
Ruthenium Complexes: An Alternative to Platinum Drugs in Colorectal Cancer Treatment





1
Department of Biochemistry and Molecular Biology, Jahangirnagar University, Savar, Dhaka 1342, Bangladesh
2
Department of Pharmacology & Toxicology, University of Otago, Dunedin 9016, New Zealand
3
Department of Biochemistry, Primeasia University, Banani, Dhaka 1213, Bangladesh
4
Department of Haematology, School of Medical Sciences, Universiti Sains Malaysia, Kubang Kerian 16150, Malaysia
*
Authors to whom correspondence should be addressed.
Pharmaceutics 2021, 13(8), 1295; https://doi.org/10.3390/pharmaceutics13081295
Submission received: 23 July 2021
/
Revised: 13 August 2021
/
Accepted: 16 August 2021
/
Published: 19 August 2021
(This article belongs to the Special Issue Beyond the Platinum in Metal-Based Cancer Therapy)
Abstract
:Colorectal cancer (CRC) is one of the intimidating causes of death around the world. CRC originated from mutations of tumor suppressor genes, proto-oncogenes and DNA repair genes. Though platinum (Pt)-based anticancer drugs have been widely used in the treatment of cancer, their toxicity and CRC cells’ resistance to Pt drugs has piqued interest in the search for alternative metal-based drugs. Ruthenium (Ru)-based compounds displayed promising anticancer activity due to their unique chemical properties. Ru-complexes are reported to exert their anticancer activities in CRC cells by regulating different cell signaling pathways that are either directly or indirectly associated with cell growth, division, proliferation, and migration. Additionally, some Ru-based drug candidates showed higher potency compared to commercially available Pt-based anticancer drugs in CRC cell line models. Meanwhile Ru nanoparticles coupled with photosensitizers or anticancer agents have also shown theranostic potential towards CRC. Ru-nanoformulations improve drug efficacy, targeted drug delivery, immune activation, and biocompatibility, and therefore may be capable of overcoming some of the existing chemotherapeutic limitations. Among the potential Ru-based compounds, only Ru (III)-based drug NKP-1339 has undergone phase-Ib clinical trials in CRC treatment.
1. Introduction
Colorectal cancer (CRC) is a type of malignant neoplasm of the colon or rectum epithelial cell lining [1,2], which is recognized as the third most prevalent cancer worldwide and is the fourth leading cause of death [3,4]. It also accounts for about 10% of all yearly diagnosed cancers and cancer-related deaths globally [5]. Moreover, CRC has been documented as the second and third most common cancer in women and men, respectively [5,6]. CRC occurrence rate is high in most of the developed countries, whereas the rate is increasing rapidly in developing countries [6]. In 2020, more than 1.9 million individuals were estimated to be diagnosed, where 935,000 individuals would die among the CRC-diagnosed patients [7]. About 2.5 million people are predicted to be diagnosed with CRC by 2035 [5].
The conventional treatment strategies of CRC consist of surgical resection, radiation, and chemotherapy, which may extend the survival rate by only five years in 90% of stage I patients to 10% of stage IV patients [8,9]. Even though surgery has been an integral part of CRC treatment, it comes out with post-operative complications such as occurrence or acceleration in recurrence of tumor cells and/or development of liver metastasis [8]. Long-term use of chemotherapeutics and radiation induces peripheral neuropathy [10] and bowel dysfunction accompanied by increased frequency and urgency problems [11]. The limitations of the existing treatment strategies encourage researchers to develop effective therapeutic alternatives.
Over the past few decades, transition metal-based compounds have been extensively used in the anticancer medicinal chemistry area [12,13,14,15]. Platinum (Pt)-based medications such as cisplatin (CIS) and its analogs carboplatin (CAR) and oxaliplatin (OXA) (Figure 1) have been used worldwide in cancer treatment [16]. Additionally, some other Pt-based drugs, for example, miriplatin (Japan), nedaplatin (Japan), lobaplatin (China), and heptaplatin (Korea) (Figure 1) are used regionally in cancer treatment (Figure 1) [17]. However, only OXA has been approved by the Food and Drug Administration (FDA) in CRC treatment [18] and stands out as the first-line therapy against CRC [19]. Despite being highly efficient, OXA has severe side effects [20] and drug resistance [21]. Such limitations inspire the search for alternative metal-based anticancer drugs.
Among other transition metals, ruthenium (Ru) is a better alternative to Pt [22]. Ru displays both early and late transition metal properties due to its central position in the second row of the transition metal series [22]. The 4d subshell of Ru is partially filled and it contains many valencies that enable Ru to form a wide range of complexes via π bond formation, which can perform as anticancer agents against various tumor cell lines [23]. Ru-complexes showed promising anti-proliferative activity in vitro, in vivo, and in chemical model systems [24,25,26]. Moreover, the Ru-complex showed synergistic activity when combined with established anticancer agents and drugs [25,27]. Furthermore, Ru-complexes are widely used as phototherapeutic agents, biomolecular probes, and bioimaging reagents [28]. Luminescent Ru-complexes can differentiate DNA structures and have the potential to be used as molecular light switches for DNA [29]. Additionally, Ru nanoparticles (RuNPs) can be used as a cancer theranostic agent for the early diagnosis and treatment of CRC [30,31]. Nanostructured Ru-complexes offer improved anticancer activity under their targeted drug delivery and reduced side effects [32].
To overcome the limitation of Pt-drugs, Ru-complexes could be used as an alternative to Pt-based chemotherapeutic drugs in CRC. In this review, we scrutinized the potential of ruthenium-based drugs, drug candidates, and ruthenium nanoparticles in the treatment of CRC. Additionally, the molecular mechanism of action(s) such as effects on nucleic acids, cell proliferative pathways, and cell cycle are summarized and compared their efficiency with Pt-based drugs and other chemotherapeutic drugs i.e., 5-Fluorouracil (5-FLU), Doxorubicin (DOX), and Etoposide (ETP).
2. Colorectal Cancer and Pt-Based Drugs
Colorectal cancer is caused by chromosomal instability, microsatellite instability (MSI), and the CpG island methylator phenotype (CIMP), which may occur alone or in combination [33,34]. Chromosomal instability is responsible for most of the genetic instability in CRC, which is characterized by significant gain or loss of entire or large portions of chromosomes [33]. The chromosomal instability pathway starts with the mutation of the APC gene, followed by the mutation of oncogene KRAS and inactivation of tumor suppressor gene, TP53 [35]. The CIMP pathway is involved in hypermethylation of the promoter region of tumor suppressor genes, mostly MGMT and MLH1. However, this hypermethylation is linked to BRAF mutation and MSI [36]. The MSI pathway refers to the inactivation of DNA mismatch repair genes through genetic alteration in short repeated sequences and hypermethylation of these mismatch repair genes. The MSI pathway is often found to be connected to the CIMP pathway [34].
Several genes have been mutated to induce CRC; Figure 2 shows the ten most frequent genes according to the cBioPortal database (https://www.cbioportal.org/, accessed on 15 July 2021) [37,38] that calculated published data on CRC [39,40,41,42,43,44,45]. Mutations of these genes could be linked with survival, CRC progression, and therapeutic outcome.
Pt-based drugs are used in the treatment of various types of cancers including CRC [47]. Although Pt-based drugs have been playing a pivotal role as anticancer drugs, some irresistible drawbacks limit their use in cancer treatment. Like other conventional chemotherapeutic drugs, Pt-based drugs including CIS, OXA, and CAR display poor cancer cells’ selectivity index [48,49,50]. Due to low selectivity, patients often experience drug-induced complications, some of which are fatal [51].
Among the Pt-based drugs, only OXA has been used in the treatment of CRC. OXA in combination with leucovorin (LEU) and 5-FLU (FOLFOX) is administered in adjuvant or neoadjuvant treatment of CRC. However, the co-treatment increases all grades of anemia significantly compared to the individual treatment with LEU and 5-FLU [52]. Furthermore, OXA induced several side effects in CRC treatment including peripheral neuropathy, fatigue, diarrhea, nausea, and stomatitis [53,54]. Acute and chronic neurosensory symptoms are also observed after OXA treatment [55]. Additionally, OXA mediates neutropenia, the most common serious hematological toxicity, in CRC patients [53]. Besides, continued use of OXA develops hypersensitivity reactions (type-I or IgE mediated reactions) in 10% of patients which is characterized by pruritus, flushing, urticarial, hypotension, and possible angioedema of the larynx, face, and/or extremities [54]. Moreover, OXA is reported to induce hepatic sinusoidal injury in CRC patients [56] as well as enlargement of spleen size in stage II or III CRC which are the potential cause of persistent thrombocytopenia [57].
3. Features of Ru-Complexes
Among numerous transition elements, Ru is found to be the best alternative to Pt [12,22,58]. The advantages of using Ru over Pt include lower toxicity, a broader range of oxidation states (2+, 3+, and 4+), a slow rate of ligand exchange, and the ability to mimic iron that facilitates its binding to human serum transferrin and other proteins [59,60]. Ru offers octahedral coordination geometry instead of square-planar geometry of Pt(II) complexes which provide a different mode of action and reactivity than CIS [61]. Furthermore, compared to typical Pt-based drugs, many Ru-based compounds have better water solubility in the biological environment, resulting in improved effectiveness against Pt-drug resistant tumor cells [62]. This increase in water solubility may aid in balancing the hydrophilicity and hydrophobicity of Ru-complexes, resulting in increased absorption in cancer cells [63,64].
Ru(IV) is unstable because of the higher oxidation state. This limits the antitumor effects and further development of Ru(IV)-complexes [65]. Nevertheless, Ru(II) and Ru(III) have antitumor activity [66]. Ru(III)-complexes possess stable thermodynamics and kinetics and are efficient in acting as a prodrug to work under hypoxic and acidic conditions [67]. However, Ru(III) is considered to be more inert than Ru(II), which might be due to a higher effective nuclear charge [68]. Thus, Ru(II)-complexes are more reactive than Ru(III)-complexes [69]. Ru(III)-complexes are reduced to the more active form, Ru(II), by the “activation by reduction” mechanism [70]. This mechanism is influenced by cellular reducing agents such as ascorbate, glutathione, and hypoxic tumor microenvironment [23,71]. Reduction of Ru(III) to Ru(II) enervates π bond with donor ligand and elevates ligand substitution rates [23]. However, the “activation by reduction” hypothesis is still a controversial issue, as some Ru(III)-complexes remained at 3+ oxidation state after 24 h of intravenous administration [72].
4. Underlying Mechanisms of Ru-Complexes in Targeting CRC
Different types of Ru-complexes are reported to target DNA, different fundamental enzymes like topoisomerase II, thioredoxin reductase, and various biomolecules linked with growth, angiogenesis, migration, metastasis, and apoptosis of CRC cells (Figure 3) [27,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88]. Furthermore, Ru-complexes induce apoptosis through overproducing reactive oxygen species (ROS) [89] and compromising cellular organelles that are required for cell survival [90,91]. Moreover, Ru-complexes also cause apoptosis in CRC cells through photodynamic activity [92]. In this section, we will compare the potency of some Ru-based complexes with Pt-drugs along with another standard drug 5-FLU, and investigate the mechanism of action(s), and promise of Ru-based complexes in CRC treatment. Herein, we considered comparing 5-FLU alongside Pt-based drugs since it has been used as a first-line treatment against CRC [93].
4.1. DNA Damage Mediated Apoptosis
Many Ru-complexes control the proliferation of cancer cells through DNA damage [94,95]. Some of the Ru-complexes bind with DNA via electrostatic attraction, major or minor grooves binding, intercalative binding mode, or by a combination of these two or more [96,97]. DNA binding modes of Ru-complexes can be confirmed by UV-Vis spectroscopy, viscosity investigation, and fluorescence spectrometry [97]. Intercalative binding opens a gap between the DNA base pair and inserts planar aromatic molecule of anticancer drug above and below the bases [98]. This results in unwinding and lengthening of the helix structure of DNA (Figure 3→I) [97,99]. Ru(II)-complexes bind preferentially to N7 of guanosine and N3 of thymidine, but insubstantially to N3 of cytidine, and little to adenosine [100]. On the other hand, Ru(III)-complexes preferentially bind to phosphate groups of the DNA backbone. This is attributed to the strong electrostatic interaction between tricationic Ru(III) fragment and anionic phosphate groups [101]. Ru(III)-complexes also have been reported to bind to the N7 site of guanine [102] but in a less pronounced way than phosphate [101]. However, both Ru(II) and (III)-complexes have a common tendency to bind to the N7 site of guanine which is similar to CIS [103].
Some Ru-complexes such as Ru(III)-PTA compound trans-[RuCl4(1,3,5-triaza-7-phosphaadamantane protonated at one N atom)2]Cl (1a), η6-arene Ru-complexes particularly Ru-5-chloro-3-((5-(3-(4-methyl-1,4-diazepane-1-carbonyl)phenyl)furan-2-yl)methylene)indolin-2-one (1b), Ru(II) arene complexes including [(η6-fluorene)RuII(ethylenediamine)Cl]+ (1c), and [(η6-9,10-dihydrophenanthrene)RuII(ethylenediamine)Cl]+ (1d) mediated apoptosis in CRC cells by damaging DNA (Figure 4) [75,76,77]. 1b, 1c, and 1d bound with guanine residue of DNA, causing DNA fragmentation [76,77]. Though the compounds share the same mechanism of action(s), they are not equally potent towards different CRC cells in terms of IC5O values (Table 1). 1a with an IC50 value of ˃100 µM displayed moderate anti-proliferative activity against HCT116 cells following 24 h of treatment [75]. Likewise, 1b showed cytotoxicity in LoVo cells (IC50 = 8.1 µmol/L) and LS174T (IC50 = 7.7 µmol/L) after six days of treatment [76]. However, both 1a and 1b were less potent compared to CIS (Table 1) [75,76]. In contrast, 1c and 1d exhibited synergistic action with ionizing radiation (IR) and were more potent than OXA against DLD1 cells (Table 1) [77]. OXA act as an alkylating agent on DNA, forming intra-strand cross-links between two adjacent guanine or two adjacent guanine-adenine and inhibit DNA synthesis through disrupting replication and transcription [104,105]. Like OXA, 1c or 1d form adducts with DNA which leads to apoptosis. Experimental results indicate that 1c or 1d was more potent than OXA towards CRC cell lines, therefore these drug candidates could be used as an alternative to OXA.
4.2. Inhibition of Topoisomerase II Enzyme
Topoisomerase II is involved in modulating topological problems related to DNA replication, transcription, chromatin remodeling, and recombination by single- or double-strand breaks in the DNA [106]. Half-sandwich Ru-arene complexes with thiosemicarbazones including [(η6-p-cymene)Ru(piperonal-N(4)-ethylthiosemicarbazone)Cl]Cl (2a), [(η6-p-cymene)Ru(piperonal-N(4)-phenylthiosemicarbazone)Cl]Cl (2b), and mixed-ligand diimine-piperonal thiosemicarbazone complexes of Ru(II) [(1,10-phenanthroline)2Ru(2-(benzo[d][1,3]dioxol-5-ylmethylene)-N-methylhydrazinecarbothioamide)](hexafluorophosphate)2 (2c) (Figure 4) act as topoisomerase II inhibitor by disrupting enzyme’s catalytic cycle which leads to apoptosis of CRC cells by inhibiting replication (Figure 3→II) [73,74]. Both 2a and 2b exhibited less anti-proliferative activity compared to CIS and ETP [73] while 2c was more potent than ETP against HCT116 and Caco-2 cells (Table 1) [74]. Although ETP has been widely used as an anti-proliferative agent [107], it is ineffective against advanced CRC [108,109]. The combination of CIS and ETP showed low anticancer activity against advanced CRC [110]. Thus, it is not prudent to use only EPT while comparing anticancer activity against CRC.
