
77Se-Enriched Selenoglycoside Enables Significant Enhancement in NMR Spectroscopic Monitoring of Glycan–Protein Interactions

 and
and
Abstract
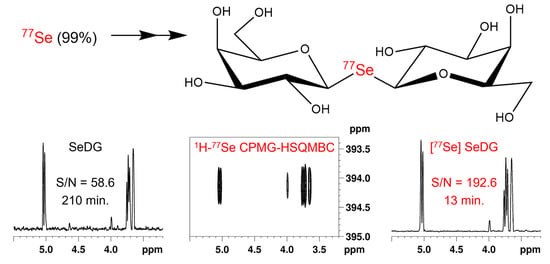
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Synthetic Procedures
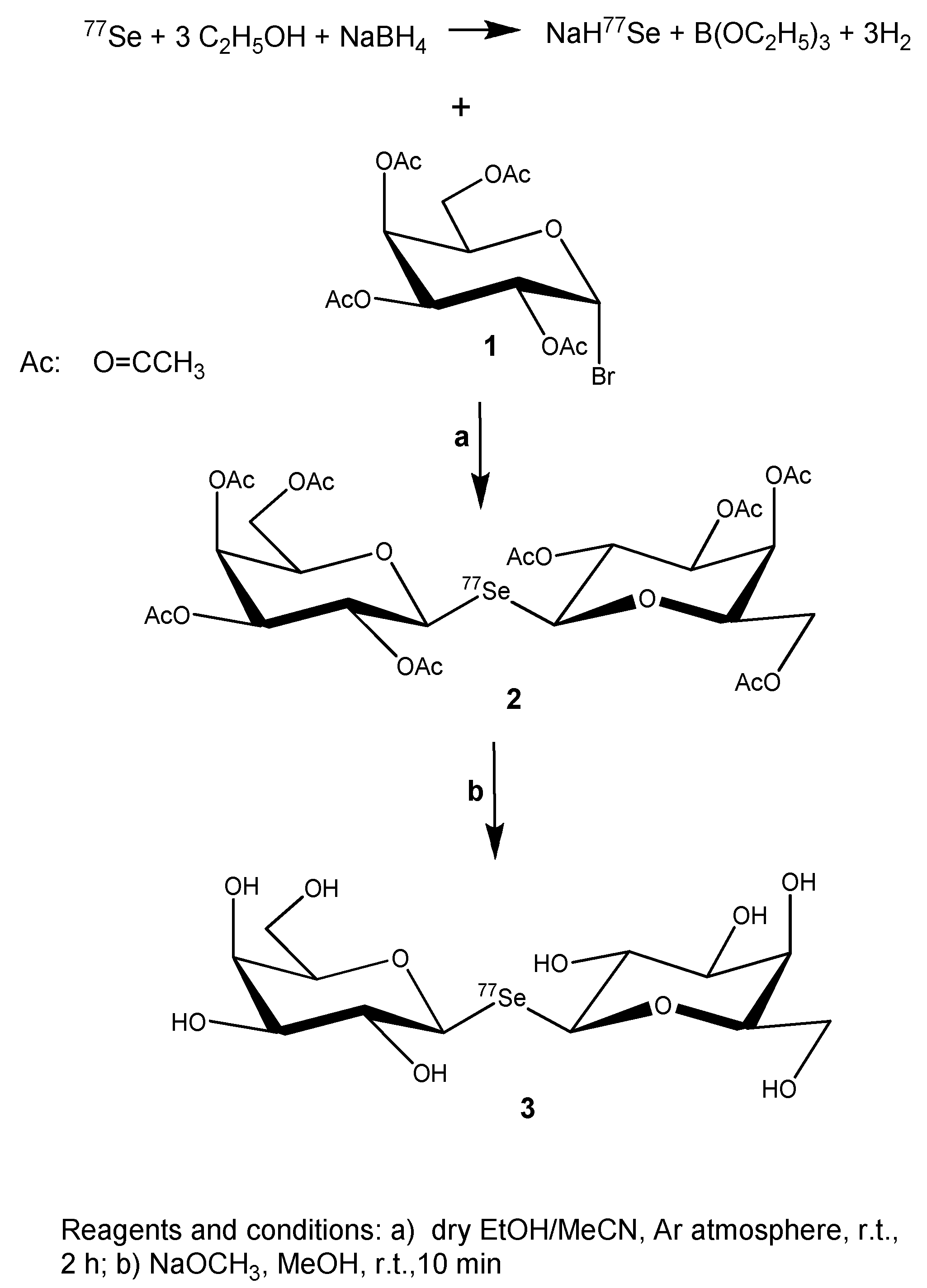
2.1.1. 77Se-Enriched Di(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)selenide (2)
2.1.2. 77Se-Enriched Di(β-D-galactopyranosyl)selenide (3)
2.2. Sample Preparation for NMR Measurements
2.3. NMR Measurements
3. Results and Discussion
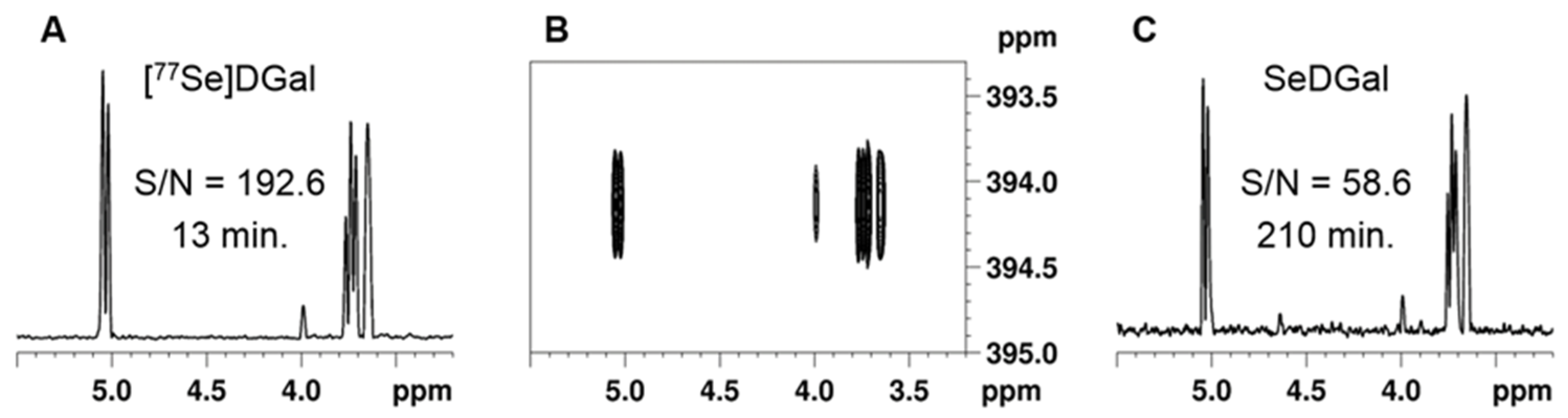
3.1. Synthesis of 77Se-Enriched Di(β-D-galactopyranosyl)selenide, [77Se]DGal
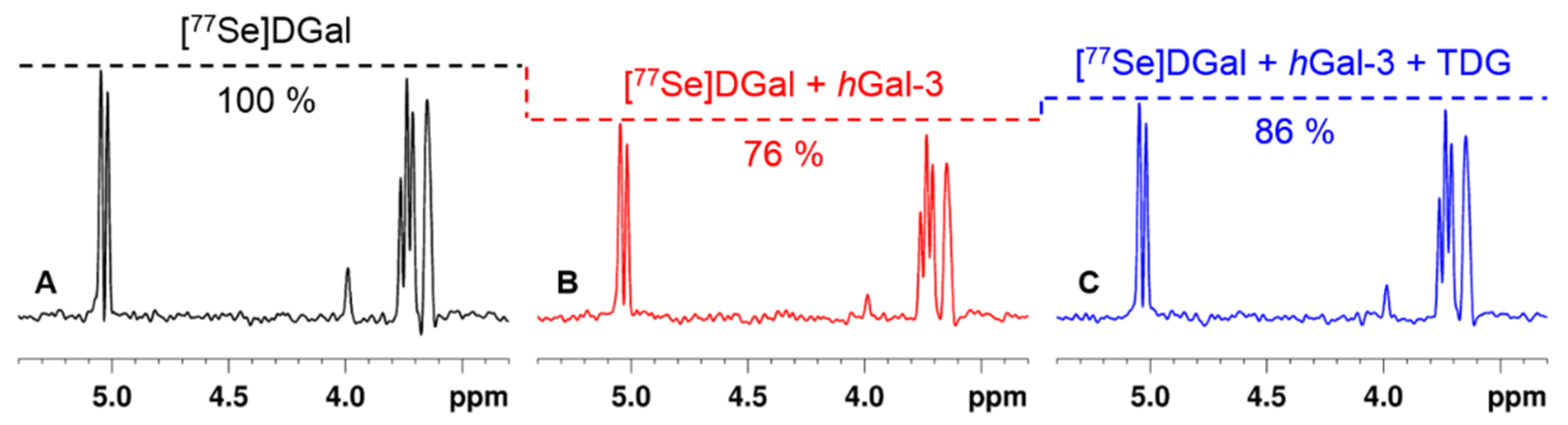
3.2. NMR Experiments
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gabius, H.-J. (Ed.) The Sugar Code. Fundamentals of Glycosciences; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; p. 569. [Google Scholar]
- Delaine, T.; Collins, P.; MacKinnon, A.; Sharma, G.; Stegmayr, J.; Rajput, V.K.; Mandal, S.; Cumpstey, I.; Larumbe, A.; Salameh, B.A.; et al. Galectin-3-Binding glycomimetics that strongly reduce bleomycin-induced lung fibrosis and modulate intracellular glycan recognition. ChemBioChem 2016, 17, 1759–1770. [Google Scholar] [CrossRef]
- Liu, F.T.; Rabinovich, G.A. Galectins: Regulators of acute and chronic inflammation. Ann. N. Y. Acad. Sci. 2019, 1183, 158–182. [Google Scholar] [CrossRef] [PubMed]
- Bartolazzi, A. Galectins in cancer and translational medicine. Int. J. Mol. Sci. 2018, 19, 2934. [Google Scholar] [CrossRef] [Green Version]
- Chou, F.-C.; Chen, H.-Y.; Kuo, C.-C.; Sytwu, H.-K. Role of galectins in tumors and in clinical immunotherapy. Int. J. Mol. Sci. 2018, 19, 430. [Google Scholar] [CrossRef] [Green Version]
- Mirandola, L.; Yu, Y.F.; Cannon, M.J.; Jenkins, M.R.; Rahman, R.L.; Nguyen, D.D.; Grizzi, F.; Cobos, E.; Figueroa, J.A.; Chiriva-Internati, M. Galectin-3 inhibition suppresses drug resistance, motility, invasion and angiogenic potential in ovarian cancer. Gynecol. Oncol. 2014, 135, 573–579. [Google Scholar] [CrossRef]
