
Receptor Specificity Engineering of TNF Superfamily Ligands


Department of Chemical and Pharmaceutical Biology, Groningen Research Institute of Pharmacy, University of Groningen, Antonius Deusinglaan 1, 9713 AV Groningen, The Netherlands
*
Author to whom correspondence should be addressed.
Pharmaceutics 2022, 14(1), 181; https://doi.org/10.3390/pharmaceutics14010181
Submission received: 30 November 2021
/
Revised: 21 December 2021
/
Accepted: 6 January 2022
/
Published: 13 January 2022
(This article belongs to the Collection Drug Delivery in The Netherlands)
Abstract
:The tumor necrosis factor (TNF) ligand family has nine ligands that show promiscuity in binding multiple receptors. As different receptors transduce into diverse pathways, the study on the functional role of natural ligands is very complex. In this review, we discuss the TNF ligands engineering for receptor specificity and summarize the performance of the ligand variants in vivo and in vitro. Those variants have an increased binding affinity to specific receptors to enhance the cell signal conduction and have reduced side effects due to a lowered binding to untargeted receptors. Refining receptor specificity is a promising research strategy for improving the application of multi-receptor ligands. Further, the settled variants also provide experimental guidance for engineering receptor specificity on other proteins with multiple receptors.
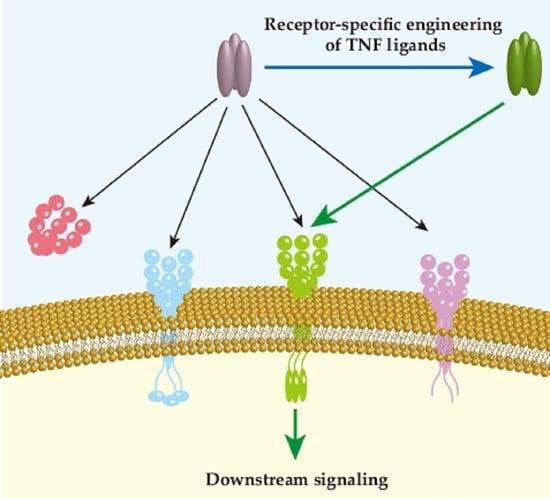
1. Introduction
The tumor necrosis factor (TNF) ligand family includes 19 ligands, which each contain a C-terminal TNF homology domain (THD). Each ligand binds to one or multiple TNF receptors (TNFR), containing extracellular cysteine-rich domains (CRD) for ligand binding [1]. The 9 TNF ligands that bind to multiple TNF receptors are TNF-α, LT, FasL, TRAIL, RANKL, APRIL, BAFF, LIGHT, and VEGI [2]. Communication pathways between TNFs and TNFRs are essential for numerous cellular processes, such as apoptosis, immune activation, inflammatory responses, and bone homeostasis, rendering the TNF family of great importance for targeted therapy [3]. Currently, clinical studies on TNF superfamily cytokines are ongoing, such as TNF-α, TNF-related apoptosis-inducing ligand (TRAIL), and receptor activator of NF-kB ligand (RANKL). These studies focus on a diversity of diseases, such as arthritis, malignant tumors, bone metastasis, and osteoporosis [4,5,6,7]. As some of the ligands can bind to different receptors, leading to undesired cross-talk or side-effects, receptor-specific engineering on ligands is crucial for unraveling the mechanism of TNF-related pathways and targeting the ligands to their pharmaceutically relevant receptor.
This review provides an overview of the engineering of promiscuous TNF ligands towards monoreceptor specificity. From the nine multiple-receptor TNF ligands, six of them are reported to have enhanced receptor specificity by engineering approaches (Figure 1).
2. Tumor Necrosis Factor and Lymphotoxin
Tumor necrosis factor (TNF or TNF-α) and lymphotoxin (LT) are the first two cytokines that have been characterized as TNF superfamily members. They have a homologous amino acid sequence and were both discovered based on their anti-tumor effects [8]. Moreover, both ligands bind to TNF receptor type 1 and type 2 (TNFR1 and TNFR2) and mediate similar cell signaling transduction. Thus, it is necessary to understand and compare the role of TNF-α and LT in pathways and diseases in order to discriminate the TNFR1- or TNFR2-specific treatments and their medical effects.
2.1. Tumor Necrosis Factor and Lymphotoxin
TNF-α was first identified in 1975 based on its ability to induce necrosis in transplanted tumors and the absence of toxicity to embryo in mice [9]. It is mainly expressed by activated macrophages, T lymphocytes, and natural killer (NK) cells [10]. It is synthesized as a cell surface transmembrane protein with 233 amino acids (26 kDa) forming a stable homotrimer, which contains a TNF family homology domain in the extracellular part, a transmembrane domain, a spacer stalk and an intracellular domain [11]. The transmembrane TNF-α (mTNF-α) is subsequently proteolytically cleaved on the spacer stalk by TNF-α-converting enzyme (TACE, or ADAM17) resulting in the release of a 17 kDa soluble TNF-α (sTNF-α) [12]. Both forms are biologically active as homotrimers and capable of binding with TNFR1 and TNFR2, whereas sTNF-α shows a lower affinity to TNFR2 compared with mTNF-α and only activates TNFR1 to mediate cellular signal transduction [13].
LT, a cytokine secreted by lymphocytes, was first identified in 1968 based on its in vitro anti-tumor activity [14,15]. Due to its similar effects of LT-α on tumor cells to TNF-α, it was renamed TNF-β in 1985 [16]. However, the discovery of the subunit LT-β revealed its additional functions other than TNF-α and led to the reversion of the name to LT. LT-α is the only TNF ligand without a transmembrane domain and is directly secreted into the cell exterior, while LT-β expresses as a transmembrane protein [17]. Three LT-α subunits form a homotrimer LTα3 that binds to TNFR1, TNFR2, and the herpes virus entry mediator (HVEM). LT-α and LT-β can also assemble the cell surface-bound heterotrimer LTα1β2, which is the predominant form that binds with the lymphotoxin β receptor (LTβR), and the LTα2β1, which binds to TNFR1 and TNFR2 with a minor physiological effect [17,18]. In this section, the LT-related TNFR1 and TNFR2 pathways are discussed, while the HVEM and LTβR pathways are discussed in the LIGHT section.
It has been demonstrated that TNF-α and LTα3 have similar affinities to TNFR1 and are able to transduce downstream signaling in a similar way [19]. Interestingly, sTNF-α cannot initiate TNFR2, but LTα3, as a soluble form, is able to transduce signaling through TNFR2 [20]. As they have similar amino acid sequences in their TNF homology domain (THD), their different function may be due to differences in their tertiary structure within subunits or the modular assembly mode in trimer formation.
TNF-α and LTα3 both bind to TNFR1 or TNFR2 to initiate similar signaling transduction; therefore, we will mainly focus on TNF-α-related research with a tacit understanding that LTα3 shows comparable performance. TNFR1 and TNFR2 mediate common and exclusive pathways to induce cell proliferation, inflammation, apoptosis, or necroptosis [21]. Thus, it is important to understand the different roles of TNFR1 and TNFR2 in cell signal transduction.
2.2. The TNFR1 and TNFR2 Induced Signal Transduction
TNFR1 protein expression is detected in almost all human nucleated cells, whereas TNFR2 is mainly expressed in the immune system and endothelial tissue [22], such as macrophages [23], T cells [24], monocytes [25], endothelial progenitor cells [26], and mesenchymal stem cells [27]. Both TNFR1 and TNFR2 are transmembrane glycoproteins with four N-terminal cysteine-rich domains (CRDs) in their extracellular domain and a transmembrane domain [28]. Both receptors are capable of activating the nuclear factor kappa B (NF-κB) and activator protein-1 (AP-1) pathway, but TNFR1 also has the capacity to induce cell necroptosis and apoptosis due to the presence of the death domain (DD) [29]. TNF-α/LTα3 binds to TNFR1 to recruit TNFR1-associated death domain protein (TRADD), the receptor-interacting protein 1 (RIP1), the TNF receptor-associated actor 2 (TRAF2) and the cellular inhibitor of apoptosis proteins 1/2 (cIAP1/2). For TNFR2, only TRAF2 and cIAP1/2 are recruited. The complex formation initiates the downstream cascades and ultimately translocates NF-κB and AP-1 to the nucleus to trigger their downstream gene expression, such as FLICE-like inhibitory protein (FLIP). Occasionally, when there is insufficient FLIP, the internalized TNF-α/TNFR1 complex recruits Fas-associated death domain protein (FADD) and procaspase-8 to form the death-inducing signaling complex (DISC) and generate apoptosis signaling [30,31]. Under other circumstances, the deficiency of active caspase-8 results in the binding of RIP3 to RIP1 to assemble necroptosome and induce necroptosis [32].
2.3. Engineering TNFR1-Specific Ligands
Due to the different binding affinities between TNF-α and its two receptors, as well as the diverse pathways they trigger, various attempts at specifically targeting the TNFR1 receptor have been made to achieve precise drug delivery, increase effectivity, and reduce TNF-α-related inflammation (Table 1). The research was initially focusing on the construction of mutated TNF-α to target either TNFR1 or TNFR2. In 1993, Ostade et al. constructed two TNF-α muteins, L29S and R32W, which are specific to TNFR1 [33]. Research showed that even though the artificial R32W had lower affinity to both receptors, it still showed equivalent cytotoxicity to colon and laryngeal cancer cell lines compared with wildtype (WT) TNF-α [33,34]. Then, to regain the full binding capacity of R32W to TNFR1, a double mutant R32W-S86T was created, with none binding to TNFR2 [35]. R32W-S86T was widely analyzed and proved to be lethal to tumor cells with low pro-inflammatory effects [34]. Nevertheless, these aforesaid mutants have similar shortages as observed for WT TNF-α, which cause coronary vasoconstriction, hypotension, anti-lipogenesis in vivo, and a short half-life [36,37]. Thus, more efforts were made to solve these problems. By introducing triple site mutations in position 5, 6, and 29 in TNF-α, Atarashi et al. created a mutein, F4614, which exhibited higher anti-tumor effects and reduced hypotensive risk more effectively than WT TNF-α [38,39]. Another TNFR1-specific mutein, M3S, was made to obtain a higher thermal stability, two-fold prolonged half-life and lower systematic cytotoxicity in vivo. However, the multiple site mutations also resulted in a lower binding affinity to TNFR1 and the M3S did not perform well in animal assays [40]. Recently, a mutTNF G4 with mutations from sites 84 to 89 was identified, showing higher affinity to TNFR1 than WT TNF-α [41]. The intravenous injection of mutTNF G4 induces the permeabilization of the blood–brain barrier (BBB), which successfully enhances the delivery of the therapeutic reagent into brain tumor.
Investigations of TNF-α antibodies, including both antagonist and agonist antibodies, have been carried out for decades. The antagonist antibody binds to the target protein and blocks its immune responses; conversely, the agonist antibody activates the checkpoint protein and initiates the signaling transduction [43]. FDA-approved antibodies are now commercially available, including infliximab, adalimumab, certolizumab pegol, and golimumab, which are all TNF-α antagonist antibodies [44]. However, the complete blockage of the TNF-α pathway leads to poor results with fatal side effects and low resistance to infections [45]. Therefore, apart from engineering TNF-α variants, the antagonistic antibody specifically targeting TNFR1 attracted more attention. To understand the receptor-specific functions in mice model, HS1097 and DMS5540 targeting and inhibiting TNFR1 were made and commercialized [46,47]. In mice model, the application of HS1097 reduces experimental autoimmune encephalomyelitis symptoms, and DMS5540 effectively suppresses collagen-induced arthritis (CIA) without additional effector T cell activation and inflammation reaction [48,49]. Further, a mouse TNFR1 antibody was humanized and named ATROSAB. It has a similar affinity to both rhesus and human TNFR1, which makes it a potential agent for the preclinical trials in the CIA model of rhesus monkeys [50]. ATROSAB binds to TNFR1 without signal activation and blocks the activity of both TNF-α and LTα3, leaving the TNFR2 pathway intact [51].
In addition to antibodies, small molecules that inhibit specific receptors are gaining popularity due to their low cost and convenient drug administration. Chen et al. utilized pharmacophore model filtering and molecular docking to obtain 10 virtual hits and evaluated their binding affinity to TNF-α and TNFR1 [52]. Three compounds out of ten suppress TNF-α induced cytotoxicity in the noncancerous cell line L929 in a dose-dependent manner. Nevertheless, all the selective compounds inhibit both TNF-α and TNFR1 and thus are lacking receptor specificity. Based on the crystal structure of TNF-α-TNFR1 complex, the TNF-α inhibitor ZINC09609430, the TNFR1 inhibitor ZINC02968981, and the TNF-α–TNFR1 complex inhibitor ZINC05462670 were selected [53]. However, the mentioned compounds only underwent simulation prediction; therefore, more biological experiments need to be done to confirm their efficiency. Another strategy was employed to retrieve seven promising compounds targeting TNFR1 by the high-throughput screening of the ChemBridge DIVERSet library. They proved that the selected noncompetitive inhibitors DS41 and DSA114 significantly block TNF-α/LTα3-induced NF-κB activation in a TNFR1-specific pattern through perturbing the conformation of TNFR1 without interfering in ligand-receptor assembly [54]. Compared with the development of TNFR1-specific mutants or antibodies, the small molecule inhibitors are still in the early progression stage.
