Culture
2.7. Gene Editing Efficacy Assays
2.7.1. Stoplight Gene Editing Assay
2.7.2. EGFP–BFP Mutation Assay
2.8. T7 Endonuclease Assay
3. Results
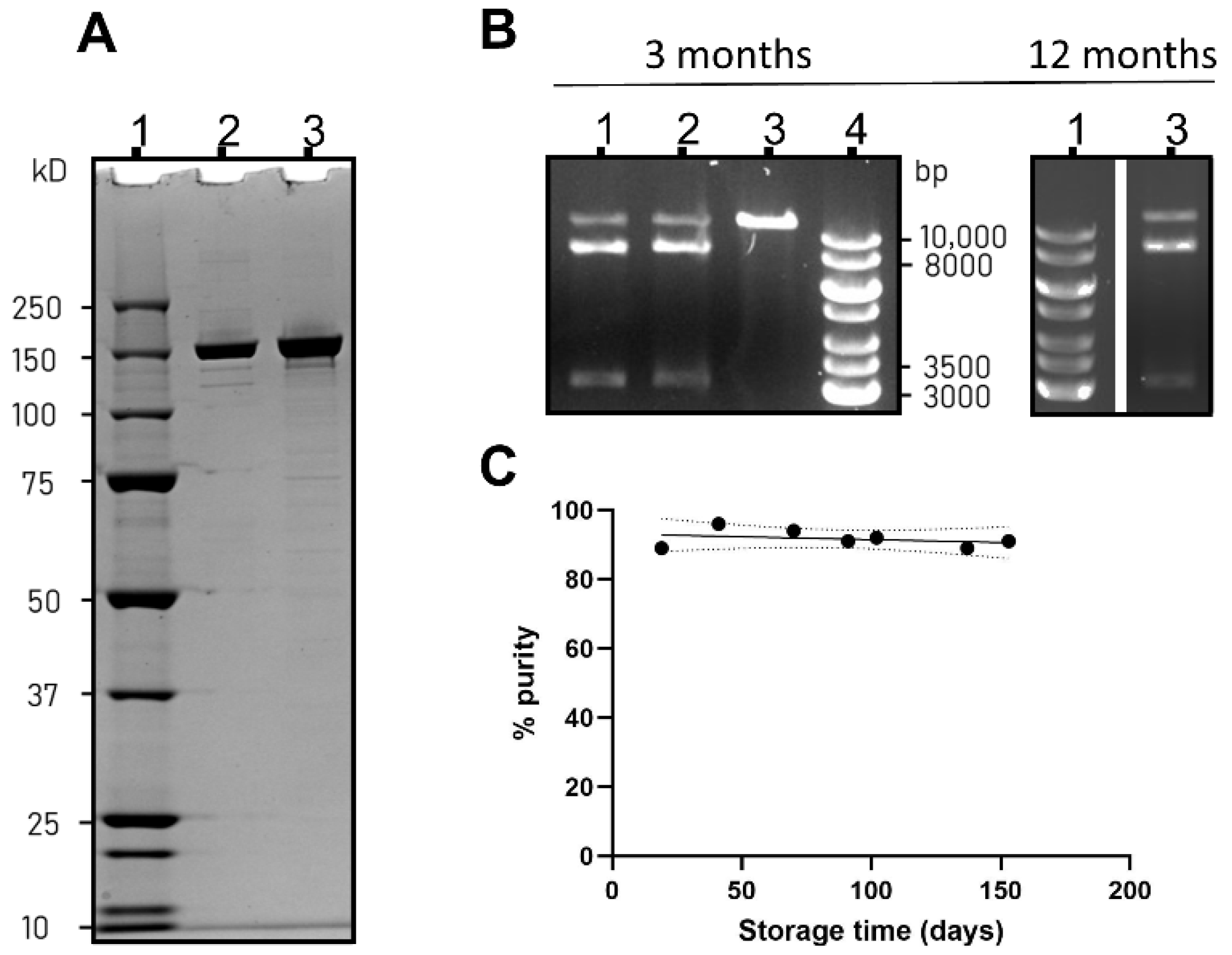
3.1. SpCas9 Production, Characterization and Stability in Storage