4.3. MAPK Signaling Pathway
Mitogen-activated protein kinase (MAPK) signaling pathway regulates apoptosis in CRC cells through controlling three MAPK family proteins—extracellular signal-regulated kinase (ERK) [111], c-Jun N-terminal kinases (JNK/SAPK) [112] and p38 MAPK [113]. Some Ru-complexes including Ru(II) naphthalimide N-Heterocyclic Carbene compounds (3a), [Ru(piplartine)(1,1-bis(diphenylphosphino)ferrocene)(2,2′-bipyridine)](hexafluorophosphate)2 (3b), [Ru(piplartine)(1,4-bis(diphenylphosphino)butane)(2,2′-bipyridine)](hexafluorophosphate)2 (3c), and Ru(II)-thymine complex [Ru(triphenylphosphine)2(thyminate)(2,2′-bipyridine)]hexafluorophosphate (3d) (Figure 4) triggered apoptosis in CRC cells through MAPK (JNK, p38 MAPK, and ERK1/2) signaling pathway (Figure 3→III) [79,80,81]. 24-48 h post-treatment of HCT116 cells with 3a (12 µM), 3b (2.5 µM), 3c (5 µM), and 3d (4 µM) induced apoptosis of HCT116 cells by controlling MAPK (JNK, p38 MAPK, and ERK1/2) signaling pathways [79,80,81]. Furthermore, 3b (2.5 µM), 3c (5 µM), and 3d (4 µM) increased apoptosis of HCT116 cells by 19%, 23%, and 51%, respectively compared to chemotherapeutic drug DOX (1 µM). Afterward, in vivo study showed that intraperitoneal injections of 3b (15 µmol/kg/day), 3c (15 µmol/kg/day), and 3d (1–2 mg/kg/day) for 15 consecutive days reduced tumor mass weight by 1.55, 1.42, and 1.47 to 1.67-fold, respectively compared to negative control in C.B-17 SCID mice model engrafted with HCT116 cells [80,81]. It should be noted that 3d (1–2 mg/kg/day) displayed 32.6–40.1% tumor mass inhibition rate while established drug 5-FLU (15 mg/kg/day) showed 62.7% [81]. Since 3d showed higher potency compared to 5-FLU, thus 3d could be used as an alternative to 5-FLU in KRAS-mutated CRC treatment.
Figure 4.
Schematic representation of some Ru-based drug candidates. Different Ru-complexes mediated CRC cells apoptosis through DNA damage (1a–d), inhibiting DNA topoisomerase II enzyme (2a–c), and regulating MAPK signaling pathway (3a–d).
Figure 4.
Schematic representation of some Ru-based drug candidates. Different Ru-complexes mediated CRC cells apoptosis through DNA damage (1a–d), inhibiting DNA topoisomerase II enzyme (2a–c), and regulating MAPK signaling pathway (3a–d).

4.4. p53 Dependent Caspase-3 Mediated Signaling
p53 is a tumor suppressor protein linked with cell cycle, apoptosis, senescence, and autophagy [114]. Several Ru-complexes including Ru-based 5-FLU complex [Ru(5-fluorouracil)(triphenylphosphine)2(2,2′-bipyridine)]hexafluorophosphate (4a), Ru-Quercetin (4b), Ru-Phloretin (4c), Ru-Baicalein (4d), and [Ru(biphenyl)Cl(1,2-ethylenediamine)]+ with hexafluorophosphate (4e) (Figure 5) induced apoptosis of colon cancer cells via p53 dependent caspase-3 mediated apoptosis (Figure 3→IV) [27,82,83,84,85]. 4b, 4c, and 4d upregulated the expression of p53, and anti-apoptotic protein Bax while downregulated Bcl-2 expression, resulting in activation of apoptotic protein caspase-3 to induce apoptosis and arrested HT29 cells at G0/G1 phase in a concentration-dependent manner [82,83,84]. Moreover, these compounds also displayed apoptotic activity in the 1,2-dimethylhydrazine (DMH) and dextran sulfate sodium (DSS) induced CRC in male Wistar rats [82,83] and Swiss albino mice [84]. Treatment with 4b (200 mg/kg) increased the expressions of p53 (3.47-fold) and Bax (3.77-fold) while suppressing the expression of Bcl-2 (2.72-fold) compared to the DMH and DSS treated group [83]. Similarly, the same concentration of 4d and 4c upregulated Bax expression (2.45 to 2.6-fold) while downregulated Bcl-2 expression (2.38 to 3.47-fold) compared to the DMH and DSS treated group. The upregulation and downregulation of proteins (i.e., p53, Bax, and Bcl-2) expression were determined by immunohistochemical analysis [82,84]. Additionally, 4b, 4c, and 4d diminished the cellular level of proliferating cell nuclear antigen (PCNA) which is actively controlled by p53 and subsequently induced apoptosis by minimizing cell proliferation [82,83,84].
Treating HCT116 cells with 4e (15–60 µM) for 48 h increased the expression of p53, Bax and cell cycle inhibitor p21/WAF1 (Table 2). Protein expression levels were determined by western blotting. However, p53/Bax-null HCT116 cells did not undergo significant apoptosis in the same condition. This implies the importance of p53 and Bax proteins in 4e mediated apoptosis of CRC cells. Furthermore, treatment with 4e brought about a long-term loss of cellular replication in a p53, Bax, and p21/WAF1 independent manner [85]. Silva et al. [27] developed a novel Ru-based complex 4a, where the addition of Ru increased the cytotoxic potential of 5-FLU. 4a was more potent than 5-FLU (2.73-fold) and OXA (2.87) while less potent than DOX (3-fold) (Table 1). Furthermore, trypan blue exclusion (TBE) assay pointed that treatment of HCT116 cells with 4a (4 µM) for 48 h increased caspase-3 level (3 and 3.75-fold) along with mitochondrial membrane depolarization (2.2 and 2.29-fold) compared to OXA (2.5 µM) and 5-FLU (4 µM), respectively [27]. Another Ru-based complex [(η5-C5H5)Ru(1,2-Bis(diphenylphosphino)ethane)]+ bearing a galactose ligands (4f) (Figure 5) also triggered caspase-3 and caspase-7 activities level and thus induced apoptosis. A study by Florindo et al. [115] reported that treatment of HCT116 cells with 4f (1–2 µM) reduced cell viability (40% to 16%) and increased cell death (1.5 to 2.7-fold) compared to control. Though 4f was equally potent to OXA with an IC50 value of 0.45 µM in HCT116 cells. However, at 2 µM concentration, 4f was 25% more cytotoxic than OXA. 4f (0.45 µM) significantly increased caspase-3 and caspase-7 activity by 1.4-fold than control, while OXA (0.45 µM) increased the level of caspase-3 and caspase-7 by 1.27-fold. Besides, 4f induced 30% more apoptosis in HCT116 cells than OXA at an equal concentration of 2 µM [115]. Based on the IC50 values, 4a and 4f were more or equally potent compared to 5-FLU or OXA against HCT116 cells. Therefore, 4a and 4f could be used in CRC treatment in place of 5-FLU and OXA.
4.5. Upregulation of APC and p53 Gene
APC and p53 genes are reported to acquire genetic alteration in CRC [121]. About 80% APC mutation and 60% p53 mutation are observed in CRC [122,123]. APC performs antitumorigenic activity by regulating β-catenin levels [123]. Mutation of APC causes accretion of β-catenin which in turn translocates to the nucleus and influences transcription factor Tcf/Lef to transcribe cyclin D1, C-Myc, and CRD-RB that play role in cell-cycle progression, growth, and proliferation [124]. p53 regulates genes associated with DNA repair, cell cycle arrest, and apoptosis [125]. Some Ru-based metalla-bowl compounds showed anticancer activity among which [Ru4(p-cymene)4-(5,8-dioxydo-1,4-naphthaquinonato-)2(2,6-bis(N-(4-pyridyl)carbamoyl)pyridine)2][4CF3SO3] (5a) (Figure 5) was most potent and mediated CRC cells apoptosis by upregulating APC and p53 genes expression (Figure 3→V). After 24 h, 5a exhibited higher anti-proliferative activity against HT-15 cells compared to CIS and DOX (Table 1) [86]. Additionally, 5a (2 µM) upregulated the expression of APC mRNA (2.9-fold) and p53 mRNA (4.1-fold) in HCT116 cells compared to the untreated control group [86]. Results indicate that 5a displayed anti-proliferative activity via the upregulation of APC and p53 genes expression. As the tested control drugs, DOX and CIS are not used in CRC treatment, experimentation using OXA or 5-FLU could be used in further studies.
4.6. p53 Independent Activity
Ru-complexes including [Ru(η6-p-cym)(7-(4-(Decanoyl)piperazin-1-yl)-ciprofloxacin-H)Cl] (6a) and Ru-arene Schiff-base complexes particularly [(η6-1,3,5-triisopropylbenzene)RuCl(4-methoxy-N(2-quinolinylmethylene)aniline)]Cl (6b) (Figure 5) exhibited p53 independent anticancer property against CRC cells that are resistant to Pt-based anticancer drugs [116,117]. CRC cells overexpress organic cation transporter (OCT) proteins that are responsible for resistance towards CIS. Conversely, OXA contains hydrophobic 1,2-diaminocyclohexane (DACH), which produces cationic species. These cationic species act as OCT1/2 substrates [126]. Similarly, 6b bear hydrophobic ligands and are constantly cationic. Therefore, it acts as potential substrates of OCT1/2 and is effective against CRC cells [117]. It was evident that 6a and 6b were more potent compared to CIS and OXA against HCT116 cells (Table 1) [116,117]. Furthermore, 6a was reported to arrest the S phase followed by the G2/M phase of cell cycle while 6b arrested cell cycle at both G0/G1 (2.5 µM) and G2/M (10 µM) phase in a dose-dependent manner [116,117]. Considering their IC50 values, 6b was more potent than OXA against HCT116 cells and could be used as an alternative to OXA in treating CRC with dysfunctional p53.
4.7. Inhibition of Proteasome
Proteasome activity is crucial for various cellular processes like cell-cycle regulation, cell differentiation, angiogenesis, and apoptosis [127,128,129]. Thus, inhibition of proteasome activity is a potential way to mediate apoptosis in cancer cells [130]. Curcumin, a biocompatible compound is reported to induce apoptosis in CRC cells through inhibiting proteasome activity [131]. Ru(II) arene complexes with curcumin including [Ru(p-cymene)(curcumin)Cl] (7a), [(Benzene)Ru(curcumin)Cl] (7b), and [Ru(hexamethylbenzene)(curcumin)Cl] (7c) (Figure 6) induced DNA fragmentation leading to apoptosis via proteasome inhibition in CRC cells [90]. Generally, p53 and Poly-(ADP)-ribose polymerase (PARP) increases upon DNA damage and are engaged in repairing damaged DNA [132,133]. However, extensive fragmentation of DNA leads caspase-3 to cleave 116 kDa PARP into 85 kDa fragment, resulting in the inactivation of ubiquitin protease function and triggering apoptosis (Figure 3→VII) [90,133]. Among three curcumin-based Ru-complexes, 7b was most potent. Treatment of HCT116 cells with 7b (10 µM) decreased cell viability (20%–30%) after 4–24 h and reduced 26S proteasome ChT-L activity (37%–25%) after 4 h. The same concentration of 7b complex also elevated p53 (3.2-fold), caspase-3 activity (25%) while decreased PARP level (2-fold) and 26S proteasomes activities (~30%) compared to control after 24 h in HCT116 cells (Table 2) [90]. The obtained results showed that 7b inhibited CRC cells proliferation by reducing proteasome activity. As Bonfili et al. did not use any control drug(s) (e.g., OXA), further studies are required to compare the potency of 7b with standard drug(s).
4.8. ROS-Mediated Apoptosis
ROS are chemically reactive derivatives of oxygen that can exist independently carrying an unpaired electron and show conducive or detrimental effects depending upon its cellular concentration [134,135]. Cellular ROS levels are linked with cancer cell apoptosis, and excessive levels are reported to have anticancer activity [136]. Different Ru-complexes have been reported to modulate apoptosis in CRC cells by elevation of ROS level. Half sandwich Ru(II) complexes [η6-p-cymene)RuCl(2-(5,6-dichloro-1H-benzo[d]imidazole-2-yl)quinolone)] (8a) (Figure 6) produced ROS, which in turn damaged DNA via oxidative stress and induced apoptosis in CRC cells (Figure 3→VIII). Considering their IC50 values, 8a displayed 2.90-fold better anti-proliferative activity against Caco-2 cells compared to CIS (Table 1) [118]. Treatment of HT29 cells for 24 h with 8a (5–6.8 µM) increased ROS level (around 20%–65%) as well as arrest cell cycle more than 65% of cells at G0/G1 phase [118].
Oxidative stress plays a central role in Ru(III)-based drug trans-[tetrachloro-bis(1H-indazole)ruthenate(III)] or KP-1019 (8b) (Figure 6) mediated apoptosis in CRC cells. Kapitza et al. [119] reported that 8b induced H2O2 formation in CRC cells which further reacts with mitochondrial membrane-embedded unsaturated fatty acids to induce depolarization of the mitochondrial membrane and mediated caspase-3 dependent PARP cleavage. 8b induced cytotoxicity in both SW480 and LT97 cells with an IC50 value of 30 and 50 µM, respectively. In contrast, antioxidant N-acetylcysteine (NAC) (5 µM) decreased their potency, as evident from the increase of IC50 values to 55 and 88 µM towards SW480 and LT97, respectively. This finding confirmed that ROS is involved in 8b-mediated apoptosis [119]. In vivo activity of 8b was evaluated by the chemoresistant MAC15A colon carcinoma in a rat model, closely similar to human colon cancer. Treatment of 8b (13 mg/kg) two times a week for 10 weeks reduced 8% of tumor size, where 5-FLU (40 mg/kg) reduces tumor size down to only 40% and at the same time, another Pt-based established drug CIS did not show any activity [137,138].
Figure 6.
Structures of some Ru-complexes using in CRC treatment. Different Ru-complexes mediated apoptosis of CRC cells via inhibiting proteasome activity (7a–c), ROS-mediated apoptosis (8a–c), inhibiting thioredoxin reductase activity (10a), inhibiting HIF-1 pathway (11a), anti-metastasis activity (12a), and lysosomal dysfunction (13a).
Figure 6.
Structures of some Ru-complexes using in CRC treatment. Different Ru-complexes mediated apoptosis of CRC cells via inhibiting proteasome activity (7a–c), ROS-mediated apoptosis (8a–c), inhibiting thioredoxin reductase activity (10a), inhibiting HIF-1 pathway (11a), anti-metastasis activity (12a), and lysosomal dysfunction (13a).

Sodium salt of 8b, i.e., sodium trans-[tetrachloride-bis(1H-indazole)ruthenate(III)] or NKP-1339 (8c) (Figure 6) was prepared by Keppler et al. [139]. 8c was reported to increase the ROS level and, therefore, induced ER stress-mediated apoptosis in CRC cells (Figure 3→VIII) [89]. Exposure of 8c (200 μM) to HCT116 and SW480 CRC cells elevated ROS concentration by 2-fold and 2.5-fold, respectively, after 1 h compared to control and led to apoptosis through ER stress [89]. ROS causes potential damage to proteins that piled up in the ER. Since cancer cells tend to demonstrate an increased level of oxidative stress and ER stress due to having enhanced and fast metabolic activity, hence excessively accumulated misfolded proteins led the ER to start unfolded protein response (UPR) which induced apoptosis after exceeding a certain threshold level (Figure 3→VIII) [89,140]. The underlying mechanism is associated with three transmembrane receptors namely, PrKr-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring protein 1α (IRE1α) which are bound by an ER-resident chaperone, glucose-regulated protein (GRP78), which has high affinity towards misfolded protein [89,140,141]. Upon release from GRP78, PERK phosphorylated eukaryotic translation initiation factor 2α (eIF2α), which increased the cap-independent translation of activating transcription factor 4 (ATF4) [140]. ATF4 consequently translocated to the nucleus and induced transcription factor C/eBP homologous protein (CHOP), which is involved in apoptosis (Figure 3→VIII) (Table 2) [141]. Besides, treatment of both HCT116 and SW480 with 8c (200 μM) for 6 h mediated translocation of transcription factor Nrf2 from the cytoplasm into the nucleus, which induced different genes containing an antioxidant response element (ARE) in their promoter site to exert antioxidant response [89]. The GRP78 chaperone was found to be regulated on the protein level but only had a slight influence on the mRNA level recommending involvement of ER-associated protein degradation (ERAD) in the mode of action of 8c [89,141]. Considering all observations, it can be said that ROS plays a vital role in inducing apoptosis in the CRC cells. Among three complexes (8a–c), 8a can be considered as the most potent in terms of IC50 values. However, experimentation using standard chemotherapeutics drug(s) (e.g., OXA, 5-FLU) could make the study more significant.
