
Nanoparticles to Target and Treat Macrophages: The Ockham’s Concept?


Abstract
: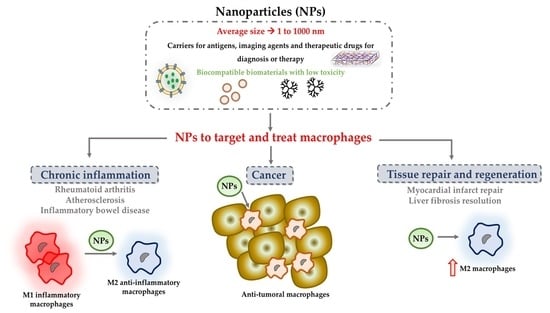
1. Introduction
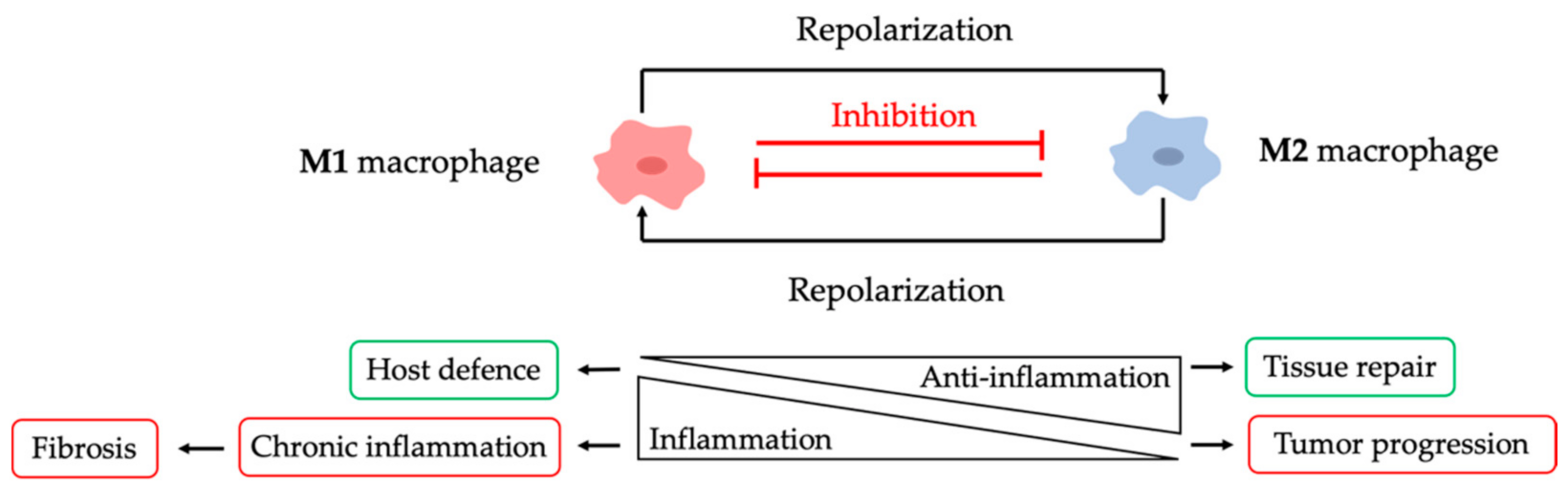
2. NPs to Modulate Macrophages in Chronic Inflammation
2.1. NPs to Induce Anti-Inflammatory Macrophage Switch
2.2. NPs Modulating Macrophages in Rheumatoid Arthritis
2.3. NPs Modulating Macrophages in Inflammatory Bowel Disease (IBD)
2.4. NPs Modulating Macrophages in Atherosclerosis
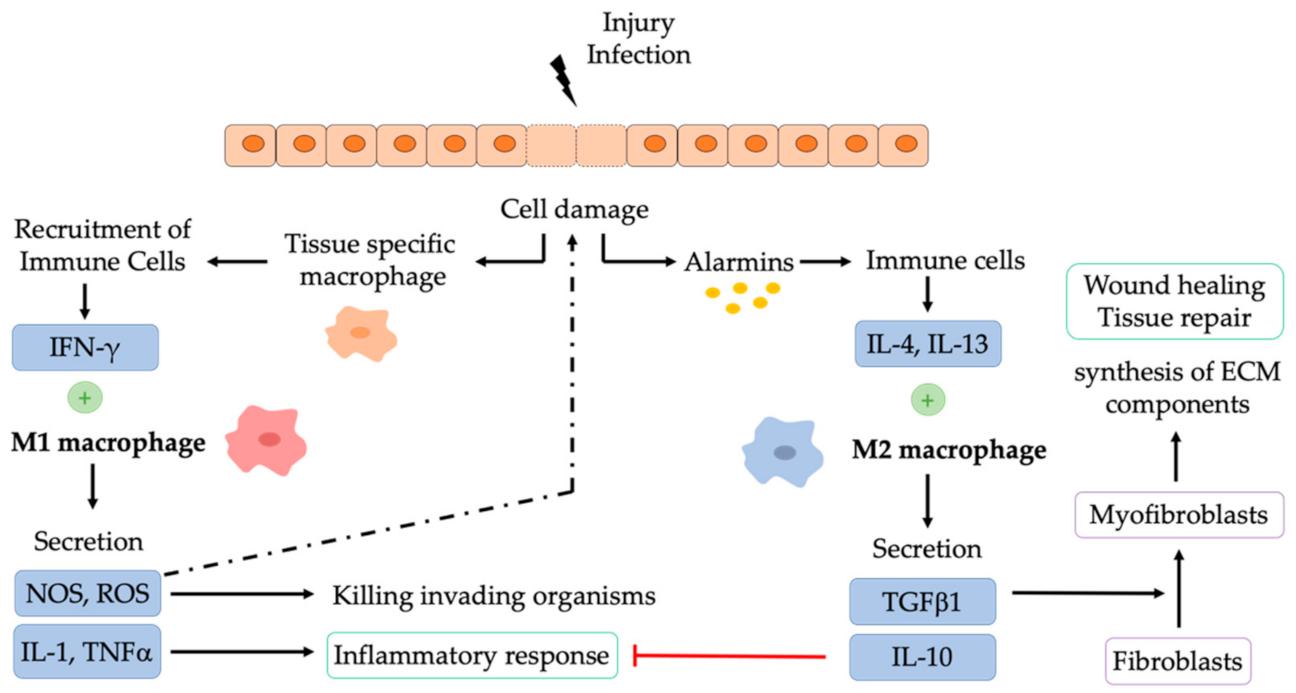
3. NPs to Stimulate Tissue Repair and Regeneration
3.1. NPs Modulating Macrophages in Tissue Regeneration
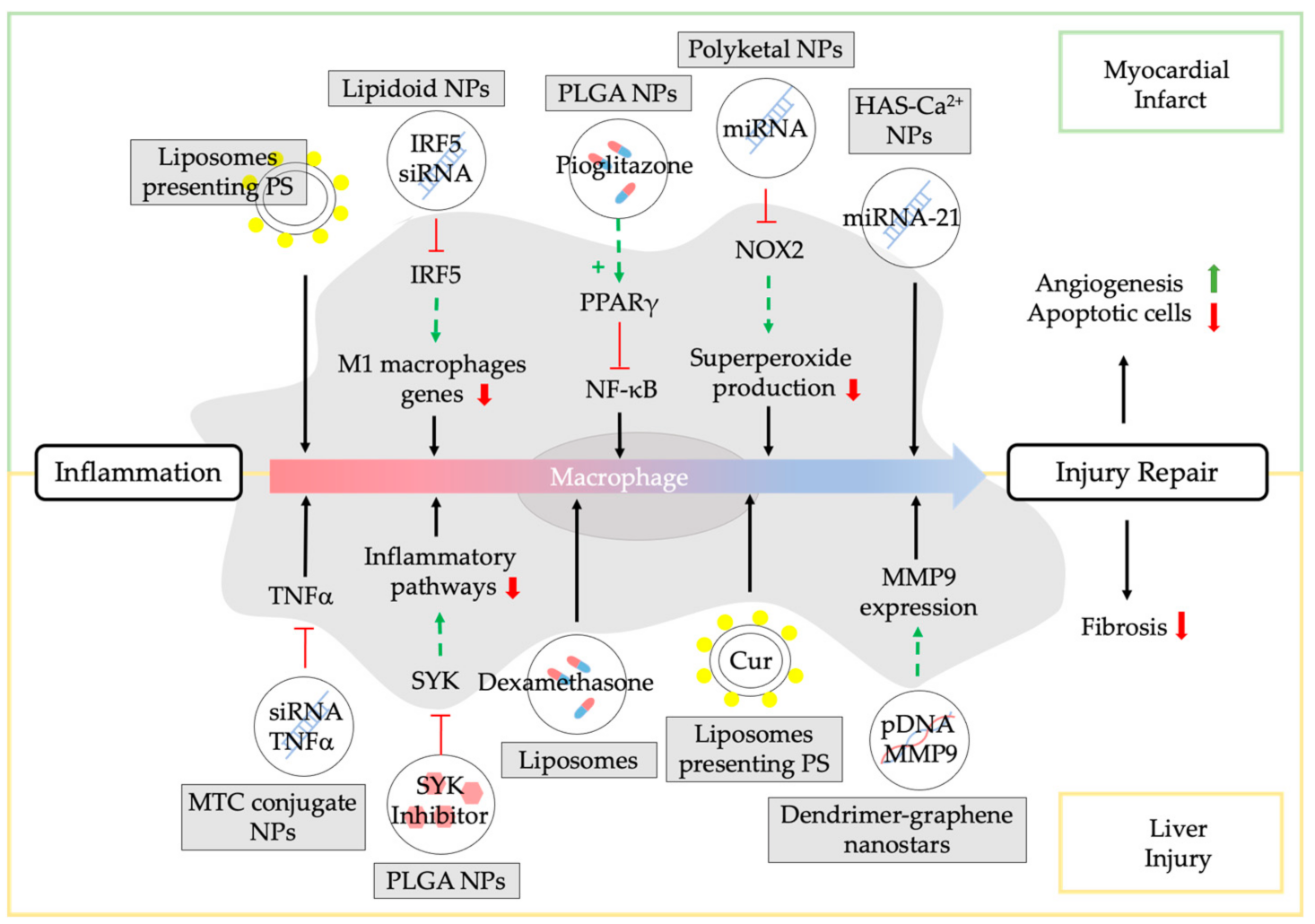
3.2. NPs Modulating Macrophages in Myocardial Infarct Repair
3.3. NPs Modulating Macrophages in Chronic Liver Injury
4. Nanoparticles to Target and Treat Tumour-Associated Macrophages
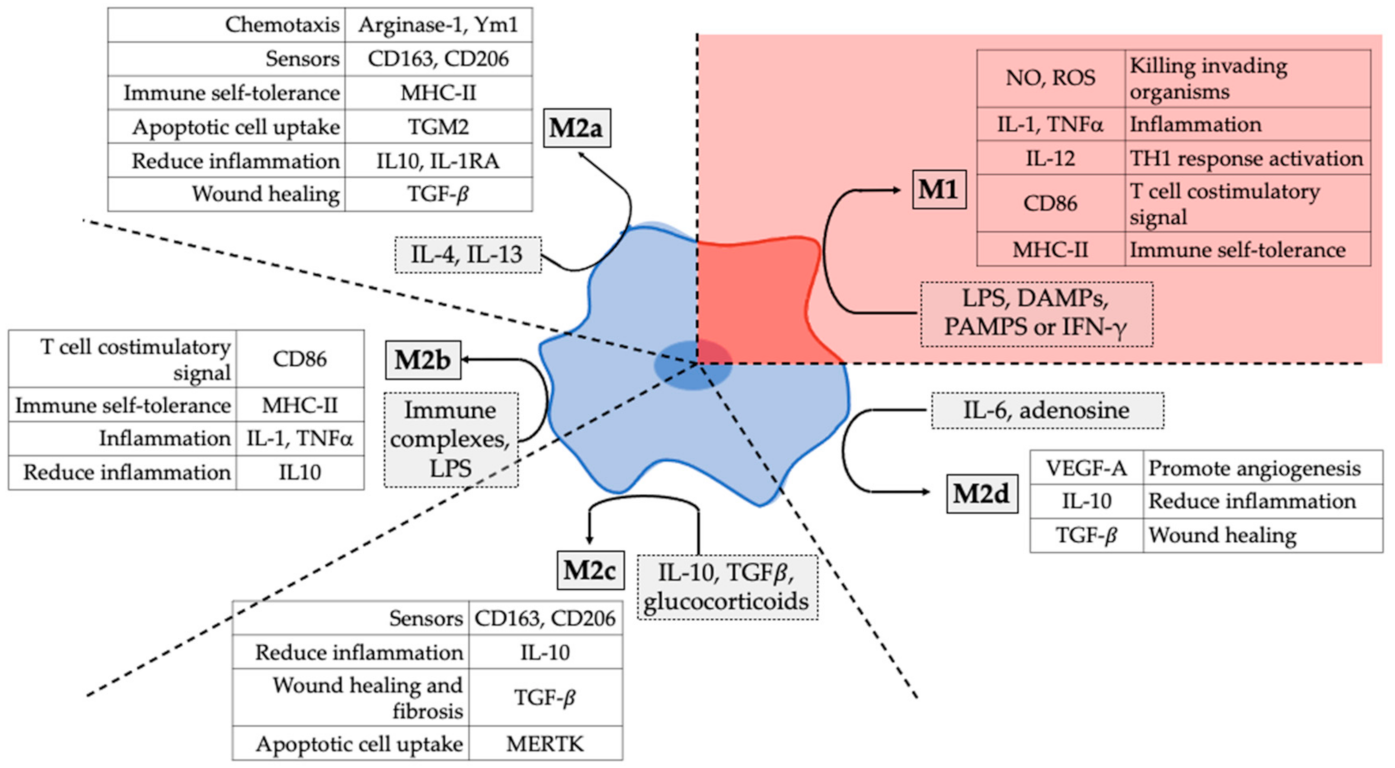
4.1. Specific Differential Phenotype of Tumour-Associated Macrophages
4.2. NPs to Target TAM for Cancer Diagnostics and Prognosis
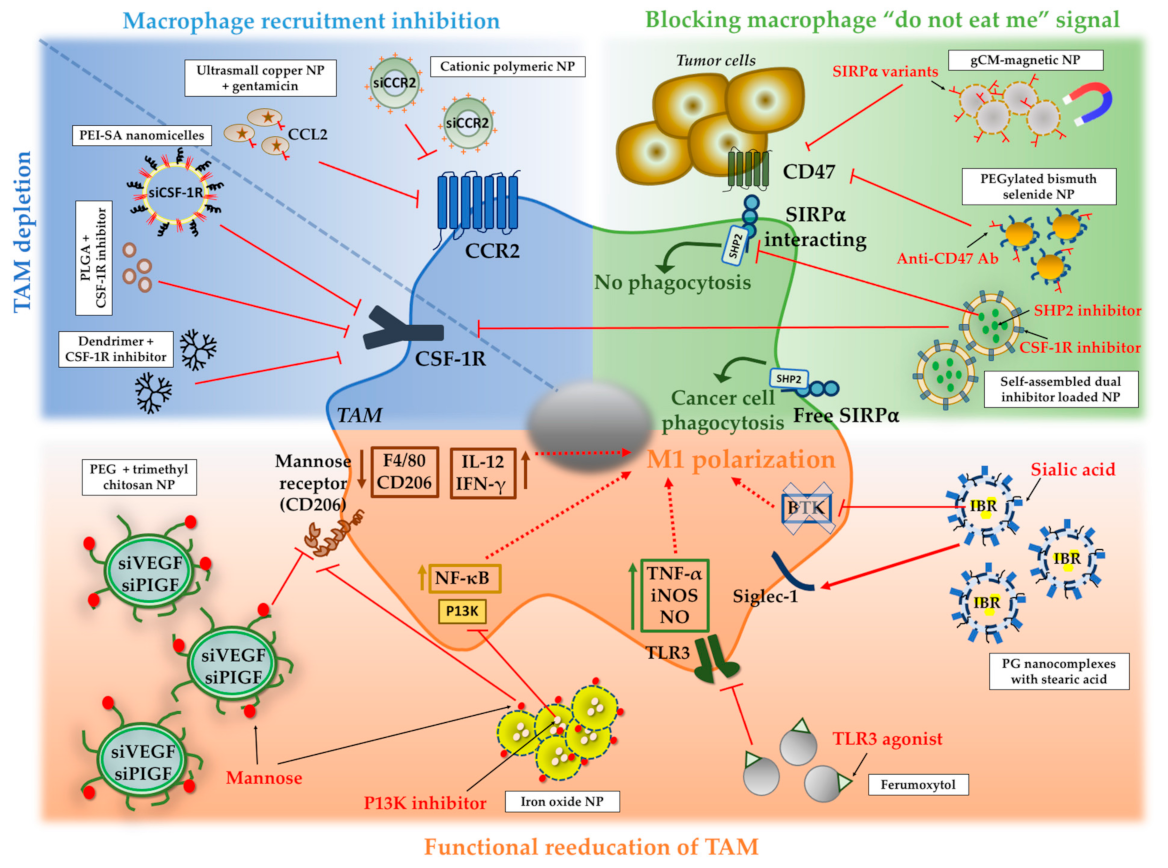
4.3. NPs to Inhibit Macrophage Recruitment and to Deplete TAM in Tumors
4.4. NPs to Block the Macrophage “Do Not Eat Me” Signal
4.5. NPs to Switch TAM to an Antitumor “M1-Like” Phenotype
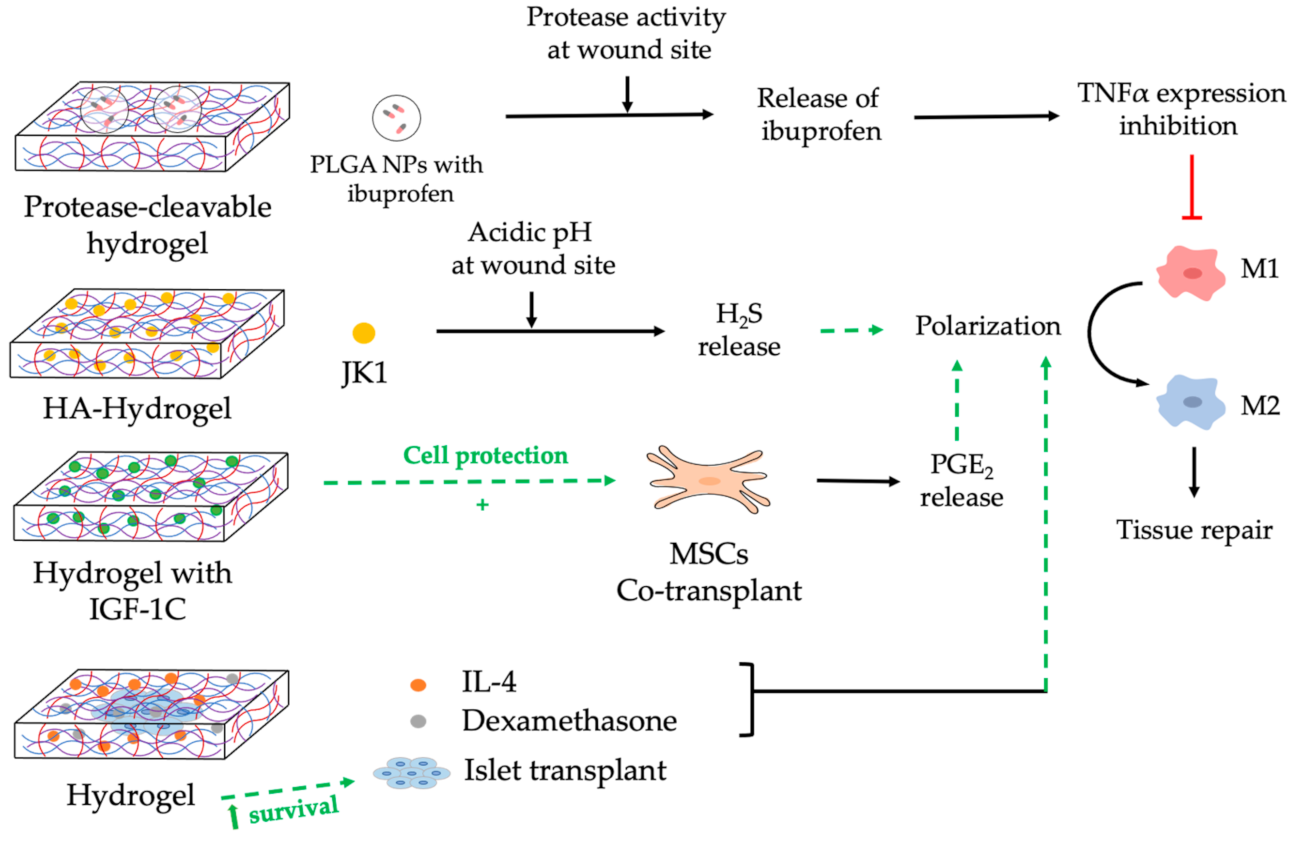
5. Nanocomposite Hydrogels to Modulate Macrophages
6. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Ponzoni, M.; Pastorino, F.; Di Paolo, D.; Perri, P.; Brignole, C. Targeting macrophages as a potential therapeutic intervention: Impact on inflammatory diseases and cancer. Int. J. Mol. Sci. 2018, 19, 1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Zang, X.; Cheng, M.; Zhang, X.; Chen, X. Targeting macrophages using nanoparticles: A potential therapeutic strategy for atherosclerosis. J. Mater. Chem. B 2021, 9, 3284–3294. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Leng, X.; Zhang, Q. The current state of nanoparticle-induced macrophage polarization and reprogramming research. Int. J. Mol. Sci. 2017, 18, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.X.; Zhang, S.X.; Wu, H.J.; Rong, X.L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, C.J.; Leibovich, S.J. Regulation of Macrophage Polarization and Wound Healing. Adv. Wound Care 2012, 1, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Hu, G.; Guo, M.; Xu, J.; Wu, F.; Fan, J.; Huang, Q.; Yang, G.; Lv, Z.; Wang, X.; Jin, Y. Nanoparticles targeting macrophages as potential clinical therapeutic agents against cancer and inflammation. Front. Immunol. 2019, 10, 1998. [Google Scholar] [CrossRef]
- Nakkala, J.R.; Li, Z.; Ahmad, W.; Wang, K.; Gao, C. Immunomodulatory biomaterials and their application in therapies for chronic inflammation-related diseases. Acta Biomater. 2021, 123, 1–30. [Google Scholar] [CrossRef]
- Garash, R.; Bajpai, A.; Marcinkiewicz, B.M.; Spiller, K.L. Drug delivery strategies to control macrophages for tissue repair and regeneration. Exp. Biol. Med. 2016, 241, 1054–1063. [Google Scholar] [CrossRef] [Green Version]
- Talamini, L.; Matsuura, E.; De Cola, L.; Muller, S. Immunologically Inert Nanostructures as Selective Therapeutic Tools in Inflammatory Diseases. Cells 2021, 10, 707. [Google Scholar] [CrossRef] [PubMed]
- Chellat, F.; Merhi, Y.; Moreau, A.; Yahia, L. Therapeutic potential of nanoparticulate systems for macrophage targeting. Biomaterials 2005, 26, 7260–7275. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.H.; Rastogi, R.; Shelke, J.; Amiji, M.M. Modulation of Macrophage Functional Polarity towards Anti-Inflammatory Phenotype with Plasmid DNA Delivery in CD44 Targeting Hyaluronic Acid Nanoparticles. Sci. Rep. 2015, 5, 16632. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.S.; Lau, C.M.; Thomas, S.N.; Gray Jerome, W.; Maron, D.J.; Dickerson, J.H.; Hubbell, J.A.; Giorgio, T.D. Size- and charge-dependent non-specific uptake of PEGylated nanoparticles by macrophages. Int. J. Nanomed. 2012, 7, 799–813. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, M.M.; Liu, J.C.; Trujillo-de Santiago, G.; Cha, B.H.; Vishwakarma, A.; Ghaemmaghami, A.M.; Khademhosseini, A. Delivery strategies to control inflammatory response: Modulating M1–M2 polarization in tissue engineering applications. J. Control Release 2016, 240, 349–363. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Tran, T.H.; Amiji, M. Macrophage repolarization with targeted alginate nanoparticles containing IL-10 plasmid DNA for the treatment of experimental arthritis. Biomaterials 2015, 61, 162–177. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kim, H.Y.; Song, S.Y.; Go, S.H.; Sohn, H.S.; Baik, S.; Soh, M.; Kim, K.; Kim, D.; Kim, H.C.; et al. Synergistic Oxygen Generation and Reactive Oxygen Species Scavenging by Manganese Ferrite/Ceria Co-decorated Nanoparticles for Rheumatoid Arthritis Treatment. ACS Nano 2019, 13, 3206–3217. [Google Scholar] [CrossRef]
- Howard, K.A.; Paludan, S.R.; Behlke, M.A.; Besenbacher, F.; Deleuran, B.; Kjems, J. Chitosan/siRNA nanoparticle-mediated TNF-α knockdown in peritoneal macrophages for anti-inflammatory treatment in a murine arthritis model. Mol. Ther. 2009, 17, 162–168. [Google Scholar] [CrossRef]
- Fernandes, J.C.; Wang, H.; Jreyssaty, C.; Benderdour, M.; Lavigne, P.; Qiu, X.; Winnik, F.M.; Zhang, X.; Dai, K.; Shi, Q. Bone-protective effects of nonviral gene therapy with folate-chitosan DNA nanoparticle containing interleukin-1 receptor antagonist gene in rats with adjuvant-induced arthritis. Mol. Ther. 2008, 16, 1243–1251. [Google Scholar] [CrossRef]
