
Predicting Drug Release from 3D Printed Oral Medicines Based on the Surface Area to Volume Ratio of Tablet Geometry


Abstract
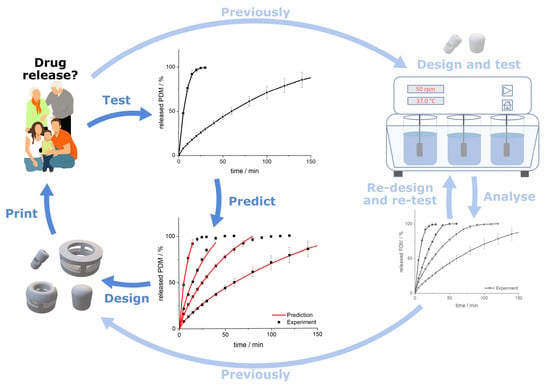
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Hot Melt Extrusion
2.2.2. 3D Printing of Tablets
2.2.3. Dissolution Test
2.2.4. Mathematical Description
Release Modeling
Prediction
Comparison of the Dissolution Profiles
2.2.5. Characterization of the Printed Tablets
3. Results
3.1. Characterization of the Printed Tablets
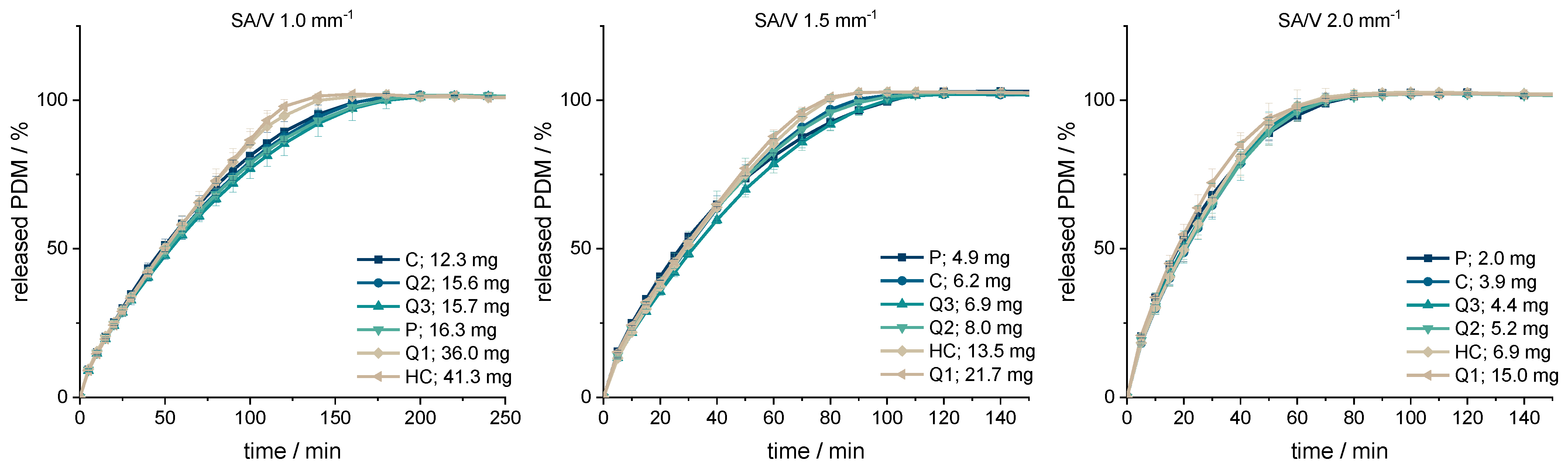
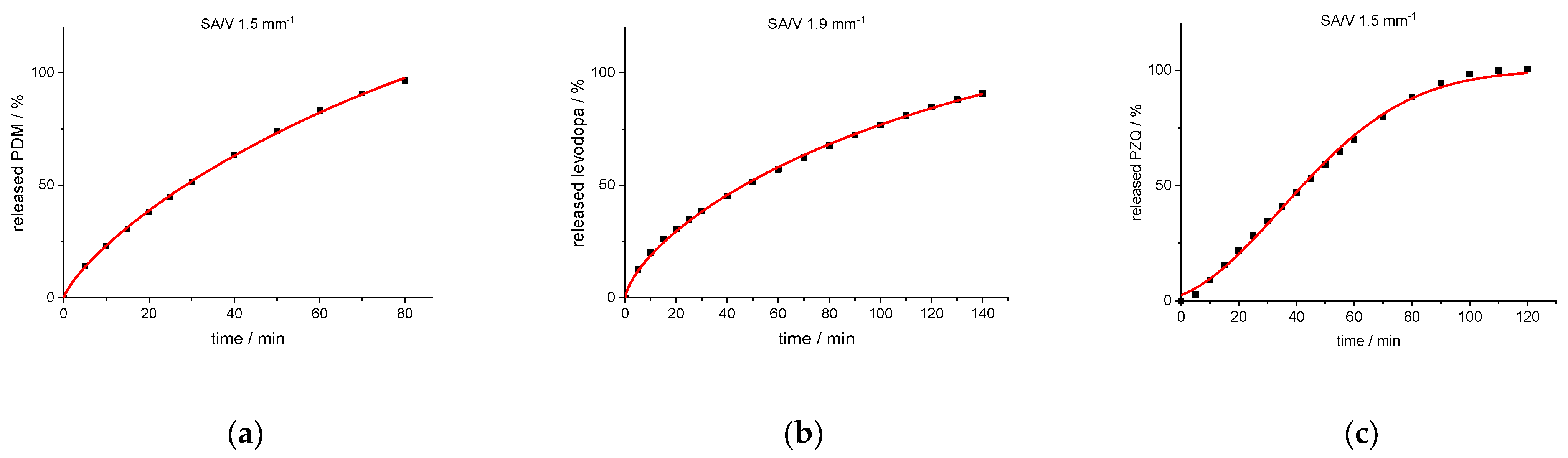
3.2. Drug Release from Dosage Forms with Defined SA/V Ratios
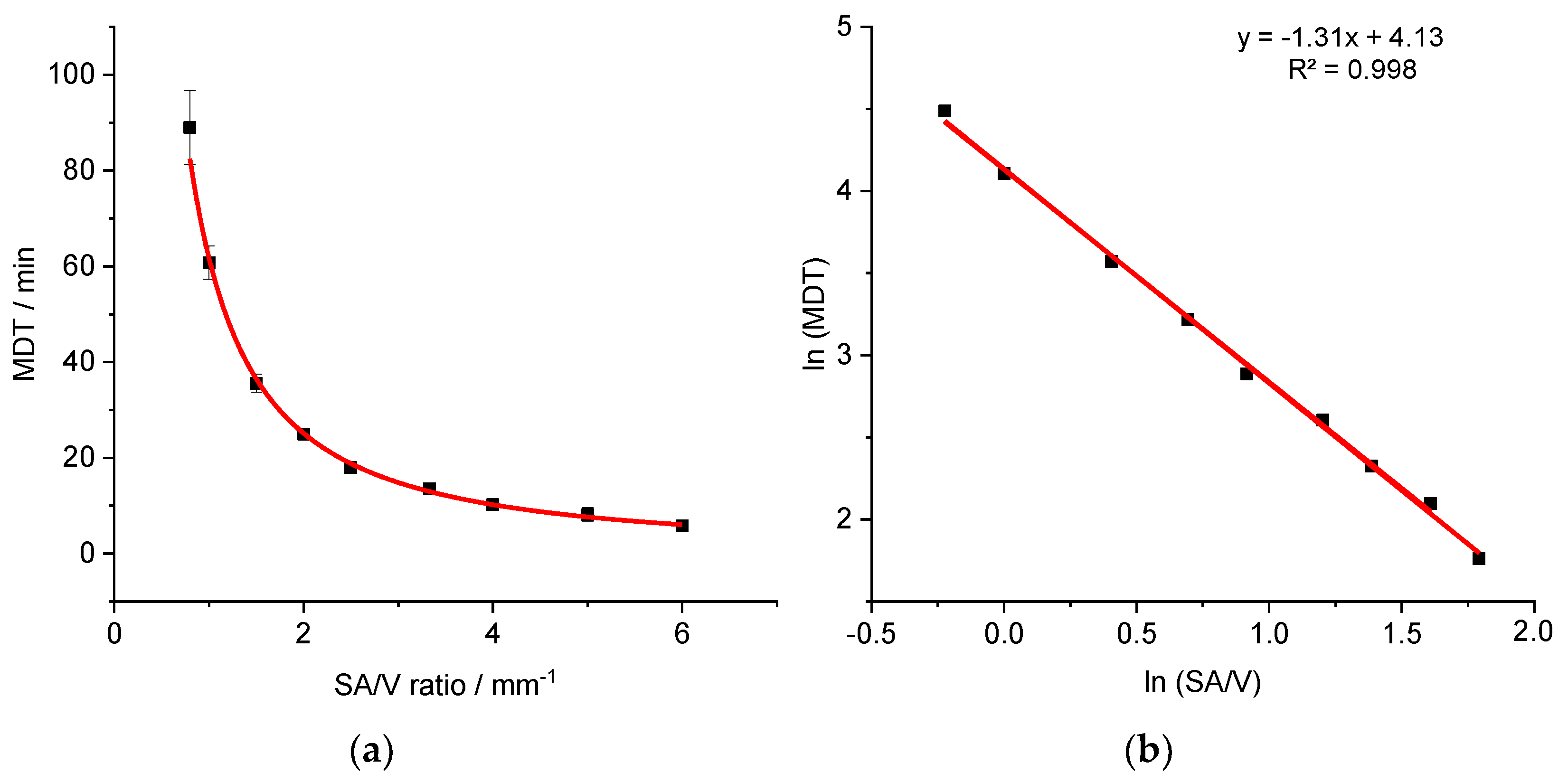
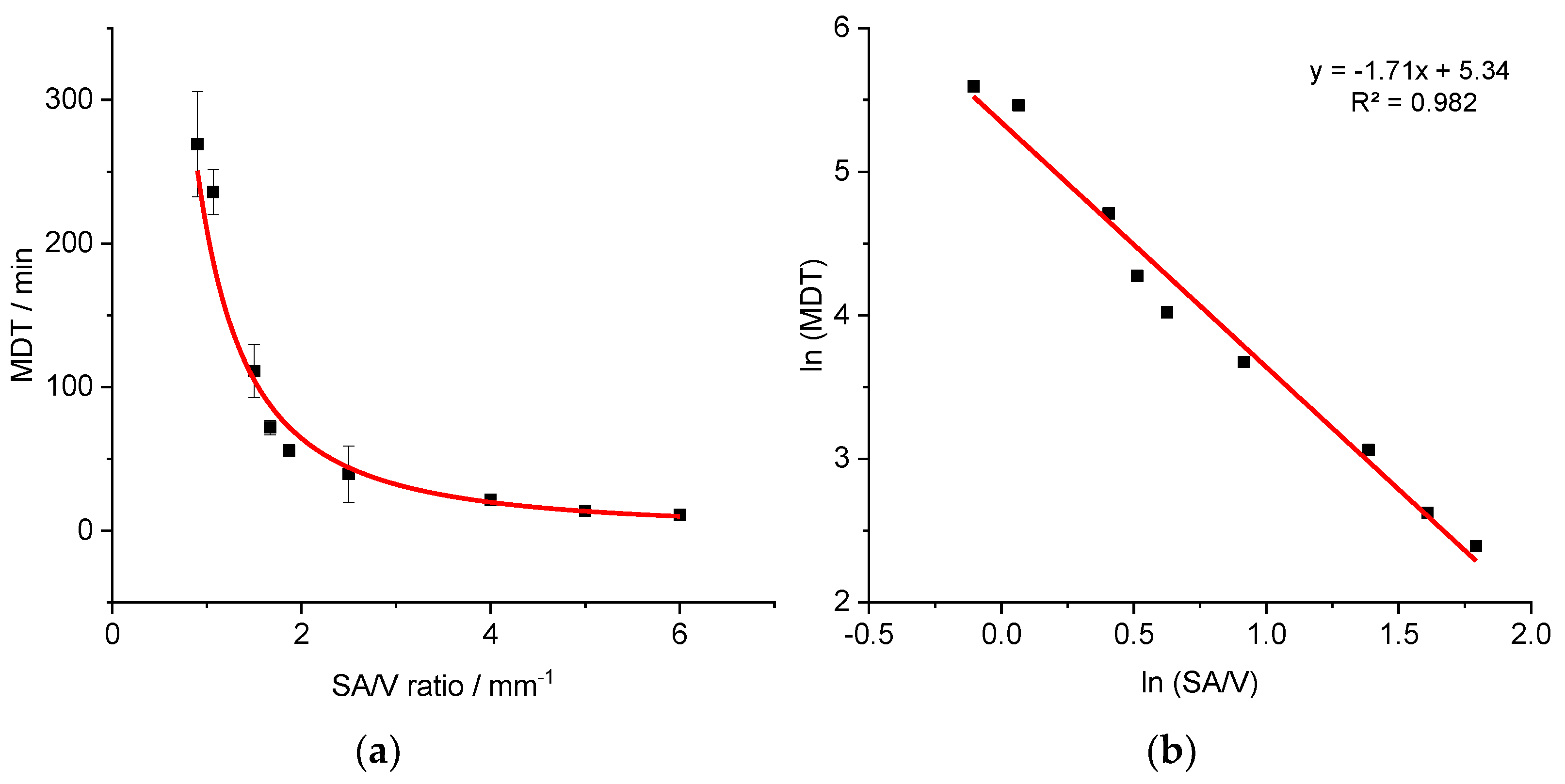
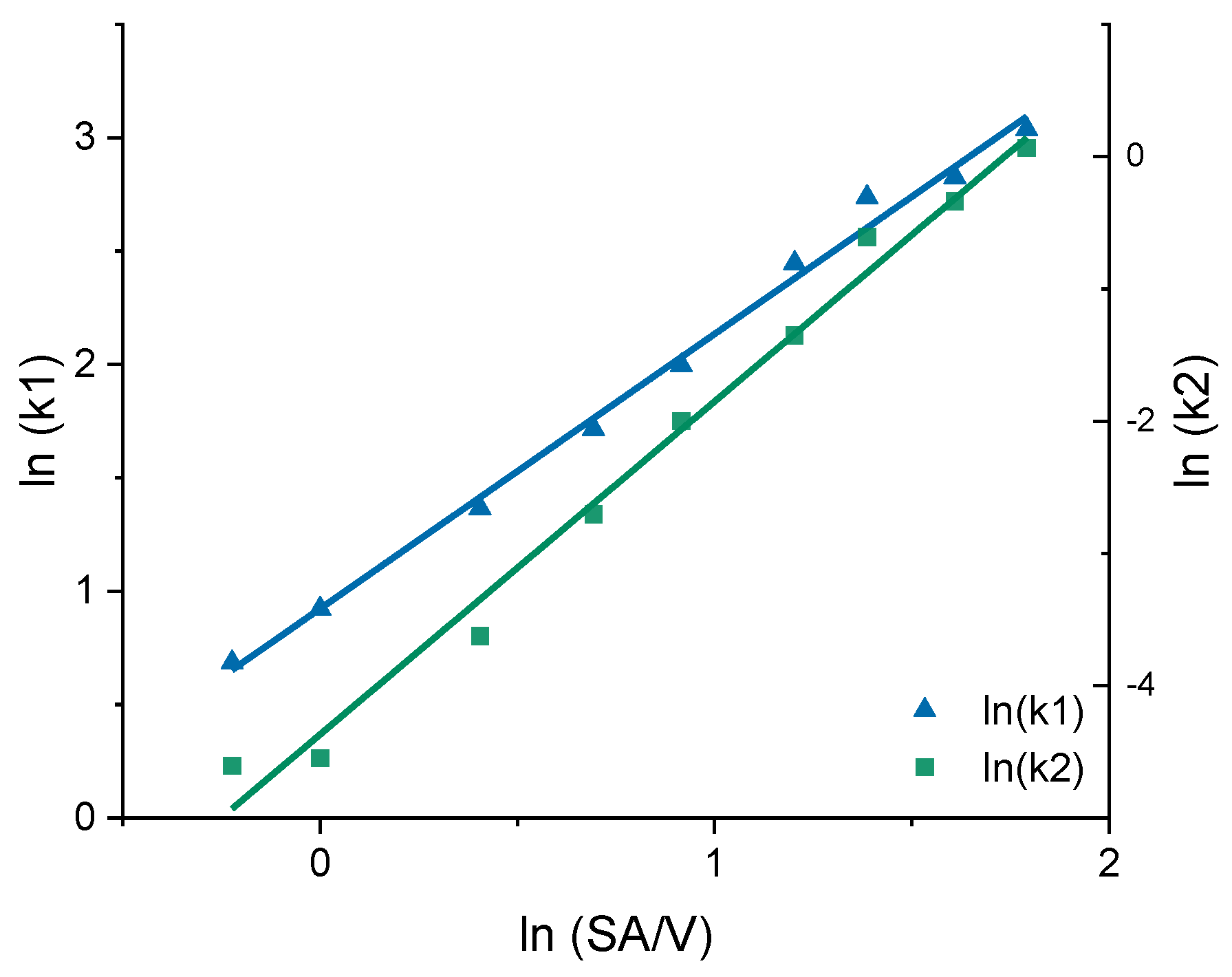
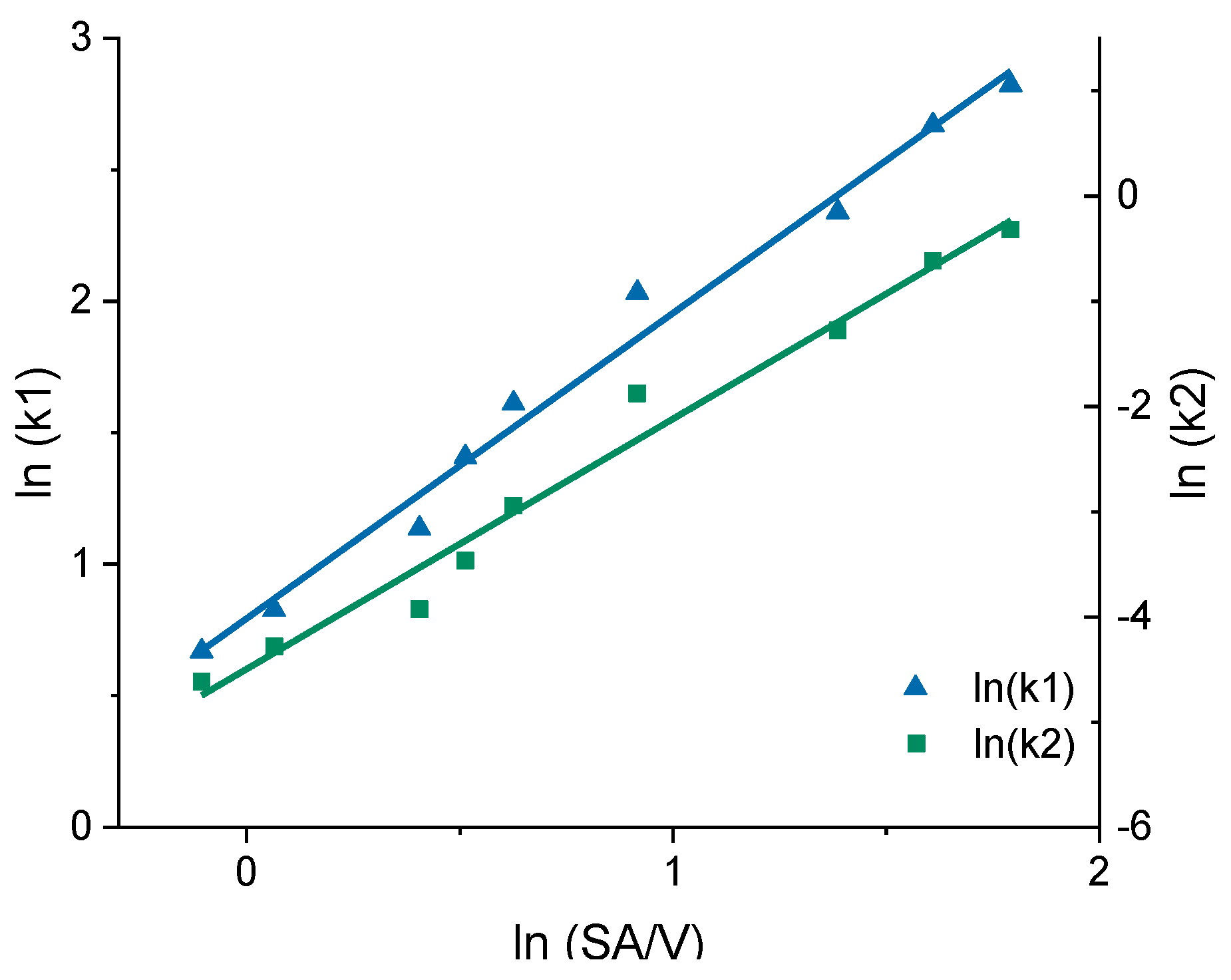
3.3. Correlation between MDT and SA/V Ratio
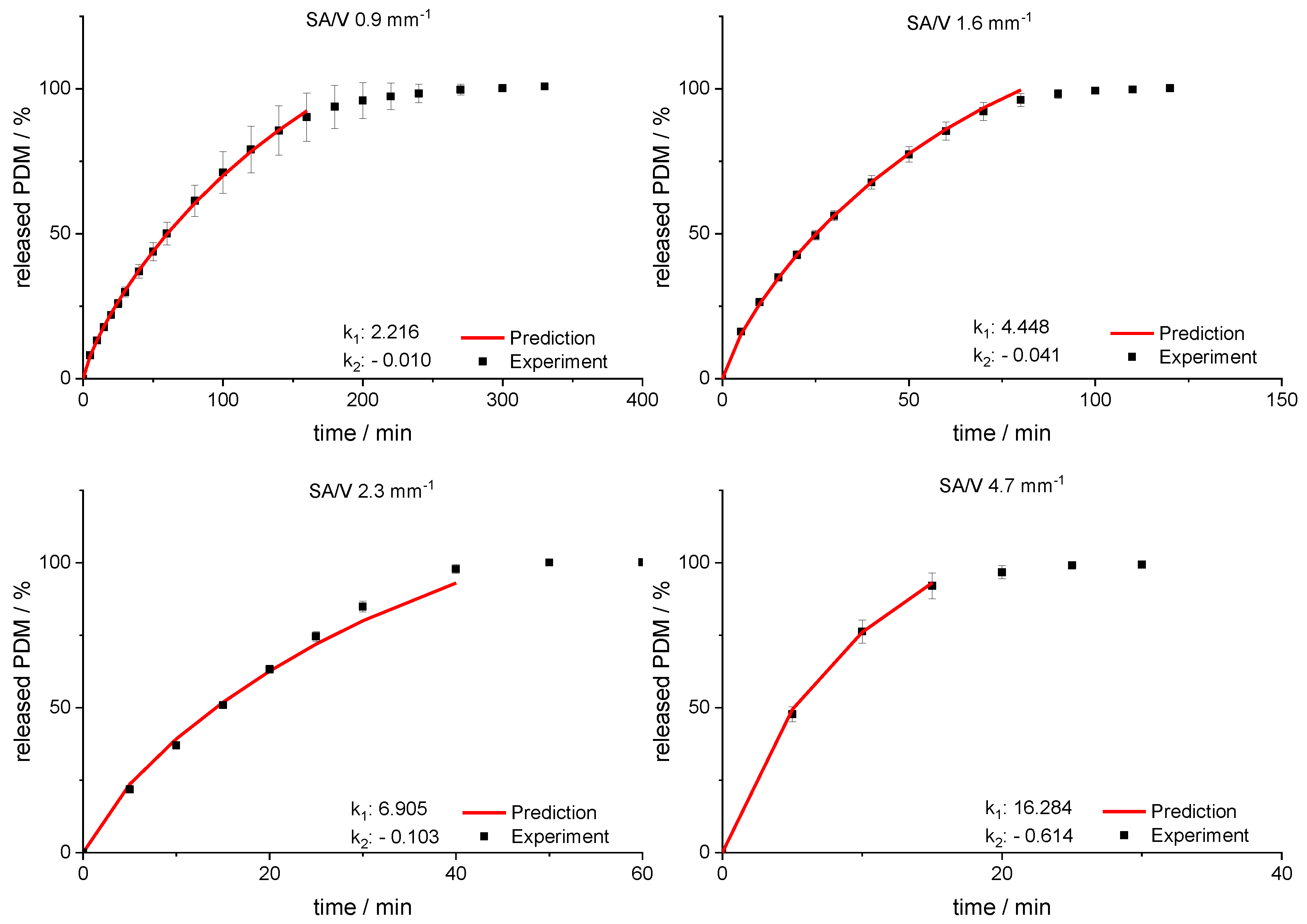
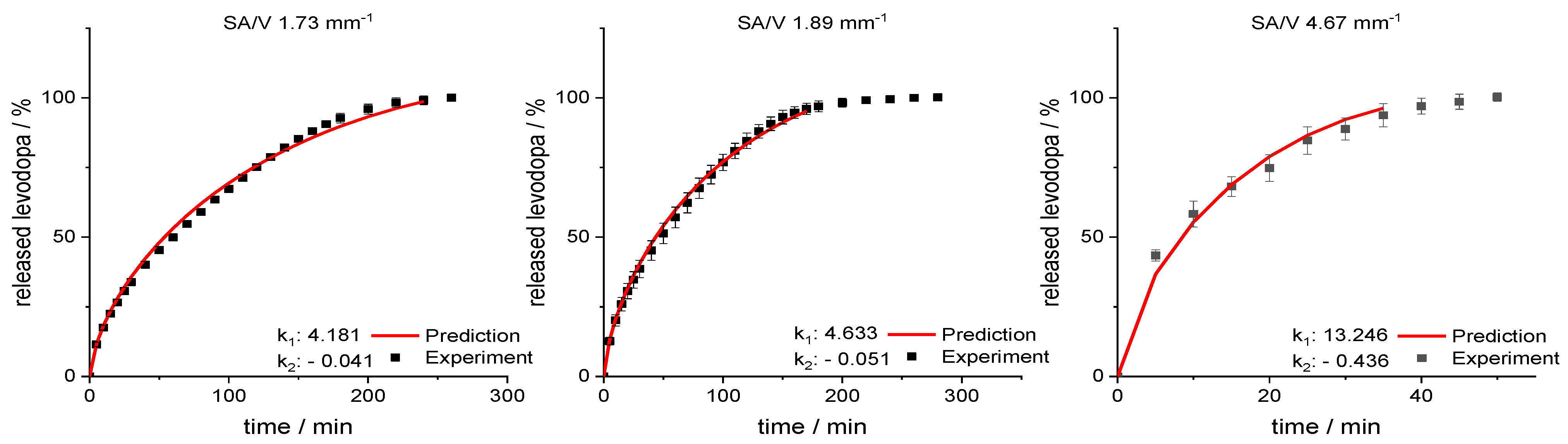
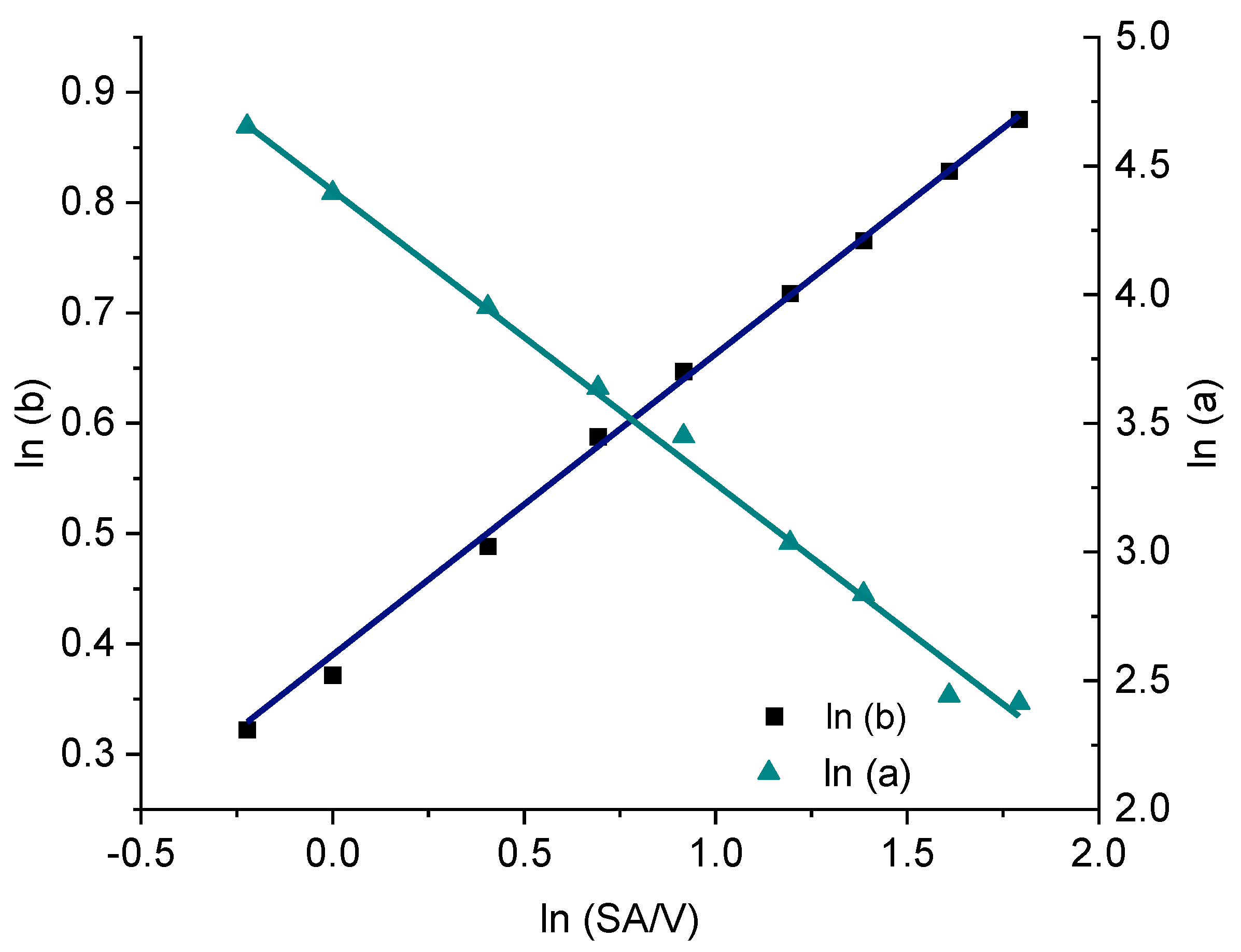
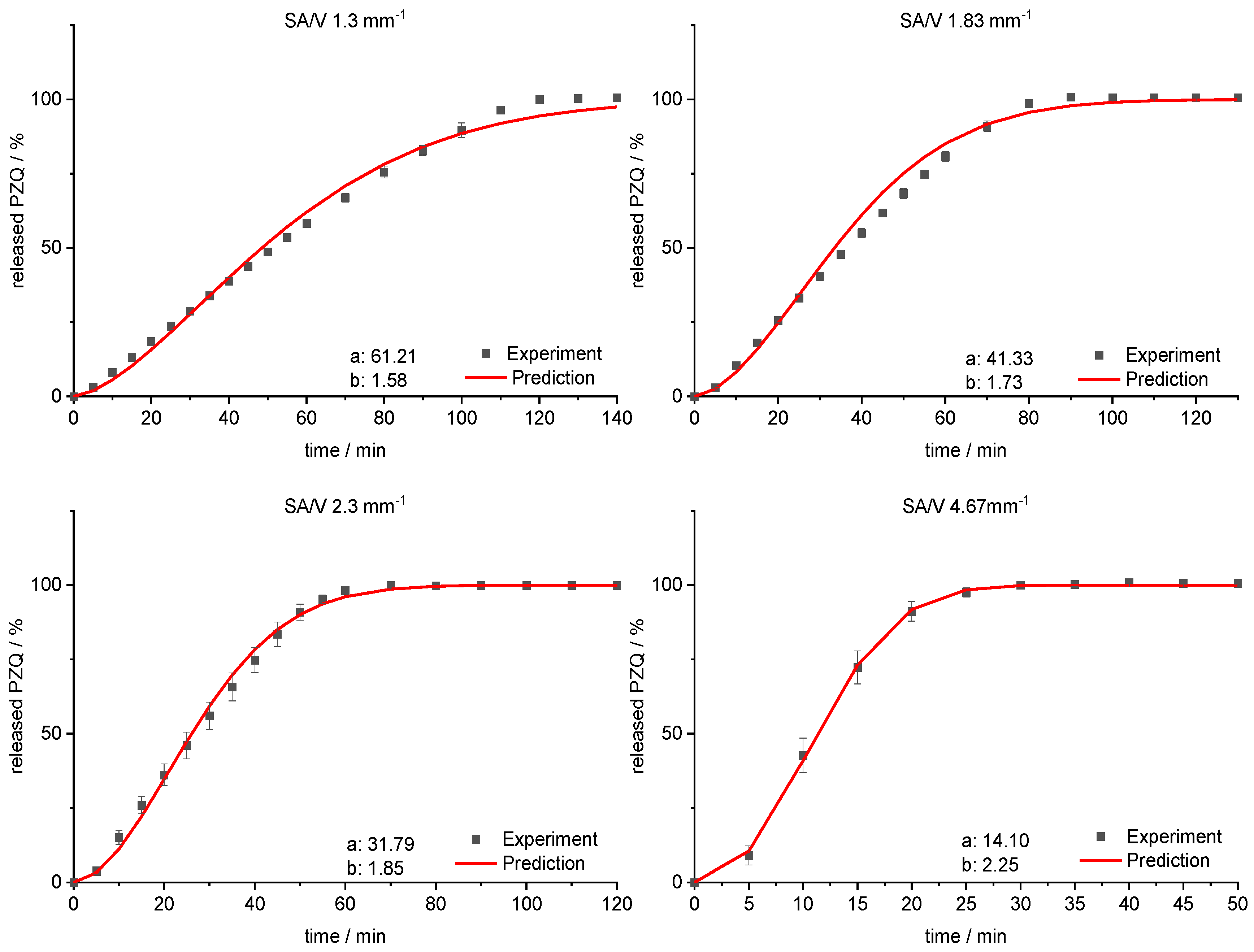
3.4. Modeling and Prediction of Release Profiles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hamburg, M.A.; Collins, F.S. The path to personalized medicine. N. Engl. J. Med. 2010, 363, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Lee, S.; Son, S.; Kim, S.H.; Leary, J.F.; Choi, K.; Kwon, I.C. Theranostic nanoparticles for future personalized medicine. J. Control. Release 2014, 190, 477–484. [Google Scholar] [CrossRef]
- Tan, D.K.; Maniruzzaman, M.; Nokhodchi, A. Advanced pharmaceutical applications of hot-melt extrusion coupled with Fused Deposition Modelling (FDM) 3D printing for personalised drug delivery. Pharmaceutics 2018, 10, 203. [Google Scholar] [CrossRef] [Green Version]
- Breitkreutz, J.; Boos, J. Paediatric and geriatric drug delivery. Expert Opin. Drug Deliv. 2007, 4, 37–45. [Google Scholar] [CrossRef]
- Collins, F.S.; Varmus, H. A New initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodson, R. Precision medicine. Nature 2016, 537, S49. [Google Scholar] [CrossRef] [Green Version]
- van Santen, E.; Barends, D.M.; Frijlink, H.W. Breaking of scored tablets: A review. Eur. J. Pharm. Biopharm. 2002, 53, 139–145. [Google Scholar] [CrossRef]
- Quinzler, R.; Gasse, C.; Schneider, A.; Kaufmann-Kolle, P.; Szecsenyi, J.; Haefeli, W.E. The frequency of inappropriate tablet splitting in primary care. Eur. J. Clin. Pharmacol. 2006, 62, 1065–1073. [Google Scholar] [CrossRef]
- Wening, K.; Breitkreutz, J. Oral drug delivery in personalized medicine: Unmet needs and novel approaches. Int. J. Pharm. 2011, 404, 1–9. [Google Scholar] [CrossRef]