4.9. Immunogenic Cell Death
8c is responsible to mediate ER stress that induces a cascade of events leading to CRC cells death along with providing critical signals to visualize dying cancer cells to the immune system. This consequently introduces sustained immune response against the CRC; a phenomenon termed immunogenic cell death (ICD) [120]. ICD is characterized by secretion of immune-modulatory damage-associated molecular patterns (DAMPs), such as pre-apoptotic calreticulin (CRT) surface-exposure, extracellular adenosine triphosphate (ATP), and high mobility group box 1 (HMGB-1) [142,143]. The ER stress triggers a cascade of reactions that activate PERK. This activated PERK phosphorylates eIF2α which in turn translocate CRT to the cell membrane that is generally located at the lumen of the endoplasmic reticulum of colon cancer cells [144]. Exposure of CRT on cell membrane elicits an “eat me” signal which induces maturation of phagocyte dendritic cell (DC) as well as uptake tumor antigens (Figure 3→IX) [145,146,147]. The other DAMP, HMGB-1 protein, residing in the nucleus moves to the extracellular space in the course of ICD and attach to pattern recognition receptors (PRRs) such as toll-like receptor 4 (TLR-4), receptor for advanced glycation end products (RAGE), and nuclear factor-κB (NF-κB) of DCs and presented antigens from dying tumor cells. This also accelerates DC maturation and migration. HMGB-1 functions as a crucial DAMP showing immune-stimulatory and pro-inflammatory effects [147,148] Lastly, extracellular adenosine triphosphate (ATP), released from dying tumor cell expresses a “find me” signal [147]. Released ATP binds to the purinergic P2RX7 receptors of dendritic cells and activated the (NOD)-like receptor protein 3 (NLRP3) inflammasome. This, in turn, stimulates tumor-specific cytotoxic T cells to secrete IFN-γ [120,147]. IFN-γ have pro-apoptotic and anti-proliferative functions such as inhibiting tumor angiogenesis, inducing regulatory T-cell apoptosis, and influencing M1 pro-inflammatory macrophages activity to suppress tumor progression [149].
Wernitznig et al. [120] described that treatment with 8c (100 µM) for 24 h upsurged CRT expression by approximately 7%, 10%, and 7% as well as extracellular ATP level by around 3%, 2.5%, and 2.6% in HCT116, HT15, and HT29 cell membrane, respectively, compared to the control. However, 8c increased the CRT level by 3.75 and 1.25-fold compared to CIS and OXA, respectively, as well as ATP level than both CIS and OXA by 1.36-fold in HT29 cells. Furthermore, the same concentration in HCT116 enhanced the release of HMGB-1 into the cytoplasm (Table 2) [120]. These findings consolidate that 8c could inhibit CRC cells proliferation via immunogenic death as well as ROS-mediated apoptosis and could be used as an alternative to OXA.
4.10. Inhibition of Thioredoxin Reductase Activity
Thioredoxin reductase (TrRx) is associated with redox-regulation and cell signaling [150]. Overexpression of TrRx is observed in CRC cells [151,152] and is considered to play a role in resisting CIS [78]. Heterobimetallic Ru(II)–gold(I) complexes [Ru(p-cymene)Cl2(µ-1,1-bis(diphenylphosphino)methane)Au(IMes)]-ClO4 (10a) (Figure 6) inhibited TrRx activity. 10a (IC50 = 5.22 µM) inhibited TrRx activity of HCT116 cells after 72 h where CIS was unable to induce any effect [78]. As CIS is not an approved drug in CRC treatment, studies using OXA would better reflect experimental findings.
4.11. Inhibition of HIF-1 Pathway
Hypoxia-inducible factor-1 (HIF-1) plays a fundamental role in tumor growth, angiogenesis, survival, and energy metabolism [153,154]. Decreased expression of HIF-1 protein level affects in downregulating HIF-1 target genes including vascular endothelial growth factor (VEGF) which modulate tumor angiogenesis [153,155], glucose transporter 1 (GLUT1) that mediate glucose uptake [156], and alpha-enolase (ENO1) which is crucial for glucose metabolism acting as a key catalyzing enzyme in the glycolysis (Figure 3→XI) [157]. All of these genes are responsible for the progression of CRC [158,159,160]. The HIF-1 pathway is regulated by the redox enzyme prolyl hydroxylase 2 (PHD2) [161]. Ru derived compound 11 (11a) (Figure 6) was synthesized by Leyva et al. [162]. Vidimar et al. [87], reported that 11a showed higher anti-proliferative activity than CIS against HCT116 cells (2.7-fold) (Table 1). 11a interfered with the HIF1 pathway by upregulating PHD2, which consequently decreased the HIF-1α protein level. This, in turn, caused reduced angiogenesis and altered glucose metabolism in CRC cells by decreasing VEGF expression, GLUT1 and ENO1 (Figure 3→XI). Western blotting showed that exposure of HCT116 cells to 11a (5µM) under hypoxic conditions (1% O2) elevated the level of PHD2 enzyme after 6 h. Additionally, treatment of HCT116 cells with 11a at the same concentration and under the same experimental conditions reduced the expression of VEGF RNA, GLUT1 RNA, and ENO1 RNA by 240%, 320%, and 70%, respectively, while CIS reduced the expression of these genes by 150%, 350%, and 10%, respectively. This reflected better therapeutic efficiency of 11a than CIS. Moreover, 11a (11 µmole/Kg) impeded CRC progression in C57BL/6 female mice xenografted human colon tumors by reducing VEGF mRNA expression (30%), GLUT1 mRNA expression (50%), vascularization (30%), and tumor size (90%) compared to OXA after 21 days [87]. Since 11a showed better potency than OXA through blocking HIF-1 Pathway of CRC cells. Therefore, 11a can be used in place of OXA in CRC treatment.
4.12. Anti-Metastasis Activity
Novel Ru(III)-based drug, Imidazolium-trans-tetrachloro(dimethylsulfoxide)imidazoleruthenium(III) or NAMI-A (12a) (Figure 6) was synthesized by Alessio et al. [163]. 12a displayed anti-proliferative property against CRC cells by virtue of its anti-metastasis activity. Unlike other Ru-based drugs, 12a focuses on the tumor microenvironment instead of showing direct cytotoxicity to the cell [164,165,166]. 12a interact with actin-like proteins of the cell surface and collagens of the extracellular matrix and reduced invasive cancer cells mobility [167]. 12a selectively targets surface adhesion receptor α5β1 integrin of colon cancer cells [88]. Highly invasive colon cell lines tend to express an increased level of α5β1 integrin [168], which is responsible for adhesion and migration of colon cancer cells through interacting with extracellular matrix proteins (ECM) [88,169,170]. According to Pelillo et al. [88], about 78% of cell adhesion is reduced by blocking α5β1 integrin site of HCT116 cells. 12a blocked both the steps of adhesion and migration of the tumor cells by impairing the contact between α5β1 integrin and fibronectin of HCT116 cells in an inverse concentration-dependent manner (Figure 3→XII). 12a (1–10 µM) inversely decreased the attachment of fibronectin with α5β1 integrin by 38–25%. 12a (10–100 μM) also reduced the adhesion rate by 58–82%. Molecular insight of CRC revealed that 12a altered the expression of the genes encoding the α5 and β1 subunit and, therefore, decreased the number of α5β1 integrin receptor molecules (Table 2). 12a at a concentration of 1 μM downregulated α5 subunit encoding gene ITGA5 while 100 μM concentration upregulated ITGA5 expression up to 3.5-fold. Nonetheless, β1 subunit encoding gene ITGA1 did not respond to the alteration of concentrations [88]. Besides, the binding event activated autophosphorylation at the Tyr 397 site of the intracellular focal adhesion kinase (FAK) [170], which not only mediated tumor cell proliferation, survival, and migration [171] but also regulated the binding strength between integrins and ECM proteins [88]. 12a (1–0 μM) decreased nearly 70–15% level of p-Tyr 397 FAK [88].
The metastasis of CRC cells is influenced by the hepatic microenvironment [170]. Bergamo et al. [169] reported that normal epithelial colon cells and hepatocytes release different soluble factors involved in the transcription of genes of the tumor cells associated with tumor growth, invasion, and migration. 12a prevented transcription of those genes, thus inhibit the growth and dissemination of CRC cells. VEGF or MCP-1 either alone or in combination increased the migration ability of HCT116 cells. Exposure to 12a decreased VEGF or MCP-1 induced migration of HCT116 cells [169]. As Pelillo et al. did not use any standard chemotherapeutics drug(s) (e.g., OXA, 5-FLU), further investigations are required to determine the potency of 12a compared to standard drugs.
4.13. Lysosomal Dysfunction
Lysosomes contain various hydrolytic enzymes which degrade damaged proteins and organelles to regulate cellular functions [172]. However, releasing these hydrolase enzymes from lysosomes degrade other cytoplasmic organelles and lead to cell death [173]. Some Ru-complexes can be localized inside the lysosome, specifically where they cause lysosomal dysfunction [91,174,175]. Lysosomal dysfunction can be identified by unusual instigation of lysosomal enzymes, reduced lysosome-associated membrane proteins (LAMPs) expression as well as the permeability of lysosomes [176]. Disintegration of the lysosome induces the release of lysosomal hydrolases like cathepsin B from the lysosomal lumen to the cytosol, which makes the cells prone to lysosome-induced cell death (Figure 3→XIII) [173].
According to Xu et al. [91], half-sandwich Ru(II)-complexes bearing aryl- bis(imino)acenaphthene chelating ligands with fluorine group (13a) (Figure 6) induced lysosome mediated CRC cells death in vitro. 13a displayed higher anti-proliferative activity compared to CIS against HT29 cells (5.91-fold), HCT116 cells (15.85-fold), and CT-26 cells (11.13-fold) in terms of IC50 values (Table 1). Moreover, 13a suppressed tumor growth in CT-26 cells xenografted BALB/c mice model. Treatment of CT-26 cells with 13a (0.575–2.3 µM) for 24 h elevated permeability of lysosomes and released cathepsin B [91]. 13a downregulated LAMPs expression (20–60%) compared to control, thereby suppress metastasis [91,177]. Additionally, 13a (0.575–1.15 µM) also increased ROS production (25–75%) in CT-26 cells [91]. Elevation of ROS level induced lipid peroxidation to rapture lysosome via destabilizing lysosomal membrane that leads to cell death [178].
4.14. Photodynamic Therapy
Photodynamic therapy (PDT) has emerged as a potential cancer therapy that is either used as a sole treatment or in combination with chemotherapy, surgery, and/or radiation [179]. PDT uses lights with appropriate wavelength to stimulate photosensitizer (PS) which mediates photochemical reaction to produce ROS and consequently kill tumors (Figure 6) [180].
Ru(II) PSs [Ru(2,2′-bipyridine)2(2-(2′,2″:5″,2′′′-terthiophene)-imidazo[4,5-f][1,10]phenanthroline)]2+ AKA TLD1411 (14a) and [Ru(4,4′-dimethyl-2,2′-bipyridine)2(2-(2′,2″:5″,2′′′-terthiophene)-imidazo[4,5-f][1,10]phenanthroline)]2+ AKA TLD1433 (14b) (Figure 7A) contain both photo-biological and photo-physical properties [92]. 14b was originally synthesized by Sherri MacFarland and this Ru(II)-based photosensitizer entered clinical trial to treat bladder cancer through PDT. A phase I clinical study was conducted with 14b (at 0.70 mgcm−2 dose) on six non-muscle-invasive bladder cancer patients (NCT03053635) and tumor relapse was not observed up to 180 days [181].
Ru(II) in 14a, and 14b makes the complexes specific towards cancer cells rather than normal cells and upon light irradiation increased singlet oxygen (1O2) quantum yield [182]. 14a and 14b induced fragmentation of DNA via photocleavage activities (Figure 7B) [92,183]. According to Fong et al. [92], 14a (4 µM) and 14b (1 µM) exhibited photodynamic effects which cause photon-mediated complete death of CT-26 cells. However, both 14a (10 µM) and 14b (10 µM) showed minimal toxicity (less than 10%) in the dark. The maximum tolerated dose was recorded to 36 and 103 mg/kg for 14a and 14b, respectively. Besides, in vivo treatment of 14a and 14b modulated tumor cell regression. Four hours post-intrathecal administration of 14a (36 and 2 mg/kg), and 14b (53 and 5 mg/kg) in BALB/c mice, followed by irradiation with a continuous wave or pulsed light sources (λ = 525–530 nm, H = 192 Jcm−2) for 30 min (with 30 s on/off cycle) exhibited a higher tumor growth reducing efficacy after 24 h. 14a (2 mg/kg) and 14b (5 mg/kg) delayed the tumor growth for 8 and 9 days, respectively. Both the compounds increased survival time in a dose-dependent manner. However, 14b extended survival time by 5-fold compared to 14a [92]. Treatment with 14b also induced antitumor immunity in the colon cancer-containing mouse model [181]. Considering all observations, 14b could be considered as a promising PDT agent in CRC treatment.
The Pt-based drug, Pt(II) 2,6-dipyrido-4-methyl-benzenechloride (PMB) also induced PDT-mediated DNA damage of CRC cells in a similar way [184]. Since 14b and PMB were not investigated under similar experimental conditions, thus the potency of 14b and PMB could not be compared.
5. Ru-Nanocomplexes in CRC Theranostics
5.1. CRC Diagnosis
RuNPs-based nanoformulations could facilitate the early diagnosis of CRC [30,185,186]. Xu et al. [30] constructed hollow mesoporous RuNPs (HMRuNPs) which are efficient in in vivo tumor imaging, drug loading, and combined treatment for CRC. Hollow mesoporous Ru containing fluorescent complex with anticancer activity (RBT) and bispecific antibodies (SS-Fc, anti-CD16, and anti-CEA) (15a) selectively accumulated into CRC cells by both active and passive targeting. Active targeting is mediated through antibody SS-Fc which can detect carcinoembryonic (CEA) antigen on CRC cell lines (i.e., HCT116, SW480, HT29, and Lovo) and attach with natural killer (NK) cells to induce an immune response. Passive targeting facilitates drug accumulation by the EPR effect. Moreover, the same compounds also have therapeutic effects. Treating BALB/c mice containing systemically administrated CT26-CEA tumor with 15a (5 mg/kg) for every three days for a total of three treatments released RBT that generated ROS and appointed NK cells to initiate immune response, which in turn led to apoptosis and necrosis in CRC [30].
5.2. CRC Treatment
Conventional chemotherapy, as well as radiation therapy, conveys side effects and other limitations such as drug resistance [187]. The application of nanoformulations could overcome such shortcomings [188,189]. The advent of RuNPs offers excellent anticancer activity due to the high photothermal conversion rate, multiple oxidation states, and valence states [30]. Heffeter et al. [190] reported that micelle-like carriers (MC-8b) and nanoform of the established drug 8b sidestepped the limitations of 8b aqueous solutions undergo rapid hydrolysis to yield water-insoluble 8b aqua complex, [mer,trans-[Ru(III)Cl3(Hind)2(H2O)]. Nevertheless, MC-8b (0.3 mg/mL 8b) solutions were stable at 4°C for three months regarding precipitation. After 72 h of incubation, MC-8b was found to be more active than 8b against HCT116 cells (3.91-fold) and Lovo cells (4.88-fold) in terms of their IC50 values. Moreover, MC-8b (IC50 = 41 µM) provided rapid onset of anticancer activity than 8b (IC50 value of 135 μM) within only 1 h in HCT116 cells. Additionally, treatment of HCT116 cells with MC-8b (25 µM) exhibited higher apoptosis potential than 8b by 6-fold. Western blotting indicated that MC-8b increased the expression of p53, phosphorylated P38, and JNK while decreased caspase-7 and PARP expression [190].
Besides, Zhu et al. [31] developed other Ru-based nanozymes, hollow Ru@CeO2 yolk-shell nanozymes in conjugation with antitumor drug Ru-complex (RBT) along with resveratrol (Res) and coated with DEPG (16a) which exhibited anti-metastasis and anti-tumor activity in orthotopic CRC through dual-chemotherapy/Photothermal therapy (PTT) with in situ oxygen supply [31]. Moreover, recurrence of more than 60% of post-surgical colorectal tumors is associated with the liver while more than 35% of all metastases occur solely in the liver [191,192]. 16a with near infrared (NIR) efficiently inhibited intestinal, lung, and liver metastasis. 16a contains antitumor Ru drugs, RBT, and Res, which exerted dual chemotherapeutic efficiency, while Ru@CeO2 holds efficient light-to-heat conversion potency. At the same time, 16a catalyzed H2O2 to O2 in the tumor microenvironment (TME) and thereby overcame hypoxia by achieving in situ O2 supply and reduce HIF-1α hypoxic staining signal. Treatment with 16a (5 mg/kg) for every three days for a total of three treatments overcame tumor hypoxia and obtained dual-chemotherapy/PTT in BALB/c mice bearing CT-26 cells. The excellent biocompatibility of the nanozyme is achieved due to the DPEG coating that prevented the occurrence of hemolysis even at a high dose concentration [31].