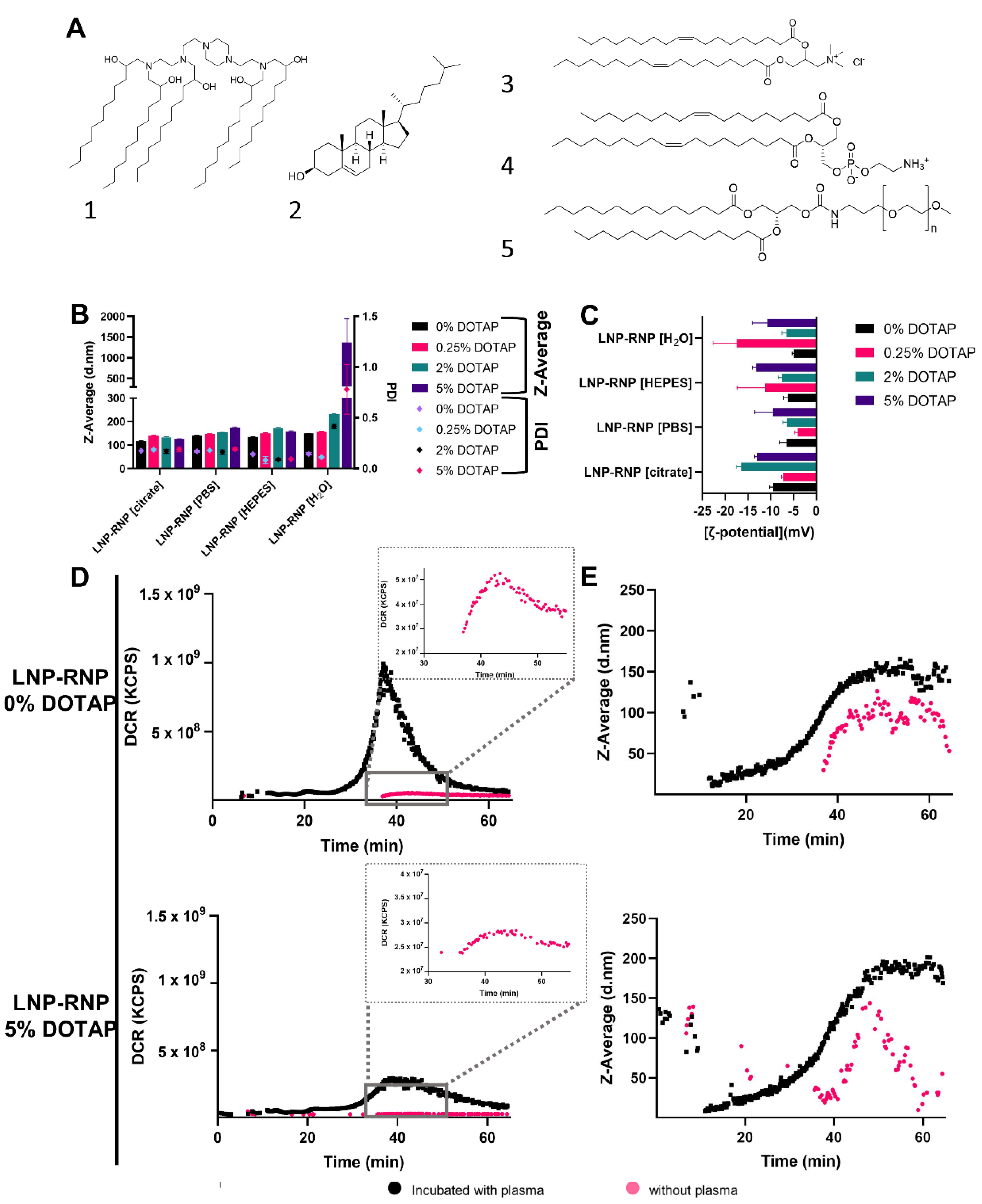
3.2. Characterization and Efficacy of LNP Formulations for Gene Knock-Out (LNP-RNP)
3.3. Determination of Gene Knock Out Efficiency of Different LNP-RNP Formulations
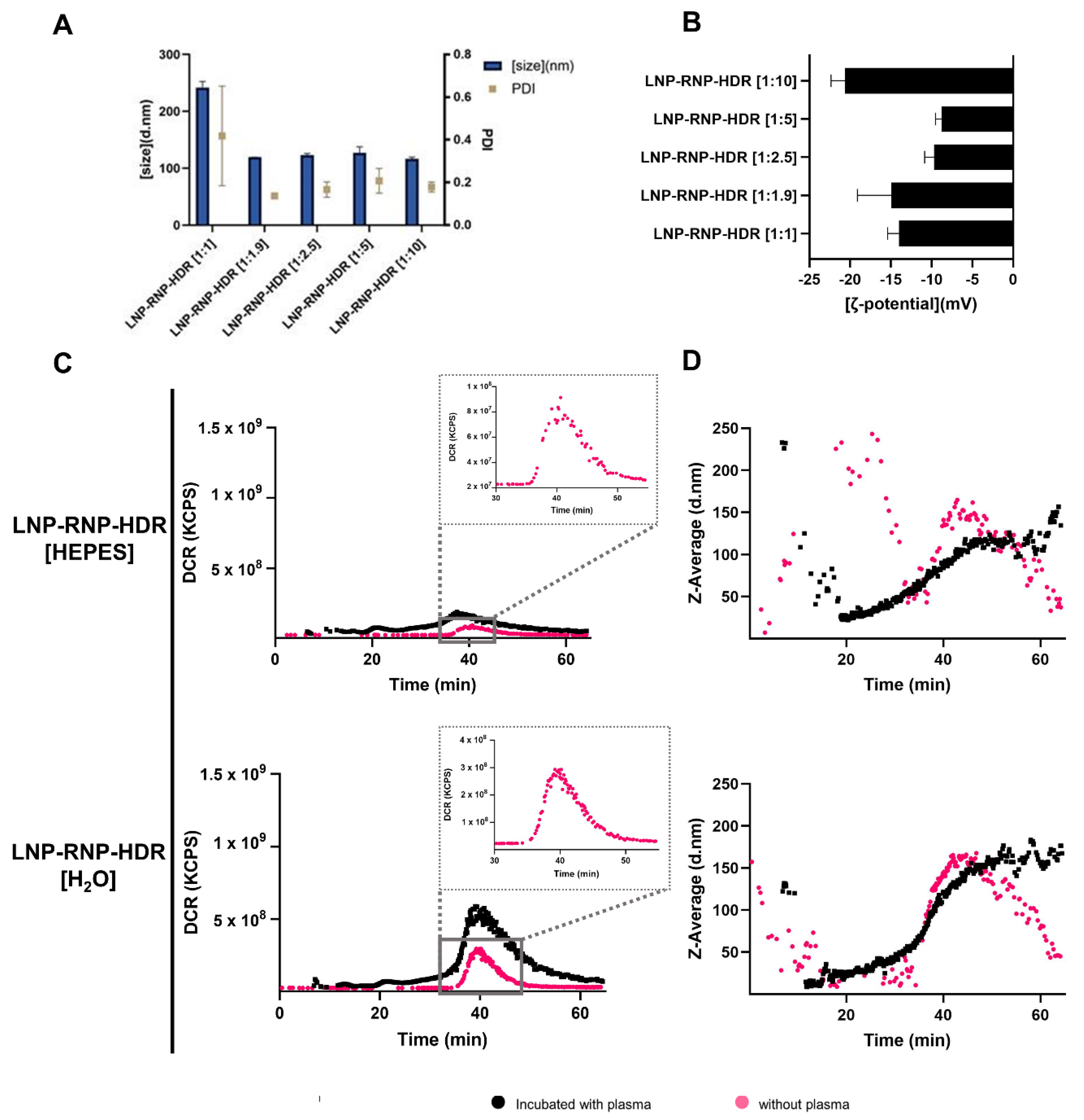