- Goyanes, A.; Det-Amornrat, U.; Wang, J.; Basit, A.W.; Gaisford, S. 3D Scanning and 3D printing as innovative technologies for fabricating personalized topical drug delivery systems. J. Control. Release 2016, 234, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Alhnan, M.A.; Okwuosa, T.C.; Sadia, M.; Wan, K.W.; Ahmed, W.; Arafat, B. Emergence of 3D printed dosage forms: Opportunities and challenges. Pharm. Res. 2016, 33, 1817–1832. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; Barnes, M.; McMillin, R.; Cook, D.W.; Smith, S.; Halquist, M.; Wijesinghe, D.; Roper, T.D. 3D printing of Metformin HCl PVA tablets by fused deposition modeling: Drug loading, tablet design, and dissolution studies. AAPS PharmSciTech 2019, 20, 195. [Google Scholar] [CrossRef] [PubMed]
- Rahman, J.; Quodbach, J. Versatility on demand—The case for semi-solid micro-extrusion in pharmaceutics. Adv. Drug Deliv. Rev. 2021, 172, 104–126. [Google Scholar] [CrossRef] [PubMed]
- Seoane-Viaño, I.; Januskaite, P.; Alvarez-Lorenzo, C.; Basit, A.W.; Goyanes, A. Semi-solid extrusion 3D printing in drug delivery and biomedicine: Personalised solutions for healthcare challenges. J. Control. Release 2021, 332, 367–389. [Google Scholar] [CrossRef]
- el Aita, I.; Rahman, J.; Breitkreutz, J.; Quodbach, J. 3D-printing with precise layer-wise dose adjustments for paediatric use via pressure-assisted microsyringe printing. Eur. J. Pharm. Biopharm. 2020, 157, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Korte, C.; Quodbach, J. Formulation development and process analysis of drug-loaded filaments manufactured via hot-melt extrusion for 3D-printing of medicines. Pharm. Dev. Technol. 2018, 23, 1117–1127. [Google Scholar] [CrossRef]
- Melocchi, A.; Parietti, F.; Maroni, A.; Foppoli, A.; Gazzaniga, A.; Zema, L. Hot-melt extruded filaments based on pharmaceutical grade polymers for 3D printing by fused deposition modeling. Int. J. Pharm. 2016, 509, 255–263. [Google Scholar] [CrossRef]
- Ponsar, H.; Wiedey, R.; Quodbach, J. Hot-melt extrusion process fluctuations and their impact on critical quality attributes of filaments and 3D-printed dosage forms. Pharmaceutics 2020, 12, 511. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, X.; Patil, H.; Tiwari, R.V.; Repka, M.A. Coupling 3D printing with hot-melt extrusion to produce controlled-release tablets. Int. J. Pharm. 2017, 519, 186–197. [Google Scholar] [CrossRef]
