
An Adaptive Biosystems Engineering Approach towards Modeling the Soluble-to-Insoluble Phase Transition of Clofazimine

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
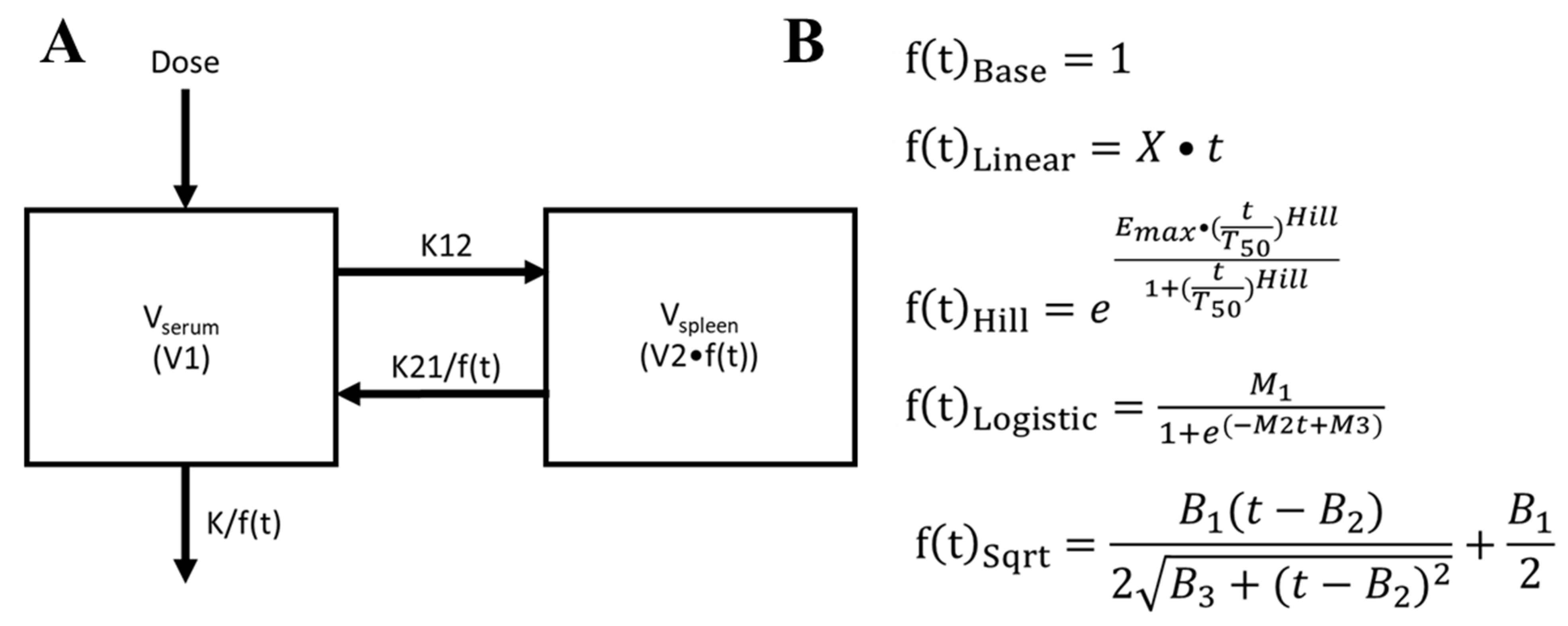
2.1. Data Acquisition and Compartmental Pharmacokinetic Modeling