Like RuNPs, Pt-based nanoparticles (PtNPs) are also used in cancer diagnosis, drug delivery, treatment and can induce PTT-mediated apoptosis [193,194,195,196,197]. For example, Pt-based nanostructure, DPMNPs, was synthesized via coating dichloro(1,2-diaminocyclohexane)platinum(II) with poly[2-(N,N-dimethylamino)ethyl methacrylate]-poly(ε-caprolactone). DPMNPs exhibited a similar PTT-mediated anticancer effect in CRC cells [198].
6. Phase I Dose-Escalation Studies (Phase Ib Clinical Trials)
Dose escalation is an integral part of the phase I study which carefully looks for the optimal dose of a new drug to avoid therapeutic overdoses. In a dose-escalation study, the dosage of a drug is gradually increased until the side effects appear. Such a study is conducted on humans to assess the pharmacokinetics, pharmacodynamics, and safety of a new drug [199]. Rademaker-Lakhai et al. [200] conducted a phase I dose-escalation study with 12a on seven CRC patients (n = 24, having solid tumors). 12a at a dose concentration of 2.4–38.4 mg/m2/day caused no drug-induced toxicity. However, a dose concentration of 76.8–115 mg/m2/day resulted in causing diarrhea, phlebitis, and fatigue, while 400–500 mg/m2/day dose caused skin blisters lasting up to several months, which caused extreme pain. Considering all these data, the prescribed dose was set as 300 mg/m2 /day. However, this study did not provide effects of 12a specifically on CRC patients rather gave a generalized overview of the effects of 12a on 24 patients. Furthermore, in the phase I dose-escalation study 12a stabilized disease for 21 weeks in a patient with lung cancer which prompted to organize a phase I/II trial on 32 non-small cell lung cancer patients in combination with gemcitabine [200,201]. Lentz et al. [202] performed a phase I dose-escalation study with 8b on two CRC patients (n = 7, having various types of solid tumors). Intravenous administration of 8b escalating from 25 to 600 mg (equivalent to 5–120.8 mg of Ru) twice weekly over 3 weeks caused no dose-limiting toxicity. Nonetheless, 8b comes up with limitations regarding low solubility that makes it challenging to obtain proper dosage. Hence, its analogous sodium salt 8c is largely used in clinical trials, which provides 35-fold better solubility [138]. In the study of Thompson et al. [203], 34 patients having solid tumors were treated with 8c among whom 10 CRC patients were reported. 8c (20–780 mg/m2) was infused in intravenous route on day 1, 8, and 15 of 28 days cycles. The maximum tolerated dose (MTD) was reported 625 mg/m2 with minor side effects [203,204]. However, none of the aforementioned phase I dose-escalation studies of 12a, 8a, and 8b are listed on the clinicaltrial.gov website (www.clinicaltrial.gov) [205].
The only registered study was conducted by Burris et al. (trial registration number: NCT01415297) [206]. The phase Ib clinical trial was conducted to investigate MTD of 8c (20–780 mg/m2) on 11 CRC patients (n = 46 patients having advanced solid tumors) and found similar MTD (625 mg/m2). However, ≥20% of the patients experienced adverse events that emerged from treatment which include fatigue, nausea, vomiting, dehydration, and anemia. In total, 59% of patients experienced ≤grade 2 and 37% of patients experienced grade 3 adverse effects though no patient was reported to have grade 4 adverse effects. It should be noted that both the studies did not present specific descriptions of the adverse effects on CRC patients [206].
Pt-based drugs, CIS, CAR, and picoplatin (PIC) have been used in combination with other chemotherapeutic drugs in clinical trials of CRC [205]. A Phase I clinical trial (NCT00465725) on various solid tumors, including CRC was studied using PIC only as an anti-proliferative agent [205]. A phase I clinical and pharmacological study with PIC revealed the MTD (150 mg/m2). Moderate level of anorexia, nausea, vomiting and transient metallic taste was evident and there was no significant sign of alopecia [207]. Peripheral neuropathy is the main disadvantage of OXA-based chemotherapy [208,209,210], whereas some Ru-based complexes (mentioned in this section) or PIC did not show such neurotoxic effects [211]. Therefore, Ru-based complexes or PIC could be used as an alternative to OXA in CRC patients with compromised neuronal function.
7. Toxicity of Ru-Drug Candidates
Pt-based drugs have toxic side effects which include neurotoxicity, nephrotoxicity, hepatotoxicity, ototoxicity, skin toxicity, myelosuppression, alopecia, diarrhea, fatigue, nausea, and vomiting [48,212,213]. Though Ru-based complexes exhibit parallel or better therapeutic efficacy than conventional Pt-based drugs, Ru-based complexes are promising for their lower toxicity [214]. [Ru(L-methionine)(2,2-bipyridine)(1,4-bis(diphenylphosphino)butane)]hexafluorophosphate (17a) and [Ru(l-tryptophan)(2,2-bipyridine)(1,4-bis(diphenylphosphino)butane)hexafluorophosphate (17b) displayed lower mutagenicity and genotoxicity in male Swiss mice compared to CIS at an equal concentration of 2 mg/kg body weight [215]. Furthermore, the in vivo biocompatibility of Ru-based drugs is another concern. 4b, 4c, 4d, 17a, and 17b were reported to be safe at <300 mg/kg in vivo Swiss albino mice and Wistar rat models [82,83,84,215]. However, a therapeutic dose of >300 mg/kg elevated serum ALT, AST, ALP, BUN, creatinine, and glucose. At the same time, architectural alteration of kidney and liver was also evident [82,83,84]. Administering [Ru(Cl-terpyridine)(ethylenediamine)Cl][Cl] (18a) and [Ru(Cl-terpyridine)(1,2-diaminocyclohexane)Cl][Cl] (18b) at 2 mg/kg dose concentration in BALB/c mice bearing CT26 mouse colon carcinoma exhibited moderate histological changes in kidney, lung, and liver but no significant changes were found in heart architecture. However, there was a lack of changes in serum creatinine, urea, AST, and ALT level [62]. Furthermore, Ru-based drugs 12a damaged kidneys by altering the glomeruli structure [216,217] and 8b showed toxicity in bone marrow besides kidney [62].
Koch et al. [218] reported that some Ru-complexes (i.e., bipyridine, terpyridine, and phenanthroline Ru-complexes) acted as cholinesterase inhibitors in vitro and induced hind limb paralysis, respiratory distress, and death in respiratory failure as well as block curariform at the neuromuscular junction in in vivo mice model [218]. Furthermore, Ru is reported to inhibit Ca2+ uptake by mitochondria which possibly contributed to β-adrenergic and neuromuscular blocking [219,220,221]. Kruszyna et al. [221] described that some Ru-nitrosyl complexes at a concentration of (55–63 mg/kg) induced rapid death (after 10 min) while the rest of the complexes mediated death after 4-7 days a concentration ranging from 8.9 to 127 mg/kg. Ru is also retained in muscle and bone, rising concern about their long-term effect [221]. Thus, the toxicity of Ru-based drug candidates is a considerable issue before clinical applications. Ru-complexes including 1c, 1d, 4a, 4f, 6a, and 6b, displayed higher cytotoxic potential than OXA [27,77,87,115,116,117] while 5-FLU were found to be less potent than 4a and 4f in vitro and 3d and 8b in vivo [27,81,115,138]. Although the anticancer activity of Ru-complexes has been explored largely, their extended toxicity studies were not investigated. Therefore, preclinical chronic toxicity studies should be performed before considering these as potential drugs of CRC.
8. Conclusions
Ru-complexes displayed promising anticancer activities in the treatment of CRC. Ru(III) and Ru(II) were the most investigated oxidation states against all cancer cells including CRC cells, where the former one acts as a prodrug and converts to Ru(II) in the tumor microenvironment. Ru(II)-complexes displayed more reactivity compared to Ru(III) complexes. Many Ru-complexes were found to be more efficient than the Pt-based drugs; therefore, Ru could be used as an alternative to Pt-based drugs. Moreover, some Ru-complexes were reported to be more effective compared to the conventional chemotherapeutic drug (5-FLU) which has been used as first-line treatment against CRC. Though Ru conjugation with organic molecules could enhance anticancer activity through a synergistic effect [82,83,84], sometimes Ru-complexes are found to be less potent compared to parent organic molecules against CRC cells [90,222]. While conjugation of Ru-compounds with RuNps enhanced cellular uptake, selectivity, and drug delivery in CRC cells. Therefore, higher attention should be given to this field. Finally, extensive preclinical studies should be formed to confirm the efficacy, elucidating the potential mechanism of action(s), and toxicity of Ru-complexes or Ru-nanoformulations before considering them as potent drug candidates against CRC.
Author Contributions
Conceptualization, M.S.S. and M.A.I.; writing—original draft preparation, K.M.M., M.S.S. and M.S.N.; writing—review and editing, M.S.S. and M.A.I.; project administration, M.S.S and M.A.I. All authors have read and agreed to the published version of the manuscript.
Funding
The APC was funded by the Research Creativity and Management (RCMO), Universiti Sains Malaysia (USM), and the School of Medical Sciences, USM.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data reported in Figure 2 are available at cBioportal database (https://www.cbioportal.org/) (accessed on 15 July 2021).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Centelles, J.J. General aspects of colorectal cancer. Int. Sch. Res. Not. 2012, 2012, 139268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manne, U.; Shanmugam, C.; Katkoori, V.R.; Bumpers, H.L.; Grizzle, W.E. Development and progression of colorectal neoplasia. Cancer Biomark. 2011, 9, 235–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeriouh, W.; Nani, A.; Belarbi, M.; Dumont, A.; de Rosny, C.; Aboura, I.; Ghanemi, F.Z.; Murtaza, B.; Patoli, D.; Thomas, C.; et al. Phenolic extract from oleaster (Olea europaea var. Sylvestris) leaves reduces colon cancer growth and induces caspase-dependent apoptosis in colon cancer cells via the mitochondrial apoptotic pathway. PLoS ONE 2017, 12, e0170823. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.; Oosterling, S.J.; van Egmond, M. Surgery for Colorectal Cancer: A Trigger for Liver Metastases Development? New Insights into the Underlying Mechanisms. Biomedicines 2021, 9, 177. [Google Scholar] [CrossRef]
- Colin, D.J.; Limagne, E.; Ragot, K.; Lizard, G.; Ghiringhelli, F.; Solary, É.; Chauffert, B.; Latruffe, N.; Delmas, D. The role of reactive oxygen species and subsequent DNA-damage response in the emergence of resistance towards resveratrol in colon cancer models. Cell Death Dis. 2014, 5, e1533. [Google Scholar] [CrossRef] [Green Version]
- Denlinger, C.S.; Barsevick, A.M. The challenges of colorectal cancer survivorship. J. Natl. Compr. Canc. Netw. 2009, 7, 883–894. [Google Scholar] [CrossRef]
- Knowles, G.; Haigh, R.; McLean, C.; Phillips, H.A.; Dunlop, M.G.; Din, F.V.N. Long term effect of surgery and radiotherapy for colorectal cancer on defecatory function and quality of life. Eur. J. Oncol. Nurs. 2013, 17, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Benjamin Garbutcheon-Singh, K.; P Grant, M.; W Harper, B.; M Krause-Heuer, A.; Manohar, M.; Orkey, N.R.; Aldrich-Wright, J. Transition Metal Based Anticancer Drugs. Curr. Top. Med. Chem. 2011, 11, 521–542. [Google Scholar] [CrossRef]
- Ndagi, U.; Mhlongo, N.; Soliman, M.E. Metal complexes in cancer therapy—An update from drug design perspective. Drug Des. Dev. Ther. 2017, 11, 599–616. [Google Scholar] [CrossRef] [Green Version]
- Parveen, S. Recent advances in anticancer ruthenium Schiff base complexes. Appl. Organomet. Chem. 2020, 34, e5687. [Google Scholar] [CrossRef]
- Raymond, E.; Chaney, S.; Taamma, A.; Cvitkovic, E. Oxaliplatin: A review of preclinical and clinical studies. Ann. Oncol. 1998, 9, 1053–1071. [Google Scholar] [CrossRef] [PubMed]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef] [PubMed]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Lippard, S.J. Direct cellular responses to platinum-induced DNA damage. Chem. Rev. 2007, 107, 1387–1407. [Google Scholar] [CrossRef]
- Hsu, H.H.; Chen, M.C.; Baskaran, R.; Lin, Y.M.; Day, C.H.; Lin, Y.J.; Tu, C.C.; Vijaya Padma, V.; Kuo, W.W.; Huang, C.Y. Oxaliplatin resistance in colorectal cancer cells is mediated via activation of ABCG2 to alleviate ER stress induced apoptosis. J. Cell. Physiol. 2018, 233, 5458–5467. [Google Scholar] [CrossRef] [PubMed]
- Drott, J.; Fomichov, V.; Starkhammar, H.; Börjeson, S.; Kjellgren, K.; Berterö, C. Oxaliplatin-Induced Neurotoxic Side Effects and Their Impact on Daily Activities: A Longitudinal Study among Patients with Colorectal Cancer. Cancer Nurs. 2019, 42, E40–E48. [Google Scholar] [CrossRef] [Green Version]
- Virag, P.; Fischer-Fodor, E.; Perde-Schrepler, M.; Brie, I.; Tatomir, C.; Balacescu, L.; Berindan-Neagoe, I.; Victor, B.; Balacescu, O. Oxaliplatin induces different cellular and molecular chemoresistance patterns in colorectal cancer cell lines of identical origins. BMC Genomics 2013, 14, 480. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Kim, C.Y.; Nam, T.-G. Ruthenium Complexes as Anticancer Agents: A Brief History and Perspectives. Drug Des. Dev. Ther. 2020, 14, 5375–5392. [Google Scholar] [CrossRef]
- Kostova, I. Ruthenium Complexes as Anticancer Agents. Curr. Med. Chem. 2006, 13, 1085–1107. [Google Scholar] [CrossRef] [PubMed]
- Dougan, S.J.; Habtemariam, A.; McHale, S.E.; Parsons, S.; Sadler, P.J. Catalytic organometallic anticancer complexes. Proc. Natl. Acad. Sci. USA 2008, 105, 11628–11633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandioller, W.; Balsano, E.; Meier, S.M.; Jungwirth, U.; Göschl, S.; Roller, A.; Jakupec, M.A.; Berger, W.; Keppler, B.K.; Hartinger, C.G. Organometallic anticancer complexes of lapachol: Metal centre-dependent formation of reactive oxygen species and correlation with cytotoxicity. Chem. Commun. 2013, 49, 3348–3350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Sadler, P.J. Advances in the design of organometallic anticancer complexes. J. Organomet. Chem. 2017, 839, 5–14. [Google Scholar] [CrossRef]
- Silva, V.R.; Corrêa, R.S.; Santos, L.D.S.; Soares, M.B.P.; Batista, A.A.; Bezerra, D.P. A ruthenium-based 5-fluorouracil complex with enhanced cytotoxicity and apoptosis induction action in HCT116 cells. Sci. Rep. 2018, 8, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shum, J.; Leung, P.K.-K.; Lo, K.K.-W. Luminescent Ruthenium(II) Polypyridine Complexes for a Wide Variety of Biomolecular and Cellular Applications. Inorg. Chem. 2019, 58, 2231–2247. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.-P.; Zhong, Y.-M.; Ji, L.-N.; Mao, Z.-W. Phosphorescent metal complexes as theranostic anticancer agents: Combining imaging and therapy in a single molecule. Chem. Sci. 2021, 12, 2357–2367. [Google Scholar] [CrossRef]
- Xu, M.; Wen, Y.; Liu, Y.; Tan, X.; Chen, X.; Zhu, X.; Wei, C.; Chen, L.; Wang, Z.; Liu, J. Hollow mesoporous ruthenium nanoparticles conjugated bispecific antibody for targeted anti-colorectal cancer response of combination therapy. Nanoscale 2019, 11, 9661–9678. [Google Scholar] [CrossRef]
- Zhu, X.; Gong, Y.; Liu, Y.; Yang, C.; Wu, S.; Yuan, G.; Guo, X.; Liu, J.; Qin, X. Ru@CeO2 yolk shell nanozymes: Oxygen supply in situ enhanced dual chemotherapy combined with photothermal therapy for orthotopic/subcutaneous colorectal cancer. Biomaterials 2020, 242, 119923. [Google Scholar] [CrossRef]
- Thangavel, P.; Viswanath, B.; Kim, S. Recent developments in the nanostructured materials functionalized with ruthenium complexes for targeted drug delivery to tumors. Int. J. Nanomed. 2017, 12, 2749–2758. [Google Scholar] [CrossRef] [Green Version]
- Malki, A.; ElRuz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.-E. Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements. Int. J. Mol. Sci. 2021, 22, 130. [Google Scholar] [CrossRef]
- Tariq, K.; Ghias, K. Colorectal cancer carcinogenesis: A review of mechanisms. Cancer Biol.Med. 2016, 13, 120–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Guda, K.; Veigl, M.L.; Varadan, V.; Nosrati, A.; Ravi, L.; Lutterbaugh, J.; Beard, L.; Willson, J.K.V.; Sedwick, W.D.; Wang, Z.J.; et al. Novel recurrently mutated genes in African American colon cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 1149–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasaikar, S.; Huang, C.; Wang, X.; Petyuk, V.A.; Savage, S.R.; Wen, B.; Dou, Y.; Zhang, Y.; Shi, Z.; Arshad, O.A.; et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177, 1035–1049. [Google Scholar] [CrossRef] [Green Version]
- Giannakis, M.; Mu, J.X.; Shukla, A.S.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Brannon, A.R.; Vakiani, E.; Sylvester, B.E.; Scott, S.N.; McDermott, G.; Shah, R.H.; Kania, K.; Viale, A.; Oschwald, D.M.; Vacic, V.; et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014, 15, 454. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- cBioPortal Colon Cancer. Available online: https://bit.ly/2Od7xhB (accessed on 15 July 2021).