- Gültekin, H.E.; Tort, S.; Acartürk, F. An effective technology for the development of immediate release solid dosage forms containing low-dose drug: Fused deposition modeling 3D printing. Pharm. Res. 2019, 36, 128. [Google Scholar] [CrossRef]
- Kollamaram, G.; Croker, D.M.; Walker, G.M.; Goyanes, A.; Basit, A.W.; Gaisford, S. Low temperature Fused Deposition Modeling (FDM) 3D printing of thermolabile drugs. Int. J. Pharm. 2018, 545, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, T.D.; Mitchell, S.A.; Balwinski, K.M. Investigation of the effect of tablet surface area/volume on drug release from hydroxypropylmethylcellulose controlled-release matrix tablets. Drug Dev. Ind. Pharm. 2002, 28, 457–466. [Google Scholar] [CrossRef]
- Goyanes, A.; Robles Martinez, P.; Buanz, A.; Basit, A.W.; Gaisford, S. Effect of geometry on drug release from 3D printed tablets. Int. J. Pharm. 2015, 494, 657–663. [Google Scholar] [CrossRef]
- Sadia, M.; Arafat, B.; Ahmed, W.; Forbes, R.T.; Alhnan, M.A. Channelled tablets: An innovative approach to accelerating drug release from 3D printed tablets. J. Control. Release 2018, 269, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Gorkem Buyukgoz, G.; Soffer, D.; Defendre, J.; Pizzano, G.M.; Davé, R.N. Exploring tablet design options for tailoring drug release and dose via Fused Deposition Modeling (FDM) 3D printing. Int. J. Pharm. 2020, 591, 119987. [Google Scholar] [CrossRef] [PubMed]
- Narayana Raju, P. Effect of tablet surface area and surface area/volume on drug release from lamivudine extended release matrix tablets. Int. J. Pharm. Sci. Nanotechnol. 2010, 3, 872–876. [Google Scholar] [CrossRef]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug delivery. Int. J. Pharm. 2008, 364, 328–343. [Google Scholar] [CrossRef]
- Peppas, N.A.; Sahlin, J.J. A simple equation for the description of solute release. III. Coupling of diffusion and relaxation. Int. J. Pharm. 1989, 57, 169–172. [Google Scholar] [CrossRef]
- Siepmann, J.; Kranz, H.; Peppas, N.A.; Bodmeier, R. Calculation of the required size and shape of hydroxypropyl methylcellulose matrices to achieve desired drug release profiles. Int. J. Pharm. 2000, 201, 151–164. [Google Scholar] [CrossRef]
- Siepmann, J.; Streubel, A.; Peppas, N.A. Understanding and predicting drug delivery from hydrophilic matrix tablets using the “Sequential Layer” model. Pharm. Res. 2002, 3, 306–314. [Google Scholar] [CrossRef]
