
Comprehensive Investigation of Stereoselective Food Drug Interaction Potential of Resveratrol on Nine P450 and Six UGT Isoforms in Human Liver Microsomes



Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Stereoselective Inhibition of Resveratrol against Cytochrome P450 Activity
2.3. Stereoselective Inhibition of Resveratrol against Uridine 5′-Diphosphoglucuronosyl Transferase Activity
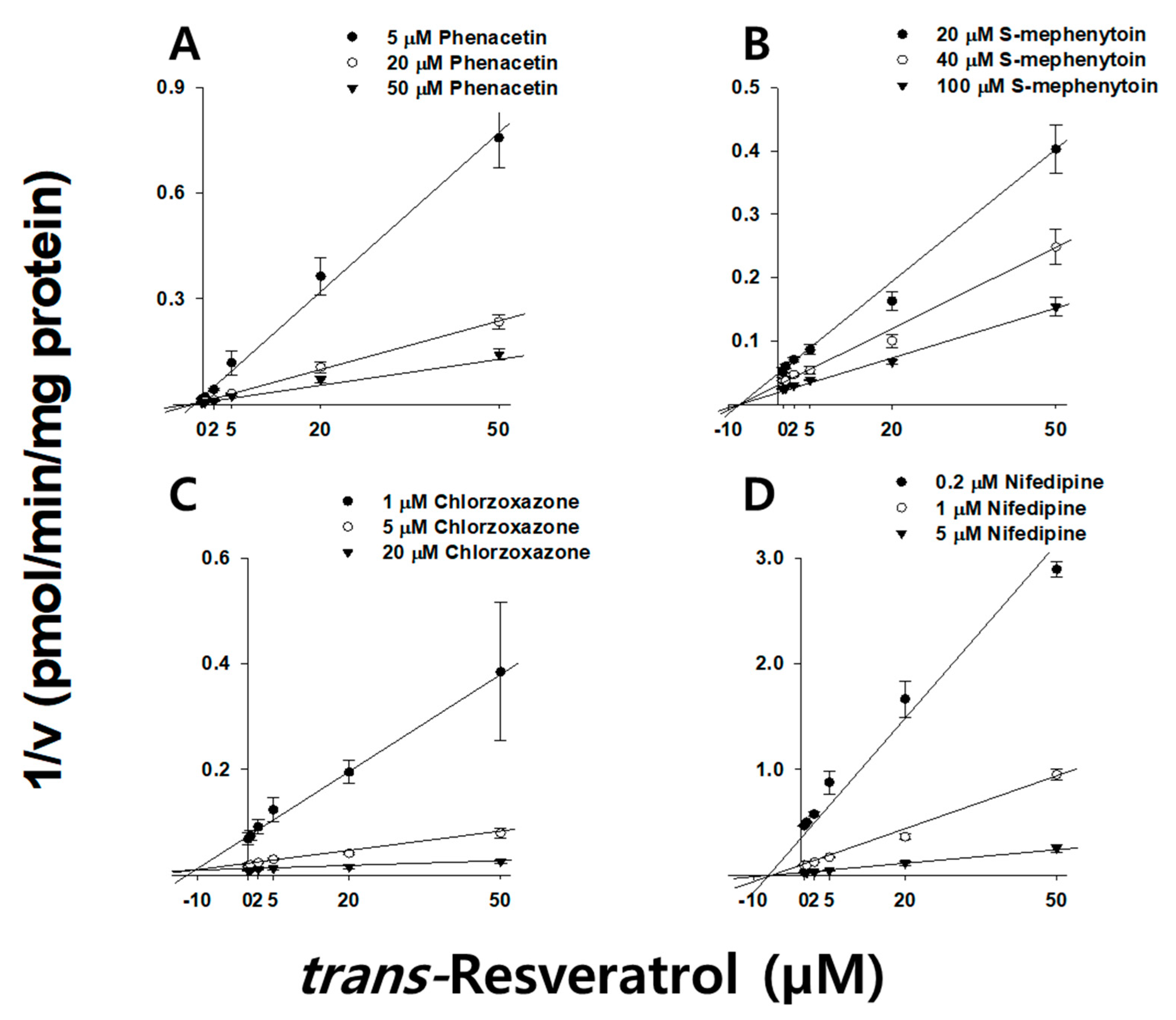
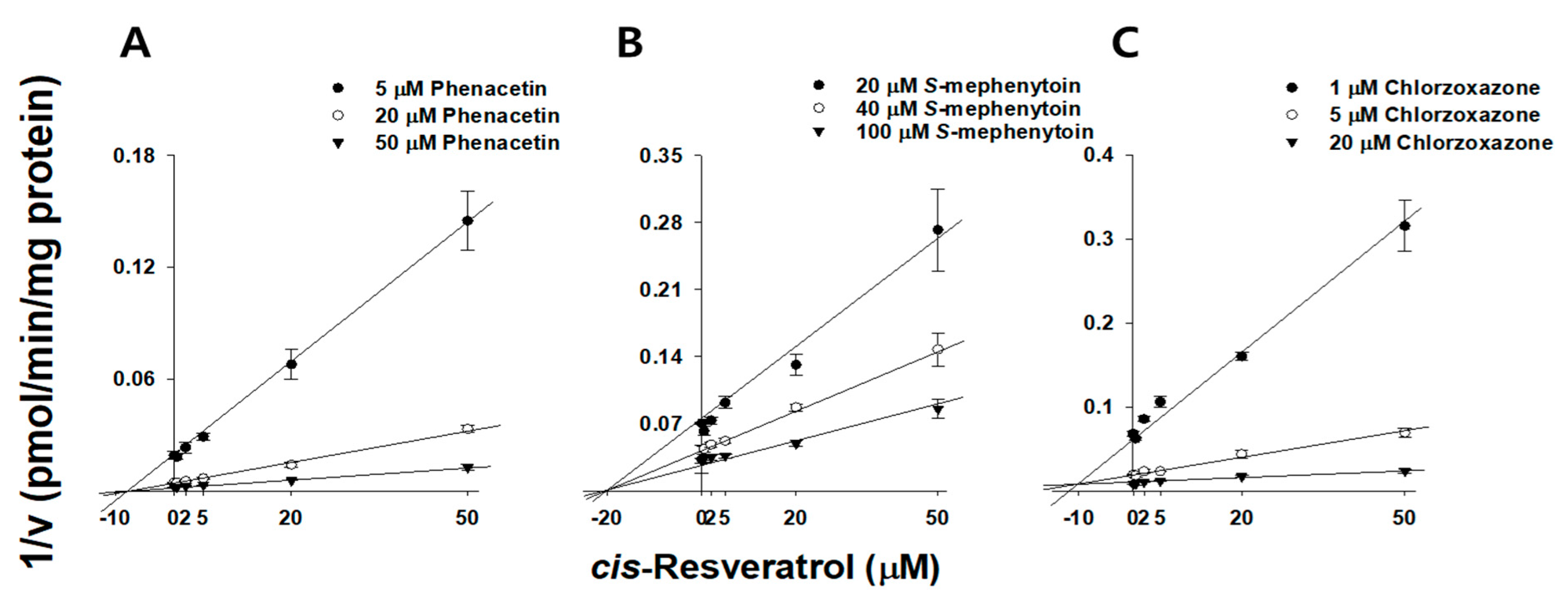
2.4. Kinetic Characterization of Resveratrol on CYP3A, CYP2E1, CYP2C19, and CYP1A2 in HLMs
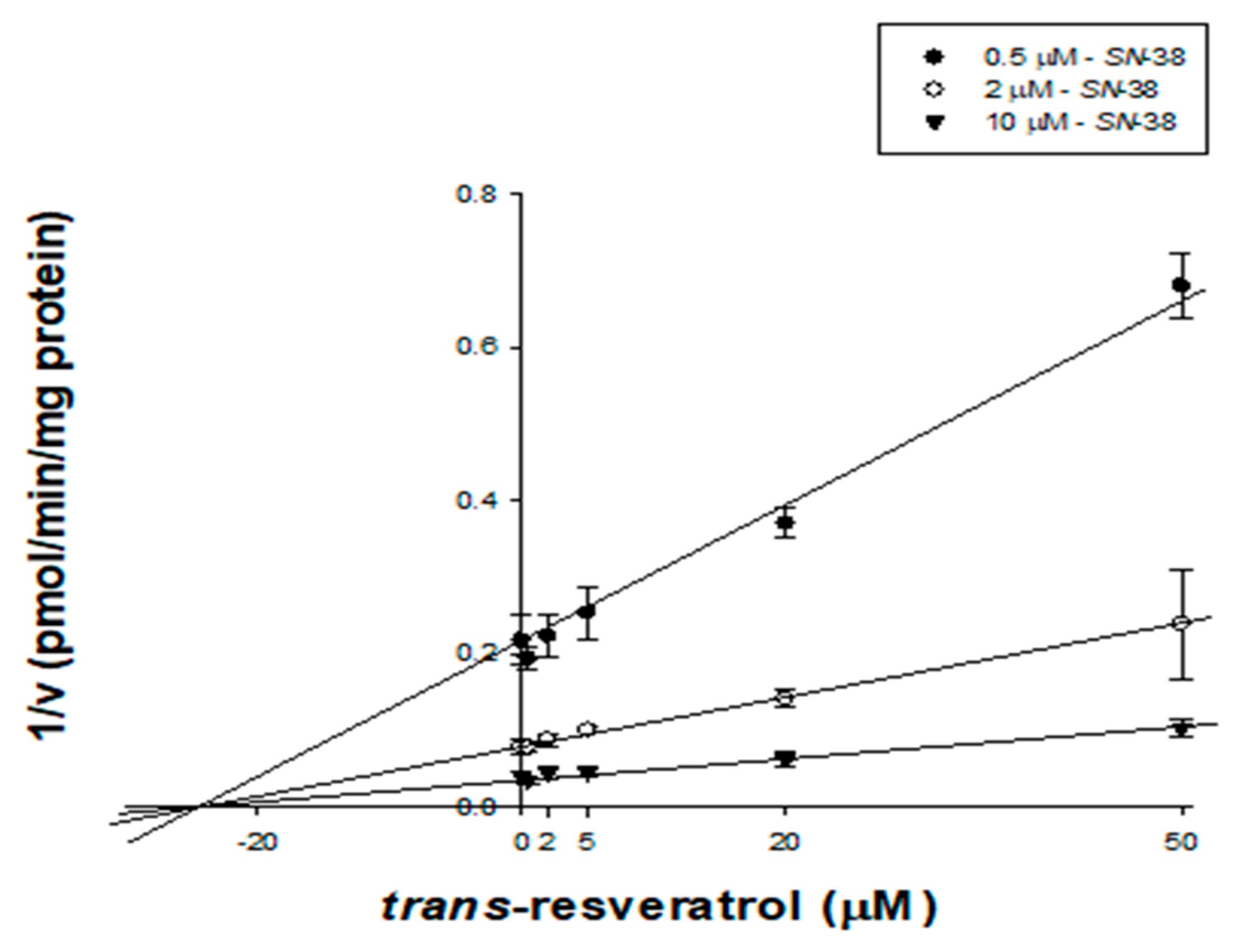
2.5. Kinetic Characterization of trans-Resveratrol on UGT1A1 in HLMs
2.6. Characterization of Glutathione Conjugates of Resveratrol in Recombinant Cytochrome P450 Isoforms
2.7. LC-MS/MS Analysis
2.8. Data Analysis
3. Results and Discussion
3.1. Stereoselective Inhibition of Cytochrome P450 Isoform Activities by Resveratrol
3.2. Stereoselective Inhibition of Uridine 5′-Diphosphoglucuronosyl Transferase Isoform Activities by Resveratrol
3.3. Kinetic Characterization of trans-Resveratrol and cis-Resveratrol Inhibition against CYP1A2, CYP2C19, CYP2E1, CYP3A, and UGT1A1 in HLMs