2.4. TNFR2-Specific Applications
TNFR2 has different features to TNFR1 because of its absence of a DD in the intracellular part. The TNFR2-specific mutant protein was mainly used to investigate the receptor-specific signaling pathways and pathogenesis (Table 1), such as, the D143N-A145R with extremely low binding to TNFR1 [35]. The mutation sites were selected based on a library of site-directed mutants of human TNF-α, and it was shown that sites 143–145 are responsible for binding with TNFR1 but not with TNFR2. This mutein is frequently used in a comparison with R32W-S86T to explore the functions of different receptors [55,56,57]. Even though these recombinant proteins were made to target TNFR2, more investigations of TNF-α and its receptors have indicated that TNFR2 is incapable of being activated by binding with sTNF-α.
In contrast to TNFR1, TNFR2 is crucial for the generation and proper functioning of regulatory T cells (Tregs). It regulates immune suppression and promotes apoptosis of autoreactive T cells in multiple diseases [58,59]. Due to the key role of TNFR2 in the immune system, researchers have attempted to develop specific TNFR2 agonists by mimicking mTNF-α. Therefore, based on the achieved TNFR2-specific mutated TNF-α, it was fused with other protein domains to form spontaneous oligomer and to increase the avidity. The fusion protein STAR2 is constructed by fusing a mutated single-chain mouse TNF-α trimer (D221N-A223R) with one domain of chicken tenascin C (TNC) and three of the fused complexes were trimerized to form a nonameric molecule [60]. STAR2 induced the expansion of Tregs without pro-inflammatory side effects [60]. However, it also showed a mixed result in myocardial infarction mice, which improved the left ventricular function yet decreased the survival rate [47]. In addition, a human TNFR2-specific fusion protein (TNC-scTNFR2) was created with a single-chain human TNF-α mutant (D143N-A145R) and a human TNC instead of a chicken derivative, which yielded a similar 3D structure to STAR2. It established a neuroprotective effect through preventing oxidative stress-induced cell death from H2O2 and catecholaminergic in human dopaminergic neuronal cell line LUHMES [61]. Along with using TNC, the dimerization domain EHD2 derived from the heavy chain domain CH2 of IgE was also fused with human TNF-α (D143N-A145R) to form a dimer (EHD2-scTNFR2) under nonreducing conditions. It was shown that EHD2-scTNFR2 protects mice from acute neurodegeneration and memory impairment, and the murine ortholog EHD2-sc-mTNFR2 induces the expansion of Tregs with anti-inflammatory responses [58,59]. Unlike TNFR1-specific application, the research into TNFR2 related treatments is still in the early phase. With greater recognition of the crucial role of TNFR2 in neuroprotection and immune system counterpoise, more research is expected to be seen in the future.
3. TRAIL
TRAIL, also known as Apo2 ligand (Apo2L), is a homotrimeric transmembrane protein, which has 28% sequence homology with FASL and 23% homology with TNF-α [62]. The membrane-bound TRAIL can be cleaved by proteases into soluble TRAIL that is released into the intercellular space [63]. Moreover, TRAIL selectively induces cell apoptosis in cancer cells and not in normal cells, which causes less side effects as compared to radiotherapy and chemotherapy.
3.1. TRAIL Induced Signaling Conduction
There are five receptors known for TRAIL: death receptor 4 (DR4/TRAIL-R1), death receptor 5 (DR5/TRAIL-R2), decoy receptor 1 (DcR1/TRAIL-R3), decoy receptor 2 (DcR2/TRAIL-R4), and the soluble receptor fragment osteoprotegerin (OPG). When TRAIL binds to DR4 or DR5, it recruits FADD to the DR4/DR5 death domain, then DISC is formed to activate caspase 8 through cleaving procaspase 8, to initiate the caspase cascade for cell apoptosis. It can also trigger the intrinsic apoptosis pathway via mitochondrial factors [64]. However, some cells are resistant to TRAIL, which can be attributed to the following: (i) cellular anti-apoptosis proteins, (ii) instability and ubiquitination of caspase protein, (iii) methylation of DR4 and DR5 genes, and (iv) overexpression of decoy receptors DcR1, DcR2, and OPG. Although DcR1 and DcR2 show close homology to DR4 and DR5, they cannot induce apoptosis, because both of them lack an intact or functional death domain. OPG acts as a soluble receptor that is able to sequester extracellularly available TRAIL from DR4 and DR5, thereby also antagonizing the induction of apoptosis.
To evaluate the utility of TRAIL as a cancer therapeutic, human TRAIL (Dulanermin/AMG 951/RG3639) was created (Table 2), which induces cell death in glioma, melanoma, Kaposi’s sarcoma, and breast cancer [65,66,67]. It also suppressed tumor growth, which resulted in prolonged survival of CB.17 (SCID) mice bearing breast cancer or colon carcinoma on systemic administration [68,69]. More importantly, it did not cause side effects like neural tissue toxicity in either mice or cynomolgus monkeys and chimpanzees, which could be due to the inability of dulanermin to cross the blood–brain barrier [69,70]. Even though showed promise in preclinical trials, the antitumor effects were not observed in patients. It was speculated that the short distribution half-life and elimination half-life [68,71], the low binding affinity to DR5, and the interference of the decoy receptor are the reasons for the poor performance of the recombinant protein TRAIL in the clinical phase [72]. Engineering longer half-life TRAIL variants that can specifically bind to DR4/5 with lower binding to decoy receptors would solve the problem.
3.2. TRAIL Variants with Enhanced Binding to Both DR4 and DR5
TRAIL G131R showed enhanced binding affinity to DR4 (2.9-fold KD) and DR5 (2.3-fold KD) compared to TRAIL WT. Although it also binds to decoy receptors, it still increases the apoptotic activity compared to TRAIL WT in both DR4- and DR5-responsive tumor cells [73]. Another strategy that helped to increase binding to DR4 and DR5 is the membrane-penetrating peptide-alike (TMPPA) technique. Amino acids 114–121 (VRERGPQR) were replaced by RRRRRRRR (named as TRAIL-Mu3), allowing faster penetration of pancreatic cancer cells (AsPC-1, BxPC-1, PANC-1) membrane and thereby enhancing apoptosis signaling through stimulating the death receptors distributed on the interior of the cells [74].
3.3. TRAIL Variants with Specific Binding to DR4 or DR5
Phage display technology was used to obtain the DR4 or DR5-selective rTRAIL variants Apo2L.DR4–8 and Apo2L.DR5–8. This was the first time that phage display technology was applied successfully for TNF family (Figure 2). In this study, they described Gln-205 and Tyr-216 as important residues for the binding of TRAIL and the five receptors. Compared with TRAIL WT, Apo2L.DR4–8 showed a similar affinity to DR4 and a lower affinity to DR5, while Apo2L.DR5–8 showed the opposite behavior. However, Apo2L.DR5–8 performed better induction of apoptosis in colon carcinoma cell line (Colo205, Colo320) and breast cancer cell line (MDA-MB-231) than TRAIL WT, but Apo2L.DR4–8 did not. This led to the conclusion that DR5 contributes more to cell death signaling than DR4 [75]. Interestingly, it was observed that Apo2L.DR5–8 has lower binding affinity to decoy receptors than Apo2L.DR4–8, which may also be the reason that Apo2L.DR5–8 performs better. Nevertheless, in the same year, another group synthesized TRAIL.R1-6 and TRAIL.R2-6, which are DR4- and DR5-selective rhTRAIL variants. Unlike the previous two variants, the DR4-selective variant TRAIL.R1-6 showed significant apoptosis induction in chronic lymphocytic leukemia and mantle cell lymphoma cells, but not in TRAIL.R2-6 [76]. However, they did not show binding affinity to decoy receptors. These two studies made clear that TRAIL-mediated apoptosis shows preference for DR4 or DR5 signaling depending on the cancer types.
The second approach is to construct variants that have specific binding to DR4 or DR5 and lower binding to decoy receptors. Our group used the automatic design algorithm FOLD-X approach to successfully obtain DR5-specific TRAIL D269H/E195R (DHER), which has KD = 2.9 nM with DR4 binding, while the TRAIL WT has an 0.17 nM KD towards DR4. DHER maintains high binding to DR5 (0.012 nM) compared to TRAIL WT (0.030 nM). In addition, this mutant shows lowered binding to DcR1, DcR2, and OPG. This variant is also very potent in inducing cell death with a lower median effective dose (ED50) in A2789 cells and in Colo205 cells than TRAIL WT [79,81,83]. Further, the combination treatment of subtoxic concentrations of the endoplasmic reticulum (ER) stress-inducing agent 2,5-dimethyl-celecoxib (DMC) and DHER increased TRAIL-induced caspase-8 activation in TRAIL sensitive and insensitive glioblastoma multiforme cell lines (A172 and U87) [84]. Cancer cells treated with artemisinin have higher DR5 expression, which allows better synergy effects of DHER in inducing cell apoptosis [85,86]. DHER also showed promising pro-apoptotic activity in therapy-induced senescent cancer cells or activated hepatic stellate cells [87,88]. For in vivo study, intraperitoneal administration of DHER strongly enhanced DR5 surface expression and caspase-3 activation and delayed A2780 tumor progression with an average reduction of 68.3% [79]. Later, two more DR5 specific variants, DR5-A and DR5-B, were established with the mixed mutation sites from Apo2L.DR5-8 and D269H. They were demonstrated to be highly selective to DR5 and to not bind to DR4 and DcR1 receptors, also low binding to DcR2 and OPG. Cellular assays showed promising antitumor efficacy independent of the decoy receptor expression level [82].
These findings inspired other researchers to combine the preliminary computational screening of proteins to identify positions necessary for TRAIL receptor interactions, which lead to the engineering of DR4 specific TRAIL variant by our group: D218H and D218Y. These two variants showed a 3.5-fold increase in apparent Kd to DR5 than TRAIL WT, while maintaining a relatively consistent Kd value to DR4. As for the decoy receptors, D218H and D218Y had a lower binding affinity to DcR2 and OPG, while having a modestly decreased affinity to DcR1. However, both variants caused less cell death in DR4-sensitive cells EM-2 and ML-1 compared with TRAIL WT [77]. Another single mutation rhTRAILDR4 reversed the TRAIL resistance caused by abundant X-linked inhibitor of apoptosis protein, and accelerated the apoptosis of pancreatic cancer cells [78]. Later, based on the engineering strategy of rhTRAILDR4, our group designed a six-site mutated DR4 specific variant 4C7, which has a higher affinity for DR4 and a lower affinity for DR5. Notably, 4C7 can promote the faster and stronger activation of caspase to induce apoptosis not only in TRAIL-sensitive human colon adenocarcinoma (Colo205, SW948, DLD-1, HCT-15 and CL-34), Burkitt’s lymphoma (BJAB) and the human ovarian carcinoma OVCAR-3, but also the TRAIL-resistant cancer cell line: pancreatic carcinoma (PANC-1) and breast cancer MCF-7 [79]. Further, our group found that HDAC inhibitor can enhance DR4 expression in the DLD-1 cell line, indicating that the combination treatment with 4C7 induces more cell apoptosis [89]. This creates potential for TRAIL as a more general anticancer drug. Later, rhTRAIL-C3 was created with triple mutations of G131R, N199R, and K201H. Of these mutations, G131R was described as having enhanced binding affinity to both death receptors, and N199Rand K201H were described as leading to lower affinity to DR5. RhTRAIL-C3 was a strong activator of pro-caspase 8 and enhanced the loss of mitochondrial membrane potential, resulting in DR4-mediated apoptosis in TRAIL-insensitive acute myelogenous leukemia and primary blast cell lines at a lower dose (1/6) compared to rhTRAIL WT [80]. This indicates that even though AML cells are completely resistant to rhTRAIL WT, they are sensitive to DR4-specific TRAIL variants, which is similar to the CLL cells described above [76].
Specific binding variants can be used to distinguish which death receptor the primary cells isolated from tumor tissues are more sensitive to. Subsequently, this can be used in the clinic to establish a therapy with a specific variant to target the tumor of the individual patient, which is a step towards personalized medicine.
4. RANKL
4.1. RANKL Induced Signaling Conduction
RANKL is a soluble protein secreted by bone-producing osteoblasts and can bind to its receptor RANK, which is expressed on bone resorbing osteoclast precursor cells. RANKL can also bind to the soluble OPG and same as in TRAIL, OPG will compete for the binding between RANKL and RANK [90]. The RANKL–RANK–OPG system was first found to play a major role in bone remodeling systems to control the bone producing and resorbing process, where the binding of RANKL recruits TNF receptor-associated factors 1, -2, -3, -5 and -6 to intracellular domain of RANK, leading to the expression of osteoclast-specific genes, like NF-kB, tartrate-resistant acid phosphate (TRAP), cathepsin K, and matrix metallopeptidase 9 (MMP-9). This facilitates the fusion of the osteoclast precursors into osteoclast, resulting in bone resorption [91,92,93]. If the RANKL and RANK pathways are overactivated, excessive osteoclasts leads to osteoporosis. In addition, MMP-9 is a significant enzyme to degrade extracellular matrix (ECM), which is the main composition of fibrotic tissue [94,95]. Interestingly, the binding of RANKL and RANK contributes to the degradation of ECM and prevents fibrosis [96].