- McQuade, R.M.; Stojanovska, V.; Bornstein, J.C.; Nurgali, K. PARP inhibition in platinum-based chemotherapy: Chemopotentiation and neuroprotection. Pharmacol. Res. 2018, 137, 104–113. [Google Scholar] [CrossRef]
- Apps, M.G.; Choi, E.H.Y.; Wheate, N.J. The state-of-play and future of platinum drugs Endocr. Relat. Cancer 2015, 22, R219–R233. [Google Scholar] [CrossRef] [Green Version]
- Mehmood, R.K. Review of Cisplatin and oxaliplatin in current immunogenic and monoclonal antibody treatments. Oncol. Rev. 2014, 8, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coverdale, J.P.C.; Laroiya-McCarron, T.; Romero-Canelón, I. Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates? Inorganics 2019, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Chiorazzi, A.; Semperboni, S.; Marmiroli, P. Current View in Platinum Drug Mechanisms of Peripheral Neurotoxicity. Toxics 2015, 3, 304–321. [Google Scholar] [CrossRef] [Green Version]
- André, T.; Boni, C.; Mounedji-Boudiaf, L.; Navarro, M.; Tabernero, J.; Hickish, T.; Topham, C.; Zaninelli, M.; Clingan, P.; Bridgewater, J.; et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N. Engl. J. Med. 2004, 350, 2343–2351. [Google Scholar] [CrossRef] [Green Version]
- Rosati, G.; Cordio, S.; Reggiardo, G.; Aprile, G.; Butera, A.; Avallone, A.; Tucci, A.; Novello, G.; Blanco, G.; Caputo, G.J.C. Oxaliplatin-based chemotherapy in patients with metastatic colorectal cancer aged at least 75 Years: A post-hoc subgroup analysis of three phase II trials. Cancers 2019, 11, 578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forcello, N.P.; Khubchandani, S.; Patel, S.J.; Brahaj, D. Oxaliplatin-induced immune-mediated cytopenias: A case report and literature review. J. Oncol. Pharm. Pract. 2015, 21, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Haller, D.G. Safety of oxaliplatin in the treatment of colorectal cancer. Oncology 2000, 14, 15–20. [Google Scholar]
- Rubbia-Brandt, L.; Audard, V.; Sartoretti, P.; Roth, A.; Brezault, C.; Le Charpentier, M.; Dousset, B.; Morel, P.; Soubrane, O.; Chaussade, S.J. Severe hepatic sinusoidal obstruction associated with oxaliplatin-based chemotherapy in patients with metastatic colorectal cancer. Ann. Oncol. 2004, 15, 460–466. [Google Scholar] [CrossRef]
- Overman, M.J.; Maru, D.M.; Charnsangavej, C.; Loyer, E.M.; Wang, H.; Pathak, P.; Eng, C.; Hoff, P.M.; Vauthey, J.-N.; Wolff, R.; et al. Oxaliplatin-mediated increase in spleen size as a biomarker for the development of hepatic sinusoidal injury. J. Clin. Oncol. 2010, 28, 2549–2555. [Google Scholar] [CrossRef] [PubMed]
- Süss-Fink, G. Arene ruthenium complexes as anticancer agents. Dalton Trans. 2010, 39, 1673–1688. [Google Scholar] [CrossRef] [PubMed]
- Jakupec, M.A.; Galanski, M.; Arion, V.B.; Hartinger, C.G.; Keppler, B.K. Antitumour metal compounds: More than theme and variations. Dalton Trans. 2008, 14, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Ang, W.H.; Dyson, P.J. Classical and non-classical ruthenium-based anticancer drugs: Towards targeted chemotherapy. Eur. J. Inorg. Chem. 2006, 2006, 4003–4018. [Google Scholar] [CrossRef]
- Motswainyana, W.M.; Ajibade, P.A. Anticancer Activities of Mononuclear Ruthenium(II) Coordination Complexes. Adv. Chem. 2015, 2015, 859730. [Google Scholar] [CrossRef] [Green Version]
- Savic, M.; Arsenijevic, A.; Milovanovic, J.; Stojanovic, B.; Stankovic, V.; Rilak Simovic, A.; Lazic, D.; Arsenijevic, N.; Milovanovic, M. Antitumor Activity of Ruthenium(II) Terpyridine Complexes towards Colon Cancer Cells In Vitro and In Vivo. Molecules 2020, 25, 4699. [Google Scholar] [CrossRef]
- Fukushi, S.; Yoshino, H.; Yoshizawa, A.; Kashiwakura, I. p53-independent structure-activity relationships of 3-ring mesogenic compounds’ activity as cytotoxic effects against human non-small cell lung cancer lines. BMC Cancer 2016, 16, 521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, E.; Zhu, M.; Liu, L.; Huang, Y.; Wang, L.; Shi, C.; Zhang, W.; Sun, Y. Impact of the carbon chain length of novel palladium (II) complexes on interaction with DNA and cytotoxic activity. Inorg. Chem. 2010, 49, 3261–3270. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Fischer, A.; Xu, Y.; Sun, L. Isolated Seven-Coordinate Ru(IV) Dimer Complex with [HOHOH]—Bridging Ligand as an Intermediate for Catalytic Water Oxidation. J. Am. Chem. Soc. 2009, 131, 10397–10399. [Google Scholar] [CrossRef] [PubMed]
- Jabłońska-Wawrzycka, A.; Rogala, P.; Michałkiewicz, S.; Hodorowicz, M.; Barszcz, B. Ruthenium complexes in different oxidation states: Synthesis, crystal structure, spectra and redox properties. Dalton Trans. 2013, 42, 6092–6101. [Google Scholar] [CrossRef]
- Lin, K.; Zhao, Z.-Z.; Bo, H.-B.; Hao, X.-J.; Wang, J.-Q. Applications of Ruthenium Complex in Tumor Diagnosis and Therapy. Front. Pharmacol. 2018, 9, 1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allardyce, C.S.; Dyson, P.J. Ruthenium in medicine: Current clinical uses and future prospects. Platin. Metals Rev. 2001, 45, 62. [Google Scholar]
- Riccardi, C.; Musumeci, D.; Trifuoggi, M.; Irace, C.; Paduano, L.; Montesarchio, D. Anticancer Ruthenium(III) Complexes and Ru(III)-Containing Nanoformulations: An Update on the Mechanism of Action and Biological Activity. Pharmaceuticals 2019, 12, 146. [Google Scholar] [CrossRef] [Green Version]
- Schluga, P.; Hartinger, C.G.; Egger, A.; Reisner, E.; Galanski, M.; Jakupec, M.A.; Keppler, B.K. Redox behavior of tumor-inhibiting ruthenium(III) complexes and effects of physiological reductants on their binding to GMP. Dalton Trans. 2006, 14, 1796–1802. [Google Scholar] [CrossRef]
- Wiśniewska, J.; Fandzloch, M.; Łakomska, I. The reduction of ruthenium(III) complexes with triazolopyrimidine ligands by ascorbic acid and mechanistic insight into their action in anticancer therapy. Inorg. Chim. Acta 2019, 484, 305–310. [Google Scholar] [CrossRef]
- Blazevic, A.; Hummer, A.A.; Heffeter, P.; Berger, W.; Filipits, M.; Cibin, G.; Keppler, B.K.; Rompel, A. Electronic State of Sodium trans-[Tetrachloridobis (1 H-indazole) ruthenate (III)](NKP-1339) in Tumor, Liver and Kidney Tissue of a SW480-bearing Mouse. Sci. Rep. 2017, 7, 40966. [Google Scholar] [CrossRef] [PubMed]
- Beckford, F.; Dourth, D.; Shaloski, M.; Didion, J.; Thessing, J.; Woods, J.; Crowell, V.; Gerasimchuk, N.; Gonzalez-Sarrías, A.; Seeram, N.P. Half-sandwich ruthenium–arene complexes with thiosemicarbazones: Synthesis and biological evaluation of [(η6-p-cymene)Ru(piperonal thiosemicarbazones)Cl]Cl complexes. J. Inorg. Biochem. 2011, 105, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Beckford, F.A.; Thessing, J.; Shaloski, M.; Mbarushimana, P.C.; Brock, A.; Didion, J.; Woods, J.; Gonzalez-Sarrías, A.; Seeram, N.P. Synthesis and characterization of mixed-ligand diimine-piperonal thiosemicarbazone complexes of ruthenium(II): Biophysical investigations and biological evaluation as anticancer and antibacterial agents. J. Mol. Struct. 2011, 992, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battistin, F.; Scaletti, F.; Balducci, G.; Pillozzi, S.; Arcangeli, A.; Messori, L.; Alessio, E. Water-soluble Ru(II)- and Ru(III)-halide-PTA complexes (PTA=1,3,5-triaza-7-phosphaadamantane): Chemical and biological properties. J. Inorg. Biochem. 2016, 160, 180–188. [Google Scholar] [CrossRef]
- Camm, K.D.; El-Sokkary, A.; Gott, A.L.; Stockley, P.G.; Belyaeva, T.; McGowan, P.C. Synthesis, molecular structure and evaluation of new organometallic ruthenium anticancer agents. Dalton Trans. 2009, 48, 10914–10925. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.; Westhorpe, A.; Romero, M.J.; Habtemariam, A.; Gallevo, C.R.; Bark, Y.; Menezes, N.; Sadler, P.J.; Sharma, R.A. Radiosensitisation of human colorectal cancer cells by ruthenium(II) arene anticancer complexes. Sci. Rep. 2016, 6, 20596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Gallardo, J.; Elie, B.T.; Sanaú, M.; Contel, M. Versatile synthesis of cationic N-heterocyclic carbene–Gold(I) complexes containing a second ancillary ligand. Design of heterobimetallic ruthenium–gold anticancer agents. Chem. Commun. 2016, 52, 3155–3158. [Google Scholar] [CrossRef] [Green Version]
- Dabiri, Y.; Schmid, A.; Theobald, J.; Blagojevic, B.; Streciwilk, W.; Ott, I.; Wölfl, S.; Cheng, X. A Ruthenium(II) N-Heterocyclic Carbene (NHC) Complex with Naphthalimide Ligand Triggers Apoptosis in Colorectal Cancer Cells via Activating the ROS-p38 MAPK Pathway. Int. J. Mol. Sci. 2018, 19, 3964. [Google Scholar] [CrossRef] [Green Version]
- Baliza, I.R.S.; Silva, S.L.R.; Santos, L.D.S.; Neto, J.H.A.; Dias, R.B.; Sales, C.B.S.; Rocha, C.A.G.; Soares, M.B.P.; Batista, A.A.; Bezerra, D.P. Ruthenium Complexes With Piplartine Cause Apoptosis Through MAPK Signaling by a p53-Dependent Pathway in Human Colon Carcinoma Cells and Inhibit Tumor Development in a Xenograft Model. Front. Oncol. 2019, 9, 582. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.L.R.; Baliza, I.R.S.; Dias, R.B.; Sales, C.B.S.; Rocha, C.A.G.; Soares, M.B.P.; Correa, R.S.; Batista, A.A.; Bezerra, D.P. Ru(II)-thymine complex causes DNA damage and apoptotic cell death in human colon carcinoma HCT116 cells mediated by JNK/p38/ERK1/2 via a p53-independent signaling. Sci. Rep. 2019, 9, 11094. [Google Scholar] [CrossRef] [Green Version]
- Jin, G.; Zhao, Z.; Chakraborty, T.; Mandal, A.; Roy, A.; Roy, S.; Guo, Z. Decrypting the Molecular Mechanistic Pathways Delineating the Chemotherapeutic Potential of Ruthenium-Phloretin Complex in Colon Carcinoma Correlated with the Oxidative Status and Increased Apoptotic Events. Oxid. Med. Cell. Longev. 2020, 2020, 7690845. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Das, R.; Ghosh, B.; Chakraborty, T. Deciphering the biochemical and molecular mechanism underlying the in vitro and in vivo chemotherapeutic efficacy of ruthenium quercetin complex in colon cancer. Mol. Carcinog. 2018, 57, 700–721. [Google Scholar] [CrossRef]
- Wang, Y.; Bian, L.; Chakraborty, T.; Ghosh, T.; Chanda, P.; Roy, S. Construing the Biochemical and Molecular Mechanism Underlying the in Vivo and in Vitro Chemotherapeutic Efficacy of Ruthenium-Baicalein Complex in Colon Cancer. Int. J. Biol. Sci. 2019, 15, 1052–1071. [Google Scholar] [CrossRef] [Green Version]
- Hayward, R.L.; Schornagel, Q.C.; Tente, R.; Macpherson, J.S.; Aird, R.E.; Guichard, S.; Habtemariam, A.; Sadler, P.; Jodrell, D.I. Investigation of the role of Bax, p21/Waf1 and p53 as determinants of cellular responses in HCT116 colorectal cancer cells exposed to the novel cytotoxic ruthenium(II) organometallic agent, RM175. Cancer Chemother. Pharmacol. 2005, 55, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Jeong, Y.J.; Jo, J.-H.; Kang, S.C.; Lah, M.S.; Chi, K.-W. Anticancer Potency Studies of Coordination Driven Self-Assembled Arene–Ru-Based Metalla-Bowls. ChemBioChem 2014, 15, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Vidimar, V.; Licona, C.; Cerón-Camacho, R.; Guerin, E.; Coliat, P.; Venkatasamy, A.; Ali, M.; Guenot, D.; Le Lagadec, R.; Jung, A.C.; et al. A redox ruthenium compound directly targets PHD2 and inhibits the HIF1 pathway to reduce tumor angiogenesis independently of p53. Cancer Lett. 2019, 440–441, 145–155. [Google Scholar] [CrossRef]
- Pelillo, C.; Mollica, H.; Eble, J.A.; Grosche, J.; Herzog, L.; Codan, B.; Sava, G.; Bergamo, A. Inhibition of adhesion, migration and of α5β1 integrin in the HCT-116 colorectal cancer cells treated with the ruthenium drug NAMI-A. J. Inorg. Biochem. 2016, 160, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Flocke, L.S.; Trondl, R.; Jakupec, M.A.; Keppler, B.K. Molecular mode of action of NKP-1339—A clinically investigated ruthenium-based drug—Involves ER-and ROS-related effects in colon carcinoma cell lines. Investig. New Drugs 2016, 34, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonfili, L.; Pettinari, R.; Cuccioloni, M.; Cecarini, V.; Mozzicafreddo, M.; Angeletti, M.; Lupidi, G.; Marchetti, F.; Pettinari, C.; Eleuteri, A.M. Arene—RuII Complexes of Curcumin Exert Antitumor Activity via Proteasome Inhibition and Apoptosis Induction. ChemMedChem 2012, 7, 2010–2020. [Google Scholar] [CrossRef]
- Xu, Z.; Huang, J.; Kong, D.; Yang, Y.; Guo, L.; Jia, X.; Zhong, G.; Liu, Z. Potent half-sandwich Ru(Ⅱ) N^N (aryl-BIAN) complexes: Lysosome-mediated apoptosis, in vitro and in vivo anticancer activities. Eur. J. Med. Chem. 2020, 207, 112763. [Google Scholar] [CrossRef]
- Fong, J.; Kasimova, K.; Arenas, Y.; Kaspler, P.; Lazic, S.; Mandel, A.; Lilge, L. A novel class of ruthenium-based photosensitizers effectively kills in vitro cancer cells and in vivo tumors. Photochem. Photobiol. Sci. 2015, 14, 2014–2023. [Google Scholar] [CrossRef]
- Cho, Y.-H.; Ro, E.J.; Yoon, J.-S.; Mizutani, T.; Kang, D.-W.; Park, J.-C.; Il Kim, T.; Clevers, H.; Choi, K.-Y. 5-FU promotes stemness of colorectal cancer via p53-mediated WNT/β-catenin pathway activation. Nat. Commun. 2020, 11, 5321. [Google Scholar] [CrossRef]
- Su, W.; Qian, Q.; Li, P.; Lei, X.; Xiao, Q.; Huang, S.; Huang, C.; Cui, J. Synthesis, Characterization, and Anticancer Activity of a Series of Ketone-N4-Substituted Thiosemicarbazones and Their Ruthenium(II) Arene Complexes. Inorg. Chem. 2013, 52, 12440–12449. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, P.; Yu, B.; Chen, Y.; Wang, J.; Ji, L.; Chao, H. Targeting Nucleus DNA with a Cyclometalated Dipyridophenazineruthenium(II) Complex. J. Med. Chem. 2014, 57, 8971–8983. [Google Scholar] [CrossRef] [PubMed]
- Coury, J.E.; McFail-Isom, L.; Williams, L.D.; Bottomley, L.A. A novel assay for drug-DNA binding mode, affinity, and exclusion number: Scanning force microscopy. Proc. Natl. Acad. Sci. USA 1996, 93, 12283–12286. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Liang, Y.; Huang, C.; Su, W.; Lei, X.; Liu, Y.; Xiao, Q. Systematical investigation of binding interaction between novel ruthenium(II) arene complex with curcumin analogs and ctDNA. Luminescence 2016, 31, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-K.; Parkinson, J.A.; Bella, J.; Wang, F.; Sadler, P.J. Penetrative DNA intercalation and G-base selectivity of an organometallic tetrahydroanthracene RuII anticancer complex. Chem. Sci. 2010, 1, 258–270. [Google Scholar] [CrossRef] [Green Version]