3.4. Characterization of Glutathione Conjugates of trans-Resveratrol in Recombinant Cytochrome P450 Isoforms
3.5. Evaluation of Food Drug Interaction Potential of Resveratrol
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hyrsova, L.; Vanduchova, A.; Dusek, J.; Smutny, T.; Carazo, A.; Maresova, V.; Trejtnar, F.; Barta, P.; Anzenbacher, P.; Dvorak, Z.; et al. Trans-resveratrol, but not other natural stilbenes occurring in food, carries the risk of drug-food interaction via inhibition of cytochrome P450 enzymes or interaction with xenosensor receptors. Toxicol. Lett. 2019, 300, 81–91. [Google Scholar] [CrossRef]
- Bavaresco, L.; Lucini, L.; Busconi, M.; Flamini, R.; De Rosso, M. Wine Resveratrol: From the Ground Up. Nutrients 2016, 8, 222. [Google Scholar] [CrossRef] [Green Version]
- Silva, P.; Sureda, A.; Tur, J.A.; Andreoletti, P.; Cherkaoui-Malki, M.; Latruffe, N. How efficient is resveratrol as an antioxidant of the Mediterranean diet, towards alterations during the aging process? Free Radic. Res. 2019, 53, 1101–1112. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Rafiei, H.; Mohammadinejad, R.; Farkhondeh, T.; Samarghandian, S. Anti-tumor activity of resveratrol against gastric cancer: A review of recent advances with an emphasis on molecular pathways. Cancer Cell Int. 2021, 21, 1–10. [Google Scholar] [CrossRef]
- Tome-Carneiro, J.; Gonzalvez, M.; Larrosa, M.; Yanez-Gascon, M.J.; Garcia-Almagro, F.J.; Ruiz-Ros, J.A.; Tomas-Barberan, F.A.; Garcia-Conesa, M.T.; Espin, J.C. Resveratrol in primary and secondary prevention of cardiovascular disease: A dietary and clinical perspective. Ann. N. Y. Acad. Sci. 2013, 1290, 37–51. [Google Scholar] [CrossRef]
- Gomes, B.A.Q.; Silva, J.P.B.; Romeiro, C.F.R.; Dos Santos, S.M.; Rodrigues, C.A.; Goncalves, P.R.; Sakai, J.T.; Mendes, P.F.S.; Varela, E.L.P.; Monteiro, M.C. Neuroprotective Mechanisms of Resveratrol in Alzheimer’s Disease: Role of SIRT1. Oxid. Med. Cell Longev. 2018, 2018, 8152373. [Google Scholar] [CrossRef] [PubMed]
- AMERICANHS.ORG. Best Erectile Dysfunction Pills on the Market in 2020. Available online: https://www.americanhs.org/best-ed-pills/ (accessed on 27 August 2021).