- Girotti, M.R.; Salatino, M.; Dalotto-Moreno, T.; Rabinovich, G.A. Sweetening the hallmarks of cancer: Galectins as multifunctional mediators of tumor progression. J. Exp. Med. 2020, 217, e20182041. [Google Scholar] [CrossRef]
- Compagno, D.; Tiraboschi, C.; Garcia, J.D.; Rondón, Y.; Corapi, E.; Velazquez, C.; Laderach, D.J. Galectins as checkpoints of the immune system in cancers, their clinical relevance, and implication in clinical trials. Biomolecules 2020, 10, 750. [Google Scholar] [CrossRef]
- Bertuzzi, S.; Quintana, J.I.; Arda, A.; Gimeno, A.; Jimenez-Barbero, J. Targeting galectins with glycomimetics. Front. Chem. 2020, 8, 593. [Google Scholar] [CrossRef]
- Denavit, V.; Laine, D.; Tremblay, T.; St-Gelais, J.; Giguere, D. Synthetic inhibitors of galectins: Structures and syntheses. Trends Glycosci. Glycotechnol. 2018, 30, SE21–SE40. [Google Scholar] [CrossRef] [Green Version]
- Stegmayr, J.; Zetterberg, F.; Carlsson, M.C.; Huang, X.L.; Sharma, G.; Kahl-Knutson, B.; Schambye, H.; Nilsson, U.J.; Oredsson, S.; Leffler, H. Extracellular and intracellular small-molecule galectin-3 inhibitors. Sci. Rep. 2019, 9, 2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arda, A.; Jimenez-Barbero, J. The recognition of glycans by protein receptors. Insights from NMR spectroscopy. Chem. Commun. 2018, 54, 4761–4769. [Google Scholar] [CrossRef] [PubMed]
- Valverde, P.; Quintana, J.I.; Santos, J.I.; Ardá, A.; Jiménez-Barbero, J. Novel NMR avenues to explore the conformation and interactions of glycans. ACS Omega 2019, 4, 13618–13630. [Google Scholar] [CrossRef] [Green Version]
- Cala, O.; Guilliere, F.; Krimm, I. NMR-based analysis of protein-ligand interactions. Anal. Bioanal. Chem. 2014, 406, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, J.L.; Taylor, S.L.; Howard, M.J. Recent developments and applications of saturation transfer difference nuclear magnetic resonance (STD NMR) spectroscopy. Mol. Biosyst. 2013, 9, 571–577. [Google Scholar] [CrossRef]