- Tsunematsu, H.; Hifumi, H.; Kitamura, R.; Hirai, D.; Takeuchi, M.; Ohara, M.; Itai, S.; Iwao, Y. Analysis of available surface area can predict the long-term dissolution profile of tablets using short-term stability studies. Int. J. Pharm. 2020, 586, 119504. [Google Scholar] [CrossRef] [PubMed]
- Baishya, H. Application of mathematical models in drug release kinetics of carbidopa and levodopa ER tablets. J. Dev. Drugs 2017, 6, 1–8. [Google Scholar] [CrossRef]
- Petru, J.; Zamostny, P. Prediction of dissolution behavior of final dosage forms prepared by different granulation methods. Procedia Eng. 2012, 42, 1463–1473. [Google Scholar] [CrossRef] [Green Version]
- Madzarevic, M.; Medarevic, D.; Vulovic, A.; Sustersic, T.; Djuris, J.; Filipovic, N.; Ibric, S. Optimization and prediction of ibuprofen release from 3D DLP printlets using artificial neural networks. Pharmaceutics 2019, 11, 544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siepmann, J. A new model combining diffusion, swelling, and dissolutions mechanisms and predicting the release kinetics. Pharm. Res. 1999, 16, 1748–1756. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Peppas, N.A. Hydrophilic matrices for controlled drug delivery: An improved mathematical model to predict the resulting drug release kinetics the “Sequential Layer” model). Pharm. Res. 2000, 17, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Borgquist, P.; Körner, A.; Piculell, L.; Larsson, A.; Axelsson, A. A model for the drug release from a polymer matrix tablet-effects of swelling and dissolution. J. Control. Release 2006, 113, 216–225. [Google Scholar] [CrossRef]
- Narasimhan, B. Mathematical models describing polymer dissolution: Consequences for drug delivery. Adv. Drug Deliv. Rev. 2001, 48, 195–210. [Google Scholar] [CrossRef]
- Siepmann, J.; Siegel, R.A.; Siepmann, F. Diffusion controlled drug delivery systems. In Fundamentals and Applications of Controlled Release Drug Delivery; Siepmann, J., Siegel, R.A., Eds.; Springer: New York, NY, USA, 2012; pp. 127–152. ISBN 9781461408819. [Google Scholar]
- Siepmann, J.; Siepmann, F. Swelling controlled drug delivery systems. In Fundamentals and Applications of Controlled Release Drug Delivery; Springer: New York, NY, USA, 2012; pp. 153–170. ISBN 9781461408819. [Google Scholar]
- Korte, C.; Quodbach, J. 3D-printed network structures as controlled-release drug delivery systems: Dose adjustment, API release analysis and prediction. AAPS PharmSciTech 2018, 19, 3333–3342. [Google Scholar] [CrossRef]