- Lin, J.H.; Lu, A.Y. Inhibition and induction of cytochrome P450 and the clinical implications. Clin. Pharmacokinet. 1998, 35, 361–390. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.G.; Dresser, G.K.; Bend, J.R. Bergamottin, lime juice, and red wine as inhibitors of cytochrome P450 3A4 activity: Comparison with grapefruit juice. Clin. Pharmacol. Ther. 2003, 73, 529–537. [Google Scholar] [CrossRef]
- Jiang, W.; Wang, X.; Xu, X.; Kong, L. Effect of Schisandra sphenanthera extract on the concentration of tacrolimus in the blood of liver transplant patients. Int. J. Clin. Pharmacol. Ther. 2010, 48, 224–229. [Google Scholar] [CrossRef]
- Piver, B.; Berthou, F.; Dreano, Y.; Lucas, D. Differential inhibition of human cytochrome P450 enzymes by epsilon-viniferin, the dimer of resveratrol: Comparison with resveratrol and polyphenols from alcoholized beverages. Life Sci. 2003, 73, 1199–1213. [Google Scholar] [CrossRef]
- Yu, C.; Shin, Y.G.; Kosmeder, J.W.; Pezzuto, J.M.; van Breemen, R.B. Liquid chromatography/tandem mass spectrometric determination of inhibition of human cytochrome P450 isozymes by resveratrol and resveratrol-3-sulfate. Rapid Commun. Mass Spectrom. 2003, 17, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Detampel, P.; Beck, M.; Krahenbuhl, S.; Huwyler, J. Drug interaction potential of resveratrol. Drug Metab. Rev. 2012, 44, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.K.; Chen, J.; Lee, W.B. Differential inhibition and inactivation of human CYP1 enzymes by trans-resveratrol: Evidence for mechanism-based inactivation of CYP1A2. J. Pharmacol. Exp. Ther. 2001, 299, 874–882. [Google Scholar] [PubMed]
- Seki, H.; Akiyoshi, T.; Imaoka, A.; Ohtani, H. Inhibitory kinetics of fruit components on CYP2C19 activity. Drug Metab. Pharmacokinet. 2019, 34, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Mikstacka, R.; Gnojkowski, J.; Baer-Dubowska, W. Effect of natural phenols on the catalytic activity of cytochrome P450 2E1. Acta Biochim. Pol. 2002, 49, 917–925. [Google Scholar] [CrossRef] [Green Version]
- Piver, B.; Berthou, F.; Dreano, Y.; Lucas, D. Inhibition of CYP3A, CYP1A and CYP2E1 activities by resveratrol and other non volatile red wine components. Toxicol. Lett. 2001, 125, 83–91. [Google Scholar] [CrossRef]
- Bedada, S.K.; Nearati, P. Effect of resveratrol on the pharmacokinetics of carbamazepine in healthy human volunteers. Phytother. Res. 2015, 29, 701–706. [Google Scholar] [CrossRef]
- Bedada, S.K.; Neerati, P. Resveratrol Pretreatment Affects CYP2E1 Activity of Chlorzoxazone in Healthy Human Volunteers. Phytother. Res. 2016, 30, 463–468. [Google Scholar] [CrossRef]
- Brill, S.S.; Furimsky, A.M.; Ho, M.N.; Furniss, M.J.; Li, Y.; Green, A.G.; Bradford, W.W.; Green, C.E.; Kapetanovic, I.M.; Iyer, L.V. Glucuronidation of trans-resveratrol by human liver and intestinal microsomes and UGT isoforms. J. Pharm. Pharmacol. 2006, 58, 469–479. [Google Scholar] [CrossRef]
- Francioso, A.; Lastovickova, L.; Mosca, L.; Boffi, A.; Bonamore, A.; Macone, A. Gas Chromatographic-Mass Spectrometric Method for the Simultaneous Determination of Resveratrol Isomers and 2,4,6-Trihydroxyphenanthrene in Red Wines Exposed to UV-Light. J. Agric. Food Chem. 2019, 67, 11752–11757. [Google Scholar] [CrossRef]
- Paulo, L.; Domingues, F.; Queiroz, J.A.; Gallardo, E. Development and validation of an analytical method for the determination of trans- and cis-resveratrol in wine: Analysis of its contents in 186 Portuguese red wines. J. Agric. Food Chem. 2011, 59, 2157–2168. [Google Scholar] [CrossRef]
- Stark, T.; Wollmann, N.; Losch, S.; Hofmann, T. Quantitation of resveratrol in red wines by means of stable isotope dilution analysis-ultra-performance liquid chromatography-Quan-time-of-flight mass spectrometry and cross validation. Anal. Chem. 2011, 83, 3398–3405. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.; Gardner, P.T.; O’Neil, J.; Crawford, S.; Morecroft, I.; McPhail, D.B.; Lister, C.; Matthews, D.; MacLean, M.R.; Lean, M.E.; et al. Relationship among antioxidant activity, vasodilation capacity, and phenolic content of red wines. J. Agric. Food Chem. 2000, 48, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Ferlito, V.; Xu, C.; Flockhart, D.A.; Caccamese, S. Enantiomers of naringenin as pleiotropic, stereoselective inhibitors of cytochrome P450 isoforms. Chirality 2011, 23, 891–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.Y.; Wang, Y.Q.; Li, L.P.; Wang, L.; Zeng, S.; Zhou, H.; Jiang, H.D. Stereoselective interaction between tetrahydropalmatine enantiomers and CYP enzymes in human liver microsomes. Chirality 2013, 25, 43–47. [Google Scholar] [CrossRef]