- Thomas, T.P.; Goonewardena, S.N.; Majoros, I.J.; Kotlyar, A.; Cao, Z.; Leroueil, P.R.; Baker, J.R. Folate-targeted nanoparticles show efficacy in the treatment of inflammatory arthritis. Arthritis Rheumatol. 2011, 63, 2671–2680. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhao, M.; Yu, C.; Zhang, X.; Liu, J.; Cheng, X.; Lee, R.J.; Sun, F.; Teng, L.; Li, Y. Multifunctional folate receptor-targeting and pH-responsive nanocarriers loaded with methotrexate for treatment of rheumatoid arthritis. Int. J. Nanomed. 2017, 12, 6735–6746. [Google Scholar] [CrossRef] [Green Version]
- Espinosa-Cano, E.; Aguilar, M.R.; Portilla, Y.; Barber, D.F.; Román, J.S. Anti-inflammatory polymeric nanoparticles based on ketoprofen and dexamethasone. Pharmaceutics 2020, 12, 723. [Google Scholar] [CrossRef]
- Ni, R.; Song, G.; Fu, X.; Song, R.; Li, L.; Pu, W.; Gao, J.; Hu, J.; Liu, Q.; He, F.; et al. Reactive oxygen species-responsive dexamethasone-loaded nanoparticles for targeted treatment of rheumatoid arthritis via suppressing the iRhom2/TNF-α/BAFF signaling pathway. Biomaterials 2020, 232, 119730. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Laroui, H.; Ayyadurai, S.; Viennois, E.; Charania, M.A.; Zhang, Y.; Merlin, D. Mannosylated bioreducible nanoparticle-mediated macrophage-specific TNF-α RNA interference for IBD therapy. Biomaterials 2013, 34, 7471–7482. [Google Scholar] [CrossRef] [Green Version]
- Laroui, H.; Viennois, E.; Xiao, B.; Canup, B.S.B.; Geem, D.; Denning, T.L.; Merlin, D. Fab’-bearing siRNA TNFα-loaded nanoparticles targeted to colonic macrophages offer an effective therapy for experimental colitis. J. Control Release 2014, 186, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, D.S.; Dalmasso, G.; Wang, L.; Sitaraman, S.V.; Merlin, D.; Murthy, N. Orally delivered thioketal nanoparticles loaded with TNF-α-siRNA target inflammation and inhibit gene expression in the intestines. Nat. Mater. 2010, 9, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Guo, J.; Gui, S. Orally targeted galactosylated chitosan poly(lactic-co-glycolic acid) nanoparticles loaded with TNF-ɑ siRNA provide a novel strategy for the experimental treatment of ulcerative colitis. Eur. J. Pharm. Sci. 2018, 125, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Kriegel, C.; Amiji, M.M. Dual TNF-α/Cyclin D1 gene silencing with an oral polymeric microparticle system as a novel strategy for the treatment of inflammatory bowel disease. Clin. Transl. Gastroenterol. 2011, 2, e2. [Google Scholar] [CrossRef]
- Aouadi, M.; Tesz, G.J.; Nicoloro, S.M.; Wang, M.; Chouinard, M.; Soto, E.; Ostroff, G.R.; Czech, M.P. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature 2009, 458, 1180–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Wang, J.; Yannie, P.J.; Korzun, W.J.; Yang, H.; Ghosh, S. Nanoparticle-based “Two-pronged” approach to regress atherosclerosis by simultaneous modulation of cholesterol influx and efflux. Biomaterials 2020, 260, 120333. [Google Scholar] [CrossRef]
- Zhao, Y.; He, Z.; Gao, H.; Tang, H.; He, J.; Guo, Q.; Zhang, W.; Liu, J. Fine Tuning of Core-Shell Structure of Hyaluronic Acid/Cell-Penetrating Peptides/siRNA Nanoparticles for Enhanced Gene Delivery to Macrophages in Antiatherosclerotic Therapy. Biomacromolecules 2018, 19, 2944–2956. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, F.; Dutta, P.; Gorbatov, R.; Novobrantseva, T.I.; Donahoe, J.S.; Courties, G.; Lee, K.M.; Kim, J.I.; Markmann, J.F.; Marinelli, B.; et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat. Biotechnol. 2011, 29, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Chen, C.; Luo, J.; Davis, J.R.J.; Zhang, B.; Tang, L.; Shi, W.; Liao, D. EGFP-EGF1-conjugated poly (lactic-co-glycolic acid) nanoparticles as a carrier for the delivery of CCR2− shRNA to atherosclerotic macrophage in vitro. Sci. Rep. 2020, 10, 19636. [Google Scholar] [CrossRef]
- Tao, W.; Yurdagul, A.; Kong, N.; Li, W.; Wang, X.; Doran, A.C.; Feng, C.; Wang, J.; Islam, M.A.; Farokhzad, O.C.; et al. SiRNA nanoparticles targeting CaMKIIγ in lesional macrophages improve atherosclerotic plaque stability in mice. Sci. Transl. Med. 2020, 12, eaay1063. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ye, J.; Hosseini-Nassab, N.; Flores, A.; Kalashnikova, I.; Paluri, S.L.; Lotfi, M.; Leeper, N.J.; Smith, B.R. Macrophage-targeted single walled carbon nanotubes stimulate phagocytosis via pH-dependent drug release. Nano Res. 2021, 14, 762–769. [Google Scholar] [CrossRef]
- Alvarado-Vazquez, P.A.; Bernal, L.; Paige, C.A.; Grosick, R.L.; Moracho Vilrriales, C.; Ferreira, D.W.; Ulecia-Morón, C.; Romero-Sandoval, E.A. Macrophage-specific nanotechnology-driven CD163 overexpression in human macrophages results in an M2 phenotype under inflammatory conditions. Immunobiology 2017, 222, 900–912. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.H.; Krishnan, S.; Amiji, M.M. MicroRNA-223 induced repolarization of peritoneal macrophages using CD44 targeting hyaluronic acid nanoparticles for anti-inflammatory effects. PLoS ONE 2016, 11, e152024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef]