- Kyobula, M.; Adedeji, A.; Alexander, M.R.; Saleh, E.; Wildman, R.; Ashcroft, I.; Gellert, P.R.; Roberts, C.J. 3D inkjet printing of tablets exploiting bespoke complex geometries for controlled and tuneable drug release. J. Control. Release 2017, 261, 207–215. [Google Scholar] [CrossRef]
- Pishnamazi, M.; Ismail, H.Y.; Shirazian, S.; Iqbal, J.; Walker, G.M.; Collins, M.N. Application of lignin in controlled release: Development of predictive model based on artificial neural network for API release. Cellulose 2019, 26, 6165–6178. [Google Scholar] [CrossRef]
- Takayama, K.; Kawai, S.; Obata, Y.; Todo, H.; Sugibayashi, K. Prediction of dissolution data integrated in tablet database using four-layered artificial neural networks. Chem. Pharm. Bull. 2017, 65, 967–972. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Geraldrajan, M.; Chen, Q.; Sun, Y.; Johnson, J.R.; Shukla, A.J. Prediction of dissolution profiles of acetaminophen beads using artificial neural networks. Pharm. Dev. Technol. 2006, 11, 337–349. [Google Scholar] [CrossRef]
- Ghennam, A.; Rebouh, S.; Bouhedda, M. Application of artificial neural network-genetic algorithm model in the prediction of ibuprofen release from microcapsules and tablets based on plant protein and its derivatives. Lect. Notes Netw. Syst. 2021, 174, 625–634. [Google Scholar] [CrossRef]
- Novák, M.; Boleslavská, T.; Grof, Z.; Waněk, A.; Zadražil, A.; Beránek, J.; Kovačík, P.; Štěpánek, F. Virtual prototyping and parametric design of 3D-printed tablets based on the solution of inverse problem. AAPS PharmSciTech 2018, 19, 3414–3424. [Google Scholar] [CrossRef]
- Komal, C.; Dhara, B.; Sandeep, S.; Shantanu, D.; Priti, M.J. Dissolution-controlled salt of pramipexole for parenteral administration: In Vitro assessment and mathematical modeling. Dissolution Technol. 2019, 26, 28–36. [Google Scholar] [CrossRef]
- Tzankov, B.; Voycheva, C.; Yordanov, Y.; Aluani, D.; Spassova, I.; Kovacheva, D. Development and In Vitro safety evaluation of pramipexole-loaded Hollow Mesoporous Silica (HMS) particles. Biotechnol. Biotechnol. Equip. 2019, 33, 1204–1215. [Google Scholar] [CrossRef] [Green Version]
- Krisai, K.; Charnvanich, D.; Chongcharoen, W. Increasing the solubility of levodopa and carbidopa using ionization approach. Thai J. Pharm. Sci. TJPS 2020, 44, 251–255. [Google Scholar]
- Liu, Y.; Wang, T.; Ding, W.; Dong, C.; Wang, X.; Chen, J.; Li, Y. Dissolution and oral bioavailability enhancement of praziquantel by solid dispersions. Drug Deliv. Transl. Res. 2018, 8, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Dinora, G.E.; Julio, R.; Nelly, C.; Lilian, Y.M.; Cook, H.J. In Vitro characterization of some biopharmaceutical properties of Praziquantel. Int. J. Pharm. 2005, 295, 93–99. [Google Scholar] [CrossRef]