- Kobayashi, S.; Murray, S.; Watson, D.; Sesardic, D.; Davies, D.S.; Boobis, A.R. The specificity of inhibition of debrisoquine 4-hydroxylase activity by quinidine and quinine in the rat is the inverse of that in man. Biochem. Pharmacol. 1989, 38, 2795–2799. [Google Scholar] [CrossRef]
- Granvil, C.P.; Krausz, K.W.; Gelboin, H.V.; Idle, J.R.; Gonzalez, F.J. 4-Hydroxylation of debrisoquine by human CYP1A1 and its inhibition by quinidine and quinine. J. Pharmacol. Exp. Ther. 2002, 301, 1025–1032. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Kim, H.; Cha, I.J.; Park, J.S.; Shon, J.H.; Liu, K.H.; Shin, J.G. High-throughput screening of inhibitory potential of nine cytochrome P450 enzymes in vitro using liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 2651–2658. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, H.; Ji, H.K.; Lee, T.; Liu, K.H. Screening of ten cytochrome P450 enzyme activities with 12 probe substrates in human liver microsomes using cocktail incubation and liquid chromatography-tandem mass spectrometry. Biopharm. Drug Dispos. 2019, 40, 101–111. [Google Scholar] [CrossRef]
- Perloff, E.S.; Mason, A.K.; Dehal, S.S.; Blanchard, A.P.; Morgan, L.; Ho, T.; Dandeneau, A.; Crocker, R.M.; Chandler, C.M.; Boily, N.; et al. Validation of cytochrome P450 time-dependent inhibition assays: A two-time point IC50 shift approach facilitates kinact assay design. Xenobiotica 2009, 39, 99–112. [Google Scholar] [CrossRef]
- Joo, J.; Lee, B.; Lee, T.; Liu, K.H. Screening of six UGT enzyme activities in human liver microsomes using liquid chromatography/triple quadrupole mass spectrometry. Rapid Commun. Mass Spectrom. 2014, 28, 2405–2414. [Google Scholar] [CrossRef]
- Joo, J.; Kim, Y.W.; Wu, Z.; Shin, J.H.; Lee, B.; Shon, J.C.; Lee, E.Y.; Phuc, N.M.; Liu, K.H. Screening of non-steroidal anti-inflammatory drugs for inhibitory effects on the activities of six UDP-glucuronosyltransferases (UGT1A1, 1A3, 1A4, 1A6, 1A9 and 2B7) using LC-MS/MS. Biopharm. Drug Dispos. 2015, 36, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Steenwyk, R.C.; Tan, B. In vitro evidence for the formation of reactive intermediates of resveratrol in human liver microsomes. Xenobiotica 2010, 40, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Nguyen, P.H.; Kim, G.; Jang, S.N.; Lee, G.H.; Phuc, N.M.; Wu, Z.; Liu, K.H. Strong and Selective Inhibitory Effects of the Biflavonoid Selamariscina A against CYP2C8 and CYP2C9 Enzyme Activities in Human Liver Microsomes. Pharmaceutics 2020, 12, 343. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.G.; Soukhova, N.; Flockhart, D.A. Effect of antipsychotic drugs on human liver cytochrome P-450 (CYP) isoforms in vitro: Preferential inhibition of CYP2D6. Drug Metab. Dispos. 1999, 27, 1078–1084. [Google Scholar] [PubMed]
- Seo, H.-J.; Ji, S.-B.; Kim, S.-E.; Lee, G.-M.; Park, S.-Y.; Wu, Z.; Jang, D.S.; Liu, K.-H. Inhibitory Effects of Schisandra Lignans on Cytochrome P450s and Uridine 5′-Diphospho-Glucuronosyl Transferases in Human Liver Microsomes. Pharmaceutics 2021, 13, 371. [Google Scholar] [CrossRef]
- Awortwe, C.; Manda, V.K.; Avonto, C.; Khan, S.I.; Khan, I.A.; Walker, L.A.; Bouic, P.J.; Rosenkranz, B. In Vitro Evaluation of Reversible and Time-Dependent Inhibitory Effects of Kalanchoe crenata on CYP2C19 and CYP3A4 Activities. Drug Metab. Lett. 2015, 9, 48–62. [Google Scholar] [CrossRef] [Green Version]
- Fairman, D.A.; Collins, C.; Chapple, S. Progress curve analysis of CYP1A2 inhibition: A more informative approach to the assessment of mechanism-based inactivation? Drug Metab. Dispos. 2007, 35, 2159–2165. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.S.; Zhao, Z.Q.; Qin, Z.S.; Wu, K.; Xia, T.F.; Pang, L.Q. Herb-drug interaction between irinotecan and psoralidin-containing herbs. Eur. J. Drug Metab. Pharmacokinet. 2015, 40, 481–484. [Google Scholar] [CrossRef]
- Ma, J.; Zheng, L.; Deng, T.; Li, C.L.; He, Y.S.; Li, H.J.; Li, P. Stilbene glucoside inhibits the glucuronidation of emodin in rats through the down-regulation of UDP-glucuronosyltransferases 1A8: Application to a drug-drug interaction study in Radix Polygoni Multiflori. J. Ethnopharmacol. 2013, 147, 335–340. [Google Scholar] [CrossRef]