2.2. Optimizing Compartmental Pharmacokinetics Modeling by Incorporating an Adaptive Vd Function
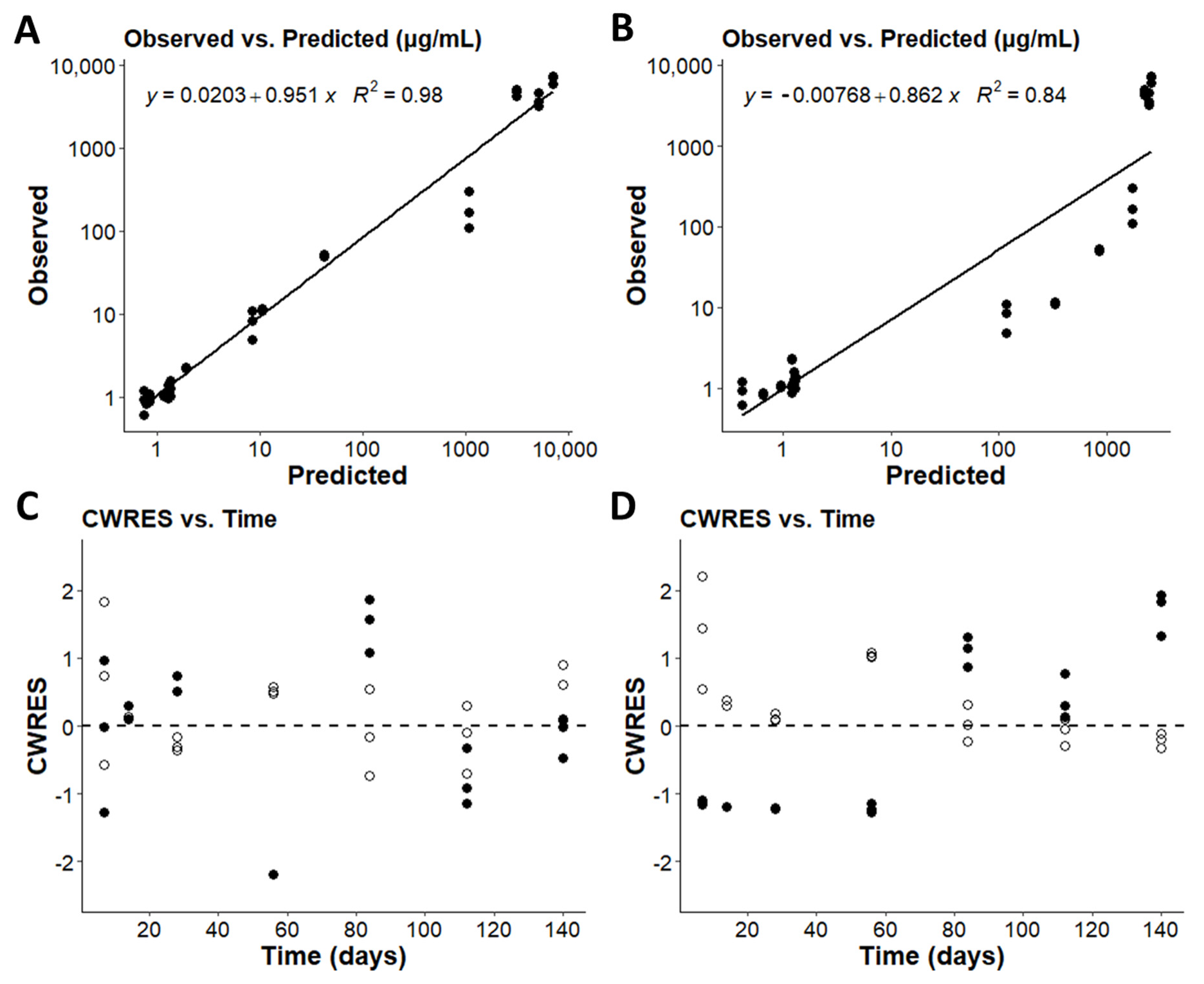
2.3. Model Validation
2.4. Variable Clofazimine (CFZ) Mass Sequestration
2.5. Statistical Analysis of Experimental Data