- Fehér, K.; Groves, P.; Batta, G.; Jimenez-Barbero, J.; Muhle-Goll, C.; Kövér, K.E. Competition saturation transfer difference experiments improved with isotope editing and filtering schemes in NMR-based screening. J. Am. Chem. Soc. 2008, 130, 17148–17153. [Google Scholar] [CrossRef] [PubMed]
- Fittolani, G.; Shanina, E.; Guberman, M.; Seeberger, P.H.; Rademacher, C.; Delbianco, M. Automated glycan assembly of F-19-labeled glycan probes enables high-throughput NMR studies of protein-glycan interactions. Angew. Chem. Int. Ed. 2021, 60, 13302–13309. [Google Scholar] [CrossRef]
- Martinez, J.D.; Manzano, A.I.; Calvino, E.; de Diego, A.; de Francisco, B.R.; Romano, C.; Oscarson, S.; Millet, O.; Gabius, H.J.; Jimenez-Barbero, J.; et al. Fluorinated carbohydrates as lectin ligands: Simultaneous screening of a monosaccharide library and chemical mapping by F-19 NMR spectroscopy. J. Org. Chem. 2020, 85, 16072–16081. [Google Scholar] [CrossRef] [PubMed]
- Troelsen, N.S.; Shanina, E.; Gonzalez-Romero, D.; Dankova, D.; Jensen, I.S.A.; Sniady, K.J.; Nami, F.; Zhang, H.X.; Rademacher, C.; Cuenda, A.; et al. The 3F library: Fluorinated Fsp(3)-rich fragments for expeditious F-19 NMR based screening. Angew. Chem. Int. Ed. 2020, 59, 2204–2210. [Google Scholar] [CrossRef]
- Linclau, B.; Arda, A.; Reichardt, N.C.; Sollogoub, M.; Unione, L.; Vincent, S.P.; Jimenez-Barbero, J. Fluorinated carbohydrates as chemical probes for molecular recognition studies. Current status and perspectives. Chem. Soc. Rev. 2020, 49, 3863–3888. [Google Scholar] [CrossRef]
- Teichberg, V.I.; Silman, I.; Beitsch, D.D.; Resheff, G. A beta-D-galactoside binding protein from electric organ tissue of Electrophorus electricus. Proc. Natl. Acad. Sci. USA 1975, 72, 1383–1387. [Google Scholar] [CrossRef] [Green Version]
- Sparrow, C.P.; Leffler, H.; Barondes, S.H. Multiple soluble beta-galactoside-binding lectins from human lung. J. Biol. Chem. 1987, 262, 7383–7390. [Google Scholar] [CrossRef]