- Marahatta, A.; Bhandary, B.; Jeong, S.K.; Kim, H.R.; Chae, H.J. Soybean greatly reduces valproic acid plasma concentrations: A food-drug interaction study. Sci. Rep. 2014, 4, 4362. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.K.; Delucchi, A.B. Resveratrol, a red wine constituent, is a mechanism-based inactivator of cytochrome P450 3A4. Life Sci. 2000, 67, 3103–3112. [Google Scholar] [CrossRef]
- Zhou, S.; Koh, H.L.; Gao, Y.; Gong, Z.Y.; Lee, E.J. Herbal bioactivation: The good, the bad and the ugly. Life Sci. 2004, 74, 935–968. [Google Scholar] [CrossRef]
- Piver, B.; Fer, M.; Vitrac, X.; Merillon, J.M.; Dreano, Y.; Berthou, F.; Lucas, D. Involvement of cytochrome P450 1A2 in the biotransformation of trans-resveratrol in human liver microsomes. Biochem. Pharmacol. 2004, 68, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, X.; Huang, X.S.; Degnan, A.P.; Snyder, L.B.; Yang, F.; Huang, H.; Shu, Y.Z.; Johnson, B.M. Identification of glutathione conjugates of acetylene-containing positive allosteric modulators of metabotropic glutamate receptor subtype 5. Drug Metab. Dispos. 2015, 43, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Guo, X.; Ma, Y.; Hu, X.; Peng, Y.; Zheng, J. Identification of Quinone Methide Intermediate Resulting from Metabolic Activation of Icaritin in Vitro and in Vivo. Chem. Res. Toxicol. 2019, 32, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Berretta, M.; Bignucolo, A.; Di Francia, R.; Comello, F.; Facchini, G.; Ceccarelli, M.; Iaffaioli, R.V.; Quagliariello, V.; Maurea, N. Resveratrol in Cancer Patients: From Bench to Bedside. Int. J. Mol. Sci. 2020, 21, 2945. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Y.Y.; Liang, B.Q.; Li, X.Y.; Gu, E.M.; Dai, D.P.; Cai, J.P.; Hu, G.X. The effect of resveratrol on pharmacokinetics of aripiprazole in vivo and in vitro. Xenobiotica 2016, 46, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Surendran, S.; Sapkal, R.; Paul, D.; Nanjappan, S. Effect of resveratrol on dipeptidyl peptidase-4 inhibitors pharmacokinetics: An in vitro and in vivo approach. Chem. Biol. Interact. 2020, 315, 108909. [Google Scholar] [CrossRef]
- Chow, H.H.; Garland, L.L.; Hsu, C.H.; Vining, D.R.; Chew, W.M.; Miller, J.A.; Perloff, M.; Crowell, J.A.; Alberts, D.S. Resveratrol modulates drug- and carcinogen-metabolizing enzymes in a healthy volunteer study. Cancer Prev. Res. 2010, 3, 1168–1175. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.P.; Choi, D.H.; Choi, J.S. Effects of resveratrol on the pharmacokinetics of diltiazem and its major metabolite, desacetyldiltiazem, in rats. Cardiovasc. Ther. 2008, 26, 269–275. [Google Scholar] [CrossRef]
- Kim, K.A.; Park, P.W.; Hong, S.J.; Park, J.Y. The effect of CYP2C19 polymorphism on the pharmacokinetics and pharmacodynamics of clopidogrel: A possible mechanism for clopidogrel resistance. Clin. Pharmacol. Ther. 2008, 84, 236–242. [Google Scholar] [CrossRef]
- Zhao, W.; Leroux, S.; Biran, V.; Jacqz-Aigrain, E. Developmental pharmacogenetics of CYP2C19 in neonates and young infants: Omeprazole as a probe drug. Br. J. Clin. Pharmacol. 2018, 84, 997–1005. [Google Scholar] [CrossRef] [Green Version]
- Raucy, J.L.; Lasker, J.M.; Lieber, C.S.; Black, M. Acetaminophen activation by human liver cytochromes P450IIE1 and P450IA2. Arch. Biochem. Biophys. 1989, 271, 270–283. [Google Scholar] [CrossRef]
- Loi, C.M.; Day, J.D.; Jue, S.G.; Bush, E.D.; Costello, P.; Dewey, L.V.; Vestal, R.E. Dose-dependent inhibition of theophylline metabolism by disulfiram in recovering alcoholics. Clin. Pharmacol. Ther. 1989, 45, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Howells, L.M.; Berry, D.P.; Elliott, P.J.; Jacobson, E.W.; Hoffmann, E.; Hegarty, B.; Brown, K.; Steward, W.P.; Gescher, A.J. Phase I randomized, double-blind pilot study of micronized resveratrol (SRT501) in patients with hepatic metastases--safety, pharmacokinetics, and pharmacodynamics. Cancer Prev. Res. 2011, 4, 1419–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.Y.; Yu, C.P.; Hsieh, Y.W.; Lin, S.P.; Hou, Y.C. Resveratrol stereoselectively affected (+/-)warfarin pharmacokinetics and enhanced the anticoagulation effect. Sci. Rep. 2020, 10, 15910. [Google Scholar] [CrossRef]
- Mohamed, M.F.; Tseng, T.; Frye, R.F. Inhibitory effects of commonly used herbal extracts on UGT1A1 enzyme activity. Xenobiotica 2010, 40, 663–669. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Resveratrol. Available online: https://www.drugs.com/npp/resveratrol.html (accessed on 27 August 2021).