3. Results
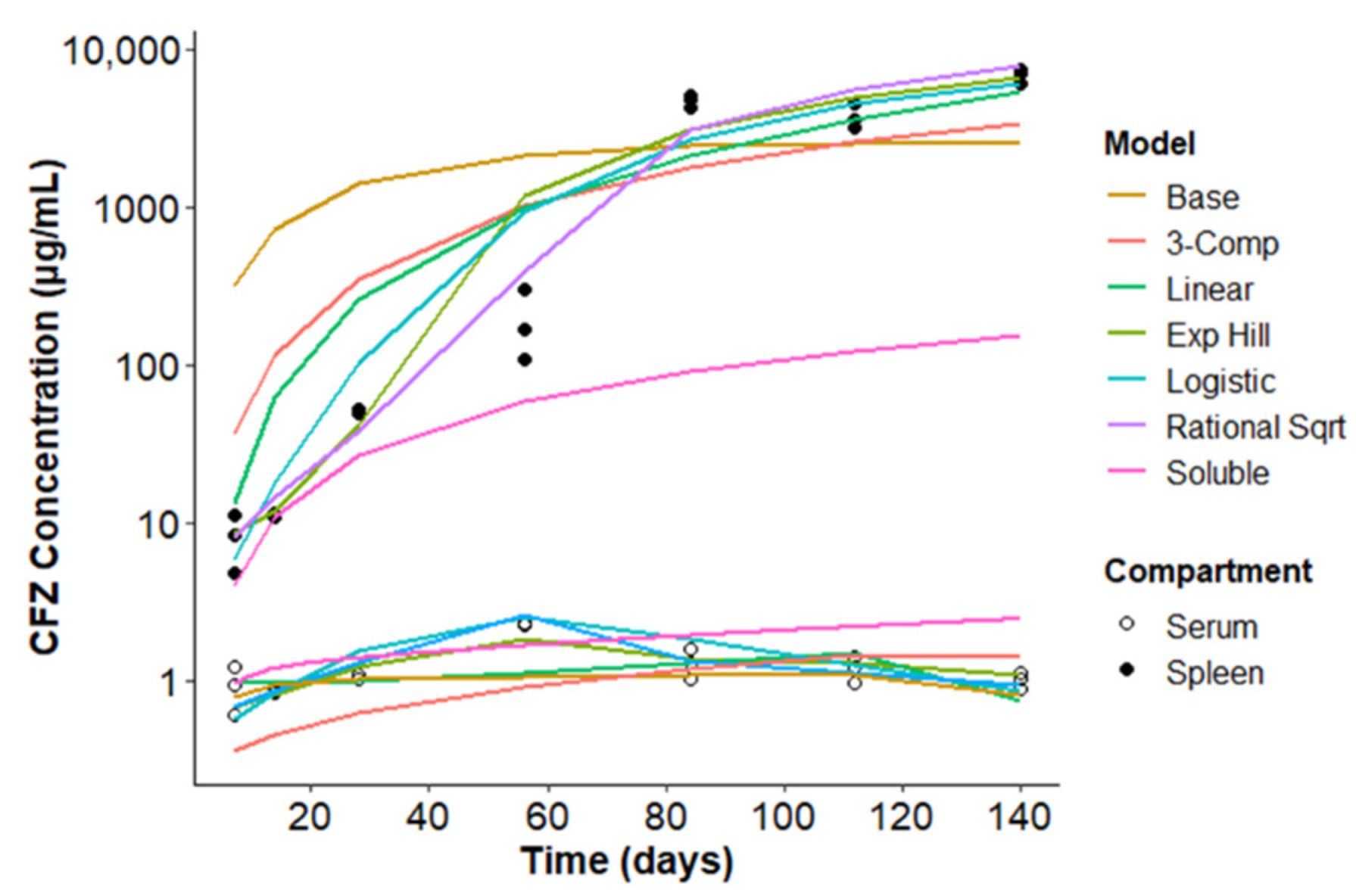
3.1. A Baseline, Compartmental Pharmacokinetics Model to Capture the Soluble-to-Insoluble Phase Transition of CFZ
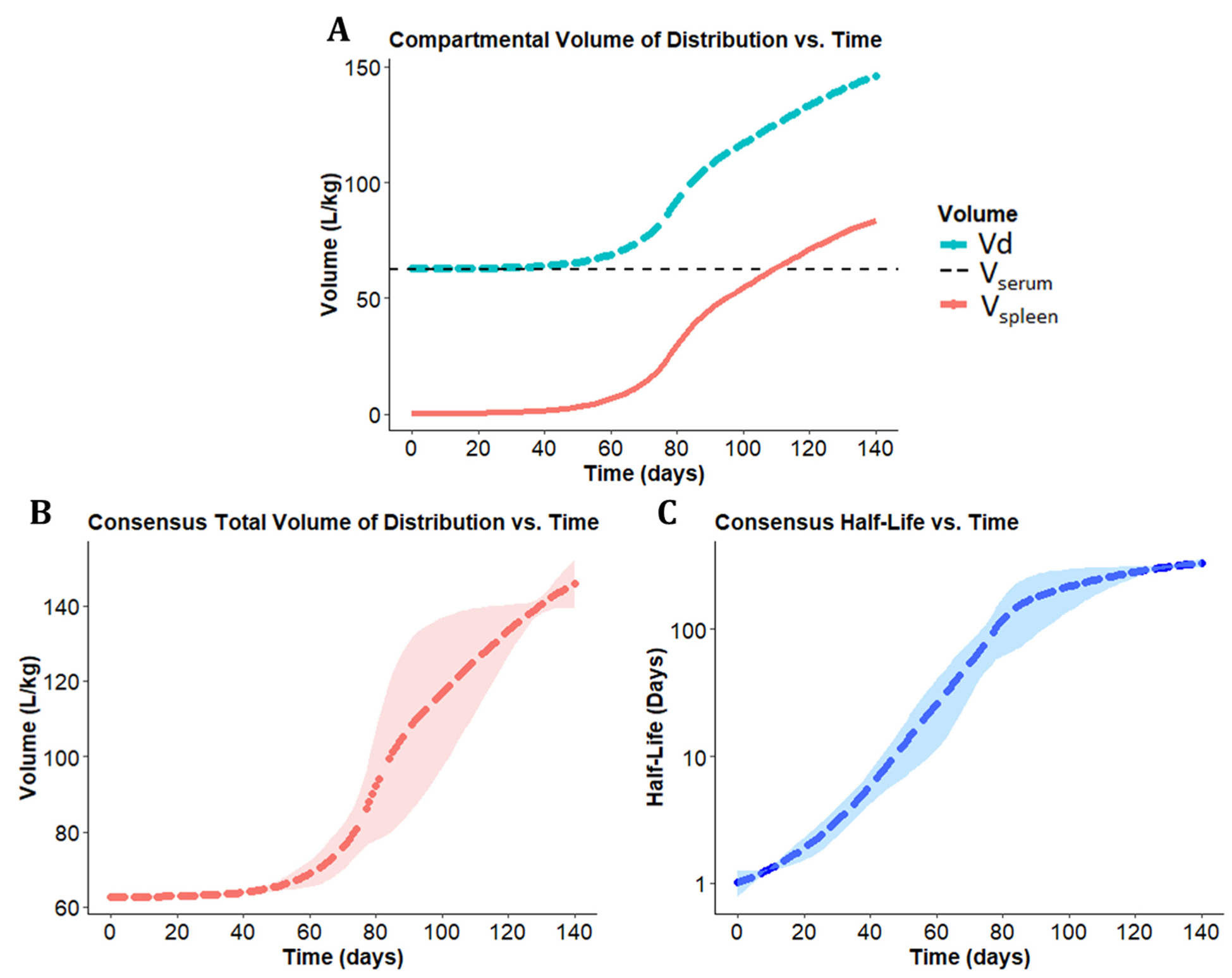
3.2. Optimizing the Two-Compartment Model by including an Adaptive Vd Expansion Function