3.4. Characterization of LNP Formulations for Gene Correction (LNP-RNP-HDR)
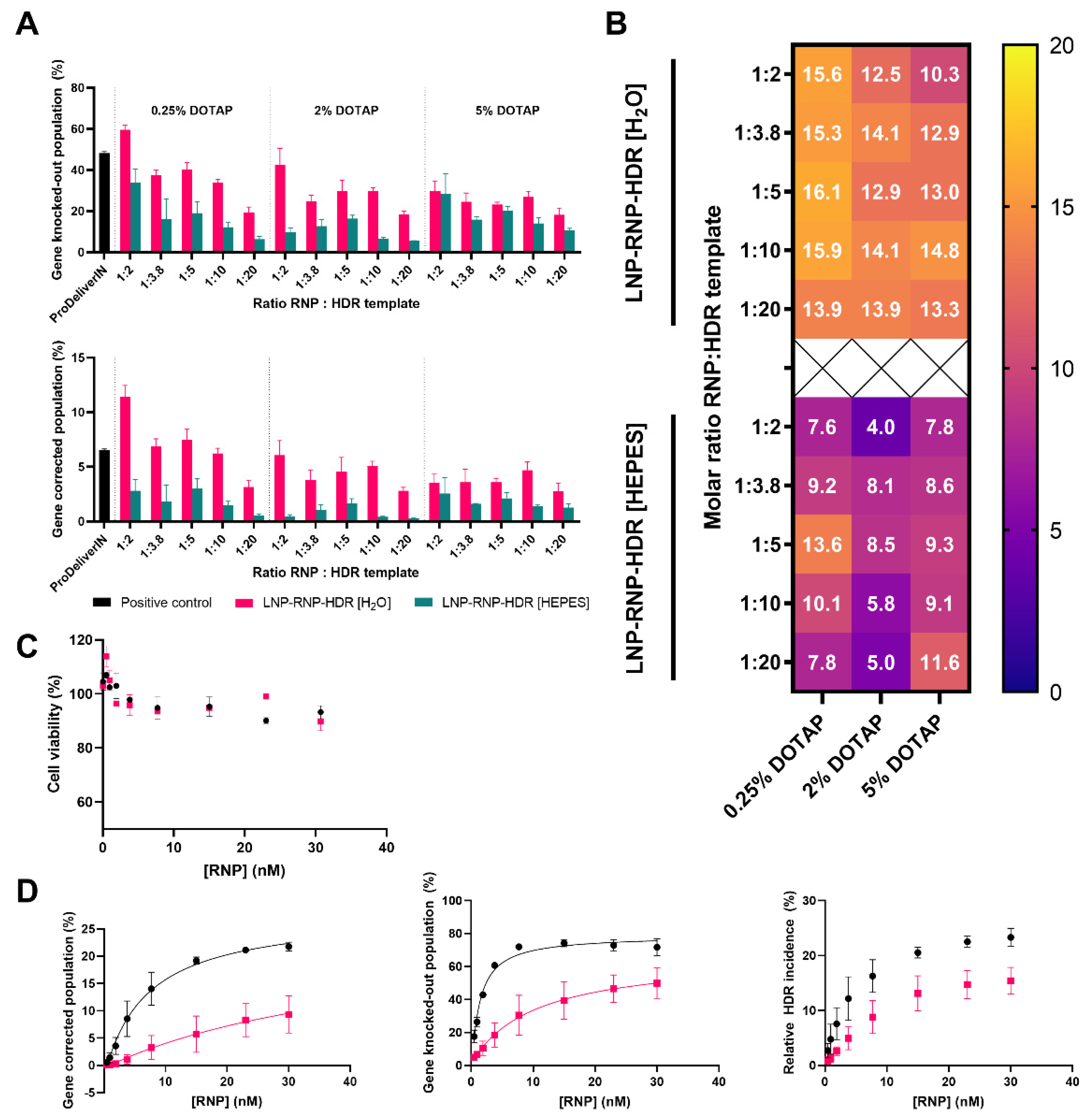
3.5. Determination of Gene Correction Efficiency of Different LNP-RNP-HDR Formulations
4. Discussion
5. Conclusions
- Preparing RNP for formulation in nuclease-free water or HEPES buffer yielded superior gene editing results as compared with PBS or citrate buffer, due to inadequate formation of an active RNP complex in the latter two buffers. There was no marked difference in encapsulation efficiency of Cas9 between these tested systems.