- Bjornsson, T.D.; Callaghan, J.T.; Einolf, H.J.; Fischer, V.; Gan, L.; Grimm, S.; Kao, J.; King, S.P.; Miwa, G.; Ni, L.; et al. Manufacturers of America Drug Metabolism/Clinical Pharmacology Technical Working G, Evaluation, FDACfD, and Research (2003). The conduct of in vitro and in vivo drug-drug interaction studies: A Pharmaceutical Research and Manufacturers of America (PhRMA) perspective. Drug Metab. Dispos. 2003, 31, 815–832. [Google Scholar]
- Boocock, D.J.; Faust, G.E.; Patel, K.R.; Schinas, A.M.; Brown, V.A.; Ducharme, M.P.; Booth, T.D.; Crowell, J.A.; Perloff, M.; Gescher, A.J.; et al. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1246–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Substrate * | Concentration (µM) | Metabolite | SRM Transition (m/z) | Collision Energy (eV) | Polarity ** |
|---|---|---|---|---|---|---|
| CYP1A2 | Phenacetin | 20 | Acetaminophen | 152 > 110 | 25 | ESI+ |
| CYP2A6 | Coumarin | 1 | 7-Hydroxycoumarin | 163 > 107 | 17 | ESI+ |
| CYP2B6 | Bupropion | 3 | Hydroxybupropion | 256 > 238 | 10 | ESI+ |
| CYP2C8 | Amodiaquine | 0.1 | N-Desethylamodiaquine | 328 > 283 | 13 | ESI+ |
| CYP2C9 | Diclofenac | 1 | 4-Hydroxydiclofenac | 312 > 231 | 15 | ESI+ |
| CYP2C19 | S-Mephenytoin | 40 | 4-Hydroxymephenytoin | 235 > 150 | 15 | ESI+ |
| CYP2D6 | Dextromethorphan | 2 | Dextrorphan | 258 > 157 | 30 | ESI+ |
| CYP2E1 | Chlorzoxazone | 5 | 6-Hydroxychlorzoxazone | 184 > 120 | 18 | ESI− |
| CYP3A | Nifedipine | 0.2 | Dehydronifedipine | 345 > 284 | 30 | ESI+ |
| Midazolam | 0.1 | 1′-Hydroxymidazolam | 342 > 203 | 28 | ESI+ | |
| IS | Trimipramine | 0.07 | - | 295 > 100 | 17 | ESI+ |
| UGT1A1 | SN-38 | 0.5 | SN-38 glucuronide | 569 > 393 | 30 | ESI+ |
| UGT1A3 | CDCA | 2 | CDCA-24 glucuronide | 567 > 391 | 20 | ESI− |
| UGT1A4 | TFP | 0.5 | TFP-β-d-glucuronide | 584 > 408 | 30 | ESI+ |
| UGT1A6 | N-ASER | 1 | N-ASER-β-d-glucuronide | 395 > 219 | 10 | ESI+ |
| UGT1A9 | MPA | 0.2 | MPA-β-d-glucuronide | 495 > 319 | 25 | ESI− |
| UGT2B7 | Naloxone | 0.2 | Naloxone-β-d-glucuronide | 504 > 310 | 30 | ESI+ |
| IS | Estrone-β-d-glucuronide | 0.25 | - | 445 > 269 | 35 | ESI− |
| P450 Enzyme | Substrate | Trans-resveratrol (µM) | Cis-resveratrol (µM) | ||||
|---|---|---|---|---|---|---|---|
| Without Preincubation | With Preincubation (30 min) | IC50 Shift | Without Preincubation | With Preincubation (30 min) | IC50 Shift | ||
| 1A2 | Phenacetin | 12.9 ± 2.50 | 1.21 ± 0.20 | 10.80 | 14.0 ± 4.10 | 7.20 ± 0.50 | 1.90 |
| 2A6 | Coumarin | >50.0 | >50.0 | – | >50.0 | 48.3 ± 17.0 | – |
| 2B6 | Bupropion | 34.8 ± 7.20 | 40.1 ± 14.9 | 0.90 | 28.1 ± 13.9 | 32.8 ± 6.10 | 0.90 |
| 2C8 | Amodiaquine | >50.0 | 35.2 ± 6.40 | >1.40 | >50.0 | >50.0 | – |