- Singh, A.; Talekar, M.; Raikar, A.; Amiji, M. Macrophage-targeted delivery systems for nucleic acid therapy of inflammatory diseases. J. Control Release 2014, 190, 515–530. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Tang, Y.; Lv, Z.; Lin, Y.; Chen, L. Nanomedicine—Advantages for their use in rheumatoid arthritis theranostics. J. Control Release 2019, 316, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijden, J.W.; Oerlemans, R.; Dijkmans, B.A.C.; Qi, H.; Van Der Laken, C.J.; Lems, W.F.; Jackman, A.L.; Kraan, M.C.; Tak, P.P.; Ratnam, M.; et al. Folate receptor β as a potential delivery route for novel folate antagonists to macrophages in the synovial tissue of rheumatoid arthritis patients. Arthritis Rheumatol. 2009, 60, 12–21. [Google Scholar] [CrossRef]
- Peer, D.; Eun, J.P.; Morishita, Y.; Carman, C.V.; Shimaoka, M. Systemic leukocyte-directed siRNA delivery revealing cyclin D1 as an anti-inflammatory target. Science 2008, 319, 627–630. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Huang, Q.; Liu, C.; Kwong, C.H.T.; Yue, L.; Wan, J.B.; Lee, S.M.Y.; Wang, R. Treatment of atherosclerosis by macrophage-biomimetic nanoparticles via targeted pharmacotherapy and sequestration of proinflammatory cytokines. Nat. Commun. 2020, 11, 2622. [Google Scholar] [CrossRef]
- Bobryshev, Y.V.; Ivanova, E.A.; Chistiakov, D.A.; Nikiforov, N.G.; Orekhov, A.N. Macrophages and Their Role in Atherosclerosis: Pathophysiology and Transcriptome Analysis. Biomed. Res. Int. 2016, 2016, 9582430. [Google Scholar] [CrossRef] [Green Version]
- Beldman, T.J.; Senders, M.L.; Alaarg, A.; Pérez-Medina, C.; Tang, J.; Zhao, Y.; Fay, F.; Deichmöller, J.; Born, B.; Desclos, E.; et al. Hyaluronan Nanoparticles Selectively Target Plaque-Associated Macrophages and Improve Plaque Stability in Atherosclerosis. ACS Nano 2017, 11, 5785–5799. [Google Scholar] [CrossRef]
- Duivenvoorden, R.; Tang, J.; Cormode, D.P.; Mieszawska, A.J.; Izquierdo-Garcia, D.; Ozcan, C.; Otten, M.J.; Zaidi, N.; Lobatto, M.E.; Van Rijs, S.M.; et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat. Commun. 2014, 5, 3065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Yuan, Q.; Bie, J.; Wallace, R.L.; Yannie, P.J.; Wang, J.; Lancina, M.G.; Zolotarskaya, O.Y.; Korzun, W.; Yang, H.; et al. Development of mannose functionalized dendrimeric nanoparticles for targeted delivery to macrophages: Use of this platform to modulate atherosclerosis. Transl. Res. 2018, 193, 13–30. [Google Scholar] [CrossRef]
- Kojima, Y.; Weissman, I.L.; Leeper, N.J. The Role of Efferocytosis in Atherosclerosis. Circulation 2017, 135, 476–489. [Google Scholar] [CrossRef] [Green Version]
- Shen, P.; Chen, Y.; Luo, S.; Fan, Z.; Wang, J.; Chang, J.; Deng, J. Applications of biomaterials for immunosuppression in tissue repair and regeneration. Acta Biomater. 2021, 126, 31–44. [Google Scholar] [CrossRef]
- Raimondo, T.M.; Mooney, D.J. Functional muscle recovery with nanoparticle-directed M2 macrophage polarization in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 10648–10653. [Google Scholar] [CrossRef] [Green Version]
- Ni, C.; Zhou, J.; Kong, N.; Bian, T.; Zhang, Y.; Huang, X.; Xiao, Y.; Yang, W.; Yan, F. Gold nanoparticles modulate the crosstalk between macrophages and periodontal ligament cells for periodontitis treatment. Biomaterials 2019, 206, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Corsi, F.; Carotenuto, F.; Di Nardo, P.; Teodori, L. Harnessing inorganic nanoparticles to direct macrophage polarization for skeletal muscle regeneration. Nanomaterials 2020, 10, 1963. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Han, S.; Shi, M.; Liu, G.; Chen, Z.; Chang, J.; Wu, C.; Xiao, Y. Immunomodulatory effects of mesoporous silica nanoparticles on osteogenesis: From nanoimmunotoxicity to nanoimmunotherapy. Appl. Mater. Today 2018, 10, 184–193. [Google Scholar] [CrossRef]
- Shi, M.; Chen, Z.; Farnaghi, S.; Friis, T.; Mao, X.; Xiao, Y.; Wu, C. Copper-doped mesoporous silica nanospheres, a promising immunomodulatory agent for inducing osteogenesis. Acta Biomater. 2016, 30, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Heo, D.N.; Kim, H.J.; Ko, W.K.; Lee, S.J.; Heo, M.; Bang, J.B.; Lee, J.B.; Hwang, D.S.; Do, S.H.; et al. Inhibition of Osteoclast Differentiation and Bone Resorption by Bisphosphonate-conjugated Gold Nanoparticles. Sci. Rep. 2016, 6, 27336. [Google Scholar] [CrossRef] [PubMed]
- Harel-Adar, T.; Mordechai, T.B.; Amsalem, Y.; Feinberg, M.S.; Leor, J.; Cohen, S. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc. Natl. Acad. Sci. USA 2011, 108, 1827–1832. [Google Scholar] [CrossRef] [Green Version]
- Bejerano, T.; Etzion, S.; Elyagon, S.; Etzion, Y.; Cohen, S. Nanoparticle Delivery of miRNA-21 Mimic to Cardiac Macrophages Improves Myocardial Remodeling after Myocardial Infarction. Nano Lett. 2018, 18, 5885–5891. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Brown, M.E.; Zhang, H.; Martinez, M.; Zhao, Z.; Bhutani, S.; Yin, S.; Trac, D.; Xi, J.J.; Davis, M.E. High-throughput screening identifies microRNAs that target Nox2 and improve function after acute myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H1002–H1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courties, G.; Heidt, T.; Sebas, M.; Iwamoto, Y.; Jeon, D.; Truelove, J.; Tricot, B.; Wojtkiewicz, G.; Dutta, P.; Sager, H.B.; et al. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J. Am. Coll. Cardiol. 2014, 63, 1556–1566. [Google Scholar] [CrossRef]