- Lombardo, F.C.; Perissutti, B.; Keiser, J. Activity and pharmacokinetics of a praziquantel crystalline polymorph in the Schistosoma Mansoni Mouse model. Eur. J. Pharm. Biopharm. 2019, 142, 240–246. [Google Scholar] [CrossRef]
- Łaszcz, M.; Trzcí Nska, K.; Kubiszewski, M.; Zenna Kosmacińska, B.; Glice, M. Stability studies and structural characterization of Pramipexole. J. Pharm. Biomed. Anal. 2010, 53, 1033–1036. [Google Scholar] [CrossRef]
- Pawar, S.M.; Khatal, L.D.; Gabhe, S.Y.; Dhaneshwar, S.R. Establishment of inherent stability of Pramipexole and development of validated stability indicating LC–UV and LC–MS method. J. Pharm. Anal. 2013, 3, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Panditrao, V.M.; Sarkate, A.P.; Sangshetti, J.N.; Wakte, P.S.; Shinde, D.B. Stability-indicating HPLC determination of Pramipexole dihydrochloride in bulk drug and pharmaceutical dosage form. J. Braz. Chem. Soc. 2011, 22, 1253–1258. [Google Scholar] [CrossRef] [Green Version]
- Ledeti, A.; Olariu, T.; Caunii, A.; Vlase, G.; Circioban, D.; Baul, B.; Ledeti, I.; Vlase, T.; Murariu, M. Evaluation of thermal stability and kinetic of degradation for levodopa in non-isothermal conditions. J. Therm. Anal. Calorim. 2018, 131, 1881–1888. [Google Scholar] [CrossRef]
- Kinetiği, D.; Vitro, I.; Çalışmaları Ve Bağlanmış, Ç.; Emisyon, P.; Spektrofluorimetri, Y.; Tayini, M. Degradation kinetics, In Vitro dissolution studies, and quantification of Praziquantel, anchored in emission intensity by spectrofluorimetry. Turk. J. Pharm. Sci. 2019, 16, 82–87. [Google Scholar] [CrossRef]
- Mainardes, R.M.; Gremião, M.P.D.; Evangelista, R.C. Thermoanalytical study of Praziquantel-loaded PLGA nanoparticles. Revista Brasileira de Ciências Farmacêuticas 2006, 42, 523–530. [Google Scholar] [CrossRef]
- Freichel, O.L. Hydrokolloidretardarzneiformen Mit Endbeschleunigter Freisetzung. Ph.D. Thesis, Heinrich-Heine-Universität, Düsseldorf, Germany, 2002. [Google Scholar]
- Genina, N.; Holländer, J.; Jukarainen, H.; Mäkilä, E.; Salonen, J.; Sandler, N. Ethylene Vinyl Acetate (EVA) as a new drug carrier for 3D printed medical drug delivery devices. Eur. J. Pharm. Sci. 2016, 90, 53–63. [Google Scholar] [CrossRef]
- European Pharmacopoeia Comission 2.9.3. Dissolution test for solid dosage forms. In European Pharmacopoeia; EDQM: Strasbourg, France, 2020; Volume 10.2, pp. 326–333.