- Martinez, R.; Chacon-Garcia, L. The Search of DNA-Intercalators as Antitumoral Drugs: What it Worked and What did not Work. Curr. Med. Chem. 2005, 12, 127–151. [Google Scholar] [CrossRef]
- Liu, S.; Liang, A.; Wu, K.; Zeng, W.; Luo, Q.; Wang, F. Binding of Organometallic Ruthenium Anticancer Complexes to DNA: Thermodynamic Base and Sequence Selectivity. Int. J. Mol. Sci. 2018, 19, 2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bešker, N.; Coletti, C.; Marrone, A.; Re, N. Binding of antitumor ruthenium complexes to DNA and proteins: A theoretical approach. J. Phys. Chem. B 2007, 111, 9955–9964. [Google Scholar] [CrossRef]
- Das, D.; Dutta, A.; Mondal, P. Interaction of aquated form of ruthenium (III) anticancer complexes with normal and mismatch base pairs: A density functional theoretical study. Comput. Theor. Chem. 2015, 1072, 28–36. [Google Scholar] [CrossRef]
- Zeng, W.; Zhang, Y.; Zheng, W.; Luo, Q.; Han, J.; Liu, J.a.; Zhao, Y.; Jia, F.; Wu, K.; Wang, F. Discovery of cisplatin binding to thymine and cytosine on a single-stranded oligodeoxynucleotide by high resolution FT-ICR mass spectrometry. Molecules 2019, 24, 1852. [Google Scholar] [CrossRef] [Green Version]
- Faivre, S.; Chan, D.; Salinas, R.; Woynarowska, B.; Woynarowski, J.M. DNA strand breaks and apoptosis induced by oxaliplatin in cancer cells. Biochem. Pharmacol. 2003, 66, 225–237. [Google Scholar] [CrossRef]
- Ray, B.; Gupta, B.; Mehrotra, R. Binding of platinum derivative, oxaliplatin to deoxyribonucleic acid: Structural insight into antitumor action. J. Biomol. Struct. Dyn. 2019, 37, 3838–3847. [Google Scholar] [CrossRef]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anti-Cancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Zaniboni, A.; Labianca, R.; Pancera, G.; Barni, S.; Frontini, L.; Marini, G.; Luporini, G. Oral Etoposide as Second-Line Chemotherapy for Colorectal Cancer: A GISCAD Study. J. Chemother. 1995, 7, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Seminara, P.; Pastore, C.; Iascone, C.; Cicconetti, F.; Nigita, G.; Ielapi, T.; Franchi, F. Mitomycin C and Etoposide in Advanced Colorectal Carcinoma. Chemotheraphy 2007, 53, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Passalacqua, R.; Bisagni, G.; Cocconi, G.; Boni, C.; Di Blasio, B.; Ceci, G. Cisplatin and etoposide in advanced colorectal carcinoma. Ann. Oncol. 1991, 2, 687–688. [Google Scholar] [CrossRef]
- Zhou, G.; Yang, J.; Song, P. Correlation of ERK/MAPK signaling pathway with proliferation and apoptosis of colon cancer cells. Oncol. Lett. 2019, 17, 2266–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhou, L.; Bao, Y.L.; Wu, Y.; Yu, C.L.; Huang, Y.X.; Sun, Y.; Zheng, L.H.; Li, Y.X. Butyrate induces cell apoptosis through activation of JNK MAP kinase pathway in human colon cancer RKO cells. Chem.-Biol. Interact. 2010, 185, 174–181. [Google Scholar] [CrossRef]
- Shin, D.Y.; Lee, W.S.; Lu, J.N.; Kang, M.H.; Ryu, C.H.; Kim, G.Y.; Kang, H.S.; Shin, S.C.; Choi, Y.H. Induction of apoptosis in human colon cancer HCT-116 cells by anthocyanins through suppression of Akt and activation of p38-MAPK. Int. J. Oncol. 2009, 35, 1499–1504. [Google Scholar]
- Zilfou, J.T.; Lowe, S.W. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Med. 2009, 1, a001883. [Google Scholar] [CrossRef] [PubMed]
- Florindo, P.R.; Pereira, D.M.; Borralho, P.M.; Rodrigues, C.M.P.; Piedade, M.F.M.; Fernandes, A.C. Cyclopentadienyl–Ruthenium(II) and Iron(II) Organometallic Compounds with Carbohydrate Derivative Ligands as Good Colorectal Anticancer Agents. J. Med. Chem. 2015, 58, 4339–4347. [Google Scholar] [CrossRef] [PubMed]
- Ude, Z.; Romero-Canelón, I.; Twamley, B.; Fitzgerald Hughes, D.; Sadler, P.J.; Marmion, C.J. A novel dual-functioning ruthenium(II)–arene complex of an anti-microbial ciprofloxacin derivative—Anti-proliferative and anti-microbial activity. J. Inorg. Biochem. 2016, 160, 210–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, M.J.; Licona, C.; Yuan Qiang Wong, D.; Pastorin, G.; Gaiddon, C.; Ang, W.H. Discovery and Investigation of Anticancer Ruthenium–Arene Schiff-Base Complexes via Water-Promoted Combinatorial Three-Component Assembly. J. Med. Chem. 2014, 57, 6043–6059. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Sen, U.; Roy, N.; Muthukumar, V.; Sahoo, S.K.; Bose, B.; Paira, P. DNA targeting half sandwich Ru(II)-p-cymene-N^N complexes as cancer cell imaging and terminating agents: Influence of regioisomers in cytotoxicity. Dalton Trans. 2021, 50, 979–997. [Google Scholar] [CrossRef] [PubMed]
- Kapitza, S.; Jakupec, M.A.; Uhl, M.; Keppler, B.K.; Marian, B. The heterocyclic ruthenium(III) complex KP1019 (FFC14A) causes DNA damage and oxidative stress in colorectal tumor cells. Cancer Lett. 2005, 226, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Wernitznig, D.; Kiakos, K.; Del Favero, G.; Harrer, N.; Machat, H.; Osswald, A.; Jakupec, M.A.; Wernitznig, A.; Sommergruber, W.; Keppler, B.K. First-in-class ruthenium anticancer drug (KP1339/IT-139) induces an immunogenic cell death signature in colorectal spheroids in vitro. Metallomics 2019, 11, 1044–1048. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, J.-S.; Lin, S.-R.; Chang, M.-Y.; Chen, F.-M.; Lu, C.-Y.; Huang, T.-J.; Huang, Y.-S.; Huang, C.-J.; Wang, J.-Y. APC, K-ras, and p53 Gene Mutations in Colorectal Cancer Patients: Correlation to Clinicopathologic Features and Postoperative Surveillance. Am. Surg. 2005, 71, 336–343. [Google Scholar] [CrossRef]
- Nakayama, M.; Oshima, M. Mutant p53 in colon cancer. J. Mol. Cell Biol. 2018, 11, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Fodde, R. The APC gene in colorectal cancer. Eur. J. Cancer 2002, 38, 867–871. [Google Scholar] [CrossRef]
- Kwong, L.N.; Dove, W.F. APC and its modifiers in colon cancer. Adv. Exp. Med. Biol. 2009, 656, 85–106. [Google Scholar] [PubMed] [Green Version]
- Kaeser, M.D.; Pebernard, S.; Iggo, R.D. Regulation of p53 Stability and Function in HCT116 Colon Cancer Cells. J. Biol. Chem. 2004, 279, 7598–7605. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Lovejoy, K.S.; Shima, J.E.; Lagpacan, L.L.; Shu, Y.; Lapuk, A.; Chen, Y.; Komori, T.; Gray, J.W.; Chen, X.; et al. Organic Cation Transporters Are Determinants of Oxaliplatin Cytotoxicity. Cancer Res. 2006, 66, 8847–8857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landis-Piwowar, K.R.; Milacic, V.; Chen, D.; Yang, H.; Zhao, Y.; Chan, T.H.; Yan, B.; Dou, Q.P. The proteasome as a potential target for novel anticancer drugs and chemosensitizers. Drug Resist. Updates 2006, 9, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Mani, A.; Gelmann, E.P. The ubiquitin-proteasome pathway and its role in cancer. J. Clin. Oncol. 2005, 23, 4776–4789. [Google Scholar] [CrossRef]
- Richardson, P.G.; Mitsiades, C.; Hideshima, T.; Anderson, K.C. Proteasome Inhibition in the Treatment of Cancer. Cell Cycle 2005, 4, 289–295. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Richardson, P.G.; Hideshima, T.; Anderson, K.C. Proteasome Inhibition As a Novel Therapeutic Target in Human Cancer. J. Clin. Oncol. 2005, 23, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Milacic, V.; Banerjee, S.; Landis-Piwowar, K.R.; Sarkar, F.H.; Majumdar, A.P.N.; Dou, Q.P. Milacic, V.; Banerjee, S.; Landis-Piwowar, K.R.; Sarkar, F.H.; et al. Curcumin inhibits the proteasome activity in human colon cancer cells in vitro and in vivo. Cancer Res. 2008, 68, 7283–7292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.B.; Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [Green Version]
- Arnold, J.; Grune, T. PARP-mediated proteasome activation: A co-ordination of DNA repair and protein degradation? BioEssays 2002, 24, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Kotsafti, A.; Scarpa, M.; Castagliuolo, I.; Scarpa, M. Reactive Oxygen Species and Antitumor Immunity—From Surveillance to Evasion. Cancers 2020, 12, 1748. [Google Scholar] [CrossRef]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updates 2004, 7, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Sreevalsan, S.; Safe, S. Reactive Oxygen Species and Colorectal Cancer. Curr. Colorectal Cancer Rep. 2013, 9, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, A New Redox-Active Anticancer Agent—Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Zorbas-Seifried, S.; Jakupec, M.A.; Kynast, B.; Zorbas, H.; Keppler, B.K. From bench to bedside—Preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891–904. [Google Scholar] [CrossRef]
- Peti, W.; Pieper, T.; Sommer, M.; Keppler, B.K.; Giester, G. Synthesis of Tumor-Inhibiting Complex Salts Containing the Anion trans-Tetrachlorobis(indazole)ruthenate(III) and Crystal Structure of the Tetraphenylphosphonium Salt. Eur. J. Inorg. Chem. 1999, 1999, 1551–1555. [Google Scholar] [CrossRef]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Wernitznig, D.; Favero, G.D.; Kiakos, K.; Harrer, N.; Machat, H.; Marko, D.; Jakupec, M.; Sommergruber, W.; Keppler, B.K. KP-1339 (IT-139) induces the hallmarks of immunogenic cell death in a colon cancer 3D model in vitro. Cancer Res. 2018, 78, 4395. [Google Scholar]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Kepp, O.; Menger, L.; Vacchelli, E.; Locher, C.; Adjemian, S.; Yamazaki, T.; Martins, I.; Sukkurwala, A.Q.; Michaud, M.; Senovilla, L.; et al. Crosstalk between ER stress and immunogenic cell death. Cytokine Growth Factor Rev. 2013, 24, 311–318. [Google Scholar] [CrossRef]
- Osman, R.; Tacnet-Delorme, P.; Kleman, J.-P.; Millet, A.; Frachet, P. Calreticulin Release at an Early Stage of Death Modulates the Clearance by Macrophages of Apoptotic Cells. Front. Immunol. 2017, 8, 1034. [Google Scholar] [CrossRef] [Green Version]
- Dudek, A.M.; Garg, A.D.; Krysko, D.V.; De Ruysscher, D.; Agostinis, P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 2013, 24, 319–333. [Google Scholar] [CrossRef]
- Ruan, H.; Leibowitz, B.J.; Zhang, L.; Yu, J. Immunogenic cell death in colon cancer prevention and therapy. Mol. Carcinog. 2020, 59, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhao, L.; Loos, F.; Iribarren, K.; Lachkar, S.; Zhou, H.; Gomes-da-Silva, L.C.; Chen, G.; Bezu, L.; Boncompain, G.; et al. Identification of pharmacological agents that induce HMGB1 release. Sci. Rep. 2017, 7, 14915. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, P.B.; Edes, K.; Nelson, C.C.; Parsawar, K.; Fitzpatrick, F.A.; Moos, P.J. Thioredoxin reductase is required for the inactivation of tumor suppressor p53 and for apoptosis induced by endogenous electrophiles. Carcinogenesis 2006, 27, 2538–2549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, S.; Müller-Ladner, U.; Neumann, E.; Spöttl, T.; Schlottmann, K.; Rüschoff, J.; Schölmerich, J.; Kullmann, F. Thioredoxin Reductase 1 Expression in Colon Cancer: Discrepancy between in Vitro and in Vivo Findings. Lab. Investig. 2003, 83, 1321–1331. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.-J.; Geng, W.-S.; Wang, Z.-Q.; Chen, L.; Zeng, X.-S. The role of thioredoxin system in cancer: Strategy for cancer therapy. Cancer Chemother. Pharmacol. 2019, 84, 453–470. [Google Scholar] [CrossRef]
- Burkitt, K.; Chun, S.Y.; Dang, D.T.; Dang, L.H. Targeting both HIF-1 and HIF-2 in human colon cancer cells improves tumor response to sunitinib treatment. Mol. Cancer Ther. 2009, 8, 1148–1156. [Google Scholar] [CrossRef] [Green Version]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharmacol. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Imamura, T.; Kikuchi, H.; Herraiz, M.-T.; Park, D.-Y.; Mizukami, Y.; Mino-Kenduson, M.; Lynch, M.P.; Rueda, B.R.; Benita, Y.; Xavier, R.J.; et al. HIF-1α and HIF-2α have divergent roles in colon cancer. Int. J. Cancer 2009, 124, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Sadlecki, P.; Bodnar, M.; Grabiec, M.; Marszalek, A.; Walentowicz, P.; Sokup, A.; Zegarska, J.; Walentowicz-Sadlecka, M. The role of Hypoxia-inducible factor-1α, glucose transporter-1,(GLUT-1) and carbon anhydrase IX in endometrial cancer patients. Biomed. Res. Int. 2014, 2014, 616850. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Jang, W.-C.; Fung, F.K.C.; Lo, A.C.Y.; Wong, I.Y.H. Up-Regulation of ENO1 by HIF-1α in Retinal Pigment Epithelial Cells after Hypoxic Challenge Is Not Involved in the Regulation of VEGF Secretion. PLoS ONE 2016, 11, e0147961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, W.; Cui, G.; Tang, C.-W.; Zhang, X.-L.; Dai, C.; Xu, Y.-Q.; Gong, H.; Xue, T.; Guo, H.-H.; Bao, Y. Role of glucose metabolism related gene GLUT1 in the occurrence and prognosis of colorectal cancer. Oncotarget 2017, 8, 56850–56857. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Wang, Y.; Zhao, S.; Liu, C.; Wang, Y.; Wen, M.; Mao, J.-H.; Wei, G.; Zhang, P. FBXW7 negatively regulates ENO1 expression and function in colorectal cancer. Lab. Investig. 2015, 95, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Takahashi, Y.; Liu, W.; Shaheen, R.M. Vascular Endothelial Growth Factor in Human Colon Cancer: Biology and Therapeutic Implications. Oncologist 2000, 5, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-Y.; He, Y.-L.; Zhu, L.-L. Possible Role of PHD Inhibitors as Hypoxia-Mimicking Agents in the Maintenance of Neural Stem Cells’ Self-Renewal Properties. Front. Cell Dev. Biol. 2018, 6, 169. [Google Scholar] [CrossRef]
- Leyva, L.; Sirlin, C.; Rubio, L.; Franco, C.; Le Lagadec, R.; Spencer, J.; Bischoff, P.; Gaiddon, C.; Loeffler, J.-P.; Pfeffer, M. Synthesis of Cycloruthenated Compounds as Potential Anticancer Agents. Eur. J. Inorg. Chem. 2007, 2007, 3055–3066. [Google Scholar] [CrossRef]