- Tokutome, M.; Matoba, T.; Nakano, Y.; Okahara, A.; Fujiwara, M.; Koga, J.I.; Nakano, K.; Tsutsui, H.; Egashira, K. Peroxisome proliferator-activated receptor-gamma targeting nanomedicine promotes cardiac healing after acute myocardial infarction by skewing monocyte/macrophage polarization in preclinical animal models. Cardiovasc. Res. 2019, 115, 419–431. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Yin, L.; Tang, C.; Yin, C. Multifunctional polymeric nanoparticles for oral delivery of TNF-α siRNA to macrophages. Biomaterials 2013, 34, 2843–2854. [Google Scholar] [CrossRef]
- Kurniawan, D.W.; Jajoriya, A.K.; Dhawan, G.; Mishra, D.; Argemi, J.; Bataller, R.; Storm, G.; Mishra, D.P.; Prakash, J.; Bansal, R. Therapeutic inhibition of spleen tyrosine kinase in inflammatory macrophages using PLGA nanoparticles for the treatment of non-alcoholic steatohepatitis. J. Control Release 2018, 288, 227–238. [Google Scholar] [CrossRef]
- Bartneck, M.; Scheyda, K.M.; Warzecha, K.T.; Rizzo, L.Y.; Hittatiya, K.; Luedde, T.; Storm, G.; Trautwein, C.; Lammers, T.; Tacke, F. Fluorescent cell-traceable dexamethasone-loaded liposomes for the treatment of inflammatory liver diseases. Biomaterials 2015, 37, 367–382. [Google Scholar] [CrossRef]
- Wanga, J.; Pana, W.; Wang, Y.; Lei, W.; Feng, B.; Du, C.; Wanga, X.J. Enhanced efficacy of curcumin with phosphatidylserine-decorated nanoparticles in the treatment of hepatic fibrosis. Drug Deliv. 2018, 25, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Melgar-Lesmes, P.; Luquero, A.; Parra-Robert, M.; Mora, A.; Ribera, J.; Edelman, E.R.; Jiménez, W. Graphene-Dendrimer Nanostars for Targeted Macrophage Overexpression of Metalloproteinase 9 and Hepatic Fibrosis Precision Therapy. Nano Lett. 2018, 18, 5839–5845. [Google Scholar] [CrossRef]
- Pöttler, M.; Cicha, I.; Unterweger, H.; Janko, C.; Friedrich, R.P.; Alexiou, C. Nanoparticles for regenerative medicine. Nanomedicine 2019, 14, 1929–1933. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Venkatesan, R.; Zhai, Z.; Muhammad, W.; Nakkala, J.R.; Gao, C. Micro- and nanoparticles-based immunoregulation of macrophages for tissue repair and regeneration. Colloids Surf. B Biointerfaces 2020, 192, 111075. [Google Scholar] [CrossRef]
- Ju, C.; Tacke, F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell. Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef] [Green Version]
- van der Heide, D.; Weiskirchen, R.; Bansal, R. Therapeutic Targeting of Hepatic Macrophages for the Treatment of Liver Diseases. Front. Immunol. 2019, 10, 2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartneck, M.; Warzecha, K.T.; Tacke, F. Therapeutic targeting of liver inflammation and fibrosis by nanomedicine. Hepatobiliary Surg. Nutr. 2014, 3, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Su, G.; Zhai, S. Recent advances in nanomedicine for the diagnosis and therapy of liver fibrosis. Nanomaterials 2020, 10, 1945. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L. Europe PMC Funders Group Tumor-Associated Macrophages as Treatment Targets in Oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Petty, A.J.; Yang, Y. Tumor-associated macrophages: Implications in cancer immunotherapy. Immunotherapy 2017, 9, 289–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Saccani, A.; Bottazzi, B.; Polentarutti, N.; Vecchi, A.; Van Damme, J.; Mantovani, A. Autocrine Production of IL-10 Mediates Defective IL-12 Production and NF-κB Activation in Tumor-Associated Macrophages. J. Immunol. 2000, 164, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Bonde, A.K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, S.A.; Gregory, S.L.; Zhang, Y.; Emmanuel M Paul, H.J.D. Tumor-Associated Macrophages Produce Interleukin 6 and Signal via STAT3 to Promote Expansion of Human Hepatocellular Carcinoma Stem Cells Shanshan. Gastroenterology 2012, 27, 2359–2372. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Li, J.; Gu, P.; Fan, X. The application of nanoparticles in cancer immunotherapy: Targeting tumor microenvironment. Bioact. Mater. 2021, 6, 1973–1987. [Google Scholar] [CrossRef]
- Leimgruber, A.; Berger, C.; Cortez-Retamozo, V.; Etzrodt, M.; Newton, A.P.; Waterman, P.; Figueiredo, J.L.; Kohler, R.H.; Elpek, N.; Mempel, T.R.; et al. Behavior of endogenous Tumor-associated macrophages assessed in vivo using a functionalized nanoparticle. Neoplasia 2009, 11, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Daldrup-Link, H.E.; Golovko, D.; Ruffell, B.; DeNardo, D.G.; Castaneda, R.; Ansari, C.; Rao, J.; Tikhomirov, G.A.; Wendland, M.F.; Corot, C.; et al. MRI of tumor-associated macrophages with clinically applicable iron oxide nanoparticles. Clin. Cancer Res. 2011, 17, 5695–5704. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.S.C.; Gunda, V.; Li, R.; Prytyskach, M.; Iwamoto, Y.; Kohler, R.H.; Parangi, S.; Weissleder, R.; Miller, M.A. Detecting immune response to therapies targeting PDL1 and BRAF by using ferumoxytol MRI and macrin in anaplastic thyroid cancer. Radiology 2020, 298, 123–132. [Google Scholar] [CrossRef]
- Baroni, S.; Ruggiero, M.R.; Bitonto, V.; Broche, L.M.; Lurie, D.J.; Aime, S.; Geninatti Crich, S. In vivo assessment of tumour associated macrophages in murine melanoma obtained by low-field relaxometry in the presence of iron oxide particles. Biomaterials 2020, 236, 119805. [Google Scholar] [CrossRef]