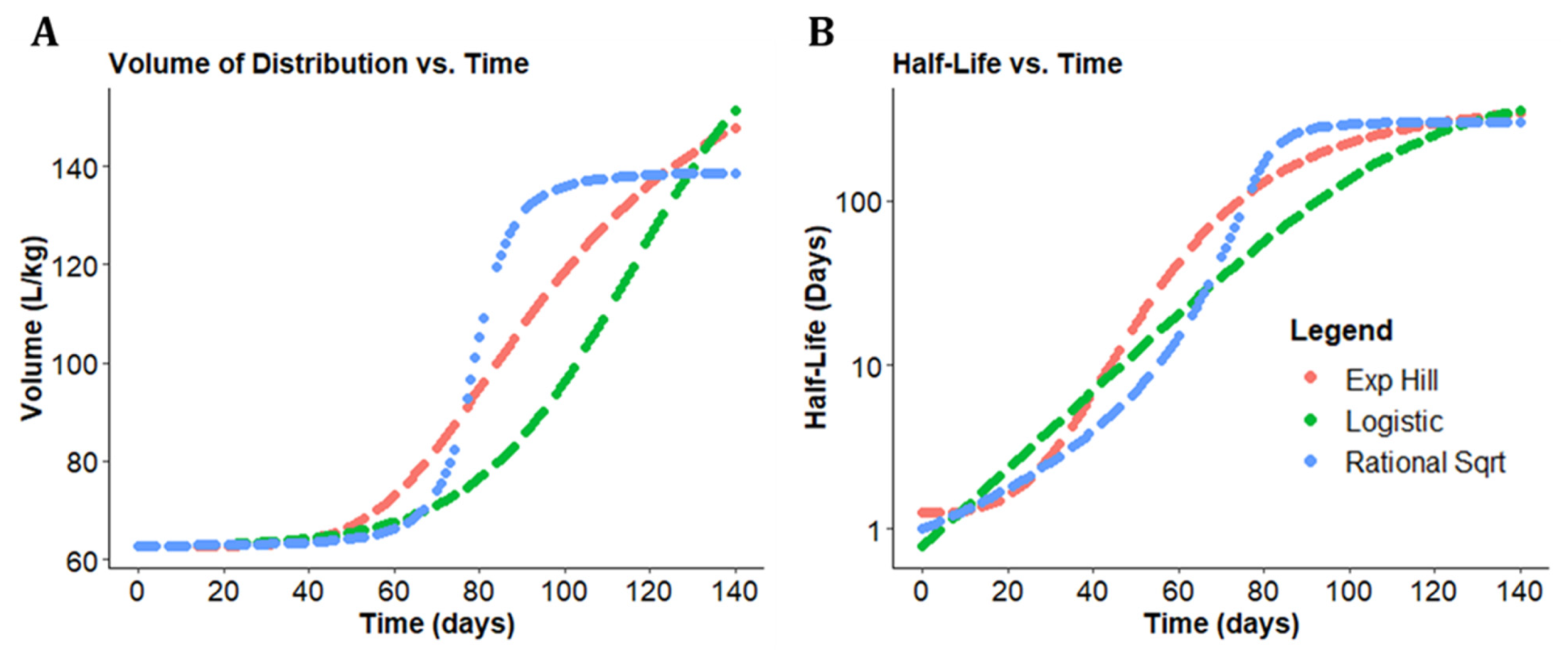
3.3. A Consensus Pharmacokinetics Model of the Expanding Vd of CFZ
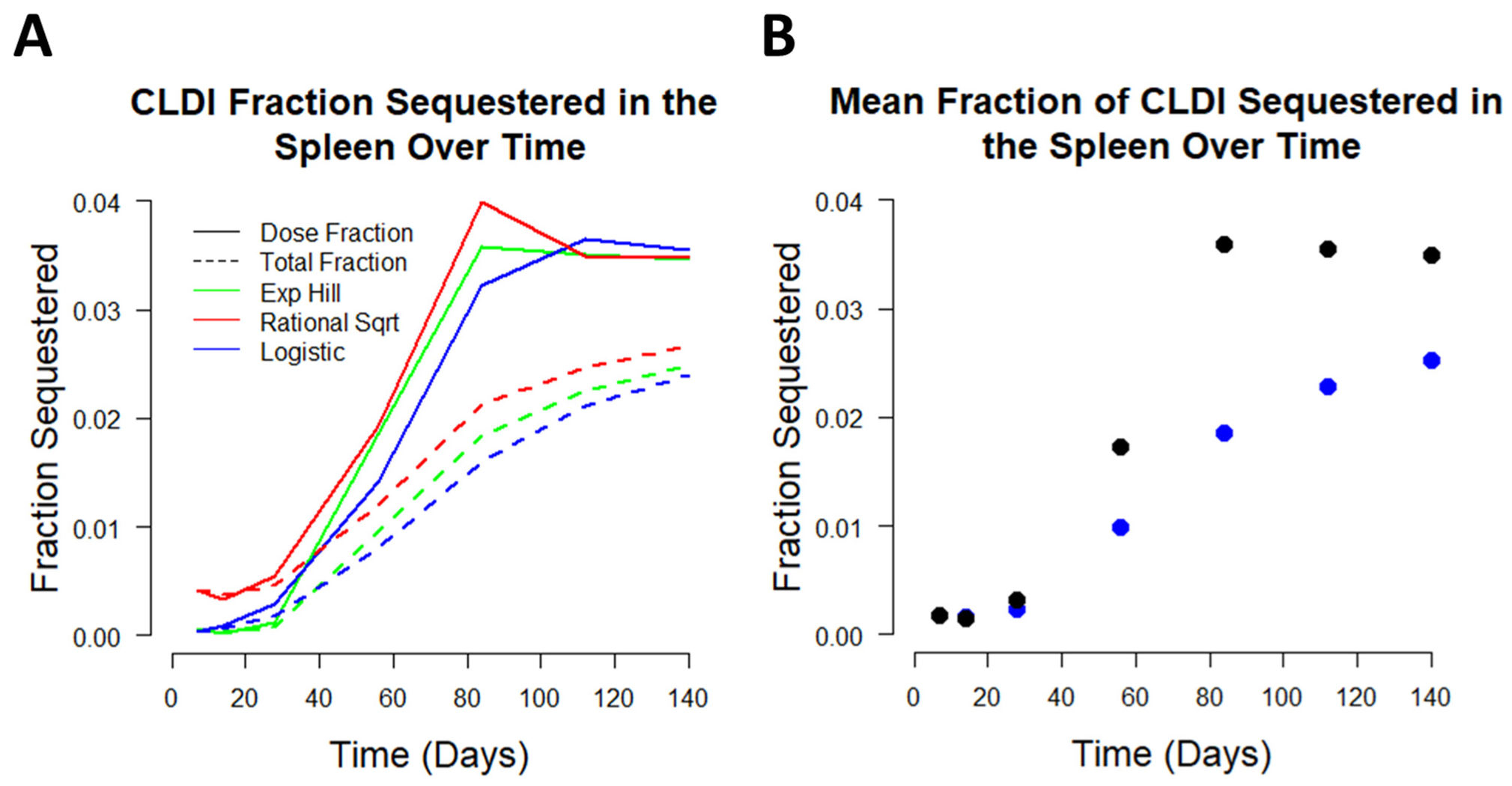
3.4. Analyzing CFZ Time-Dependent Pharmacokinetics and Mass Accumulation in the Spleen
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yuan, S.; Yin, X.; Meng, X.; Chan, J.F.-W.; Ye, Z.-W.; Riva, L.; Pache, L.; Chan, C.C.-Y.; Lai, P.-M.; Poon, V.K.-M.; et al. Clofazimine broadly inhibits coronaviruses including SARS-CoV-2. Nat. Cell Biol. 2021, 593, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef] [PubMed]
- “Dual Therapy With Interferon Beta-1b and Clofazimine for COVID-19—Full Text View.” Full Text View—ClinicalTrials.gov. Available online: Clinicaltrials.gov/ct2/show/NCT04465695 (accessed on 14 September 2021).