4.2. RANKL Variants with Lower Binding to RANK
Protein engineering has been used to develop RANKL variants with lower binding affinity to RANK to prevent osteoporosis (Table 3). Before the crystal structure of RANKL–RANK was confirmed, mutation engineering of RANKL was conducted based on the crystal structures of TNF-β–TNFR and TRAIL–DR5 [97,98]. The solvent-accessible surface loops of RANKL are unique, and are distinguished as the AA″ loop (residues 170–193), the CD loop (residues 224–233), the DE loop (residues 245–251), and the EF loop (residues 261–269). The AA″ loop and DE loop were thought to be RANKL–RANK binding related. From these loops, I248, which is located in the DE loop of RANKL, was selected. I248D showed an 8-fold decrease of osteoclast-inducing activity to RANKL WT, but no protein–protein interaction data was shown [99]. After identifying the 3D crystal structure of the RANKL–RANK complex, more precise predictions can be made for the mutation development [90]. As the hydrophilic interactions dominate the binding between the DE loop of RANKL and RANK, our group substituted other amino acids on the I248 site according to the change of binding energy and hydrophilic properties. We built up a computer-aided structure-based RANKL mutant library, in which I248K and I248Y showed higher binding association rate constant (Ka) to RANK with the respective Ka of 15.7 and 17.2 M−1s−1, while RANKL WT was 4.3 M−1s−1. Meanwhile, the dissociation rate constant (Kd) is similar to that of RANKL WT, which results in the mutants having four times the affinity of the wild type. However, higher affinity did not increase RANKL-induced osteoclastogenesis in RAW 264.7 cells; rather, they reduced it [100]. The general workflow of the computer-aided protein engineering is depicted in Figure 3. Some studies demonstrated that binding of the ligand to receptors limits the conformational freedom of the receptor and thereby hinders the intracellular signaling [101,102]. This may also apply for the case of RANKL–RANK. A follow-up study of the AA″ loop after the resolving of RANKL–RANK crystal structure was also completed. rRANKL5 is a recombinant RANKL with deletion sites from aa 246 to 318 in the AA″ loop and CD loop. SPR data showed that the mutant rRANKL5 bind less to RANK compared to RANKL WT and inhibit the osteoclastogenesis progress in RAW 264.7 cells. However, they could not determine the KD [103].
4.3. RANKL Variant with Lower Binding to OPG and Targeted to RANK
To maximize the binding of RANKL to RANK, mutants were created that are not able to bind to OPG. As mentioned above, binding of RANKL and RANK prevent fibrosis by the degradation of ECM. High level of OPG is found in several fibrotic tissues in the liver, vascular system and lung, which correlate with profibrotic effects [105,106,107,108]. From our RANKL mutant library, RANKL Q236D showed reduced binding to OPG compared to RANKL WT. The binding of Q236D to OPG showed a 3-fold decrease of Ka, and a 10-fold increase of Kd, which led to a 30-fold lowered binding affinity KD to OPG. Binding simulation performed by BIOVIA Discovery Studio 4.5 showed that changing the RANKL residue GLN to ASP at 236 position eliminates the intramolecular hydrogen bonds between residue E93 and E95 of OPG (Figure 4). Meanwhile, the binding of Q236D to RANK is as strong as RANKL WT to RANK. This mutant Q236D is still able to activate the formation of osteoclasts. Further, the mutant can significantly increase the gene expression of MMP9, which is important for ECM degradation. Therefore, the research of RANKL Q236D provides us a new therapeutics strategy against fibrosis [104].
4.4. RANKL-Related Peptides and Antibodies That Inhibit RANK Funtion
In addition, there are some studies focusing on peptides that act as an inhibitor of RANK through mimicking RANKL. Among them, MHP1, a mimic of DE loop and part of EF loop of RANKL, does not activate the NF-kB signal, blocked RANKL-induced osteoclast differentiation in a dose-dependent manner [109]. Another peptide (WP9QY) blocked bone resorption by inhibiting recruitment and activation of osteoclasts to prevent osteoclastogenesis in mice [110]. Peptide L3-3, which is based on crystal structure of extracellular domains of mouse RANK–RANKL complex, strongly bound to RANKL ectodomain to block the RANKL-induced maturation of osteoclast precursors [111]. The monoclonal antibody denosumab also inhibits RANK–RANKL interactions and limits the formation and function of osteoclasts, leading to the suppression of bone resorption [91,112]. Denosumab is currently in multiple clinical trial phases for the treatment of bone-related diseases and cancers, such as osteoporosis, breast cancer, and prostate cancer [113,114,115,116].
5. FASL
FasL (CD95L/Apo1L) is a TNF-related homotrimeric transmembrane protein, expressed on diverse immune cells like B, T, and NK cells [117]. It conducts cell apoptosis signaling after binding to a membrane-bound Fas receptor. The receptor has three CRDs in the extracellular part, which are required to bind to FasL. The intercellular death domain is crucial for conduction of apoptosis signaling [118,119]. FasL also interacts with decoy receptor 3 (DcR3). DcR3 is a soluble receptor secreted in the extracellular space, which can compete with Fas for the binding to FasL and thereby is able to suppress the FasL–Fas mediated apoptosis [120].
Binding of membrane-bound FasL to Fas recruits the adapter molecule FADD to the death domain; this, in turn, can activate procaspase 8 in a similar way to TRAIL signaling [121]. Fas is widely expressed in not only memory and effector T cells upon contact with antigen, but also in nonlymphoid tissues, such as the skin, liver, ovary, lung, and heart, and various tumor cell lines [122,123,124]. As FasL is also mainly expressed on T cells, activation of cell death signaling eliminates the redundant T cells after an immune response, pathogen infection, or in oncogenically transformed cells [125]. However, the metalloprotease-cleaved soluble form of FasL (sFasL) cannot form the DISC complex and therefore is not able to induce apoptosis signaling [126].
sFasL fails to activate Fas-induced signaling, but oligomerization of ligand could reverse this functional defect. Several fusion proteins of sFasL were engineered to overcome this problem by self-oligomerization. One such self-oligomerizing FasL fusion protein is APO010 (MegaFasL), which is a hexametric fusion protein created by the fusion of two trimeric FasL extracellular domains to a dimer-forming stalk region of human adipocyte complement-related protein (which itself has no functional activity) [127,128]. APO010 showed anticancer activity in mice carrying various types of cancer cells, such as multiple myeloma, colon, and glioma cancers without obvious side effects [129,130,131]. APO010 is currently in Phase I clinical trial in patients with solid tumor; patient recruitment is complete, but new data are not yet available [132]. Another group fusion protein is engineered with tumor-associated antigen. The first fusion protein is CTLA-4–FasL, which was designed to fuse cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) and FasL amino acids 127–281. CTLA-4 binds to membrane-bound ligand B7 on antigen-presenting cells (APC), which helps FasL act like mFasL to trigger apoptosis. CTLA-4–FasL, as a representation of “trans signal converter proteins” strategy, efficiently inhibits Jurkat cell growth [133]. Another protein–protein fusing form is CD40·FasL, in which CD40 functions as apoptotic receptor [134]. Individual CD40·FasL or the combination with CTLA-4–FasL induce apoptosis in malignant cells [135,136]. Further, FasL fused with a single-chain variable fragments (scFv) worked well. scFv is an engineered antibody targeting specific antigens that are expressed in transformed cells. Once scFv binds to specific cells, the scFv-fused FasL performs as the membrane-bound FasL and induces cell signaling transduction [137]. sc40-FasL was made by fusing with sc40, which targets fibroblast activation protein (FAP). This compound achieved the local activation of FasL to suppress FAP-positive tumor cells. This is the first described cell-surface antigen-mediated local activation of Fas in vivo [138,139]. Another three scFv fused protein, scFvRit:sFasL, scFvCD7:sFasL and cc49scFv-FasLext, also showed promising anttumor activity [140,141,142].
However, all research on the recombinant FasL has focused on mimicking the function of membrane-bound FasL to interact with receptor Fas. DcR3 has been found to be overexpressed in inflammatory diseases and cancer. Excessive DcR3 can occupy FasL to limit the FasL-induced signaling conduction. Recombinant oligomerized FasL is expected to overcome DcR3 blockage. Therefore, based on the experience of other TNF-family ligands with multireceptors, such as STAR2 and TNC-scTNFR2, we expect more studies to enhance the selection specificity towards Fas or DcR3.
6. LIGHT
6.1. LIGHT and Its Receptors
LIGHT (TNFSF14) was first identified by its binding ability to HVEM:Fc in HEK293 cells in 1998; it shows a high sequence homology with LT-β and the other TNF family ligands [143]. It is also expressed as transmembrane protein, which only forms homotrimer from 29 kDa monomers. Like TNF-α, it exists in both soluble and membrane bond forms and mainly expresses in immune cells, such as activated T cells, natural killer (NK) cells, neutrophils, and dendritic cells (DC) [144]. As well as HVEM, LIGHT also binds to LTβR and decoy receptor 3 (DcR3). These three receptors all have four CRDs in their C-terminal domain that interact with LIGHT [145,146,147].
Both HVEM and LTβR are transmembrane protein belonging to the TNFR superfamily. HVEM mainly expresses in lymphocytes, NK cells, epithelium cells, and monocytes. In contrast, LTβR is not expressed on lymphocytes, but rather on follicular dendritic cells (FDC), myeloid cells, monocytes, and DC [148]. By interacting with LIGHT or LTα3, HVEM recruits TRAF2 and TRAF5 and further induces NF-κB and AP-1 pathways [149,150]. The interaction of ligands with HVEM enhances antibacterial activity of monocytes [151], blocks glycoprotein D of herpes simplex virus (HSV) entry [152], and activates NK cells and CD8+ cells [153]. As a receptor for LIGHT and LTα1β2, the formation of a ligand-LTβR complex results in the shuttling of TRAF2, TRAF3, and TRAF5 to the locality of LTβR and mediates noncanonical NF-κB signaling, AP-1 signaling, or cell apoptosis (even without DD in its intracellular domain) [154]. Hence, LTβR signaling induces tumor cell death in a reactive oxygen species (ROS) dependent way, limits the capacity of tumor cells recruiting immune suppressor cells, assists the extravasation of leukocyte to acting locus, regulates immune cell migration, and maintains splenic microenvironments [148,155]. DcR3 is different from the other two receptors due to its lack of transmembrane domain, making it an inherent inhibitor of LIGHT [156]. Due to its divergent functions from HVEM and LTβR as well as the existence of DcR3, the receptor-specific targeting strategy is of vital importance.
6.2. Specific Targeting HVEM and LTβR Strategy
Similar to previously employed strategies, to remove the neutralizing influence of DcR3, mutated ligands were developed and their biological activity was determined. Morishige et al. constructed a phage library to select a receptor-specific LIGHT mutant with a low affinity to DcR3. Five muteins were selected, of which Clone 1 with four site mutations shows the strongest binding affinity to both HVEM and LTβR with high equilibrium dissociation constant to DcR3. Its antitumor capacity is higher than WT LIGHT in the presence of DcR3 in HT29 [157]. They also demonstrated that the G–H loop motif (222 to 229) is crucial for the interaction between LIGHT to HVEM, LTβR, but not to DcR3, providing a clear understanding of the association between them.
With the application of a receptor-specific biologic agent, the diametrically opposed functions of HVEM and LTβR have been discovered. LIGHT G119E mutant has a high affinity to HVEM, but fails to bind LTβR, cannot induce HT29 cell death [158]. In addition, LTβR agonist 31G4D8 (antibody) and LIGHT-R228E mutein induces ROS production, resulting in cancer cell apoptosis [155]. Recently, it was shown that LTβR agonistic antibody BS1 initiates apolipoprotein B mRNA editing enzyme catalytic subunit 3B (APOBEC3B) expression through noncanonical NFκB signaling to suppress hepatitis B virus (HBV) replication [159,160,161].
7. Summary
In this review, we described the engineering of six promiscuous TNF family ligands towards single receptor specificity. Studies of the remaining three ligands, BAFF, APRIL, and VEGI, are still in the early stage due to difficulties in protein expression and purification.
Currently, antibodies are also used for targeting a specific receptor. Compared with antibodies, the engineered TNF ligand variants involve fewer residue changes, which leads to less complicated interaction kinetics, reduced autoimmune response, and higher treatment safety in vivo. Further, as variants can be produced by E. coli, they are easier to produce and cost effective. However, the use of ligand variants can be hindered by drawbacks, such as expression and purification difficulties, low stability, and short half-life. Our group is making several attempts to solve these problems, such as the long-term secretion of adenovirally expressed systems.
In summary, the specificity of receptor selection is helpful to (i) investigate specific signaling pathways, such as D143N-A145R was used to study the TNFR2-specific signaling pathway, (ii) reduce side effects, such as TNFR1-specific ligand R32W-S86T, which was found to be lethal to tumor cells with low proinflammatory effects, and (iii) increase efficacy of ligands, such asDR5-specific ligand DHER, which induced more cancer cell death than TRAIL WT by escaping from the neutralization of the decoy receptor OPG. Therefore, improving receptor specificity is a promising research strategy for multireceptor ligands.