- Incorporation of DOTAP in the LNP-RNP formulation was associated with a high gene-editing efficacy overall, while for LNP-RNP-HDR, a lower concentration was optimal.
- High gene knock-out efficacies above 80% were achieved for LNP-RNP prepared in HEPES buffer, with DOTAP 5 mole%, with a clear dose-dependent relationship.
- As a highlighted result, 20% gene correction efficacy was achieved with LNP-RNP-HDR formulated in nuclease-free water, DOTAP 0.25 mole%, and a 2:1 ratio of HDR template to RNP, with a clear dose-dependent relationship as well, and high cell viability (>90%).
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9–CrRNA Ribonucleoprotein Complex Mediates Specific DNA Cleavage for Adaptive Immunity in Bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doudna, J.A.; Charpentier, E. Genome Editing. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Salsman, J.; Masson, J.-Y.; Orthwein, A.; Dellaire, G. CRISPR/Cas9 Gene Editing: From Basic Mechanisms to Improved Strategies for Enhanced Genome Engineering In Vivo. Curr. Gene Ther. 2017, 17, 263–274. [Google Scholar] [CrossRef]
- Yang, H.; Ren, S.; Yu, S.; Pan, H.; Li, T.; Ge, S.; Zhang, J.; Xia, N. Methods Favoring Homology-Directed Repair Choice in Response to Crispr/Cas9 Induced-Double Strand Breaks. Int. J. Mol. Sci. 2020, 21, 6461. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Haemmerle, M.; Genovese, G.; Draetta, G.F.; Chin, L. Post-Translational Regulation of Cas9 during G1 Enhances Homology-Directed Repair. Cell Rep. 2016, 14, 1555–1566. [Google Scholar] [CrossRef] [Green Version]
- Wilbie, D.; Walther, J.; Mastrobattista, E. Delivery Aspects of CRISPR/Cas for in Vivo Genome Editing. Acc. Chem. Res. 2019, 52, 1555–1564. [Google Scholar] [CrossRef] [Green Version]
- Legut, M.; Dolton, G.; Mian, A.A.; Ottmann, O.G.; Sewell, A.K. CRISPR-Mediated TCR Replacement Generates Superior Anticancer Transgenic T Cells. Blood 2018, 131, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-Engineered T Cells in Patients with Refractory Cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and Feasibility of CRISPR-Edited T Cells in Patients with Refractory Non-Small-Cell Lung Cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2455–2468. [Google Scholar] [CrossRef] [Green Version]
- Ledford, H. CRISPR Treatment Inserted Directly into the Body for First Time. Nature 2020, 579, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Bolinger, J.; Yu, Y.; Glass, Z.; Shi, N.; Yang, L.; Wang, M.; Xu, Q. Intracellular Delivery and Biodistribution Study of CRISPR/Cas9 Ribonucleoprotein Loaded Bioreducible Lipidoid Nanoparticles. Biomater. Sci. 2019, 7, 596–606. [Google Scholar] [CrossRef]
- Patel, P.; Ibrahim, N.M.; Cheng, K. The Importance of Apparent PKa in the Development of Nanoparticles Encapsulating SiRNA and MRNA. Trends Pharmacol. Sci. 2021, 42, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Onuma, H.; Sato, R.; Sato, Y.; Hashiba, A.; Maeki, M.; Tokeshi, M.; Kayesh, M.E.H.; Kohara, M.; Tsukiyama-Kohara, K.; et al. Lipid Nanoparticles Loaded with Ribonucleoprotein-Oligonucleotide Complexes Synthesized Using a Microfluidic Device Exhibit Robust Genome Editing and Hepatitis B Virus Inhibition. J. Control. Release Off. J. Control. Release Soc. 2021, 330, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Kouranova, E.; Forbes, K.; Zhao, G.; Warren, J.; Bartels, A.; Wu, Y.; Cui, X. CRISPRs for Optimal Targeting: Delivery of CRISPR Components as DNA, RNA, and Protein into Cultured Cells and Single-Cell Embryos. Hum. Gene Ther. 2016, 27, 464–475. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Tu, L.-C.; Naseri, A.; Huisman, M.; Zhang, S.; Grunwald, D.; Pederson, T. CRISPR-Cas9 Nuclear Dynamics and Target Recognition in Living Cells. J. Cell Biol. 2016, 214, 529–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, Z.; Yang, W.; Yan, S.; Yin, A.; Gao, J.; Liu, X.; Zheng, Y.; Zheng, J.; Li, Z.; Yang, S.; et al. Promoting Cas9 Degradation Reduces Mosaic Mutations in Non-Human Primate Embryos. Sci. Rep. 2017, 7, 42081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced Homology-Directed Human Genome Engineering by Controlled Timing of CRISPR/Cas9 Delivery. eLife 2014, 3, e04766. [Google Scholar] [CrossRef]
- Klein, M.; Eslami-Mossallam, B.; Arroyo, D.G.; Depken, M. Hybridization Kinetics Explains CRISPR-Cas Off-Targeting Rules. Cell Rep. 2018, 22, 1413–1423. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.; Cheng, Q.; Min, Y.L.; Olson, E.N.; Siegwart, D.J. Systemic Nanoparticle Delivery of CRISPR-Cas9 Ribonucleoproteins for Effective Tissue Specific Genome Editing. Nat. Commun. 2020, 11, 3232. [Google Scholar] [CrossRef]
- Manning, M.C.; Chou, D.K.; Murphy, B.M.; Payne, R.W.; Katayama, D.S. Stability of Protein Pharmaceuticals: An Update. Pharm. Res. 2010, 27, 544–575. [Google Scholar] [CrossRef]
- Love, K.T.; Mahon, K.P.; Levins, C.G.; Whitehead, K.A.; Querbes, W.; Dorkin, J.R.; Qin, J.; Cantley, W.; Qin, L.L.; Racie, T.; et al. Lipid-like Materials for Low-Dose, in Vivo Gene Silencing. Proc. Natl. Acad. Sci. USA 2010, 107, 1864–1869. [Google Scholar] [CrossRef] [Green Version]
- D’Astolfo, D.S.; Pagliero, R.J.; Pras, A.; Karthaus, W.R.; Clevers, H.; Prasad, V.; Lebbink, R.J.; Rehmann, H.; Geijsen, N. Efficient Intracellular Delivery of Native Proteins. Cell 2015, 161, 674–690. [Google Scholar] [CrossRef] [Green Version]
- Alonso Villela, S.M.; Kraïem, H.; Bouhaouala-Zahar, B.; Bideaux, C.; Aceves Lara, C.A.; Fillaudeau, L. A Protocol for Recombinant Protein Quantification by Densitometry. MicrobiologyOpen 2020, 9, 1175–1182. [Google Scholar] [CrossRef] [Green Version]
- Burger, A.; Lindsay, H.; Felker, A.; Hess, C.; Anders, C.; Chiavacci, E.; Zaugg, J.; Weber, L.M.; Catena, R.; Jinek, M.; et al. Maximizing Mutagenesis with Solubilized CRISPR-Cas9 Ribonucleoprotein Complexes. Development 2016, 143, 2025–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jong, O.G.; Murphy, D.E.; Mäger, I.; Willms, E.; Garcia-Guerra, A.; Gitz-Francois, J.J.; Lefferts, J.; Gupta, D.; Steenbeek, S.C.; van Rheenen, J.; et al. A CRISPR-Cas9-Based Reporter System for Single-Cell Detection of Extracellular Vesicle-Mediated Functional Transfer of RNA. Nat. Commun. 2020, 11, 1113. [Google Scholar] [CrossRef]