- Puig-Kröger, A.; Sierra-Filardi, E.; Domínguez-Soto, A.; Samaniego, R.; Corcuera, M.T.; Gómez-Aguado, F.; Ratnam, M.; Sánchez-Mateos, P.; Corbí, A.L. Folate receptor β is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory Macrophages. Cancer Res. 2009, 69, 9395–9403. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.H.; Park, S.H.; Kang, S.H.; Kim, S.W.; Kim, M.; Kim, D. Fluorine-19 magnetic resonance imaging and positron emission tomography of tumor-associated macrophages and tumor metabolism. Contrast Media Mol. Imaging 2017, 2017, 4896310. [Google Scholar] [CrossRef]
- Pérez-Medina, C.; Tang, J.; Abdel-Atti, D.; Hogstad, B.; Merad, M.; Fisher, E.A.; Fayad, Z.A.; Lewis, J.S.; Mulder, W.J.M.; Reiner, T. PET imaging of tumor-associated macrophages with 89Zr-labeled high-density lipoprotein nanoparticles. J. Nucl. Med. 2015, 56, 1272–1277. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Li, R.; Ng, T.S.C.; Courties, G.; Rodell, C.B.; Prytyskach, M.; Kohler, R.H.; Pittet, M.J.; Nahrendorf, M.; Weissleder, R.; et al. Quantitative Imaging of Tumor-Associated Macrophages and Their Response to Therapy Using 64 Cu-Labeled Macrin. ACS Nano 2018, 12, 12015–12029. [Google Scholar] [CrossRef] [PubMed]
- Locke, L.W.; Mayo, M.W.; Yoo, A.D.; Williams, M.B.; Berr, S.S. PET imaging of tumor associated macrophages using mannose coated 64Cu liposomes. Biomaterials 2012, 33, 7785–7793. [Google Scholar] [CrossRef]
- Lee, C.; Kim, G.R.; Yoon, J.; Kim, S.E.; Yoo, J.S.; Piao, Y. In vivo delineation of glioblastoma by targeting tumor-associated macrophages with near-infrared fluorescent silica coated iron oxide nanoparticles in orthotopic xenografts for surgical guidance. Sci. Rep. 2018, 8, 11122. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, Y.; Chen, K.G.; Luo, Y.L.; Wang, J. Cationic Polymeric Nanoparticle Delivering CCR2 siRNA to Inflammatory Monocytes for Tumor Microenvironment Modification and Cancer Therapy. Mol. Pharm. 2018, 15, 3642–3653. [Google Scholar] [CrossRef]
- Zhang, X.; Detering, L.; Sultan, D.; Luehmann, H.; Li, L.; Heo, G.S.; Zhang, X.; Lou, L.; Grierson, P.M.; Greco, S.; et al. CC Chemokine Receptor 2-Targeting Copper Nanoparticles for Positron Emission Tomography-Guided Delivery of Gemcitabine for Pancreatic Ductal Adenocarcinoma. ACS Nano 2021, 15, 1186–1198. [Google Scholar] [CrossRef]
- Li, M.; Li, M.; Yang, Y.; Liu, Y.; Xie, H.; Yu, Q.; Tian, L.; Tang, X.; Ren, K.; Li, J.; et al. Remodeling tumor immune microenvironment via targeted blockade of PI3K-γ and CSF-1/CSF-1R pathways in tumor associated macrophages for pancreatic cancer therapy. J. Control Release 2020, 321, 23–35. [Google Scholar] [CrossRef]
- Qian, Y.; Qiao, S.; Dai, Y.; Xu, G.; Dai, B.; Lu, L.; Yu, X.; Luo, Q.; Zhang, Z. Molecular-Targeted Immunotherapeutic Strategy for Melanoma via Dual-Targeting Nanoparticles Delivering Small Interfering RNA to Tumor-Associated Macrophages. ACS Nano 2017, 11, 9536–9549. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Pei, Y.; Uzunalli, G.; Hyun, H.; Lyle, L.T.; Yeo, Y. Surface Modification of Polymeric Nanoparticles with M2pep Peptide for Drug Delivery to Tumor-Associated Macrophages. Pharm. Res. 2019, 36, 65. [Google Scholar] [CrossRef]
- Liaw, K.; Reddy, R.; Sharma, A.; Li, J.; Chang, M.; Sharma, R.; Salazar, S.; Kannan, S.; Kannan, R.M. Targeted systemic dendrimer delivery of CSF-1R inhibitor to tumor-associated macrophages improves outcomes in orthotopic glioblastoma. Bioeng. Transl. Med. 2021, 6, e10205. [Google Scholar] [CrossRef]
- Wang, Y.; Luan, Z.; Zhao, C.; Bai, C.; Yang, K. Target delivery selective CSF-1R inhibitor to tumor-associated macrophages via erythrocyte-cancer cell hybrid membrane camouflaged pH-responsive copolymer micelle for cancer immunotherapy. Eur. J. Pharm. Sci. 2020, 142, 105136. [Google Scholar] [CrossRef]
- Cuccarese, M.F.; Dubach, J.M.; Pfirschke, C.; Engblom, C.; Garris, C.; Miller, M.A.; Pittet, M.J.; Weissleder, R. Heterogeneity of macrophage infiltration and therapeutic response in lung carcinoma revealed by 3D organ imaging. Nat. Commun. 2017, 8, 14293. [Google Scholar] [CrossRef]
- Ramesh, A.; Kumar, S.; Nandi, D.; Kulkarni, A. CSF1R- and SHP2-Inhibitor-Loaded Nanoparticles Enhance Cytotoxic Activity and Phagocytosis in Tumor-Associated Macrophages. Adv. Mater. 2019, 31, 1904364. [Google Scholar] [CrossRef]
- Rao, L.; Zhao, S.K.; Wen, C.; Tian, R.; Lin, L.; Cai, B.; Sun, Y.; Kang, F.; Yang, Z.; He, L.; et al. Activating Macrophage-Mediated Cancer Immunotherapy by Genetically Edited Nanoparticles. Adv. Mater. 2020, 32, 2004853. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, Y.; Zhou, H.; Zheng, K.; Wang, D.; Jia, M.; Xu, P.; Ma, K.; Cui, C.; Wang, L. CD47-targeted bismuth selenide nanoparticles actualize improved photothermal therapy by increasing macrophage phagocytosis of cancer cells. Colloids Surf. B Biointerfaces 2019, 184, 110546. [Google Scholar] [CrossRef]
- Song, Y.; Tang, C.; Yin, C. Combination antitumor immunotherapy with VEGF and PIGF siRNA via systemic delivery of multi-functionalized nanoparticles to tumor-associated macrophages and breast cancer cells. Biomaterials 2018, 185, 117–132. [Google Scholar] [CrossRef]
- Conde, J.; Bao, C.; Tan, Y.; Cui, D.; Edelman, E.R.; Azevedo, H.S.; Byrne, H.J.; Artzi, N.; Tian, F. Dual Targeted Immunotherapy via in Vivo Delivery of Biohybrid RNAi-Peptide Nanoparticles to Tumor-Associated Macrophages and Cancer Cells. Adv. Funct. Mater. 2015, 25, 4183–4194. [Google Scholar] [CrossRef] [Green Version]