Author Contributions
Writing—original draft preparation, F.S. and X.Z.; writing—review and editing, F.S., X.Z., R.S. and W.J.Q.; visualization, X.Z. supervision, W.J.Q.; funding acquisition, W.J.Q. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support was received in the form of the scholarship from China Scholarship Council (CSC) for Fengzhi Suo (201907040076) and Xinyu Zhou (201708610140).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; Siegel, R.M. Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 217–233. [Google Scholar] [CrossRef] [Green Version]
- Anti-TNFα Use During Elective Foot and Ankle Surgery in Patients With Rheumatoid Arthritis. Available online: https://clinicaltrials.gov/ct2/show/NCT02242474?term=TNF-α&draw=3&rank=11 (accessed on 5 January 2022).
- Targeted Stem Cells Expressing TRAIL as a Therapy for Lung Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03298763?term=TRAIL&draw=2&rank=7 (accessed on 5 January 2022).
- Safety, Tolerability and PK/PD of JMT103 in Patients With Bone Metastases From Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT03550508?term=RANKL&draw=2&rank=13 (accessed on 5 January 2022).
- Determination of the RANKL/Osteoprotegerin Ratio in Patients With Systemic Lupus Erythematosus. Role in Osteoporosis and Cardiovascular Calcification. Available online: https://clinicaltrials.gov/ct2/show/NCT02799173?term=RANKL&draw=2&rank=4 (accessed on 5 January 2022).
- Buhrmann, C.; Shayan, P.; Aggarwal, B.B.; Shakibaei, M. Evidence that TNF-β (lymphotoxin α) can activate the inflammatory environment in human chondrocytes. Arthritis Res. Ther. 2013, 15, R202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wakeham, J.; Harkness, R.; Xing, Z. Macrophages are a significant source of type 1 cytokines during mycobacterial infection. J. Clin. Investig. 1999, 103, 1023–1029. [Google Scholar] [CrossRef] [Green Version]
- Maskos, K.; Fernandez-Catalan, C.; Huber, R.; Bourenkov, G.P.; Bartunik, H.; Ellestad, G.A.; Reddy, P.; Wolfson, M.F.; Rauch, C.T.; Castner, B.J.; et al. Crystal structure of the catalytic domain of human tumor necrosis factor-α-converting enzyme. Proc. Natl. Acad. Sci. USA 1998, 95, 3408–3412. [Google Scholar] [CrossRef] [Green Version]
- Dittrich, G.M.; Heineke, J. TNF-α signaling: TACE inhibition to put out the burning heart. PLoS Biol. 2020, 18, e3001037. [Google Scholar] [CrossRef]
- Grell, M.; Wajant, H.; Zimmermann, G.; Scheurich, P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc. Natl. Acad. Sci. USA 1998, 95, 570–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruddle, N.H.; Waksman, B.H. Cytotoxicity mediated by soluble antigen and lymphocytes in delayed hypersensitivity. 3. Analysis of mechanism. J. Exp. Med. 1968, 128, 1267–1279. [Google Scholar] [CrossRef] [Green Version]
- Kolb, W.P.; Granger, G.A. Lymphocyte in vitro cytotoxicity: Characterization of human lymphotoxin. Proc. Natl. Acad. Sci. USA 1968, 61, 1250–1255. [Google Scholar] [CrossRef] [Green Version]
- Shalaby, M.R.; Aggarwal, B.B.; Rinderknecht, E.; Svedersky, L.P.; Finkle, B.S.; Palladino, M.A. Activation of human polymorphonuclear neutrophil functions by interferon-gamma and tumor necrosis factors. J. Immunol. 1985, 135, 2069–2073. [Google Scholar] [PubMed]
- Kucka, K.; Lang, I.; Zhang, T.; Siegmund, D.; Medler, J.; Wajant, H. Membrane lymphotoxin-α2β is a novel tumor necrosis factor (TNF) receptor 2 (TNFR2) agonist. Cell Death Dis. 2021, 12, 360. [Google Scholar] [CrossRef] [PubMed]
- Gubernatorova, E.O.; Polinova, A.I.; Petropavlovskiy, M.M.; Namakanova, O.A.; Medvedovskaya, A.D.; Zvartsev, R.V.; Telegin, G.B.; Drutskaya, M.S.; Nedospasov, S.A. Dual role of tnf and ltα in carcinogenesis as implicated by studies in mice. Cancers 2021, 13, 1775. [Google Scholar] [CrossRef]
- Roach, D.R.; Briscoe, H.; Saunders, B.; France, M.P.; Riminton, S.; Britton, W.J. Secreted lymphotoxin-alpha is essential for the control of an intracellular bacterial infection. J. Exp. Med. 2001, 193, 239–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruglov, A.A.; Grivennikov, S.I.; Kuprash, D.V.; Winsauer, C.; Prepens, S.; Seleznik, G.M.; Eberl, G.; Littman, D.R.; Heikenwalder, M.; Tumanov, A.V.; et al. Nonredundant function of soluble ltα3 produced by innate lymphoid cells in intestinal homeostasis. Science 2013, 342, 1243–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etemadi, N.; Holien, J.K.; Chau, D.; Dewson, G.; Murphy, J.M.; Alexander, W.S.; Parker, M.W.; Silke, J.; Nachbur, U. Lymphotoxin α induces apoptosis, necroptosis and inflammatory signals with the same potency as tumour necrosis factor. FEBS J. 2013, 280, 5283–5297. [Google Scholar] [CrossRef]
- Pegoretti, V.; Baron, W.; Laman, J.D.; Eisel, U.L.M. Selective Modulation of TNF–TNFRs Signaling: Insights for Multiple Sclerosis Treatment. Front. Immunol. 2018, 9, 925. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Raeiszadeh, M.; Verney, M.; Craddock, C.; Wajant, H.; Moss, P.; Chen, F. TNFR2 Is Expressed Preferentially By Late Differentiated CD8 T-Cells and Can be Triggered By TNFR2-Specific Ligand to Induce Cell Death of Recently Activated Antigen-Specific T Cells: A Possible Role of TNFR2 in T-Cell Deflation. Blood 2014, 124, 4352. [Google Scholar] [CrossRef]
- Hijdra, D.; Vorselaars, A.D.; Grutters, J.C.; Claessen, A.M.; Rijkers, G.T. Differential expression of TNFR1 (CD120a) and TNFR2 (CD120b) on subpopulations of human monocytes. J. Inflamm. 2012, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Beldi, G.; Bahiraii, S.; Lezin, C.; Nouri Barkestani, M.; Abdelgawad, M.E.; Uzan, G.; Naserian, S. TNFR2 Is a Crucial Hub Controlling Mesenchymal Stem Cell Biological and Functional Properties. Front. Cell Dev. Biol. 2020, 8, 596831. [Google Scholar] [CrossRef] [PubMed]
- Naserian, S.; Shamdani, S.; Arouche, N.; Uzan, G. Regulatory T cell induction by mesenchymal stem cells depends on the expression of TNFR2 by T cells. Stem Cell Res. Ther. 2020, 11, 534. [Google Scholar] [CrossRef] [PubMed]
- Morton, P.E.; Perrin, C.; Levitt, J.; Matthews, D.R.; Marsh, R.J.; Pike, R.; McMillan, D.; Maloney, A.; Poland, S.; Ameer-Beg, S.; et al. TNFR1 membrane reorganization promotes distinct modes of TNFα signaling. Sci. Signal. 2019, 12, 2418. [Google Scholar] [CrossRef] [PubMed]
- Cabal-Hierro, L.; Lazo, P.S. Signal transduction by tumor necrosis factor receptors. Cell. Signal. 2012, 24, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Zhao, H.; Jin, M.; Zhu, H.; Shan, B.; Geng, J.; Dziedzic, S.A.; Amin, P.; Mifflin, L.; Naito, M.G.; et al. Modulating TRADD to restore cellular homeostasis and inhibit apoptosis. Nature 2020, 587, 133–138. [Google Scholar] [CrossRef]
- Fischer, R.; Kontermann, R.E.; Pfizenmaier, K. Selective Targeting of TNF Receptors as a Novel Therapeutic Approach. Front. Cell Dev. Biol. 2020, 8, 401. [Google Scholar] [CrossRef]
- Van Ostade, X.; Vandenabeele, P.; Everaerdt, B.; Loetscher, H.; Gentz, R.; Brockhaus, M.; Lesslauer, W.; Tavernier, J.; Brouckaert, P.; Fiers, W. Human TNF mutants with selective activity on the p55 receptor. Nature 1993, 361, 266–269. [Google Scholar] [CrossRef]
- Barbara, J.A.; Smith, W.B.; Gamble, J.R.; Van Ostade, X.; Vandenabeele, P.; Tavernier, J.; Fiers, W.; Vadas, M.A.; Lopez, A.F. Dissociation of TNF-alpha cytotoxic and proinflammatory activities by p55 receptor- and p75 receptor-selective TNF-alpha mutants. EMBO J. 1994, 13, 843–850. [Google Scholar] [CrossRef]
- Loetscher, H.; Stueber, D.; Banner, D.; Mackay, F.; Lesslauer, W. Human tumor necrosis factor alpha (TNF alpha) mutants with exclusive specificity for the 55-kDa or 75-kDa TNF receptors. J. Biol. Chem. 1993, 268, 26350–26357. [Google Scholar] [CrossRef]
- Hube, F.; Hauner, H. The two tumor necrosis factor receptors mediate opposite effects on differentiation and glucose metabolism in human adipocytes in primary culture. Endocrinology 2000, 141, 2582–2588. [Google Scholar] [CrossRef]
- Lees, D.M.; Pallikaros, Z.; Corder, R. The p55 tumor necrosis factor receptor (CD120a) induces endothelin-1 synthesis in endothelial and epithelial cells. Eur. J. Pharmacol. 2000, 390, 89–94. [Google Scholar] [CrossRef]
- Shikama, H.; Miyata, K.; Sakae, N.; Mitsuishi, Y.; Nishimura, K.; Kuroda, K.; Kato, M. Novel mutein of tumor necrosis factor alpha (F4614) with reduced hypotensive effect. J. Interferon Cytokine Res. 1995, 15, 677–684. [Google Scholar] [CrossRef]
- Atarashi, Y.; Yasumura, S.; Nambu, S.; Yoshio, Y.; Murakami, J.; Takahara, T.; Higuchi, K.; Watanabe, A.; Miyata, K.; Kato, M. A novel human tumor necrosis factor alfa mutein, F4614, inhibits in vitro and in vivo growth of murine and human hepatoma: Implication for immunotherapy of human hepatocellular carcinoma. Hepatology 1998, 28, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.-S.; Kim, J.-S.; Cho, H.-S.; Shin, N.-K.; Jeong, W.; Shin, H.-C.; Kim, Y.J.; Hahn, J.H.; Oh, B.-H. High Resolution Crystal Structure of a Human Tumor Necrosis Factor-α Mutant with Low Systemic Toxicity. J. Biol. Chem. 1998, 273, 2153–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz Pinto, M.F.; Campbell, S.J.; Simoglou Karali, C.; Johanssen, V.A.; Bristow, C.; Cheng, V.W.T.; Zarghami, N.; Larkin, J.R.; Pannell, M.; Hearn, A.; et al. Selective blood-brain barrier permeabilization of brain metastases by a type 1 receptor-selective tumor necrosis factor mutein. Neuro-Oncology 2021, 24, 52–63. [Google Scholar] [CrossRef]
- Van Ostade, X.; Tavernier, J.; Prangé, T.; Fiers, W. Localization of the active site of human tumour necrosis factor (hTNF) by mutational analysis. EMBO J. 1991, 10, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Mayes, P.A.; Hance, K.W.; Hoos, A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 2018, 17, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.U.; Shin, W.; Son, J.Y.; Yoo, K.-Y.; Heo, Y.-S. Molecular Basis for the Neutralization of Tumor Necrosis Factor α by Certolizumab Pegol in the Treatment of Inflammatory Autoimmune Diseases. Int. J. Mol. Sci. 2017, 18, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, M.C.; Cohen, S.; Moreland, L.; Lium, D.; Robbins, S.; Newmark, R.; Bekker, P. Combination Therapy with Etanercept and Anakinra in the Treatment of Patients with Rheumatoid Arthritis Who Have Been Treated Unsuccessfully with Methotrexate. Arthritis Rheum. 2004, 50, 1412–1419. [Google Scholar] [CrossRef]
- Goodall, L.J.; Ovecka, M.; Rycroft, D.; Friel, S.L.; Sanderson, A.; Mistry, P.; Davies, M.L.; Stoop, A.A. Pharmacokinetic and pharmacodynamic characterisation of an anti-mouse TNF receptor 1 domain antibody formatted for in vivo half-life extension. PLoS ONE 2015, 10, e0137065. [Google Scholar] [CrossRef] [Green Version]
- Gouweleeuw, L.; Wajant, H.; Maier, O.; Eisel, U.L.M.; Blankesteijn, W.M.; Schoemaker, R.G. Effects of selective TNFR1 inhibition or TNFR2 stimulation, compared to non-selective TNF inhibition, on (neuro)inflammation and behavior after myocardial infarction in male mice. Brain Behav. Immun. 2021, 93, 156–171. [Google Scholar] [CrossRef] [PubMed]