| 2C9 | Diclofenac | 36.2 ± 3.30 | 23.8 ± 4.10 | 1.50 | >50.0 | >50.0 | – |
| 2C19 | S-Mephenytoin | 48.4 ± 10.9 | 8.13 ± 1.61 | 6.00 | 46.5 ± 16.1 | 19.8 ± 4.20 | 2.30 |
| 2D6 | Dextromethor-phan | >50.0 | 32.8 ± 3.20 | >1.50 | >50.0 | >50.0 | - |
| 2E1 | Chlorzoxazone | >50.0 | 9.82 ± 2.21 | >5.00 | >50.0 | 10.8 ± 2.90 | >4.60 |
| 3A | Midazolam | 29.2 ± 4.50 | 16.6 ± 4.00 | 1.80 | >50.0 | >50.0 | - |
| Nifedipine | 19.6 ± 2.30 | 5.62 ± 0.60 | 3.50 | >50.0 | >50.0 | - | |
| UGT Enzyme | Substrate * | Trans-Resveratrol | Cis-Resveratrol |
|---|---|---|---|
| 1A1 | SN-38 | 9.57 ± 1.60 | >50.0 |
| 1A3 | CDCA | >50.0 | >50.0 |
| 1A4 | TFP | >50.0 | >50.0 |
| 1A6 | N-ASER | >50.0 | >50.0 |
| 1A9 | MPA | >50.0 | >50.0 |
| 2B7 | Naloxone | >50.0 | >50.0 |
| Enzyme | Substrate | trans-Resveratrol | cis-Resveratrol | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Without Preincubation | With Preincubation (30 min) | Without Preincubation | With Preincubation (30 min) | ||||||
| Ki (μM) | Mode of Inhibition | Ki (μM) | Mode of Inhibition | Ki (μM) | Mode of Inhibition | Ki (μM) | Mode of Inhibition | ||
| CYP1A2 | Phenacetin | 13.8 ± 1.70 | Competitive | 1.62 ± 0.11 | Noncompetitive | 9.19 ± 1.41 | Competitive | 9.46 ± 0.63 | Noncompetitive |
| CYP2C19 | S-Mephenytoin | - | - | 9.78 ± 0.60 | Noncompetitive | - | - | 21.8 ± 6.10 | Noncompetitive |
| CYP2E1 | Chlorzoxazone | - | - | 9.11 ± 1.62 | Competitive | - | - | 8.08 ± 1.43 | Competitive |
| CYP3A | Nifedipine | 23.6 ± 1.90 | Noncompetitive | 4.60 ± 0.11 | Noncompetitive | - | - | - | - |
| Midazolam | 23.8 ± 2.30 | Noncompetitive | 1.02 ± 0.22 | Uncompetitive | - | - | - | - | |
| UGT1A1 | SN-38 * | 27.4 ± 3.60 | Noncompetitive | - | - | - | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, S.-B.; Park, S.-Y.; Bae, S.; Seo, H.-J.; Kim, S.-E.; Lee, G.-M.; Wu, Z.; Liu, K.-H. Comprehensive Investigation of Stereoselective Food Drug Interaction Potential of Resveratrol on Nine P450 and Six UGT Isoforms in Human Liver Microsomes. Pharmaceutics 2021, 13, 1419. https://doi.org/10.3390/pharmaceutics13091419
Ji S-B, Park S-Y, Bae S, Seo H-J, Kim S-E, Lee G-M, Wu Z, Liu K-H. Comprehensive Investigation of Stereoselective Food Drug Interaction Potential of Resveratrol on Nine P450 and Six UGT Isoforms in Human Liver Microsomes. Pharmaceutics. 2021; 13(9):1419. https://doi.org/10.3390/pharmaceutics13091419
Chicago/Turabian StyleJi, Seung-Bae, So-Young Park, Subin Bae, Hyung-Ju Seo, Sin-Eun Kim, Gyung-Min Lee, Zhexue Wu, and Kwang-Hyeon Liu. 2021. "Comprehensive Investigation of Stereoselective Food Drug Interaction Potential of Resveratrol on Nine P450 and Six UGT Isoforms in Human Liver Microsomes" Pharmaceutics 13, no. 9: 1419. https://doi.org/10.3390/pharmaceutics13091419