- Parayath, N.N.; Parikh, A.; Amiji, M.M. Repolarization of Tumor-Associated Macrophages in a Genetically Engineered Nonsmall Cell Lung Cancer Model by Intraperitoneal Administration of Hyaluronic Acid-Based Nanoparticles Encapsulating MicroRNA-125b. Nano Lett. 2018, 18, 3571–3579. [Google Scholar] [CrossRef]
- Hu, A.; Chen, X.; Bi, Q.; Xiang, Y.; Jin, R.; Ai, H.; Nie, Y. A parallel and cascade control system: Magnetofection of miR125b for synergistic tumor-Association macrophage polarization regulation and tumor cell suppression in breast cancer treatment. Nanoscale 2020, 12, 22615–22627. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, Z.; Xue, Y.; Wang, G.; Cheng, Y.; Pan, Y.; Zhao, S.; Hou, Y. Anti-tumor macrophages activated by ferumoxytol combined or surface-functionalized with the TLR3 agonist poly (I: C) promote melanoma regression. Theranostics 2018, 8, 6307–6321. [Google Scholar] [CrossRef]
- Li, K.; Lu, L.; Xue, C.; Liu, J.; He, Y.; Zhou, J.; Xia, Z.; Dai, L.; Luo, Z.; Mao, Y.; et al. Polarization of tumor-associated macrophage phenotype: Via porous hollow iron nanoparticles for tumor immunotherapy in vivo. Nanoscale 2020, 12, 130–144. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Y.; Guo, X.; Zhao, J.; Zhou, S. A Biomimetic Polymer Magnetic Nanocarrier Polarizing Tumor-Associated Macrophages for Potentiating Immunotherapy. Small 2020, 16, 2003543. [Google Scholar] [CrossRef]
- Shan, H.; Dou, W.; Zhang, Y.; Qi, M. Targeted ferritin nanoparticle encapsulating CpG oligodeoxynucleotides induces tumor-associated macrophage M2 phenotype polarization into M1 phenotype and inhibits tumor growth. Nanoscale 2020, 12, 22268–22280. [Google Scholar] [CrossRef]
- Qiu, Q.; Li, C.; Song, Y.; Shi, T.; Luo, X.; Zhang, H.; Hu, L.; Yan, X.; Zheng, H.; Liu, M.; et al. Targeted delivery of ibrutinib to tumor-associated macrophages by sialic acid-stearic acid conjugate modified nanocomplexes for cancer immunotherapy. Acta Biomater. 2019, 92, 184–195. [Google Scholar] [CrossRef]
- Cieslewicz, M.; Tang, J.; Yu, J.L.; Cao, H.; Zaèaljeèski, M.; Motoyama, K.; Lieber, A.; Raines, E.W.; Pun, S.H. Targeted delièery of proapoptotic peptides to tumor-associated macrophages improèes surèièal. Proc. Natl. Acad. Sci. USA 2013, 110, 15919–15924. [Google Scholar] [CrossRef] [Green Version]
- Matlung, H.L.; Szilagyi, K.; Barclay, N.A.; van den Berg, T.K. The CD47-SIRPα signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 2017, 276, 145–164. [Google Scholar] [CrossRef]
- Matozaki, T.; Murata, Y.; Okazawa, H.; Ohnishi, H. Functions and molecular mechanisms of the CD47-SIRPα signalling pathway. Trends Cell Biol. 2009, 19, 72–80. [Google Scholar] [CrossRef]
- Perisé-Barrios, A.J.; Gómez, R.; Corbí, A.L.; De La Mata, J.; Domínguez-Soto, A.; Muñoz-Fernandez, M.A. Use of carbosilane dendrimer to switch macrophage polarization for the acquisition of antitumor functions. Nanoscale 2015, 7, 3857–3866. [Google Scholar] [CrossRef]
- Liu, L.; Yi, H.; He, H.; Pan, H.; Cai, L.; Ma, Y. Tumor associated macrophage-targeted microRNA delivery with dual-responsive polypeptide nanovectors for anti-cancer therapy. Biomaterials 2017, 134, 166–179. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage M1-M2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [Green Version]
- Gong, T.; Song, X.; Yang, L.; Chen, T.; Zhao, T.; Zheng, T.; Sun, X.; Gong, T.; Zhang, Z. Spontaneously formed porous structure and M1 polarization effect of Fe3O4 nanoparticles for enhanced antitumor therapy. Int. J. Pharm. 2019, 559, 329–340. [Google Scholar] [CrossRef]
- Chen, L.; Ma, X.; Dang, M.; Dong, H.; Hu, H.; Su, X.; Liu, W.; Wang, Q.; Mou, Y.; Teng, Z. Simultaneous T Cell Activation and Macrophage Polarization to Promote Potent Tumor Suppression by Iron Oxide-Embedded Large-Pore Mesoporous Organosilica Core–Shell Nanospheres. Adv. Healthc. Mater. 2019, 8, e1900039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cao, S.; Liang, S.; Tan, C.H.; Luo, B.; Xu, X.; Saw, P.E. Differently Charged Super-Paramagnetic Iron Oxide Nanoparticles Preferentially Induced M1-Like Phenotype of Macrophages. Front. Bioeng. Biotechnol. 2020, 8, 537. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, R.S.; Ishikawa, U.; Silva, E.S.; Silva-Júnior, A.A.; Araújo, A.A.; Cruz, L.J.; Chan, A.B.; de Araújo Júnior, R.F. STAT3/NF-κB signalling disruption in M2 tumour-associated macrophages is a major target of PLGA nanocarriers/PD-L1 antibody immunomodulatory therapy in breast cancer. Br. J. Pharmacol. 2021, 178, 2284–2304. [Google Scholar] [CrossRef]
- Dorrington, M.G.; Fraser, I.D.C. NF-κB signaling in macrophages: Dynamics, crosstalk, and signal integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef]
- Wei, B.; Pan, J.; Yuan, R.; Shao, B.; Wang, Y.; Guo, X.; Zhou, S. Polarization of Tumor-Associated Macrophages by Nanoparticle-Loaded Escherichia coli Combined with Immunogenic Cell Death for Cancer Immunotherapy. Nano Lett. 2021, 21, 4231–4240. [Google Scholar] [CrossRef]
- Good, L.; Benner, B.; Carson, W.E. Bruton’s tyrosine kinase: An emerging targeted therapy in myeloid cells within the tumor microenvironment. Cancer Immunol. Immunother. 2021, 70, 2439–2451. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef]
- Narayanaswamy, R.; Torchilin, V.P. Hydrogels and their applications in targeted drug delivery. Molecules 2019, 24, 603. [Google Scholar] [CrossRef] [Green Version]