- European Pharmacopoeia Comission 5.17.1. Recommendations on dissolution testing. In European Pharmacopoeia; EDQM: Strasbourg, France, 2020; Volume 10.2, pp. 801–807.
- Tanigawara, Y.; Yamaoka, K.; Nakagawa, T.; Uno, T. New method for the evaluation of In Vitro dissolution time and disintegration time. Chem. Pharm. Bull. 1982, 30, 1088–1090. [Google Scholar] [CrossRef] [Green Version]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Modeling of drug release from delivery systems based on Hydroxypropyl Methylcellulose (HPMC). Adv. Drug Deliv. Rev. 2001, 48, 139–157. [Google Scholar] [CrossRef]
- Higuchi, T. Rate of release of medicaments from ointment bases containing drugs in suspension. J. Pharm. Sci. 1961, 50, 874–875. [Google Scholar] [CrossRef]
- Higuchi, T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Higuchi equation: Derivation, applications, use and misuse. Int. J. Pharm. 2011, 418, 6–12. [Google Scholar] [CrossRef]
- Lapidus, H.; Lordi, N.G. Drug release from compressed hydrophilic matrices. J. Pharm. Sci. 1968, 57, 1292–1301. [Google Scholar] [CrossRef]
- Lapidus, H.; Lordi, N.G. Some factors affecting the release of a water-soluble drug from a compressed hydrophilic matrix. J. Pharm. Sci. 1966, 55, 840–843. [Google Scholar] [CrossRef] [PubMed]
- Hixson, A.W.; Crowell, J.H. Dependence of reaction velocity upon surface and agitation: II—Experimental procedure in study of surface. Ind. Eng. Chem. 1931, 23, 1002–1009. [Google Scholar] [CrossRef]
- Bruschi, M.L. Strategies to Modify the Drug Release from Pharmaceutical Systems; Woodhead Publishing: Sawston, UK, 2015; pp. 63–86. [Google Scholar]
- Hopfenberg, H.B. Controlled release from erodible slabs, cylinders, and spheres. In Division of Organic Coatings and Plastics Chemistry: Preprints-Advantages and Problems; American Chemical Society: Washington, DC, USA, 1976; Volume 36, pp. 229–234. [Google Scholar]
- Katzhendler, I.; Hoffman, A.; Goldberger, A.; Friedman, M. Modeling of drug release from erodible tablets. J. Pharm. Sci. 1997, 86, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Langenbucher, F. Letters to the editor: Linearization of dissolution rate curves by the weibull distribution. J. Pharm. Pharmacol. 1972, 24, 979–981. [Google Scholar] [CrossRef] [PubMed]
- Dokoumetzidis, A.; Papadopoulou, V.; Macheras, P. Analysis of dissolution data using modified versions of Noyes-Whitney Equation and the Weibull function. Pharm. Res. 2006, 23, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Waloddi Weibull, B. A Statistical distribution function of wide applicability. J. Appl. Mech. 1951, 18, 293–297. [Google Scholar] [CrossRef]
- Kenney, J.F.; Keeping, E.S. “Root mean square”. In Mathematics of Statistics: Part 1; Van Nostrand: Princeton, NJ, USA, 1962; pp. 59–60. [Google Scholar]
- FDA. FDA Guidance for Industry—Dissolution Testing of Immediate Release Solid Oral Dosage Forms; FDA Center for Drug Evaluation and Research: Silver Spring, MD, USA, 1997; Volume 1, pp. 1–17. [Google Scholar]
- Shah, V.P.; Tsong, Y.; Sathe, P.; Liu, J.-P. In Vitro dissolution profile comparison—Statistics and analysis of the similarity factor, F2. Pharm. Res. 1998, 15, 889–896. [Google Scholar] [CrossRef]
- Skoug, J.W.; Borin, M.T.; Fleishaker, J.C.; Cooper, A.M. In Vitro and In Vivo evaluation of whole and half tablets of sustained-release Adinazolam Mesylate. Pharm. Res. Off. J. Am. Assoc. Pharm. Sci. 1991, 8, 1482–1488. [Google Scholar] [CrossRef]
- Tinke, A.P.; Vanhoutte, K.; de Maesschalck, R.; Verheyen, S.; de Winter, H. A new approach in the prediction of the dissolution behavior of suspended particles by means of their particle size distribution. J. Pharm. Biomed. Anal. 2005, 39, 900–907. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature Profile in Zone 2–10/°C | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Zone/- | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| PVA-PDM formulation/°C | 30 | 100 | 180 | 180 | 180 | 180 | 180 | 195 | 195 |
| PVA-PZQ formulation/°C | 21 | 31 | 78 | 180 | 180 | 180 | 180 | 180 | 190 |
| EVA-LD formulation/°C | 25 | 28 | 78 | 130 | 140 | 155 | 155 | 120 | 100 |
| Screw Configuration (Die–Gear) | |||||||||
| PVA/EVA formulation | die-10 CE 1 L/D-KZ: 5 × 60°-3 × 30°-5 CE 1 L/D-KZ: 4 × 90°-5 × 60°-3 × 30°-10 CE 1 L/D-2 CE 3/2 L/D-gear | ||||||||
| CE = conveying element, KZ = kneading zone | |||||||||
| n | |||
|---|---|---|---|
| Thin Film | Cylinder | Sphere | Drug Release Mechanism |
| 0.50 | 0.45 | 0.43 | Fickian diffusion |
| 0.50 < n < 1.00 | 0.45 < n < 0.89 | 0.43 < n < 0.85 | Anomalous transport |
| 1.00 | 0.89 | 0.85 | Case-II transport |
| SA/V 1 mm−1 | ||||||
|---|---|---|---|---|---|---|
| Form | SA/mm2 | V/mm3 | SA/V/mm−1 | API/mg | MDT/min | f2 Value |
| Q1 | 606.00 | 585.00 | 1.00 | 35.97 | 56.95 | 77.51 |
| Q2 | 256.00 | 256.00 | 1.00 | 15.60 | 62.91 | 87.92 |
| Q3 | 250.00 | 250.00 | 1.00 | 15.66 | 65.65 | 73.88 |
| HC | 667.59 | 667.59 | 1.00 | 41.34 | 56.84 | 71.87 |
| C | 201.06 | 201.06 | 1.00 | 12.29 | 60.67 | Reference |
| P | 273.05 | 265.97 | 1.03 | 16.25 | 64.30 | 82.89 |
| SA/V 1.5 mm−1 | ||||||
| Form | SA/mm2 | V/mm3 | SA/V/mm−1 | API/mg | MDT/min | f2 Value |
| Q1 | 546.00 | 360.00 | 1.52 | 21.74 | 32.81 | 78.20 |
| Q2 | 192.00 | 128.00 | 1.50 | 8.00 | 35.67 | 92.54 |
| Q3 | 166.00 | 110.00 | 1.51 | 6.92 | 38.07 | 73.19 |
| HC | 301.59 | 201.06 | 1.50 | 13.54 | 33.83 | 90.82 |
| C | 150.80 | 100.53 | 1.50 | 6.19 | 34.80 | Reference |