- Alessio, E.; Balducci, G.; Calligaris, M.; Costa, G.; Attia, W.M.; Mestroni, G. Synthesis, molecular structure, and chemical behavior of hydrogen trans-bis(dimethyl sulfoxide)tetrachlororuthenate(III) and mer-trichlorotris(dimethyl sulfoxide)ruthenium(III): The first fully characterized chloride-dimethyl sulfoxide-ruthenium(III) complexes. Inorg. Chem. 1991, 30, 609–618. [Google Scholar]
- Sava, G.; Zorzet, S.; Turrin, C.; Vita, F.; Soranzo, M.; Zabucchi, G.; Cocchietto, M.; Bergamo, A.; DiGiovine, S.; Pezzoni, G.; et al. Dual Action of NAMI-A in inhibition of solid tumor metastasis: Selective targeting of metastatic cells and binding to collagen. Clin. Cancer Res. 2003, 9, 1898–1905. [Google Scholar]
- Gava, B.; Zorzet, S.; Spessotto, P.; Cocchietto, M.; Sava, G. Inhibition of B16 melanoma metastases with the ruthenium complex imidazolium trans-imidazoledimethylsulfoxide-tetrachlororuthenate and down-regulation of tumor cell invasion. J. Pharmacol. Exp. Ther. 2006, 317, 284–291. [Google Scholar] [CrossRef]
- Zorzet, S.; Bergamo, A.; Cocchietto, M.; Sorc, A.; Gava, B.; Alessio, E.; Iengo, E.; Sava, G. Lack of In vitro cytotoxicity, associated to increased G(2)-M cell fraction and inhibition of matrigel invasion, may predict IN vivo-selective antimetastasis activity of ruthenium complexes. J. Pharmacol. Exp. Ther. 2000, 295, 927–933. [Google Scholar]
- Alessio, E. Thirty Years of the Drug Candidate NAMI-A and the Myths in the Field of Ruthenium Anticancer Compounds: A Personal Perspective. Eur. J. Inorg. Chem. 2017, 2017, 1549–1560. [Google Scholar] [CrossRef]
- Janouskova, H.; Ray, A.-M.; Noulet, F.; Lelong-Rebel, I.; Choulier, L.; Schaffner, F.; Lehmann, M.; Martin, S.; Teisinger, J.; Dontenwill, M. Activation of p53 pathway by Nutlin-3a inhibits the expression of the therapeutic target α5 integrin in colon cancer cells. Cancer Lett. 2013, 336, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Pelillo, C.; Chambery, A.; Sava, G. Influence of components of tumour microenvironment on the response of HCT-116 colorectal cancer to the ruthenium-based drug NAMI-A. J. Inorg. Biochem. 2017, 168, 90–97. [Google Scholar] [CrossRef] [Green Version]
- Pelillo, C.; Bergamo, A.; Mollica, H.; Bestagno, M.; Sava, G. Colorectal Cancer Metastases Settle in the Hepatic Microenvironment Through α5β1 Integrin. J. Cell. Biochem. 2015, 116, 2385–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Hochwald, S.N. The role of FAK in tumor metabolism and therapy. Pharmacol. Ther. 2014, 142, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Tian, Z.; Xu, Z.; Zhang, S.; Feng, Y.; Zhang, L.; Liu, Z. Highly potent half-sandwich iridium and ruthenium complexes as lysosome-targeted imaging and anticancer agents. Dalton Trans. 2018, 47, 15772–15782. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Li, J.; Zhang, S.; Xu, Z.; Yang, Y.; Kong, D.; Zhang, H.; Ge, X.; Zhang, J.; Liu, Z. Lysosome-Targeted Chemotherapeutics: Half-Sandwich Ruthenium(II) Complexes That Are Selectively Toxic to Cancer Cells. Inorg. Chem. 2018, 57, 10498–10502. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; Jäättelä, M. Lysosomal involvement in cell death and cancer. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 746–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piao, S.; Amaravadi, R.K. Targeting the lysosome in cancer. Ann. N. Y. Acad. Sci. 2016, 1371, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Dielschneider, R.F.; Henson, E.S.; Gibson, S.B. Lysosomes as Oxidative Targets for Cancer Therapy. Oxid. Med. Cell. Longev. 2017, 2017, 3749157. [Google Scholar] [CrossRef] [Green Version]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Niloy, M.S.; Shakil, M.S.; Hossen, M.S.; Alam, M.; Rosengren, R.J. Promise of gold nanomaterials as a lung cancer theranostic agent: A systematic review. Int. Nano Lett. 2021, 11, 93–111. [Google Scholar] [CrossRef]
- Monro, S.; Colón, K.L.; Yin, H.; Roque, J.; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2019, 119, 797–828. [Google Scholar] [CrossRef]
- Levina, A.; Mitra, A.; Lay, P.A. Recent developments in ruthenium anticancer drugs. Metallomics 2009, 1, 458–470. [Google Scholar] [CrossRef]
- Zhou, Q.-X.; Lei, W.-H.; Sun, Y.; Chen, J.-R.; Li, C.; Hou, Y.-J.; Wang, X.-S.; Zhang, B.-W. [Ru(bpy)3−n(dpb)n]2+: Unusual Photophysical Property and Efficient DNA Photocleavage Activity. Inorg. Chem. 2010, 49, 4729–4731. [Google Scholar] [CrossRef]
- Doherty, R.E.; Sazanovich, I.V.; McKenzie, L.K.; Stasheuski, A.S.; Coyle, R.; Baggaley, E.; Bottomley, S.; Weinstein, J.A.; Bryant, H.E. Photodynamic killing of cancer cells by a Platinum(II) complex with cyclometallating ligand. Sci. Rep. 2016, 6, 22668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, S.A.; Farrell, D.; Grodzinski, P. Nanotechnologies in Cancer Treatment and Diagnosis. J. Natl. Compr. Cancer Netw. 2014, 12, 1727–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampado, R.; Crotti, S.; Caliceti, P.; Pucciarelli, S.; Agostini, M. Nanovectors Design for Theranostic Applications in Colorectal Cancer. J. Oncol. 2019, 2019, 2740923. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Xu, L.; Song, G.; Liu, Z. Emerging nanomedicine approaches fighting tumor metastasis: Animal models, metastasis-targeted drug delivery, phototherapy, and immunotherapy. Chem. Soc. Rev. 2016, 45, 6250–6269. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Song, G.; Liang, C.; Yi, X.; Zhao, Q.; Cheng, L.; Yang, K.; Liu, Z. Perfluorocarbon-loaded hollow Bi2Se3 nanoparticles for timely supply of oxygen under near-infrared light to enhance the radiotherapy of cancer. Adv. Mater. 2016, 28, 2716–2723. [Google Scholar] [CrossRef]
- Heffeter, P.; Riabtseva, A.; Senkiv, Y.; Kowol, C.R.; Körner, W.; Jungwith, U.; Mitina, N.; Keppler, B.K.; Konstantinova, T.; Yanchuk, I.; et al. Nanoformulation Improves Activity of the (pre)Clinical Anticancer Ruthenium Complex KP1019. J. Biomed. Nanotechnol. 2014, 10, 877–884. [Google Scholar] [CrossRef]
- Liu, H.-J.; Luan, X.; Feng, H.-Y.; Dong, X.; Yang, S.-C.; Chen, Z.-J.; Cai, Q.-Y.; Lu, Q.; Zhang, Y.; Sun, P.; et al. Integrated Combination Treatment Using a “Smart” Chemotherapy and MicroRNA Delivery System Improves Outcomes in an Orthotopic Colorectal Cancer Model. Adv. Funct. Mater. 2018, 28, 1801118. [Google Scholar] [CrossRef]
- Tomlinson, J.S.; Jarnagin, W.R.; DeMatteo, R.P.; Fong, Y.; Kornprat, P.; Gonen, M.; Kemeny, N.; Brennan, M.F.; Blumgart, L.H.; D’Angelica, M. Actual 10-year survival after resection of colorectal liver metastases defines cure. J. Clin. Oncol. 2007, 25, 4575–4580. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Cheng, L.; Gong, F.; Dong, Z.; Liang, C.; Wang, M.; Feng, L.; Li, Y.; Liu, Z.; Li, C.; et al. Platinum nanoworms for imaging-guided combined cancer therapy in the second near-infrared window. J. Mater. Chem. B 2018, 6, 5069–5079. [Google Scholar] [CrossRef]
- Zeng, X.; Sun, J.; Li, S.; Shi, J.; Gao, H.; Sun Leong, W.; Wu, Y.; Li, M.; Liu, C.; Li, P.; et al. Blood-triggered generation of platinum nanoparticle functions as an anti-cancer agent. Nat. Commun. 2020, 11, 567. [Google Scholar] [CrossRef] [Green Version]
- Gehrke, H.; Pelka, J.; Hartinger, C.G.; Blank, H.; Bleimund, F.; Schneider, R.; Gerthsen, D.; Bräse, S.; Crone, M.; Türk, M.; et al. Platinum nanoparticles and their cellular uptake and DNA platination at non-cytotoxic concentrations. Arch. Toxicol. 2011, 85, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Porcel, E.; Liehn, S.; Remita, H.; Usami, N.; Kobayashi, K.; Furusawa, Y.; Le Sech, C.; Lacombe, S. Platinum nanoparticles: A promising material for future cancer therapy? Nanotechnology 2010, 21, 85103. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Dang, M.; Tao, J.; Li, Y.; Tang, Y. Mesoporous platinum nanoparticle-based nanoplatforms for combined chemo-photothermal breast cancer therapy. J. Colloid Interface Sci. 2020, 570, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Shieh, M.-J. Platinum(II) Drug-Loaded Gold Nanoshells for Chemo-Photothermal Therapy in Colorectal Cancer. ACS Appl. Mater. Interfaces 2020, 12, 4254–4264. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Lee, J.J.; Siu, L.L. Dose escalation methods in phase I cancer clinical trials. J. Natl. Cancer Inst. 2009, 101, 708–720. [Google Scholar] [CrossRef] [Green Version]
- Rademaker-Lakhai, J.M.; van den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H.M.; Rademaker-Lakhai, J.M.; Van Den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H. A phase I and pharmacological study with imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a novel ruthenium anticancer agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef] [Green Version]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Investig. New Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef] [Green Version]
- Lentz, F.; Drescher, A.; Lindauer, A.; Henke, M.; Hilger, R.A.; Hartinger, C.G.; Scheulen, M.E.; Dittrich, C.; Keppler, B.K.; Jaehde, U. Pharmacokinetics of a novel anticancer ruthenium complex (KP1019, FFC14A) in a phase I dose-escalation study. Anticancer Drugs 2009, 20, 97–103. [Google Scholar] [CrossRef]
- Thompson, D.S.; Weiss, G.J.; Jones, S.F.; Burris, H.A.; Ramanathan, R.K.; Infante, J.R.; Bendell, J.C.; Ogden, A.; Von Hoff, D.D. NKP-1339: Maximum tolerated dose defined for first-in-human GRP78 targeted agent. J. Clin. Oncol. 2012, 30, 3033. [Google Scholar] [CrossRef]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef] [Green Version]
- clinicaltrial.gov. Available online: https://clinicaltrials.gov (accessed on 17 July 2021).
- Burris, H.A.; Bakewell, S.; Bendell, J.C.; Infante, J.; Jones, S.F.; Spigel, D.R.; Weiss, G.J.; Ramanathan, R.K.; Ogden, A.; Von Hoff, D. Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: A first-in-human, open-label, dose-escalation phase I study with expansion cohort. ESMO Open 2016, 1, e000154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beale, P.; Judson, I.; O’Donnell, A.; Trigo, J.; Rees, C.; Raynaud, F.; Turner, A.; Simmons, L.; Etterley, L. A Phase I clinical and pharmacological study of cis-diamminedichloro(2-methylpyridine) platinum II (AMD473). Br. J. Cancer 2003, 88, 1128–1134. [Google Scholar] [CrossRef]
- Goldberg, R.M.; Sargent, D.J.; Morton, R.F.; Fuchs, C.S.; Ramanathan, R.K.; Williamson, S.K.; Findlay, B.P.; Pitot, H.C.; Alberts, S.R. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J. Clin. Oncol. 2004, 22, 23–30. [Google Scholar] [CrossRef]
- De Gramont, A.; Figer, A.; Seymour, M.; Homerin, M.; Hmissi, A.; Cassidy, J.; Boni, C.; Cortes-Funes, H.; Cervantes, A.; Freyer, G.; et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J. Clin. Oncol. 2000, 18, 2938–2947. [Google Scholar] [CrossRef]
- Cassidy, J.; Clarke, S.; Díaz-Rubio, E.; Scheithauer, W.; Figer, A.; Wong, R.; Koski, S.; Rittweger, K.; Gilberg, F.; Saltz, L. XELOX vs. FOLFOX-4 as first-line therapy for metastatic colorectal cancer: NO16966 updated results. Br. J. Cancer 2011, 105, 58–64. [Google Scholar] [CrossRef]
- Earhart, R.; Cheporov, S.; Gladkov, O.; Biakhov, M.; Breitz, H.; De Jager, R. Randomized phase II study of picoplatin in combination with 5-fluorouracil and leucovorin (FOLPI) as a neuropathy-sparing alternative to modified FOLFOX-6 as first-line therapy for colorectal cancer (CRC). J. Clin. Oncol. 2009, 27, 4026. [Google Scholar] [CrossRef]
- Hartmann, J.T.; Lipp, H.P. Toxicity of platinum compounds. Expert Opin. Pharmacother. 2003, 4, 889–901. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Park, G.Y.; Lippard, S. Understanding and improving platinum anticancer drugs–phenanthriplatin. Anticancer Res. 2014, 34, 471–476. [Google Scholar]
- Dos Santos, E.R.; Graminha, A.E.; Schultz, M.S.; Correia, I.; Selistre-de-Araújo, H.S.; Correa, R.S.; Ellena, J.; Elisângela de Paula, S.L.; Pessoa, J.C.; Batista, A.A. Cytotoxic activity and structural features of Ru(II)/phosphine/amino acid complexes. J. Inorg. Biochem. 2018, 182, 48–60. [Google Scholar] [CrossRef]
- Mello-Andrade, F.; Cardoso, C.G.; e Silva, C.R.; Chen-Chen, L.; de Melo-Reis, P.R.; de Lima, A.P.; Oliveira, R.; Ferraz, I.B.M.; Grisolia, C.K.; Almeida, M.A.P. Acute toxic effects of ruthenium (II)/amino acid/diphosphine complexes on Swiss mice and zebrafish embryos. Biomed. Pharmacother. 2018, 107, 1082–1092. [Google Scholar] [CrossRef]
- Bergamo, A.; Riedel, T.; Dyson, P.J.; Sava, G. Preclinical combination therapy of the investigational drug NAMI-A+ with doxorubicin for mammary cancer. Investig. New Drugs 2015, 33, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Gagliardi, R.; Scarcia, V.; Furlani, A.; Alessio, E.; Mestroni, G.; Sava, G. In vitro cell cycle arrest, in vivo action on solid metastasizing tumors, and host toxicity of the antimetastatic drug NAMI-A and cisplatin. J. Pharmacol. Exp. Ther. 1999, 289, 559–564. [Google Scholar] [PubMed]
- Koch, J.H.; Gyarfas, E.C.; Dwyer, F. Biological Activity of Complex Ions Mechanism of Inhibition of Acetylcholinesterase. Aust. J. Biol. Sci. 1956, 9, 371–381. [Google Scholar] [CrossRef]
- Fiskum, G.; Cockrell, R. Ruthenium red sensitive and insensitive calcium transport in rat liver and Ehrlich ascites tumor cell mitochondria. FEBS Lett. 1978, 92, 125–128. [Google Scholar] [CrossRef] [Green Version]
- Rahamimoff, R.; Alnaes, E. Inhibitory Action of Ruthenium Red on Neuromuscular Transmission. Proc. Natl. Acad. Sci. USA 1973, 70, 3613–3616. [Google Scholar] [CrossRef] [Green Version]
- Kruszyna, H.; Kruszyna, R.; Hurst, J.; Smith, R.P. Toxicology and pharmacology of some ruthenium compounds: Vascular smooth muscle relaxation by nitrosyl derivatives of ruthenium and iridium. J. Toxicol. Environ. Health 1980, 6, 757–773. [Google Scholar] [CrossRef]
- Shakil, M.S.; Parveen, S.; Rana, Z.; Walsh, F.; Movassaghi, S.; Söhnel, T.; Azam, M.; Shaheen, M.A.; Jamieson, S.M.F.; Hanif, M.; et al. High Antiproliferative Activity of Hydroxythiopyridones over Hydroxypyridones and Their Organoruthenium Complexes. Biomedicines 2021, 9, 123. [Google Scholar] [CrossRef]
Figure 1.