- Ahmad, N.; Gabius, H.J.; Kaltner, H.; Andre, S.; Kuwabara, I.; Liu, F.T.; Oscarson, S.; Norberg, T.; Brewer, C.F. Thermodynamic binding studies of cell surface carbohydrate epitopes to galectins-1,-3, and-7: Evidence for differential binding specificities. Can. J. Chem. 2002, 80, 1096–1104. [Google Scholar] [CrossRef]
- André, S.; Kövér, K.E.; Gabius, H.J.; Szilágyi, L. Thio- and selenoglycosides as ligands for biomedically relevant lectins: Valency-activity correlations for benzene-based dithiogalactoside clusters and first assessment for (di)selenodigalactosides. Bioorg. Med. Chem. Lett. 2015, 25, 931–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raics, M.; Timári, I.; Diercks, T.; Szilágyi, L.; Gabius, H.J.; Kövér, K.E. Selenoglycosides as Lectin Ligands: Se-77-Edited CPMG-HSQMBC NMR Spectroscopy To Monitor Biomedically Relevant Interactions. ChemBioChem 2019, 20, 1688–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raics, M.; Timári, I.; Szilágyi, L.; Gabius, H.-J.; Kövér, K.E. Introducing 77Se NMR Spectroscopy to Analyzing Galectin–Ligand Interaction. In Galectins: Methods and Protocols, Methods in Molecular Biology; Stowell, S.R., Arthur, C.M., Cummings, R.D., Eds.; Springer Science+Business Media, LLC, Part of Springer Nature: Berlin Germany, 2022; Volume 2442, p. 22. [Google Scholar]
- Duddeck, H. 77Se NMR Spectroscopy and Its Applications in Chemistry. In Annual Reports on NMR Spectroscopy; Webb, G.A., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2004; Volume 52, pp. 105–166. [Google Scholar]
- Diercks, T.; Medrano, F.J.; FitzGerald, F.G.; Beckwith, D.; Pedersen, M.J.; Reihill, M.; Ludwig, A.-K.; Romero, A.; Oscarson, S.; Cudic, M.; et al. Galectin–Glycan Interactions: Guidelines for Monitoring by 77Se NMR Spectroscopy, and Solvent (H2O/D2O) Impact on Binding. Chem. Eur. J. 2021, 27, 316–325. [Google Scholar] [CrossRef]