- Mantha, S.; Pillai, S.; Khayambashi, P.; Upadhyay, A.; Zhang, Y. Smart Hydrogels in Tissue Engineering and Regenerative Medicine. Materials 2019, 12, 3323. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Li, M.; Yang, C.; Yin, X.; Duan, K.; Wang, J.; Feng, B. Macrophage phenotype switch by sequential action of immunomodulatory cytokines from hydrogel layers on titania nanotubes. Colloids Surf. B Biointerfaces 2018, 163, 336–345. [Google Scholar] [CrossRef]
- Nguyen, D.T.; Soeranaya, B.H.T.; Truong, T.H.A.; Dang, T.T. Modular design of a hybrid hydrogel for protease-triggered enhancement of drug delivery to regulate TNF-α production by pro-inflammatory macrophages. Acta Biomater. 2020, 117, 167–179. [Google Scholar] [CrossRef]
- Kang, J.; Neill, D.L.; Xian, M. Phosphonothioate-based hydrogen sulfide releasing reagents: Chemistry and biological applications. Front. Pharmacol. 2017, 8, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Miao, L.; Shen, X.; Whiteman, M.; Xin, H.; Shen, Y.; Xin, X.; Moore, P.K.; Zhu, Y.Z. Hydrogen Sulfide Mitigates Myocardial Infarction via Promotion of Mitochondrial Biogenesis-Dependent M2 Polarization of Macrophages. Antioxid. Redox Signal. 2016, 25, 268–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Chen, A.; Zhou, Y.; Zheng, S.; Yang, Y.; An, Y.; Xu, K.; He, H.; Kang, J.; Luckanagul, J.A.; et al. Novel H2S-Releasing hydrogel for wound repair via in situ polarization of M2 macrophages. Biomaterials 2019, 222, 119398. [Google Scholar] [CrossRef]
- Saleh, B.; Dhaliwal, H.K.; Portillo-Lara, R.; Shirzaei Sani, E.; Abdi, R.; Amiji, M.M.; Annabi, N. Local Immunomodulation Using an Adhesive Hydrogel Loaded with miRNA-Laden Nanoparticles Promotes Wound Healing. Small 2019, 15, 1902232. [Google Scholar] [CrossRef]
- Lohmann, N.; Schirmer, L.; Atallah, P.; Wandel, E.; Ferrer, R.A.; Werner, C.; Simon, J.C.; Franz, S.; Freudenberg, U. Glycosaminoglycan-based hydrogels capture inflammatory chemokines and rescue defective wound healing in mice. Sci. Transl. Med. 2017, 9, eaai9044. [Google Scholar] [CrossRef]
- Vasandan, A.B.; Jahnavi, S.; Shashank, C.; Prasad, P.; Kumar, A.; Jyothi Prasanna, S. Human Mesenchymal stem cells program macrophage plasticity by altering their metabolic status via a PGE 2 -dependent mechanism. Sci. Rep. 2016, 6, 152. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Duan, L.; Hou, H.; Liu, Y.; Chen, S.; Zhang, S.; Liu, Y.; Wang, C.; Qi, X.; Liu, N.; et al. IGF-1C hydrogel improves the therapeutic effects of MSCs on colitis in mice through PGE2-mediated M2 macrophage polarization. Theranostics 2020, 10, 7697–7709. [Google Scholar] [CrossRef]
- Zhao, X.; Cui, K.; Li, Z. The role of biomaterials in stem cell-based regenerative medicine. Future Med. Chem. 2019, 11, 1779–1792. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Sui, B.; Hua, Y.; Zhang, Y.; Bao, B.; Lin, Q.; Liu, X.; Zhu, L.; Sun, J. A well defect-suitable and high-strength biomimetic squid type II gelatin hydrogel promoted in situ costal cartilage regeneration via dynamic immunomodulation and direct induction manners. Biomaterials 2020, 240, 119841. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Gupta, P.; Bhattacharjee, S.; Nandi, S.K.; Mandal, B.B. Immunomodulatory injectable silk hydrogels maintaining functional islets and promoting anti-inflammatory M2 macrophage polarization. Biomaterials 2018, 187, 1–17. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Type of NP | Payload | Model | Effects on Macrophages | Outcome | Ref. |
|---|---|---|---|---|---|---|
| Rheumatoid Arthritis | Tuftsin-modified non-condensing alginate-based NPs | pDNA IL-10 | In vivo | Induce IL10 expression | Polarization to M2 | [19] |
| MFC-MSNs | - | In vivo | Scavenge ROS and produce O2 | Polarization to M2 | [20] | |
| Chitosan/siRNA nanoparticles | TNFα siRNA | In vivo | Reduce TNFα expression | M1 depletion | [21] | |
| Folate–Chitosan DNA nanoparticles | IL-1Ra Gene | In vivo | Reduce IL-1 expression | [22] | ||
| FA-conjugated G5 dendrimers | Methotrexate | In vivo | Reduce cell proliferation and induce apoptosis | M1 depletion | [23] | |
| pH-responsive nanocarriers | Methotrexate | In vivo | [24] | |||
| Polymeric nanoparticles | Dexamethasone and naproxen | In vitro | Prevent IL-12 expression | Polarization to M2 | [25] | |
| FA modified ROS-responsive nanoparticles | Dexamethasone | In vivo | Interfere in the iRhom2-TNF-α-BAFF signaling pathway | [26] | ||
| Inflammatory Bowel Disease (IBD) | Mannosylated nanoparticles | TNFα siRNA | In vitro Ex vivo | Reduce TNFα expression | M1 depletion and colitis attenuation | [27] |
| NPs grafting Fab’ portion of the F4/80 antibody | TNFα siRNA | In vitro | Reduce TNFα expression | [28] | ||
| Termed thioketal nanoparticles | TNFα siRNA | In vivo | Reduce TNFα expression | [29] | ||
| GC PLGA NPs | TNFα siRNA | In vitro In vivo | Reduce TNFα expression | [30] | ||
| Nanoparticles-in-microsphere oral system (NiMOS) | TNFα siRNA | In vivo | Reduce TNFα expression | [31] | ||
| Cyclin D1 siRNA | Reduce Cyclin D1 expression | |||||
| b1,3-D-glucan- particles (GeRPs) | Map4K4 siRNA | In vivo | Reduce Map4K4 expression | [32] | ||
| Atherosclerosis | Mannose functionalized dendrimer NPs | SR-A siRNA | In vivo | Reduce LDL uptake | Plaque regression | [33] |
| LXR ligand | Stimulate cholesterol efflux | |||||