- McCann, F.E.; Perocheau, D.P.; Ruspi, G.; Blazek, K.; Davies, M.L.; Feldmann, M.; Dean, J.L.E.; Stoop, A.A.; Williams, R.O. Selective tumor necrosis factor receptor i blockade is antiinflammatory and reveals immunoregulatory role of tumor necrosis factor receptor II in collagen-induced arthritis. Arthritis Rheumatol. 2014, 66, 2728–2738. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.K.; Maier, O.; Fischer, R.; Fairless, R.; Hochmeister, S.; Stojic, A.; Pick, L.; Haar, D.; Musiol, S.; Storch, M.K.; et al. Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis. PLoS ONE 2014, 9, 90117. [Google Scholar] [CrossRef] [PubMed]
- Zettlitz, K.A.; Lorenz, V.; Landauer, K.; Münkel, S.; Herrmann, A.; Scheurich, P.; Pfizenmaier, K.; Kontermann, R. ATROSAB, a humanized antagonistic anti-tumor necrosis factor receptor one-specific antibody. MAbs 2010, 2, 639–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, F.; Liebig, T.; Guenzi, E.; Herrmann, A.; Scheurich, P.; Pfizenmaier, K.; Kontermann, R.E. Antagonistic TNF Receptor One-Specific Antibody (ATROSAB): Receptor Binding and In Vitro Bioactivity. PLoS ONE 2013, 8, e72156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Feng, Z.; Wang, Y.; Ma, S.; Hu, Z.; Yang, P.; Chai, Y.; Xie, X. Discovery of Novel Ligands for TNF-α and TNF Receptor-1 through Structure-Based Virtual Screening and Biological Assay. J. Chem. Inf. Model. 2017, 57, 1101–1111. [Google Scholar] [CrossRef]
- Saddala, M.S.; Huang, H. Identification of novel inhibitors for TNFα, TNFR1 and TNFα-TNFR1 complex using pharmacophore-based approaches. J. Transl. Med. 2019, 17, 215. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.H.; Schaaf, T.M.; Grant, B.D.; Lim, C.K.W.; Bawaskar, P.; Aldrich, C.C.; Thomas, D.D.; Sachs, J.N. Noncompetitive inhibitors of TNFR1 probe conformational activation states. Sci. Signal. 2019, 12, 5637. [Google Scholar] [CrossRef]
- Van De Kar, N.C.A.J.; Kooistra, T.; Vermeer, M.; Lesslauer, W.; Monnens, L.A.H.; Van Hinsbergh, V.W.M. Tumor necrosis factor α induces endothelial galactosyl transferase activity and verocytotoxin receptors. Role of specific tumor necrosis factor receptors and protein kinase C. Blood 1995, 85, 734–743. [Google Scholar] [CrossRef]
- Yui, J.; Hemmings, D.; Garcia-Lloret, M.; Guilbert, L.J. Expression of the human p55 and p75 tumor necrosis factor receptors in primary villous trophoblasts and their role in cytotoxic signal transduction. Biol. Reprod. 1996, 55, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Garcia-Lloret, M.; Winkler-Lowen, B.; Miller, R.; Simpson, K.; Guilbert, L.J. ICAM-1-mediated adhesion of peripheral blood monocytes to the maternal surface of placental syncytiotrophoblasts: Implications for placental villitis. Am. J. Pathol. 1997, 150, 1845–1860. [Google Scholar] [PubMed]
- Dong, Y.; Fischer, R.; Naudé, P.J.W.; Maier, O.; Nyakas, C.; Duffey, M.; Van Der Zee, E.A.; Dekens, D.; Douwenga, W.; Herrmann, A.; et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 12304–12309. [Google Scholar] [CrossRef] [Green Version]
- Fischer, R.; Proske, M.; Duffey, M.; Stangl, H.; Martinez, G.F.; Peters, N.; Kraske, A.; Straub, R.H.; Bethea, J.R.; Kontermann, R.E.; et al. Selective Activation of Tumor Necrosis Factor Receptor II Induces Antiinflammatory Responses and Alleviates Experimental Arthritis. Arthritis Rheumatol. 2018, 70, 722–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, M.; Biehl, M.; Steinfatt, T.; Brandl, A.; Kums, J.; Amich, J.; Vaeth, M.; Kuen, J.; Holtappels, R.; Podlech, J.; et al. Exogenous TNFR2 activation protects from acute GvHD via host T reg cell expansion. J. Exp. Med. 2016, 213, 1881–1900. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Maier, O.; Siegemund, M.; Wajant, H.; Scheurich, P.; Pfizenmaier, K. A TNF receptor 2 selective agonist rescues human neurons from oxidative stress-induced cell death. PLoS ONE 2011, 6, e27621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Diaz Arguello, O.A.; Haisma, H.J.; Apoptosis-Inducing, H. Apoptosis-Inducing TNF Superfamily Ligands for Cancer Therapy. Cancers 2021, 13, 1543. [Google Scholar] [CrossRef]
- Zhang, B.; van Roosmalen, I.A.M.; Reis, C.R.; Setroikromo, R.; Quax, W.J. Death receptor 5 is activated by fucosylation in colon cancer cells. FEBS J. 2019, 286, 555–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, W.; Immunology, P.H. TNF-related apoptosis-inducing ligand (TRAIL) induces apoptosis in Fas ligand-resistant melanoma cells and mediates CD4 T cell killing of target cells. Am. Assoc. Immnol. 1998, 161, 2195–2200. [Google Scholar]
- Mori, S.; Murakami-Mori, K.; Nakamura, S.; Ashkenazi, A.; Bonavida, B. Sensitization of AIDS-Kaposi’s sarcoma cells to Apo-2 ligand-induced apoptosis by actinomycin D. J. Immunol. 1999, 162, 5616–56123. [Google Scholar] [PubMed]
- Rieger, J.; Naumann, U.; Glaser, T.; Ashkenazi, A.; Weller, M. APO2 ligand: A novel lethal weapon against malignant glioma? FEBS Lett. 1998, 427, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor–related apoptosis–inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Dulanermin Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Zuch de Zafra, C.L.; Ashkenazi, A.; Darbonne, W.C.; Cheu, M.; Totpal, K.; Ortega, S.; Flores, H.; Walker, M.D.; Kabakoff, B.; Lum, B.L.; et al. Antitherapeutic antibody-mediated hepatotoxicity of recombinant human Apo2L/TRAIL in the cynomolgus monkey. Cell Death Dis. 2016, 7, e2338. [Google Scholar] [CrossRef] [Green Version]
- Kelley, S.K.; Harris, L.A.; Xie, D.; Deforge, L.; Totpal, K.; Bussiere, J.; Fox, J.A. Preclinical studies to predict the disposition of Apo2L/tumor necrosis factor-related apoptosis-inducing ligand in humans: Characterization of in vivo efficacy, pharmacokinetics, and safety. J. Pharmacol. Exp. Ther. 2001, 299, 31–38. [Google Scholar] [PubMed]
- Snajdauf, M.; Havlova, K.; Vachtenheim, J.; Ozaniak, A.; Lischke, R.; Bartunkova, J.; Smrz, D.; Strizova, Z. The TRAIL in the Treatment of Human Cancer: An Update on Clinical Trials. Front. Mol. Biosci. 2021, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Reis, C.R.; van der Sloot, A.M.; Szegezdi, E.; Natoni, A.; Tur, V.; Cool, R.H.; Samali, A.; Serrano, L.; Quax, W.J. Enhancement of Antitumor Properties of rhTRAIL by Affinity Increase toward Its Death Receptors. Biochemistry 2009, 48, 2180–2191. [Google Scholar] [CrossRef]
- Huang, M.; Zhu, H.; Yi, C.; Yan, J.; Wei, L.; Yang, X.; Chen, S.; Huang, Y. A novel TRAIL mutant-TRAIL-Mu3 enhances the antitumor effects by the increased affinity and the up-expression of DR5 in pancreatic cancer. Cancer Chemother. Pharmacol. 2018, 82, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.F.; Totpal, K.; Lindstrom, S.H.; Mathieu, M.; Billeci, K.; DeForge, L.; Pai, R.; Hymowitz, S.G.; Ashkenazi, A. Receptor-selective mutants of apoptosis-inducing ligand 2/tumor necrosis factor-related apoptosis-inducing ligand reveal a greater contribution of Death Receptor (DR) 5 than DR4 to apoptosis signaling. J. Biol. Chem. 2005, 280, 2205–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacFarlane, M.; Kohlhaas, S.L.; Sutcliffe, M.J.; Dyer, M.J.S.; Cohen, G.M. TRAIL receptor-selective mutants signal to apoptosis via TRAIL-R1 in primary lymphoid malignancies. Cancer Res. 2005, 65, 11265–11270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tur, V.; van der Sloot, A.M.; Reis, C.R.; Szegezdi, E.; Cool, R.H.; Samali, A.; Serrano, L.; Quax, W.J. DR4-selective tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) variants obtained by structure-based design. J. Biol. Chem. 2008, 283, 20560–20568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.; Albarenque, S.M.; Cool, R.H.; Quax, W.J.; Mohr, A.; Zwacka, R.M. DR4 specific TRAIL variants are more efficacious than wild-type TRAIL in pancreatic cancer. Cancer Biol. Ther. 2014, 15, 1658–1666. [Google Scholar] [CrossRef] [Green Version]
- Reis, C.R.; Van Der Sloot, A.M.; Natoni, A.; Szegezdi, E.; Setroikromo, R.; Meijer, M.; Sjollema, K.; Stricher, F.; Cool, R.H.; Samali, A.; et al. Rapid and efficient cancer cell killing mediated by high-affinity death receptor homotrimerizing TRAIL variants. Cell Death Dis. 2010, 1, e83. [Google Scholar] [CrossRef] [Green Version]
- Szegezdi, E.; Reis, C.R.; Der Sloot, A.M.V.; Natoni, A.; O’Reilly, A.; Reeve, J.; Cool, R.H.; O’Dwyer, M.; Knapper, S.; Serrano, L.; et al. Targeting AML through DR4 with a novel variant of rhTRAIL. J. Cell. Mol. Med. 2011, 15, 2216–2231. [Google Scholar] [CrossRef] [Green Version]
- Reis, C.R.; van Assen, A.H.G.; Quax, W.J.; Cool, R.H. Unraveling the Binding Mechanism of Trivalent Tumor Necrosis Factor Ligands and Their Receptors. Mol. Cell. Proteom. 2011, 10, M110.002808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasparian, M.E.; Chernyak, B.V.; Dolgikh, D.A.; Yagolovich, A.V.; Popova, E.N.; Sycheva, A.M.; Moshkovskii, S.A.; Kirpichnikov, M.P. Generation of new TRAIL mutants DR5-A and DR5-B with improved selectivity to death receptor 5. Apoptosis 2009, 14, 778–787. [Google Scholar] [CrossRef]
- Van Der Sloot, A.M.; Tur, V.; Szegezdi, E.; Mullally, M.M.; Cool, R.H.; Samali, A.; Serrano, L.; Quax, W.J. Designed tumor necrosis factor-related apoptosis-inducing ligand variants initiating apoptosis exclusively via the DR5 receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 8634–8639. [Google Scholar] [CrossRef] [Green Version]
- van Roosmalen, I.A.M.; Reis, C.R.; Setroikromo, R.; Yuvaraj, S.; Joseph, J.V.; Tepper, P.G.; Kruyt, F.A.E.; Quax, W.J. The ER stress inducer DMC enhances TRAIL-induced apoptosis in glioblastoma. Springerplus 2014, 3, 495. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Zijlstra, S.N.; Soto-Gamez, A.; Setroikromo, R.; Quax, W.J. Artemisinin Derivatives Stimulate DR5-Specific TRAIL-Induced Apoptosis by Regulating Wildtype P53. Cancers 2020, 12, 2514. [Google Scholar] [CrossRef]
- Zhou, X.; Soto-Gamez, A.; Nijdam, F.; Setroikromo, R.; Quax, W.J. Dihydroartemisinin-transferrin adducts enhance TRAIL-induced apoptosis in triple-negative breast cancer in a P53-independent and ROS-dependent manner. Front. Oncol. 2022, 11, 5452. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Wang, Y.; Zhou, X.; Seras, L.; Quax, W.; Demaria, M. Enhanced extrinsic apoptosis of therapy-induced senescent cancer cells using a death receptor 5 (DR5) selective agonist. Cancer Lett. 2022, 525, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Arabpour, M.; Cool, R.H.; Faber, K.N.; Quax, W.J.; Haisma, H.J. Receptor-specific TRAIL as a means to achieve targeted elimination of activated hepatic stellate cells. J. Drug Target. 2017, 25, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, B.; Chen, D.; Setroikromo, R.; Haisma, H.J.; Quax, W.J. Histone Deacetylase Inhibitors Sensitize TRAIL-Induced Apoptosis in Colon Cancer Cells. Cancers 2019, 11, 645. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.A.; Warren, J.T.; Wang, M.W.H.; Teitelbaum, S.L.; Fremont, D.H. RANKL employs distinct binding modes to engage RANK and the osteoprotegerin decoy receptor. Structure 2012, 20, 1971–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, J.; Cronin, S.J.F.; Penninger, J.M. Targeting the RANKL/RANK/OPG Axis for Cancer Therapy. Front. Oncol. 2020, 10, 1283. [Google Scholar] [CrossRef] [PubMed]