| P | 121.92 | 80.10 | 1.52 | 4.85 | 36.92 | 75.40 |
| SA/V 2 mm−1 | ||||||
| Form | SA/mm2 | V/mm3 | SA/V/mm−1 | API/mg | MDT/min | f2 Value |
| Q1 | 516.00 | 247.50 | 2.08 | 15.04 | 22.98 | 66.45 |
| Q2 | 169.60 | 83.20 | 2.04 | 5.21 | 25.70 | 96.55 |
| Q3 | 142.00 | 70.00 | 2.03 | 4.43 | 24.90 | 91.95 |
| HC | 201.06 | 100.50 | 2.00 | 6.87 | 25.02 | 92.37 |
| C | 133.20 | 65.35 | 2.04 | 3.92 | 25.14 | Reference |
| P | 66.02 | 32.24 | 2.05 | 1.99 | 24.86 | 75.79 |
| SA/V Ratio/mm−1 | MDT Prediction/min | MDT Experiment/min | RMSEP/min |
|---|---|---|---|
| 0.90 | 71.70 | 74.06 ± 11.45 | 2.36 |
| 1.60 | 33.83 | 31.04 ± 2.20 | 2.79 |
| 2.30 | 21.07 | 16.65 ± 0.46 | 4.42 |
| 4.67 | 8.36 | 6.93 ± 0.71 | 1.43 |
| SA/V Ratio/mm−1 | MDT Prediction/min | MDT Experiment/min | RMSEP/min |
|---|---|---|---|
| 1.73 | 82.25 | 78.79 ± 7.24 | 3.46 |
| 1.89 | 70.74 | 62.60 ± 5.90 | 8.14 |
| 4.67 | 15.13 | 14.40 ± 0.77 | 0.73 |
| SA/V Ratio/mm−1 | MDT Prediction/min | MDT Experiment/min | RMSEP/min |
|---|---|---|---|
| 1.30 | 54.42 | 55.91 ± 1.11 | 1.49 |
| 1.83 | 36.79 | 38.73 ± 1.07 | 1.94 |
| 2.30 | 28.32 | 27.88 ± 2.23 | 0.44 |
| 4.67 | 12.58 | 12.16 ± 0.96 | 0.42 |
| PVA-PDM Formulation | ||||||
|---|---|---|---|---|---|---|
| SA/V mm−1 | KMP | Hixson | Peppas Sahlin n = 0.79 | Hopfenberg | Lapidus + Lordi | Weibull |
| 0.8 | 0.9837 | 0.9951 | 0.9991 | 0.9837 | 0.9837 | 0.1240 |
| 1.0 | 0.9971 | 0.9969 | 0.9989 | 0.9971 | 0.9971 | 0.1620 |
| 1.5 | 0.9981 | 0.9964 | 0.9995 | 0.9981 | 0.9981 | 0.2575 |
| 2.0 | 0.9966 | 0.9961 | 0.9996 | 0.9966 | 0.9966 | 0.9919 |
| 2.5 | 0.9783 | 0.9975 | 0.9978 | 0.9783 | 0.9783 | 0.9957 |
| 3.3 | 0.9982 | 0.9951 | 0.9997 | 0.9982 | 0.9982 | 0.9955 |
| 4.0 | 0.9949 | 0.9964 | 0.9995 | 0.9949 | 0.9949 | 0.9971 |
| 5.0 | 0.9931 | 0.9987 | 0.9999 | 0.9931 | 0.9931 | 0.9994 |
| 6.0 | 0.9970 | 0.9987 | 0.9999 | 0.9970 | 0.9970 | 0.9997 |
| PVA-PZQ Formulation | ||||||
| SA/V mm−1 | KMP | Hixson | Peppas Sahlin n= 1.1 | Hopfenberg | Lapidus + Lordi | Weibull |
| 0.8 | 0.9680 | 0.9922 | 0.9980 | 0.9680 | 0.9680 | 0.9993 |
| 1.0 | 0.9870 | 0.9832 | 0.9953 | 0.9870 | 0.9870 | 0.9972 |
| 1.5 | 0.9765 | 0.9727 | 0.9890 | 0.9765 | 0.9765 | 0.9976 |
| 2.0 | 0.9663 | 0.9646 | 0.9667 | 0.9663 | 0.9663 | 0.9973 |
| 2.5 | 0.9819 | 0.9292 | 0.9932 | 0.9819 | 0.9819 | 0.9963 |
| 3.3 | 0.9350 | 0.9408 | 0.9807 | 0.9350 | 0.9350 | 0.9980 |
| 4.0 | 0.9331 | 0.9361 | 0.9877 | 0.9331 | 0.9331 | 0.9975 |
| 5.0 | 0.9143 | 0.9787 | 0.9823 | 0.9143 | 0.9143 | 0.9994 |
| 6.0 | 0.9429 | 0.9299 | 0.9888 | 0.9429 | 0.9429 | 0.9992 |
| EVA-LD Formulation | ||||||
| SA/V mm−1 | KMP | Weibull | Peppas Sahlin n= 0.66 | Higuchi | ||
| 0.9 | 0.9941 | 0.9986 | 0.9808 | 0.8760 | ||
| 1.1 | 0.9910 | 0.9936 | 0.9853 | 0.9304 | ||
| 1.5 | 0.9961 | 0.9961 | 0.9981 | 0.9945 | ||
| 1.7 | 0.9925 | 0.9884 | 0.9966 | 0.9880 | ||
| 1.9 | 0.9949 | 0.9894 | 0.9989 | 0.9941 | ||
| 2.5 | 0.9724 | 0.9979 | 0.9993 | 0.9282 | ||
| 4.0 | 0.9979 | 0.9928 | 0.9934 | 0.9730 | ||
| 5.0 | 0.9970 | 0.9944 | 0.9956 | 0.9583 | ||
| 6.0 | 0.9974 | 0.9946 | 0.9957 | 0.9600 | ||
| PVA−PDM Formulation | |||||
|---|---|---|---|---|---|
| SA/V | k1 | k2 | ln(SA/V) | ln(k1) | ln(k2) |
| 0.8 | 1.986 | 0.010 | −0.223 | 0.686 | −4.608 |
| 1.0 | 2.516 | 0.011 | 0.000 | 0.923 | −4.549 |
| 1.5 | 3.920 | 0.027 | 0.405 | 1.366 | −3.626 |
| 2.0 | 5.561 | 0.067 | 0.693 | 1.716 | −2.704 |
| 2.5 | 7.362 | 0.135 | 0.916 | 1.996 | −2.001 |
| 3.3 | 11.555 | 0.258 | 1.203 | 2.447 | −1.353 |
| 4.0 | 15.459 | 0.545 | 1.386 | 2.738 | −0.608 |
| 5.0 | 16.888 | 0.713 | 1.609 | 2.827 | −0.338 |
| 6.0 | 20.856 | 1.069 | 1.792 | 3.038 | 0.066 |
| EVA−LD Formulation | |||||
| SA/V | k1 | k2 | ln(SA/V) | ln(k1) | ln(k2) |
| 0.9 | 1.952 | 0.010 | −0.105 | 0.669 | −4.614 |
| 1.1 | 2.286 | 0.014 | 0.065 | 0.827 | −4.282 |
| 1.5 | 3.115 | 0.020 | 0.405 | 1.136 | −3.928 |
| 1.7 | 4.087 | 0.031 | 0.513 | 1.408 | −3.465 |
| 1.9 | 5.009 | 0.053 | 0.626 | 1.611 | −2.944 |
| 2.5 | 7.636 | 0.153 | 0.916 | 2.033 | −1.876 |
| 4.0 | 10.374 | 0.279 | 1.386 | 2.339 | −1.277 |
| 5.0 | 14.444 | 0.540 | 1.609 | 2.670 | −0.617 |
| 6.0 | 16.816 | 0.727 | 1.792 | 2.822 | −0.319 |
| PVA-PZQ Formulation | |||||
|---|---|---|---|---|---|
| SA/V | a | b | ln(SA/V) | ln(a) | ln(b) |
| 0.8 | 105.0 | 1.38 | −0.223 | 4.654 | 0.322 |
| 1.0 | 81.0 | 1.45 | 0.000 | 4.394 | 0.372 |
| 1.5 | 52.0 | 1.63 | 0.405 | 3.951 | 0.489 |
| 2.0 | 38.0 | 1.80 | 0.693 | 3.638 | 0.588 |
| 2.5 | 31.5 | 1.91 | 0.916 | 3.450 | 0.647 |
| 3.3 | 20.8 | 2.05 | 1.194 | 3.035 | 0.718 |
| 4.0 | 17.1 | 2.15 | 1.386 | 2.836 | 0.765 |
| 5.0 | 11.5 | 2.29 | 1.609 | 2.442 | 0.829 |
| 6.0 | 11.2 | 2.40 | 1.792 | 2.414 | 0.875 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Windolf, H.; Chamberlain, R.; Quodbach, J. Predicting Drug Release from 3D Printed Oral Medicines Based on the Surface Area to Volume Ratio of Tablet Geometry. Pharmaceutics 2021, 13, 1453. https://doi.org/10.3390/pharmaceutics13091453
Windolf H, Chamberlain R, Quodbach J. Predicting Drug Release from 3D Printed Oral Medicines Based on the Surface Area to Volume Ratio of Tablet Geometry. Pharmaceutics. 2021; 13(9):1453. https://doi.org/10.3390/pharmaceutics13091453
Chicago/Turabian StyleWindolf, Hellen, Rebecca Chamberlain, and Julian Quodbach. 2021. "Predicting Drug Release from 3D Printed Oral Medicines Based on the Surface Area to Volume Ratio of Tablet Geometry" Pharmaceutics 13, no. 9: 1453. https://doi.org/10.3390/pharmaceutics13091453