| HA-coated cell-penetrating peptide nanocomplexes | LOX-1 siRNA | In vivo | Reduce LDL uptake | Plaque regression | [34] | |
| Lipid nanoparticle | CCR2 siRNA | In vivo | Reduce CCR2 expression | Reduce macrophage recruitment | [35] | |
| EGFP-EGF1-conjugated PLGA nanoparticles | CCR2 shRNA | In vitro | Reduce CCR2 expression | [36] | ||
| S2P-conjugated DSPE-PEG NPs | CaMKIIγ siRNA | In vivo | Unblock macrophage efferocytosis | Plaque stabilization | [37] | |
| Single-walled carbon nanotubes | TPI | In vitro | Stimulate efferocytosis | [38] |
| Disease | Type of NP | Payload | Model | Effects | Outcome | Reference |
|---|---|---|---|---|---|---|
| Ischemic injury in muscle | AuNPs | IL-4 | In vivo | Polarization to M2 | Muscle regeneration | [54] |
| Periodontitis | AuNPs | - | In vivo | Polarization to M2 | Periodontal tissue regeneration | [55] |
| Bone injury or defect | TiO2 | - | In vitro | Polarization to M2 | Bone regeneration | [56] |
| Mesoporous silica NPs (MSNs) | - | In vitro | Enhance the osteogenic differentiation of BMBMs | [57] | ||
| Copper Mesoporous silica NPs (MSNs) | - | In vitro | [58] | |||
| Alendronate conjugated GNPs | - | In vivo | Inhibit BMDMs differentiation to osteoclasts | [59] | ||
| Myocardial infarct (MI) | PS-presenting liposomes | - | In vivo | Macrophage transition to reparative state | Myocardial infarct repair | [60] |
| Hyaluronan-sulfate (HAS)-Ca2+ NPs | miRNA-21 | In vivo | Macrophage transition to reparative state | [61] | ||
| Polyketal (PK3) nanoparticles | miRNA | In vitro In vivo | Reduce Nox2 expression, leading to downregulation of inflammation | [62] | ||
| Lipidoid nanoparticles (LNPs) | IRF5 siRNA | In vivo | Attenuation of M1 macrophage polarization | [63] | ||
| PLGA NPs | Pioglitazone | In vivo | NF-κB inhibition, leading to inflammation downregulation | [64] | ||
| Chronic liver injury | MTC conjugate nanoparticles | TNFα siRNA | In vivo | Reduce TNFα expression | Reduce liver injury and fibrosis | [65] |
| PLGA NPs | SYK pathway inhibitor | In vitro In vivo | SYK inhibition | [66] | ||
| Liposomes | Dex | In vivo | Anti-inflammatory polarization of macrophages | [67] | ||
| PS-modified nanostructured lipid carriers (mNLCs) | Curcumin | In vivo | Anti-inflammatory effects | [68] | ||
| Dendrimer-Graphene nanostars | Plasmid expressing MMP9 | In vitro | Overexpression of MMP9 which lead to macrophage transition to reparative state | [69] |
| Cancer Diagnosis | ||||
|---|---|---|---|---|
| Type of NP | Diagnostic Strategy | Cancer Model | TAM Targeting | References |
| AMTA680 | Fluorescence imaging and MRI | Soft tissue sarcoma, lung carcinoma and colon adenocarcinoma | Accumulated in TAM rich areas | [88] |
| Iron oxide NP | MRI | Breast | [89] | |
| 89Zr-modified rHDL | PET | [94] | ||
| Silica-coated iron oxide NP | Fluorescence imaging | Glioblastoma | [97] | |
| 64Cu-loaded mannosylated liposomes | PET | Pulmonary adenocarcinoma | [96] | |
| 64Cu-labeled polyglucose NPs | Breast | [95] | ||
| Perfluorocarbon | MRI | Breast | [93] | |
| Ferumoxytol iron oxide NP | Anaplastic thyroid cancer | [90] | ||
| Melanoma | [91] | |||
| Cancer Therapy | ||||
| Therapeutic Approach | Type of NP | TAM Targeting | Cancer Model | References |
| Macrophage recruitment inhibition | Cationic polymeric NPs | CCR2 | Breast | [98] |
| Ultrasmall cooper NPs | Pancreatic ductal adenocarcinoma | [99] | ||
| TAM depletion | PEI-SA nanomicelles | CSF-1R | Pancreatic | [100] |
| DMCP and cholesterol oleate | Melanoma | [101] | ||
| PLGA | Melanoma | [102] | ||
| Hydroxyl dendrimer NPs | Glioblastoma | [103] | ||
| Dextran grafted poly(histidine) copolymer + erythrocyte/cancer cell membrane hybrid | Breast | [104] | ||
| Red fluorescent polymeric micelle | Pulmonary melanoma | [105] | ||
| Self-assembled colipids (DSPE, PEG and PC) | Melanoma and breast cancer | [106] | ||
| Blocking macrophage “do not eat me” signal | Magnetic NPs | CD47 | Melanoma and triple negative breast cancer | [107] |
| Bismuth selenide NPs | Breast | [108] | ||
| Self-assembled colipids (DSPE, PEG and PC) | SHP2 | Melanoma and breast cancer | [106] | |
| Functional TAM reeducation | PEG and trimethyl chitosan modified with mannose | VEGF PIGF | Breast | [109] |
| Gold core NPs decorated with thiolated-PEG-COOH polymer | VEGF | Lung | [110] | |
| Hyaluronic acid-poly (ethylenimine) NPs | miR-125b | [111] | ||
| Anionic magnetic NPs | Breast | [112] | ||
| Ferumoxytol iron oxide NPs | TLR3 | Melanoma and lung metastasis | [113] | |
| Iron oxide NPs | P13K and mannose receptor | Breast | [114] | |
| TLR7 | [115] | |||
| Human ferritin heavy chain nanocages | TLR | [116] | ||
| Phosphatidylglycerol nanocomplexes with stearic acid | BTK and Siglec-1 | Sarcoma | [117] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medrano-Bosch, M.; Moreno-Lanceta, A.; Melgar-Lesmes, P. Nanoparticles to Target and Treat Macrophages: The Ockham’s Concept? Pharmaceutics 2021, 13, 1340. https://doi.org/10.3390/pharmaceutics13091340
Medrano-Bosch M, Moreno-Lanceta A, Melgar-Lesmes P. Nanoparticles to Target and Treat Macrophages: The Ockham’s Concept? Pharmaceutics. 2021; 13(9):1340. https://doi.org/10.3390/pharmaceutics13091340
Chicago/Turabian StyleMedrano-Bosch, Mireia, Alazne Moreno-Lanceta, and Pedro Melgar-Lesmes. 2021. "Nanoparticles to Target and Treat Macrophages: The Ockham’s Concept?" Pharmaceutics 13, no. 9: 1340. https://doi.org/10.3390/pharmaceutics13091340