- Galibert, L.; Tometsko, M.E.; Andersen, D.M.; Cosman, D.; Dougall, W.C. The involvement of multiple tumor necrosis factor receptor (TNFR)- associated factors in the signaling mechanisms of receptor activator of NF- κB, a member of the TNFR superfamily. J. Biol. Chem. 1998, 273, 34120–34127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilchovska, D.; Barrow, D.M. An Overview of the NF-kB mechanism of pathophysiology in rheumatoid arthritis, investigation of the NF-kB ligand RANKL and related nutritional interventions. Autoimmun. Rev. 2021, 20, 102741. [Google Scholar] [CrossRef]
- Verma, D.; Zanetti, C.; Godavarthy, P.S.; Kumar, R.; Minciacchi, V.R.; Pfeiffer, J.; Metzler, M.; Lefort, S.; Maguer-Satta, V.; Nicolini, F.E.; et al. Bone marrow niche-derived extracellular matrix-degrading enzymes influence the progression of B-cell acute lymphoblastic leukemia. Leukemia 2020, 34, 1540–1552. [Google Scholar] [CrossRef] [PubMed]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habibie, H.; Adhyatmika, A.; Schaafsma, D.; Melgert, B.N. The role of osteoprotegerin (OPG) in fibrosis: Its potential as a biomarker and/or biological target for the treatment of fibrotic diseases. Pharmacol. Ther. 2021, 228, 107941. [Google Scholar] [CrossRef] [PubMed]
- Hymowitz, S.G.; Christinger, H.W.; Fuh, G.; Ultsch, M.; O’Connell, M.; Kelley, R.F.; Ashkenazi, A.; De Vos, A.M. Triggering cell death: The crystal structure of Apo2L/TRAIL in a complex with death receptor 5. Mol. Cell 1999, 4, 563–571. [Google Scholar] [CrossRef]
- Banner, D.W.; D’Arcy, A.; Janes, W.; Gentz, R.; Schoenfeld, H.J.; Broger, C.; Loetscher, H.; Lesslauer, W. Crystal structure of the soluble human 55 kd TNF receptor-human TNFβ complex: Implications for TNF receptor activation. Cell 1993, 73, 431–445. [Google Scholar] [CrossRef]
- Lam, J.; Nelson, C.A.; Ross, F.P.; Teitelbaum, S.L.; Fremont, D.H. Crystal structure of the TRANCE/RANKL cytokine reveals determinants of receptor-ligand specificity. J. Clin. Investig. 2001, 108, 971–979. [Google Scholar] [CrossRef]
- Wang, Y.; van Assen, A.H.G.; Reis, C.R.; Setroikromo, R.; van Merkerk, R.; Boersma, Y.L.; Cool, R.H.; Quax, W.J. Novel RANKL DE-loop mutants antagonize RANK-mediated osteoclastogenesis. FEBS J. 2017, 284, 2501–2512. [Google Scholar] [CrossRef] [Green Version]
- Wassenaar, T.A.; Quax, W.J.; Mark, A.E. The conformation of the extracellular binding domain of Death Receptor 5 in the presence and absence of the activating ligand TRAIL: A molecular dynamics study. Proteins Struct. Funct. Bioinform. 2008, 70, 333–343. [Google Scholar] [CrossRef]
- Neumann, S.; Bidon, T.; Branschädel, M.; Krippner-Heidenreich, A.; Scheurich, P.; Doszczak, M. The transmembrane domains of TNF-related apoptosis-inducing ligand (TRAIL) receptors 1 and 2 co-regulate apoptotic signaling capacity. PLoS ONE 2012, 7, e42526. [Google Scholar] [CrossRef]
- Cheng, T.; Pavlos, N.J.; Wang, C.; Tan, J.W.Y.; Lin, J.M.; Cornish, J.; Zheng, M.H.; Xu, J. Mutations within the TNF-like core domain of RANKL impair osteoclast differentiation and activation. Mol. Endocrinol. 2009, 23, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Michiels, T.; Setroikromo, R.; van Merkerk, R.; Cool, R.H.; Quax, W.J. Creation of RANKL mutants with low affinity for decoy receptor OPG and their potential anti-fibrosis activity. FEBS J. 2019, 286, 3582–3593. [Google Scholar] [CrossRef]
- Ambroszkiewicz, J.; Sands, D.; Gajewska, J.; Chelchowska, M.; Laskowska-Klita, T. Bone turnover markers, osteoprotegerin and RANKL cytokines in children with cystic fibrosis. Adv. Med. Sci. 2013, 58, 338–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Heron, L.; Guillaume, C.; Velard, F.; Braux, J.; Touqui, L.; Moriceau, S.; Sermet-Gaudelus, I.; Laurent-Maquin, D.; Jacquot, J. Cystic fibrosis transmembrane conductance regulator (CFTR) regulates the production of osteoprotegerin (OPG) and prostaglandin (PG) E2 in human bone. J. Cyst. Fibros. 2010, 9, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, B.; Pickering, R.J.; Tsorotes, D.; Wang, B.; Bernardi, S.; Kantharidis, P.; Fabris, B.; Zauli, G.; Secchiero, P.; Thomas, M.C. Osteoprotegerin promotes vascular fibrosis via a TGF-β1 autocrine loop. Atherosclerosis 2011, 218, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Adhyatmika, A.; Putri, K.S.S.S.; Beljaars, L.; Melgert, B.N. The elusive antifibrotic macrophage. Front. Med. 2015, 2, 81. [Google Scholar] [CrossRef] [Green Version]
- Kurinami, H.; Shimamura, M.; Nakagami, H.; Shimizu, H.; Koriyama, H.; Kawano, T.; Wakayama, K.; Mochizuki, H.; Rakugi, H.; Morishita, R. A Novel Therapeutic Peptide as a Partial Agonist of RANKL in Ischemic Stroke. Sci. Rep. 2016, 6, 38062. [Google Scholar] [CrossRef] [Green Version]
- Aoki, K.; Saito, H.; Itzstein, C.; Ishiguro, M.; Shibata, T.; Blanque, R.; Mian, A.H.; Takahashi, M.; Suzuki, Y.; Yoshimatsu, M.; et al. A TNF receptor loop peptide mimic blocks RANK ligand–induced signaling, bone resorption, and bone loss. J. Clin. Investig. 2006, 116, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Ta, H.M.; Nguyen, G.T.T.; Jin, H.M.; Choi, J.; Park, H.; Kim, N.; Hwang, H.Y.; Kim, K.K. Structure-based development of a receptor activator of nuclear factor-κB ligand (RANKL) inhibitor peptide and molecular basis for osteopetrosis. Proc. Natl. Acad. Sci. USA 2010, 107, 20281–20286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quax, W.J.; Van Assen, A.H.G.; Wang, Y.Z. TNF-family member Receptor Activator of NF-κB (RANK) and RANK-Ligand (RANKL) in bone remodelling. In Proceedings of the IOP Conference Series: Earth and Environmental Science, Hangzhou, China, 26–29 June 2018; IOP Publishing: Bristol, UK, 2018; Volume 185, p. 012001. [Google Scholar]
- Denosumab in Treating Patients With Bone Loss Due to Donor Stem Cell Transplant. Available online: https://clinicaltrials.gov/ct2/show/NCT03925532 (accessed on 5 January 2022).
- The Effects of 12-Months of Denosumab on Bone Density in Prevalent Kidney Transplant Recipients. Available online: https://clinicaltrials.gov/ct2/show/NCT03960554 (accessed on 5 January 2022).
- Efficacy of Denosumab on Normal BMD in Women Receiving Adjuvant Aromatase Inhibitors for Early Breast Cancer. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03324932 (accessed on 5 January 2022).
- Templeton, A.J.; Stalder, L.; Bernhard, J.; Brauchli, P.; Gillessen, S.; Hayoz, S.; Klingbiel, D.; Matter-Walstra, K.; Thurlimann, B.J.K.; Von Moos, R. Prevention of symptomatic skeletal events with denosumab administered every 4 weeks versus every 12 weeks: A noninferiority phase III trial (SAKK 96/12, REDUSE). J. Clin. Oncol. 2014, 32, TPS5095. [Google Scholar] [CrossRef]
- Yamada, A.; Arakaki, R.; Saito, M.; Kudo, Y.; Ishimaru, N. Dual role of Fas/FasL-mediated signal in peripheral immune tolerance. Front. Immunol. 2017, 8, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Q.; Fu, T.M.; Cruz, A.C.; Sengupta, P.; Thomas, S.K.; Wang, S.; Siegel, R.M.; Wu, H.; Chou, J.J. Structural Basis and Functional Role of Intramembrane Trimerization of the Fas/CD95 Death Receptor. Mol. Cell 2016, 61, 602–613. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Eberstadt, M.; Olejniczak, E.T.; Meadows, R.P.; Feslk, S.W. NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature 1996, 384, 638–641. [Google Scholar] [CrossRef]
- Liu, W.; Ramagopal, U.; Cheng, H.; Bonanno, J.B.; Toro, R.; Bhosle, R.; Zhan, C.; Almo, S.C. Crystal Structure of the Complex of Human FasL and Its Decoy Receptor DcR3. Structure 2016, 24, 2016–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Zhao, Y.; Wang, C.; Ji, H.; Yu, J.; Liu, C.; Liu, A. A novel synthetic chitosan selenate (CS) induces apoptosis in A549 lung cancer cells via the Fas/FasL pathway. Int. J. Biol. Macromol. 2020, 158, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Bellesi, S.; Metafuni, E.; Hohaus, S.; Maiolo, E.; Marchionni, F.; D’Innocenzo, S.; La Sorda, M.; Ferraironi, M.; Ramundo, F.; Fantoni, M.; et al. Increased CD95 (Fas) and PD-1 expression in peripheral blood T lymphocytes in COVID-19 patients. Br. J. Haematol. 2020, 191, 207–211. [Google Scholar] [CrossRef]
- Mitsiades, N.; Poulaki, V.; Mitsiades, C.S.; Koutras, D.A.; Chrousos, G.P. Apoptosis induced by FasL and TRAIL/Apo2L in the pathogenesis of thyroid diseases. Trends Endocrinol. Metab. 2001, 12, 384–390. [Google Scholar] [CrossRef]
- Jimbo, H.; Nagai, H.; Fujiwara, S.; Shimoura, N.; Nishigori, C. Fas-FasL interaction in cytotoxic T cell-mediated vitiligo: The role of lesional expression of tumor necrosis factor-α and interferon-γ in Fas-mediated melanocyte apoptosis. Exp. Dermatol. 2020, 29, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kouzmenko, A.; Kato, S. Targeting Fas/FasL signaling, a new strategy for maintaining bone health. Expert Opin. Ther. Targets 2011, 15, 1143–1145. [Google Scholar] [CrossRef] [PubMed]
- Malleter, M.; Tauzin, S.; Bessede, A.; Castellano, R.; Goubard, A.; Godey, F.; Levêque, J.; Jézéquel, P.; Campion, L.; Campone, M.; et al. CD95L cell surface cleavage triggers a prometastatic signaling pathway in triple-negative breast cancer. Cancer Res. 2013, 73, 6711–6721. [Google Scholar] [CrossRef] [Green Version]
- Greaney, P.; Nahimana, A.; Lagopoulos, L.; Etter, A.L.; Aubry, D.; Attinger, A.; Beltraminelli, N.; Huni, B.; Bassi, I.; Sordat, B.; et al. A Fas agonist induces high levels of apoptosis in haematological malignancies. Leuk. Res. 2006, 30, 415–426. [Google Scholar] [CrossRef]
- Tsao, T.S.; Lodish, H.F.; Fruebis, J. ACRP30, a new hormone controlling fat and glucose metabolism. Eur. J. Pharmacol. 2002, 440, 213–221. [Google Scholar] [CrossRef]
- Eisele, G.; Roth, P.; Hasenbach, K.; Aulwurm, S.; Wolpert, F.; Tabatabai, G.; Wick, W.; Weller, M. APO010, a synthetic hexameric CD95 ligand, induces human glioma cell death in vitro and in vivo. Neuro-Oncology 2011, 13, 155–164. [Google Scholar] [CrossRef]
- Ocio, E.M.; Maiso, P.; Garayoa, M.; Dupuis, M.; Pandiella, A.; Miguel, J.F.S. The Activation of Fas Receptor by APO010, a Recombinant Form of Fas Ligand, Induces In Vitro and In Vivo Antimyeloma Activity. Blood 2007, 110, 1515. [Google Scholar] [CrossRef]
- Verbrugge, I.; Wissink, E.H.J.; Rooswinkel, R.W.; Jongsma, J.; Beltraminelli, N.; Dupuis, M.; Borst, J.; Verheij, M. Combining radiotherapy with APO010 in cancer treatment. Clin. Cancer Res. 2009, 15, 2031–2038. [Google Scholar] [CrossRef] [Green Version]
- A Phase I Dose Finding Study of APO010 in Patients With Solid Tumors. Available online: http://clinicaltrials.gov/show/NCT00437736 (accessed on 5 January 2022).