- de Jong, O.G.; van Balkom, B.W.M.; Gremmels, H.; Verhaar, M.C. Exosomes from Hypoxic Endothelial Cells Have Increased Collagen Crosslinking Activity through Up-Regulation of Lysyl Oxidase-like 2. J. Cell. Mol. Med. 2016, 20, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Glaser, A.; McColl, B.; Vadolas, J. GFP to BFP Conversion: A Versatile Assay for the Quantification of CRISPR/Cas9-Mediated Genome Editing. Mol. Ther. Nucleic Acids 2016, 5, e334. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Crist, R.M.; Clogston, J.D. The Utility of Asymmetric Flow Field-Flow Fractionation for Preclinical Characterization of Nanomedicines. Anal. Bioanal. Chem. 2020, 412, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fens, M.H.A.M.; Capomaccio, R.B.; Mehn, D.; Scrivano, L.; Kok, R.J.; Oliveira, S.; Hennink, W.E.; van Nostrum, C.F. Correlation between in Vitro Stability and Pharmacokinetics of Poly(ε-Caprolactone)-Based Micelles Loaded with a Photosensitizer. J. Control. Release Off. J. Control. Release Soc. 2020, 328, 942–951. [Google Scholar] [CrossRef]
- Caputo, F.; Arnould, A.; Bacia, M.; Ling, W.L.; Rustique, E.; Texier, I.; Mello, A.P.; Couffin, A.-C. Measuring Particle Size Distribution by Asymmetric Flow Field Flow Fractionation: A Powerful Method for the Preclinical Characterization of Lipid-Based Nanoparticles. Mol. Pharm. 2019, 16, 756–767. [Google Scholar] [CrossRef] [Green Version]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro Story and the Clinical Translation of Nanomedicines Containing Nucleic Acid-Based Drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Guimaraes, P.P.G.; Zhang, R.; Spektor, R.; Tan, M.; Chung, A.; Billingsley, M.M.; El-Mayta, R.; Riley, R.S.; Wang, L.; Wilson, J.M.; et al. Ionizable Lipid Nanoparticles Encapsulating Barcoded MRNA for Accelerated in Vivo Delivery Screening. J. Control. Release 2019, 316, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, F.; Yanez Arteta, M.; Lerche, M.; Porcar, L.; Lang, C.; Bragg, R.A.; Elmore, C.S.; Krishnamurthy, V.R.; Russell, R.A.; Darwish, T.; et al. Apolipoprotein E Binding Drives Structural and Compositional Rearrangement of MRNA-Containing Lipid Nanoparticles. ACS Nano 2021, 15, 6709–6722. [Google Scholar] [CrossRef]
- Nguyen, D.N.; Roth, T.L.; Li, P.J.; Chen, P.A.; Apathy, R.; Mamedov, M.R.; Vo, L.T.; Tobin, V.R.; Goodman, D.; Shifrut, E.; et al. Polymer-Stabilized Cas9 Nanoparticles and Modified Repair Templates Increase Genome Editing Efficiency. Nat. Biotechnol. 2020, 38, 44–49. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walther, J.; Wilbie, D.; Tissingh, V.S.J.; Öktem, M.; van der Veen, H.; Lou, B.; Mastrobattista, E. Impact of Formulation Conditions on Lipid Nanoparticle Characteristics and Functional Delivery of CRISPR RNP for Gene Knock-Out and Correction. Pharmaceutics 2022, 14, 213. https://doi.org/10.3390/pharmaceutics14010213
Walther J, Wilbie D, Tissingh VSJ, Öktem M, van der Veen H, Lou B, Mastrobattista E. Impact of Formulation Conditions on Lipid Nanoparticle Characteristics and Functional Delivery of CRISPR RNP for Gene Knock-Out and Correction. Pharmaceutics. 2022; 14(1):213. https://doi.org/10.3390/pharmaceutics14010213
Chicago/Turabian StyleWalther, Johanna, Danny Wilbie, Vincent S. J. Tissingh, Mert Öktem, Heleen van der Veen, Bo Lou, and Enrico Mastrobattista. 2022. "Impact of Formulation Conditions on Lipid Nanoparticle Characteristics and Functional Delivery of CRISPR RNP for Gene Knock-Out and Correction" Pharmaceutics 14, no. 1: 213. https://doi.org/10.3390/pharmaceutics14010213