- Keswani, R.K.; Yoon, G.S.; Sud, S.; Stringer, K.A.; Rosania, G.R. A far-red fluorescent probe for flow cytometry and image-based functional studies of xenobiotic sequestering macrophages. Cytom. Part A 2015, 87, 855–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzeczycki, P.; Yoon, G.S.; Keswani, R.K.; Sud, S.; Stringer, K.A.; Rosania, G.R. Detecting ordered small molecule drug aggregates in live macrophages: A multi-parameter microscope image data acquisition and analysis strategy. Biomed. Opt. Express 2017, 8, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Rzeczycki, P.; Woldemichael, T.; Willmer, A.; Murashov, M.D.; Baik, J.; Keswani, R.; Yoon, G.S.; Stringer, K.A.; Rodriguez-Hornedo, N.; Rosania, G.R. An Expandable Mechanopharmaceutical Device (1): Measuring the Cargo Capacity of Macrophages in a Living Organism. Pharm. Res. 2018, 36, 12. [Google Scholar] [CrossRef] [PubMed]
- Logan, R.; Kong, A.C.; Axcell, E.; Krise, J.P. Amine-Containing Molecules and the Induction of an Expanded Lysosomal Volume Phenotype: A Structure–Activity Relationship Study. J. Pharm. Sci. 2014, 103, 1572–1580. [Google Scholar] [CrossRef]
- Woldemichael, T.; Keswani, R.K.; Rzeczycki, P.M.; Murashov, M.D.; LaLone, V.; Gregorka, B.; Swanson, J.A.; Stringer, K.A.; Rosania, G.R. Reverse Engineering the Intracellular Self-Assembly of a Functional Mechanopharmaceutical Device. Sci. Rep. 2018, 8, 2934. [Google Scholar] [CrossRef] [PubMed]
- Baik, J.; Stringer, K.A.; Mane, G.; Rosania, G.R. Multiscale Distribution and Bioaccumulation Analysis of Clofazimine Reveals a Massive Immune System-Mediated Xenobiotic Sequestration Response. Antimicrob. Agents Chemother. 2012, 57, 1218–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbiser, J.L.; Moschella, S.L. Clofazimine: A review of its medical uses and mechanisms of action. J. Am. Acad. Dermatol. 1995, 32, 241–247. [Google Scholar] [CrossRef]
- Cholo, M.C.; Steel, H.; Fourie, P.B.; Germishuizen, W.A.; Anderson, R. Clofazimine: Current status and future prospects. J. Antimicrob. Chemother. 2012, 67, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Abdelwahab, M.T.; Wasserman, S.; Brust, J.C.M.; Gandhi, N.R.; Meintjes, G.; Everitt, D.; Diacon, A.; Dawson, R.; Wiesner, L.; Svensson, E.M.; et al. Clofazimine pharmacokinetics in patients with TB: Dosing implications. J. Antimicrob. Chemother. 2020, 75, 3269–3277. [Google Scholar] [CrossRef] [PubMed]
- Swanson, R.; Adamson, J.; Moodley, C.; Ngcobo, B.; Ammerman, N.C.; Dorasamy, A.; Moodley, S.; Mgaga, Z.; Tapley, A.; Bester, L.A.; et al. Pharmacokinetics and Pharmacodynamics of Clofazimine in a Mouse Model of Tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 3042–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbeck, R.J.; Worthen, G.S.; Lebo, T.D.; Peloquin, C.A. Clofazimine crystals in the cytoplasm of pulmonary macrophages. Ann. Pharmacother. 1999, 33, 250. [Google Scholar] [CrossRef] [PubMed]
- Olofsen, E.; Dahan, A. Using Akaike’s information theoretic criterion in mixed-effects modeling of pharmacokinetic data: A simulation study. F1000Research 2013, 2, 71. [Google Scholar] [CrossRef]