- Huang, J.H.; Tykocinski, M.L. CTLA-4-Fas ligand functions as a trans signal converter protein in bridging antigen-presenting cells and T cells. Int. Immunol. 2001, 13, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Georgopoulos, N.T.; Steele, L.P.; Thomson, M.J.; Selby, P.J.; Southgate, J.; Trejdosiewicz, L.K. A novel mechanism of CD40-induced apoptosis of carcinoma cells involving TRAF3 and JNK/AP-1 activation. Cell Death Differ. 2006, 13, 1789–1801. [Google Scholar] [CrossRef]
- Dranitzki-Elhalel, M.; Huang, J.H.; Sasson, M.; Rachmilewitz, J.; Parnas, M.; Tykocinski, M.L. CD40·FasL inhibits human T cells: Evidence for an auto-inhibitory loop-back mechanism. Int. Immunol. 2007, 19, 355–363. [Google Scholar] [CrossRef]
- Orbach, A.; Rachmilewitz, J.; Shani, N.; Isenberg, Y.; Parnas, M.; Huang, J.H.; Tykocinski, M.L.; Dranitzki-Elhalel, M. CD40·FasL and CTLA-4·FasL fusion proteins induce apoptosis in malignant cell lines by dual signaling. Am. J. Pathol. 2010, 177, 3159–3168. [Google Scholar] [CrossRef]
- Ahamadi-Fesharaki, R.; Fateh, A.; Vaziri, F.; Solgi, G.; Siadat, S.D.; Mahboudi, F.; Rahimi-Jamnani, F. Single-Chain Variable Fragment-Based Bispecific Antibodies: Hitting Two Targets with One Sophisticated Arrow. Mol. Ther.-Oncolytics 2019, 14, 38–56. [Google Scholar] [CrossRef] [Green Version]
- Samel, D.; Müller, D.; Gerspach, J.; Assohou-Luty, C.; Sass, G.; Tiegs, G.; Pfizenmaier, K.; Wajant, H. Generation of a FasL-based proapoptotic fusion protein devoid of systemic toxicity due to cell-surface antigen-restricted activation. J. Biol. Chem. 2003, 278, 32077–32082. [Google Scholar] [CrossRef] [Green Version]
- Villa-Morales, M.; Fernández-Piqueras, J. Targeting the Fas/FasL signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 85–101. [Google Scholar] [CrossRef]
- Bremer, E.; Ten Cate, B.; Samplonius, D.F.; Mueller, N.; Wajant, H.; Stel, A.J.; Chamuleau, M.; Van De Loosdrecht, A.A.; Stieglmaier, J.; Fey, G.H.; et al. Superior activity of fusion protein scFvRit:sFasL over cotreatment with rituximab and fas agonists. Cancer Res. 2008, 68, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Bremer, E.; Ten Cate, B.; Samplonius, D.F.; De Leij, L.F.M.H.; Helfrich, W. CD7-restricted activation of Fas-mediated apoptosis: A novel therapeutic approach for acute T-cell leukemia. Blood 2006, 107, 2863–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.V.; Sharma, R.; Ju, C.-Y.A.; Roffler, S.R.; Ju, S.-T. A recombinant scFv-FasLext as a targeting cytotoxic agent against human Jurkat-Ras cancer. J. Biomed. Sci. 2013, 20, 16. [Google Scholar] [CrossRef] [Green Version]
- Mauri, D.N.; Ebner, R.; Montgomery, R.I.; Kochel, K.D.; Cheung, T.C.; Yu, G.L.; Ruben, S.; Murphy, M.; Eisenberg, R.J.; Cohen, G.H.; et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin α are ligands for herpesvirus entry mediator. Immunity 1998, 8, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, G.; Belisario, D.C.; Bortolotti, S.; Storlino, G.; Colaianni, G.; Faienza, M.F.; Sanesi, L.; Alliod, V.; Buffoni, L.; Centini, E.; et al. LIGHT/TNFSF14 Promotes Osteolytic Bone Metastases in Non-small Cell Lung Cancer Patients. J. Bone Miner. Res. 2020, 35, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zhan, C.; Cheng, H.; Kumar, P.R.; Bonanno, J.B.; Nathenson, S.G.; Almo, S.C. Mechanistic Basis for Functional Promiscuity in the TNF and TNF Receptor Superfamilies: Structure of the LIGHT:DcR3 Assembly. Structure 2014, 22, 1252–1262. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, M.T.; Dejardin, E.; dos Santos, N.R. Context-dependent roles for lymphotoxin-β receptor signaling in cancer development. Biochim. Biophys. Acta 2016, 1865, 204–219. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, R.; Garrett-Thomson, S.C.; Liu, W.; Almo, S.C.; Fiser, A. Redesigning HVEM Interface for Selective Binding to LIGHT, BTLA, and CD160. Structure 2020, 28, 1197–1205.e2. [Google Scholar] [CrossRef] [PubMed]
- Skeate, J.G.; Otsmaa, M.E.; Prins, R.; Fernandez, D.J.; Da Silva, D.M.; Kast, W.M. TNFSF14: LIGHTing the Way for Effective Cancer Immunotherapy. Front. Immunol. 2020, 11, 922. [Google Scholar] [CrossRef]
- Harrop, J.A.; McDonnell, P.C.; Brigham-Burke, M.; Lyn, S.D.; Minton, J.; Tan, K.B.; Dede, K.; Spampanato, J.; Silverman, C.; Hensley, P.; et al. Herpesvirus entry mediator ligand (HVEM-L), a novel ligand for HVEM/TR2, stimulates proliferation of T cells and inhibits HT29 cell growth. J. Biol. Chem. 1998, 273, 27548–27556. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, M.W.; Shui, J.W.; Ware, C.F.; Kronenberg, M. Regulating the mucosal immune system: The contrasting roles of LIGHT, HVEM, and their various partners. Semin. Immunopathol. 2009, 31, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, S.-K.; Ju, S.-A.; Lee, S.-C.; Park, S.-M.; Choe, S.-Y.; Kwon, B.; Kwon, B.S.; Kim, B.-S. LIGHT enhances the bactericidal activity of human monocytes and neutrophils via HVEM. J. Leukoc. Biol. 2006, 79, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Holmes, T.D.; Wilson, E.B.; Black, E.V.I.; Benest, A.V.; Vaz, C.; Tan, B.; Tanavde, V.M.; Cook, G.P. Licensed human natural killer cells aid dendritic cell maturation via TNFSF14/LIGHT. Proc. Natl. Acad. Sci. USA 2014, 111, E5688–E5696. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Yu, P.; Wang, Y.; Wang, Y.; Fu, M.L.; Liu, W.; Sun, Y.; Fu, Y.X. NK-cell activation by LIGHT triggers tumor-specific CD8 + T-cell immunity to reject established tumors. Blood 2006, 107, 1342–1351. [Google Scholar] [CrossRef]
- Schneider, K.; Potter, K.G.; Ware, C.F. Lymphotoxin and LIGHT signaling pathways and target genes. Immunol. Rev. 2004, 202, 49–66. [Google Scholar] [CrossRef]
- Chen, M.C.; Hwang, M.J.; Chou, Y.C.; Chen, W.H.; Cheng, G.; Nakano, H.; Luh, T.Y.; Mai, S.C.; Hsieh, S.L. The role of apoptosis signal-regulating kinase 1 in lymphotoxin-β receptor-mediated cell death. J. Biol. Chem. 2003, 278, 16073–16081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, H.; Liang, C.; Ren, S.; Yue, C.; Wu, J. Prognostic value of DcR3 in solid tumors: A meta-analysis. Clin. Chim. Acta 2018, 481, 126–131. [Google Scholar] [CrossRef]
- Morishige, T.; Yoshioka, Y.; Inakura, H.; Tanabe, A.; Yao, X.; Tsunoda, S.; Tsutsumi, Y.; Mukai, Y.; Okada, N.; Nakagawa, S. Creation of a LIGHT mutant with the capacity to evade the decoy receptor for cancer therapy. Biomaterials 2010, 31, 3357–3363. [Google Scholar] [CrossRef] [PubMed]
- Rooney, I.A.; Butrovich, K.D.; Glass, A.A.; Borboroglu, S.; Benedict, C.A.; Whitbeck, J.C.; Cohen, G.H.; Eisenberg, R.J.; Ware, C.F. The lymphotoxin-β receptor is necessary and sufficient for LIGHT- mediated apoptosis of tumor cells. J. Biol. Chem. 2000, 275, 14307–14315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and Nonhepatotoxic Degradation of Nuclear Hepatitis B Virus cccDNA. Science 2014, 343, 1221. [Google Scholar] [CrossRef] [PubMed]
- Riedl, T.; Faure-Dupuy, S.; Rolland, M.; Schuehle, S.; Hizir, Z.; Calderazzo, S.; Zhuang, X.; Wettengel, J.; Lopez, M.A.; Barnault, R.; et al. Hypoxia-Inducible Factor 1 Alpha–Mediated RelB/APOBEC3B Down-regulation Allows Hepatitis B Virus Persistence. Hepatology 2021, 74, 1766–1781. [Google Scholar] [CrossRef] [PubMed]
- Faure-Dupuy, S.; Riedl, T.; Rolland, M.; Hizir, Z.; Reisinger, F.; Neuhaus, K.; Schuehle, S.; Remouchamps, C.; Gillet, N.; Schönung, M.; et al. Control of APOBEC3B induction and cccDNA decay by NF-κB and miR-138-5p. JHEP Rep. 2021, 3, 100354. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Interactions between multi-receptor TNF superfamily ligands (top) and their receptors (bottom). The ligands are composed of three monomers with a jelly-roll fold. We show those ligands that have been subjected to receptor-specificity engineering approaches. TNF homology domains are shown as a cylinder, the cysteine-rich domains as oval shape, and the death domains as red box. DcR2 has a truncated death domain shown as gray box.
Figure 1.
Interactions between multi-receptor TNF superfamily ligands (top) and their receptors (bottom). The ligands are composed of three monomers with a jelly-roll fold. We show those ligands that have been subjected to receptor-specificity engineering approaches. TNF homology domains are shown as a cylinder, the cysteine-rich domains as oval shape, and the death domains as red box. DcR2 has a truncated death domain shown as gray box.

Figure 2.
Workflow of phage display, which is used for the high-throughput screening of protein interactions. DNA encoding protein of interest are cloned into the genomes of multiple helper phages. The protein of interest, which is displayed on the phage surface, binds to the immobilized receptor. After the binding and elution steps, phages fractions with high binding affinities are collected and the DNA is sequenced. Amplification and purification are followed for further analysis.
Figure 2.
Workflow of phage display, which is used for the high-throughput screening of protein interactions. DNA encoding protein of interest are cloned into the genomes of multiple helper phages. The protein of interest, which is displayed on the phage surface, binds to the immobilized receptor. After the binding and elution steps, phages fractions with high binding affinities are collected and the DNA is sequenced. Amplification and purification are followed for further analysis.

Figure 3.
General workflow of computer-aided protein engineering, which helps the design and optimization of the target protein.
Figure 3.
General workflow of computer-aided protein engineering, which helps the design and optimization of the target protein.

Figure 4.
Predicted area of interaction of RANKL and OPG receptor (PDB:4E4D) around position 236. (A) OPG (green) binds to RANKL WT (yellow); (B) OPG (green) binds to RANKL Q236D (yellow).
Figure 4.
Predicted area of interaction of RANKL and OPG receptor (PDB:4E4D) around position 236. (A) OPG (green) binds to RANKL WT (yellow); (B) OPG (green) binds to RANKL Q236D (yellow).