- Silva, M.S.; Alves, D.; Hartwig, D.; Jacob, R.G.; Perin, G.; Lenardao, E.J. Selenium-NMR Spectroscopy in Organic Synthesis: From Structural Characterization Toward New Investigations. Asian J. Org. Chem. 2021, 10, 91–128. [Google Scholar] [CrossRef]
- Hamark, C.; Landstrom, J.; Widmalm, G. SEAL by NMR: Glyco-Based Selenium-Labeled Affinity Ligands Detected by NMR Spectroscopy. Chem. Eur. J. 2014, 20, 13905–13908. [Google Scholar] [CrossRef] [Green Version]
- Perez-Victoria, I.; Boutureira, O.; Claridge, T.D.W.; Davis, B.G. Glycosyldiselenides as lectin ligands detectable by NMR in biofluids. Chem. Commun. 2015, 51, 12208–12211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehér, K.; Timári, I.; Rákosi, K.; Szolomajer, J.; Illyés, T.Z.; Bartók, A.; Varga, Z.; Panyi, G.; Tóth, G.K.; Kövér, K.E. Probing pattern and dynamics of disulfide bridges using synthesis and NMR of an ion channel blocker peptide toxin with multiple diselenide bonds. Chem. Sci. 2016, 7, 2666–2673. [Google Scholar] [CrossRef] [Green Version]
- Klayman, D.L.; Griffin, T.S. Reaction of selenium with sodium borohydride in protic solvents. A Facile Method for the introduction of selenium into organic molecules. J. Am. Chem. Soc. 1973, 95, 197–199. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timári, I.; Balla, S.; Fehér, K.; Kövér, K.E.; Szilágyi, L. 77Se-Enriched Selenoglycoside Enables Significant Enhancement in NMR Spectroscopic Monitoring of Glycan–Protein Interactions. Pharmaceutics 2022, 14, 201. https://doi.org/10.3390/pharmaceutics14010201
Timári I, Balla S, Fehér K, Kövér KE, Szilágyi L. 77Se-Enriched Selenoglycoside Enables Significant Enhancement in NMR Spectroscopic Monitoring of Glycan–Protein Interactions. Pharmaceutics. 2022; 14(1):201. https://doi.org/10.3390/pharmaceutics14010201
Chicago/Turabian StyleTimári, István, Sára Balla, Krisztina Fehér, Katalin E. Kövér, and László Szilágyi. 2022. "77Se-Enriched Selenoglycoside Enables Significant Enhancement in NMR Spectroscopic Monitoring of Glycan–Protein Interactions" Pharmaceutics 14, no. 1: 201. https://doi.org/10.3390/pharmaceutics14010201