- Desikan, K.V.; Ramanujam, K.; Ramu, G.; Balakrishnan, S. Autopsy Findings in a Case of Lepromatous Leprosy Treated with Clofazimine. Lepr. Rev. 1975, 46, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Conalty, M.L. Rimino-phenazines and the reticulo-endothelial system. Ir. J. Med Sci. 1966, 41, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Yoon, G.S.; Keswani, R.K.; Sud, S.; Rzeczycki, P.M.; Murashov, M.D.; Koehn, T.A.; Standiford, T.J.; Stringer, K.A.; Rosania, G.R. Clofazimine Biocrystal Accumulation in Macrophages Upregulates Interleukin 1 Receptor Antagonist Production To Induce a Systemic Anti-Inflammatory State. Antimicrob. Agents Chemother. 2016, 60, 3470–3479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladue, R.P.; Snider, M.E. Intracellular accumulation of azithromycin by cultured human fibroblasts. Antimicrob. Agents Chemother. 1990, 34, 1056–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honegger, U.E.; Zuehlke, R.D.; Scuntaro, I.; Schaefer, M.H.; Toplak, H.; Wiesmann, U.N. Cellular accumulation of amiodarone and desethylamiodarone in cultured human cells: Consequences of drug accumulation on cellular lipid metabolism and plasma membrane properties of chronically exposed cells. Biochem. Pharmacol. 1993, 45, 349–356. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | R2 | AIC | ^ V1 (L/kg) | V2 (L/kg) | K (1/day) | ^ K12 (1/day) | K21 (1/day) | Variable 1 | Variable 2 | Variable 3 |
|---|---|---|---|---|---|---|---|---|---|---|
| Base Model | 0.84 | 319.05 | 2.43 | 0.00516 (0%) | 0.033 (0%) | 0.183 | 0.00287 (33.8%) | N/A | N/A | N/A |
| Linear | 0.95 | 255.8 | 2.43 | 0.0119 (32.8%) | 0.275 (56.5%) | 0.183 | 0.125 (49.1%) | * 0.219 (66.7%) | N/A | N/A |
| Exponential Hill Equation | 0.98 | 211.9 | 2.43 | 0.00708 (1.61%) | 0.229 (12.6%) | 0.183 | 3.36 (1.53%) | * 6.99 (0%) | * 49 (2.68%) | 3.71 (2.59%) |
| Basic Logistic Function | 0.97 | 238.9 | 2.43 | 0.00779 (4.73%) | 0.00645 (111.3%) | 0.183 | 0.0762 (96.2%) | * 99.1 (56.2%) | * 0.0701 (2.82%) | 8.85 (3.60%) |
| Rational Square Root Sigmoid | 0.99 | 200.8 | 2.43 | 0.00533 (2.00%) | 0.107 (54.1%) | 0.183 | 2.16 (53.4%) | * 1150 (32.3%) | 80.2 (3.20%) | * 8.84 (36.5%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willmer, A.R.; Dunne, S.; Swanson, R.; Almeida, D.; Ammerman, N.C.; Stringer, K.A.; Capparelli, E.V.; Rosania, G.R. An Adaptive Biosystems Engineering Approach towards Modeling the Soluble-to-Insoluble Phase Transition of Clofazimine. Pharmaceutics 2022, 14, 17. https://doi.org/10.3390/pharmaceutics14010017
Willmer AR, Dunne S, Swanson R, Almeida D, Ammerman NC, Stringer KA, Capparelli EV, Rosania GR. An Adaptive Biosystems Engineering Approach towards Modeling the Soluble-to-Insoluble Phase Transition of Clofazimine. Pharmaceutics. 2022; 14(1):17. https://doi.org/10.3390/pharmaceutics14010017
Chicago/Turabian StyleWillmer, Andrew R., Steven Dunne, Rosemary Swanson, Deepak Almeida, Nicole C. Ammerman, Kathleen A. Stringer, Edmund V. Capparelli, and Gus R. Rosania. 2022. "An Adaptive Biosystems Engineering Approach towards Modeling the Soluble-to-Insoluble Phase Transition of Clofazimine" Pharmaceutics 14, no. 1: 17. https://doi.org/10.3390/pharmaceutics14010017