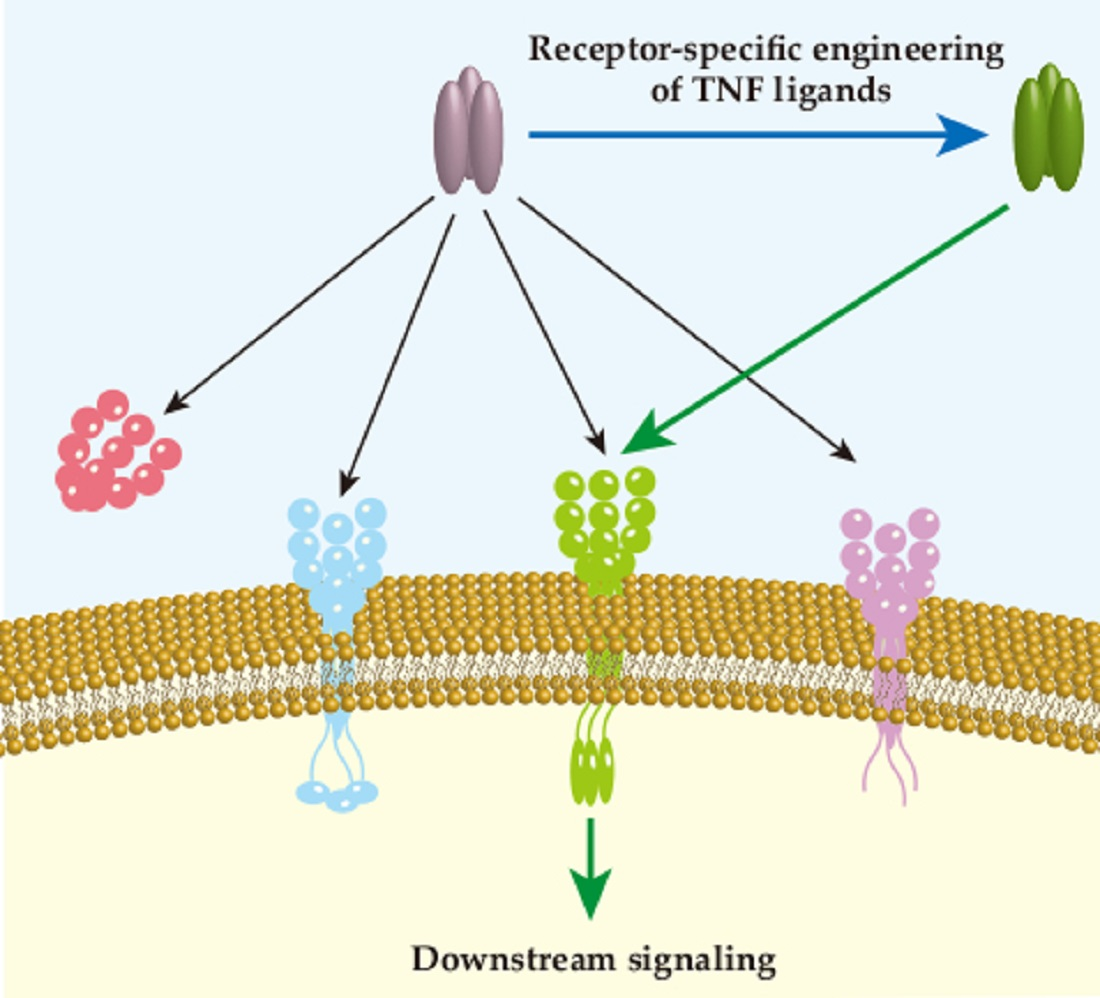{kind=link}
{kind=link}
{kind=link}
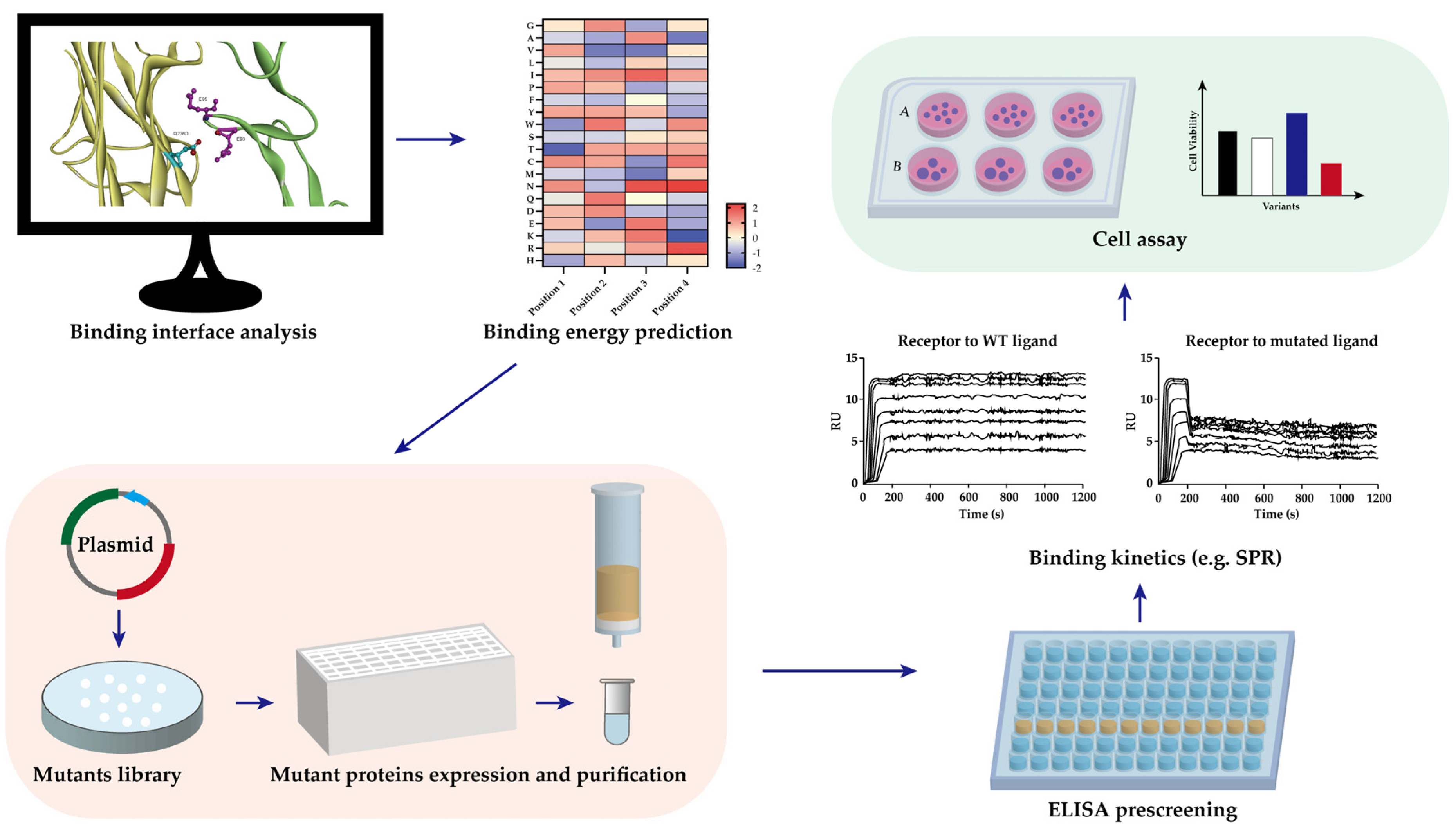{kind=link}
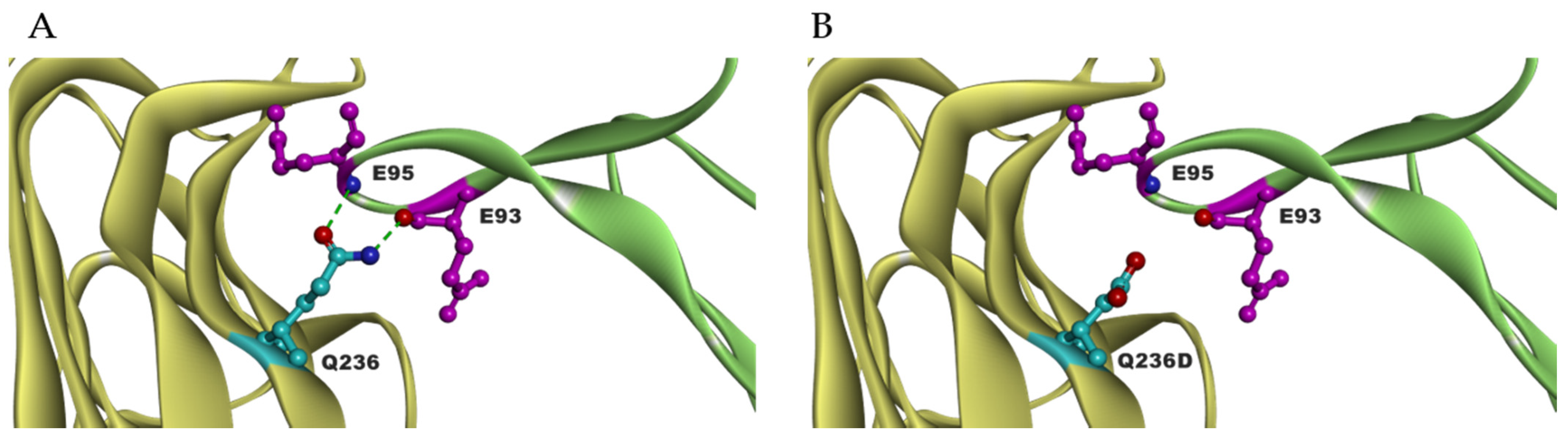{kind=link}
Table 1.
The mutation sites and binding affinity of the receptor-specific TNF-α variants.
| Ligand | Specificity | Variants | Mutation Sites | Binding Affinity (nM) | Ref. | |
|---|---|---|---|---|---|---|
| TNFR1 | TNFR2 | |||||
| TNF-α | TNFR1 &TNFR2 | WT | / | 15.8 | 35.3 | [41] |
| TNFR1 | L29S | L29S | − 1 | − | [42] | |
| R32W | R32W | − | − | [42] | ||
| R32W-S86T | R32W/S86T | 3540 | NB 2 | [42] | ||
| F4614 | T5G/P6D/R29V | − | − | [38] | ||
| M3S | L29S/S52I/Y56F and 449/455del | − | − | [40] | ||
| mutTNF G4 | A84S/V85S/S86T/ Q88N/T89P | 8.72 | NB | [41] | ||
| TNFR2 | D143N-A145R | D143N/A145R | NB 2 | 13.1 | [42] | |
1–means not available. 2 NB means no binding.
Table 2.
The mutation sites and binding affinity of the receptor-specific TRAIL variants.
| Ligand | Specificity | Variants | Mutation Sites | Binding Affinity (nM) | Ref. | |
|---|---|---|---|---|---|---|
| DR4 | DR5 | |||||
| TRAIL | DR4 & DR5 | G131R | G131R | 8.7 ± 1.0 | 7.9 ± 1.3 | [73] |
| TRAIL-Mu3 | aa 114–121 (VRERGPQR) were replaced by RRRRRRRR | − 1 | − | [74] | ||
| DR4 | Apo2L.DR4–8 | Y213W/S215D/Y189A/ Q193S/N199V/K201R | 2.3-fold to WT | NB 2 | [75] | |
| TRAIL.R1-6 | Y189A/Q193S/N199V/ K201R/Y213 W/S215N | − | − | [76] | ||
| D218H | D218H | 12.3 ± 0.6 | 28.6 ± 1.7 | [77] | ||
| D218Y | D218Y | 107 ± 0.4 | 23.3 ± 0.4 | [77] | ||
| rhTRAILDR4 | S159R | 0.37 ± 0.12 | 4.3 ± 0.9 | [78] | ||
| 4C7 | G131R/R149I/S159R/ N199R/K201H/S215D | 0.021 ± 0.01 | 7.21 ± 4.2 | [79] | ||
| rhTRAIL-C3 | G131R/N199R/K201H | − | − | [80] | ||
| DR5 | Apo2L.DR5–8 | Y189Q/R191K/Q193R/ H264R/I266L/D267Q | NB | 0.8-fold to WT | [75] | |
| TRAIL.R2-6 | Y189Q/R191K/Q193R/ H264R/I266L/D267Q | − | − | [76] | ||
| D269H/E195R | D269H/E195R | 2.9 ± 1.7 | 0.012 ± 0.005 | [81] | ||
| DR5-A | Y189N/R191K/Q193R/ H264R/I266L/D267Q/D269H | NB 2 | 0.33 ± 0.005 | [82] | ||
| DR5-B | Y189N/R191K/Q193R/ H264R/I266L/D269H | NB | 0.71 ± 0.013 | [82] | ||
1–means not available. 2 NB means no binding.
Table 3.
The mutation sites and binding affinity of the receptor-specific RANKL variants.
| Ligand | Specificity | Variants | Mutation Sites | Binding Affinity (pM) | Ref. | |
|---|---|---|---|---|---|---|
| RANK | OPG | |||||
| RANKL | RANK | I248D | I248D | − 1 | − | [99] |
| I248K | I248K | 9 ± 2 | − | [100] | ||
| I248Y | I248Y | 8 ± 3 | − | [100] | ||
| rRANKL5 | aa 246–318 deletion | − | − | [103] | ||
| Q236D | Q236D | 15.0 ± 3.2 | 112.3 ± 24.4 | [104] | ||
1–means not available.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Suo, F.; Zhou, X.; Setroikromo, R.; Quax, W.J. Receptor Specificity Engineering of TNF Superfamily Ligands. Pharmaceutics 2022, 14, 181. https://doi.org/10.3390/pharmaceutics14010181
AMA Style
Suo F, Zhou X, Setroikromo R, Quax WJ. Receptor Specificity Engineering of TNF Superfamily Ligands. Pharmaceutics. 2022; 14(1):181. https://doi.org/10.3390/pharmaceutics14010181
Chicago/Turabian StyleSuo, Fengzhi, Xinyu Zhou, Rita Setroikromo, and Wim J. Quax. 2022. "Receptor Specificity Engineering of TNF Superfamily Ligands" Pharmaceutics 14, no. 1: 181. https://doi.org/10.3390/pharmaceutics14010181
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.