Chemical structure of some Pt-based drugs. Cisplatin, Oxaliplatin, Carboplatin, and Picoplatin have been used worldwide in cancer treatment. Besides, Nedaplatin, Lobaplatin, Heptaplatin, and Miriplatin have been using regionally. Among the Pt-based drugs, only Oxaliplatin is approved by FDA in CRC treatment.
Figure 1.
Chemical structure of some Pt-based drugs. Cisplatin, Oxaliplatin, Carboplatin, and Picoplatin have been used worldwide in cancer treatment. Besides, Nedaplatin, Lobaplatin, Heptaplatin, and Miriplatin have been using regionally. Among the Pt-based drugs, only Oxaliplatin is approved by FDA in CRC treatment.

Figure 2.
The ten most frequent mutations in colorectal cancer. This frequency distribution was calculated based on the cBioPortal data on 2322 CRC patients [46]. Adenomatous polyposis coli: APC, Tumor protein p53: p53, Kirsten rat sarcoma: KRAS, Phosphatidylinositol-4,5-Bisphosphate 3-Kinase catalytic subunit alpha: PIK3CA, Titin: TTN, SMAD family member 4: SMAD4, F-Box and WD repeat domain containing 7: FBXW7, B-Raf proto-oncogene: BRAF, Spectrin repeats containing nuclear envelope protein 1: SYNE1, Mucin 16: MUC16.
Figure 2.
The ten most frequent mutations in colorectal cancer. This frequency distribution was calculated based on the cBioPortal data on 2322 CRC patients [46]. Adenomatous polyposis coli: APC, Tumor protein p53: p53, Kirsten rat sarcoma: KRAS, Phosphatidylinositol-4,5-Bisphosphate 3-Kinase catalytic subunit alpha: PIK3CA, Titin: TTN, SMAD family member 4: SMAD4, F-Box and WD repeat domain containing 7: FBXW7, B-Raf proto-oncogene: BRAF, Spectrin repeats containing nuclear envelope protein 1: SYNE1, Mucin 16: MUC16.

Figure 3.
Summary of the suggested molecular mechanisms of some representative Ru-complexes. Different Ru-complexes interact with different cellular signaling pathways of colon cancer cells, thereby suppressing cell growth, proliferation, and migration. Some Ru-complexes can selectively accumulate into subcellular organelle in CRC cells and causes dysfunction leading to cell death. Tyrosine 397 focal adhesion kinase: Tyr-397 FAK; Reactive oxygen species: ROS; Glucose-regulated protein 78: GRP 78; Activating transcription factor 6: ATF6; Inositol-requiring protein 1α: IRE1α; PrKr-like ER kinase: PERK; Eukaryotic initiation factor2α: eIF2α; Calreticulin: CRT; C/eBP homologous protein: CHOP; Adenomatous polyposis coli: APC; Tumor protein P53: p53; B-cell lymphoma: Bcl-2; Bcl-2-associated X: BAX; c-Jun N-terminal kinases: JNK; Extracellular signal-regulated protein kinase 1 and 2: ERK1/2; Mitogen-activated protein kinases: MAPK; Receptor tyrosine kinases: RTK; Phosphatidylinositol 4,5-bisphosphate: PIP2; phosphatidylinositol (3,4,5)-trisphosphate: PIP3; Phosphatase and TENsin homolog deleted on chromosome 10: PTEN; Serine/threonine kinase 1: Akt1; Mammalian target of rapamycin: mTOR; Vascular endothelial growth factor: VEGF; Hypoxia-inducible factor-1 α: HIF-1α; Prolyl hydroxylase 2: PHD2; Hypoxia-responsive element: HRE; Glucose transporter 1: GLUT1; Alpha-enolase: ENO1.
Figure 3.
Summary of the suggested molecular mechanisms of some representative Ru-complexes. Different Ru-complexes interact with different cellular signaling pathways of colon cancer cells, thereby suppressing cell growth, proliferation, and migration. Some Ru-complexes can selectively accumulate into subcellular organelle in CRC cells and causes dysfunction leading to cell death. Tyrosine 397 focal adhesion kinase: Tyr-397 FAK; Reactive oxygen species: ROS; Glucose-regulated protein 78: GRP 78; Activating transcription factor 6: ATF6; Inositol-requiring protein 1α: IRE1α; PrKr-like ER kinase: PERK; Eukaryotic initiation factor2α: eIF2α; Calreticulin: CRT; C/eBP homologous protein: CHOP; Adenomatous polyposis coli: APC; Tumor protein P53: p53; B-cell lymphoma: Bcl-2; Bcl-2-associated X: BAX; c-Jun N-terminal kinases: JNK; Extracellular signal-regulated protein kinase 1 and 2: ERK1/2; Mitogen-activated protein kinases: MAPK; Receptor tyrosine kinases: RTK; Phosphatidylinositol 4,5-bisphosphate: PIP2; phosphatidylinositol (3,4,5)-trisphosphate: PIP3; Phosphatase and TENsin homolog deleted on chromosome 10: PTEN; Serine/threonine kinase 1: Akt1; Mammalian target of rapamycin: mTOR; Vascular endothelial growth factor: VEGF; Hypoxia-inducible factor-1 α: HIF-1α; Prolyl hydroxylase 2: PHD2; Hypoxia-responsive element: HRE; Glucose transporter 1: GLUT1; Alpha-enolase: ENO1.

Figure 5.
Chemical structures of some Ru-complexes that mediated CRC cell apoptosis through p53 dependent or independent way. Some Ru-complexes induced apoptosis of CRC cells via p53 dependent caspase-3 mediated signaling (4a–f), increasing APC and p53 gene expression (5a) and p53 independent activity (6a,b).
Figure 5.
Chemical structures of some Ru-complexes that mediated CRC cell apoptosis through p53 dependent or independent way. Some Ru-complexes induced apoptosis of CRC cells via p53 dependent caspase-3 mediated signaling (4a–f), increasing APC and p53 gene expression (5a) and p53 independent activity (6a,b).

Figure 7.
Photodynamic effects of Ru-complex in colorectal cancer and normal cells. (A) Chemical structure of 14a and 14b. (B) Ru-complexes having photosensitizing properties selectively kill CRC cells. Ru-complex with photosensitizing property can selectively enter cancer cells, and with the presence of light irradiation, O2 is converted into 1O2. 1O2 interact with DNA to mediate cell death via photocleavage activity. Tryptophan receptor: TfR, Oxygen: O2, Singlet oxygen: 1O2.
Figure 7.
Photodynamic effects of Ru-complex in colorectal cancer and normal cells. (A) Chemical structure of 14a and 14b. (B) Ru-complexes having photosensitizing properties selectively kill CRC cells. Ru-complex with photosensitizing property can selectively enter cancer cells, and with the presence of light irradiation, O2 is converted into 1O2. 1O2 interact with DNA to mediate cell death via photocleavage activity. Tryptophan receptor: TfR, Oxygen: O2, Singlet oxygen: 1O2.


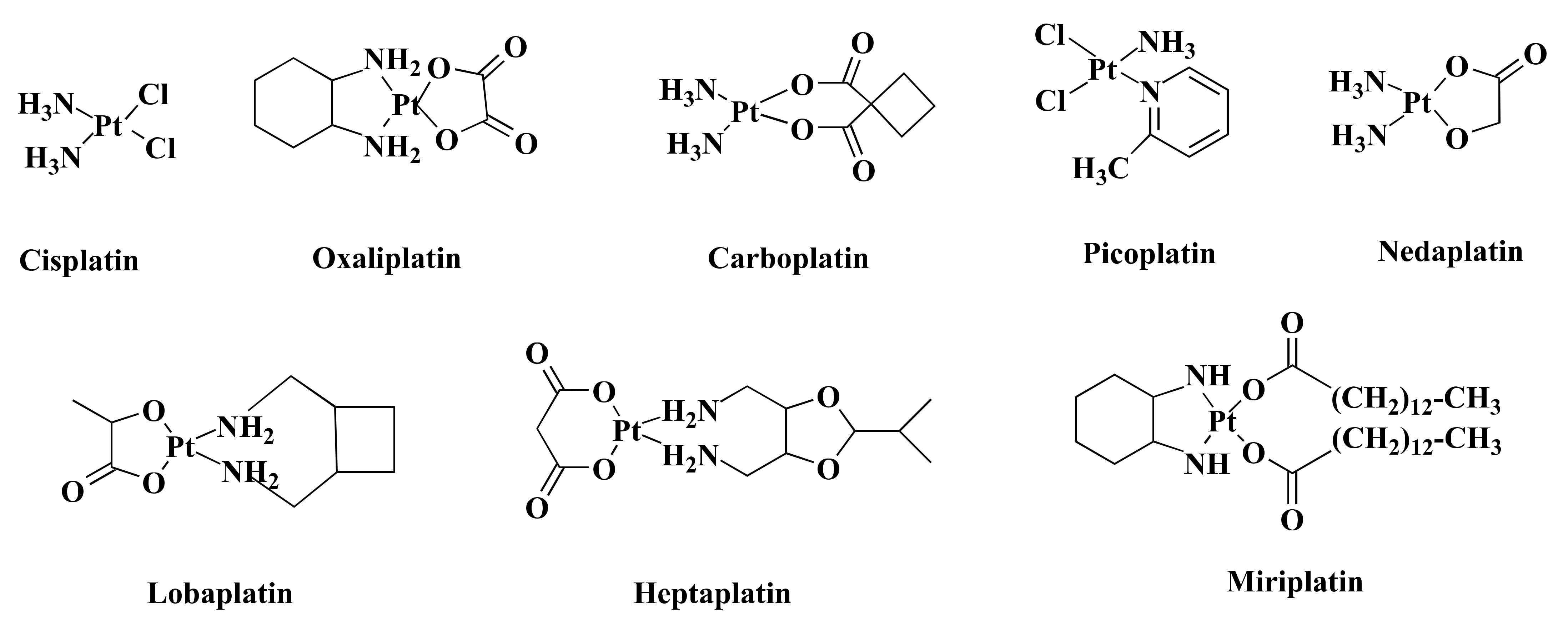{kind=link}
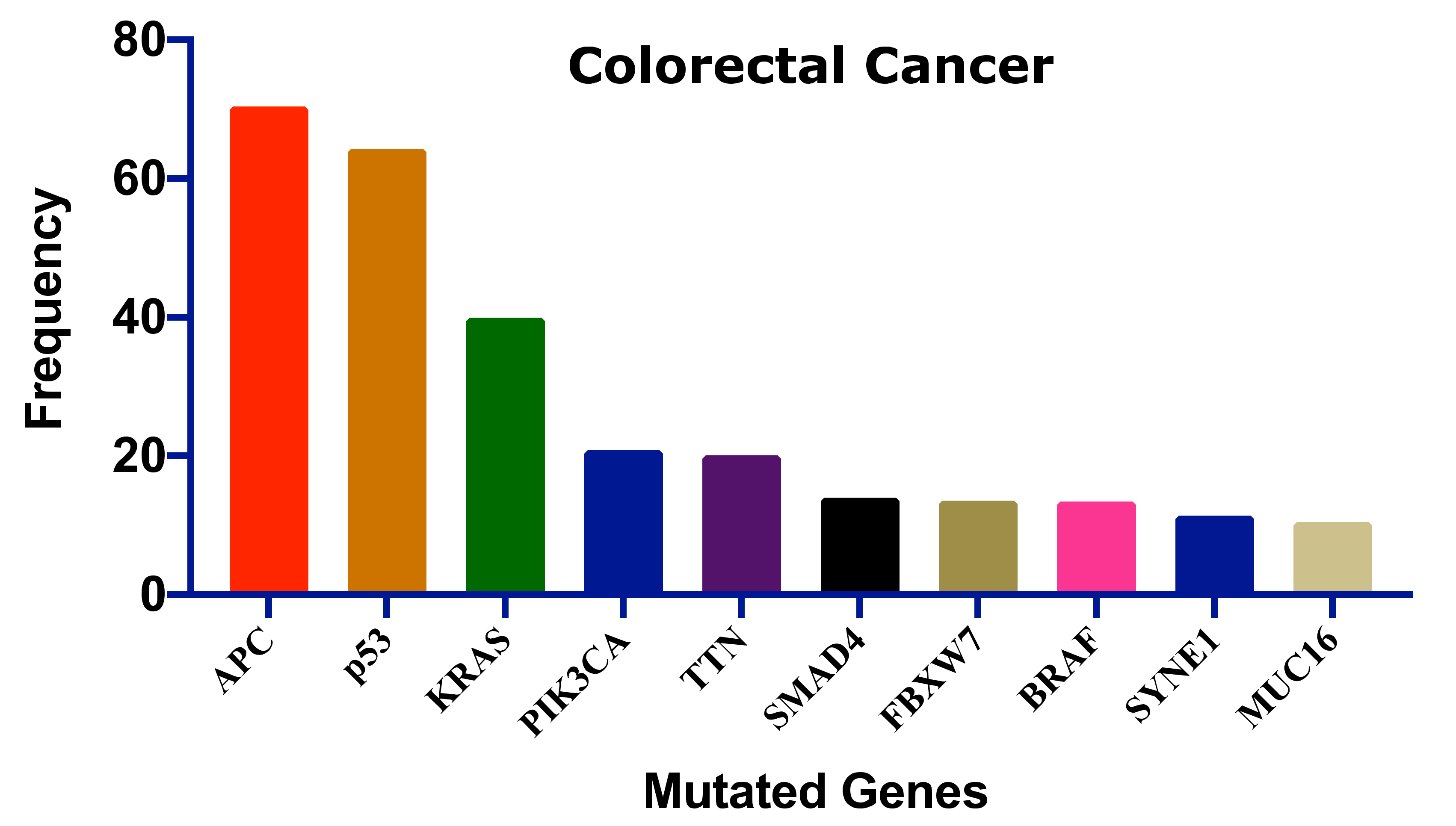{kind=link}
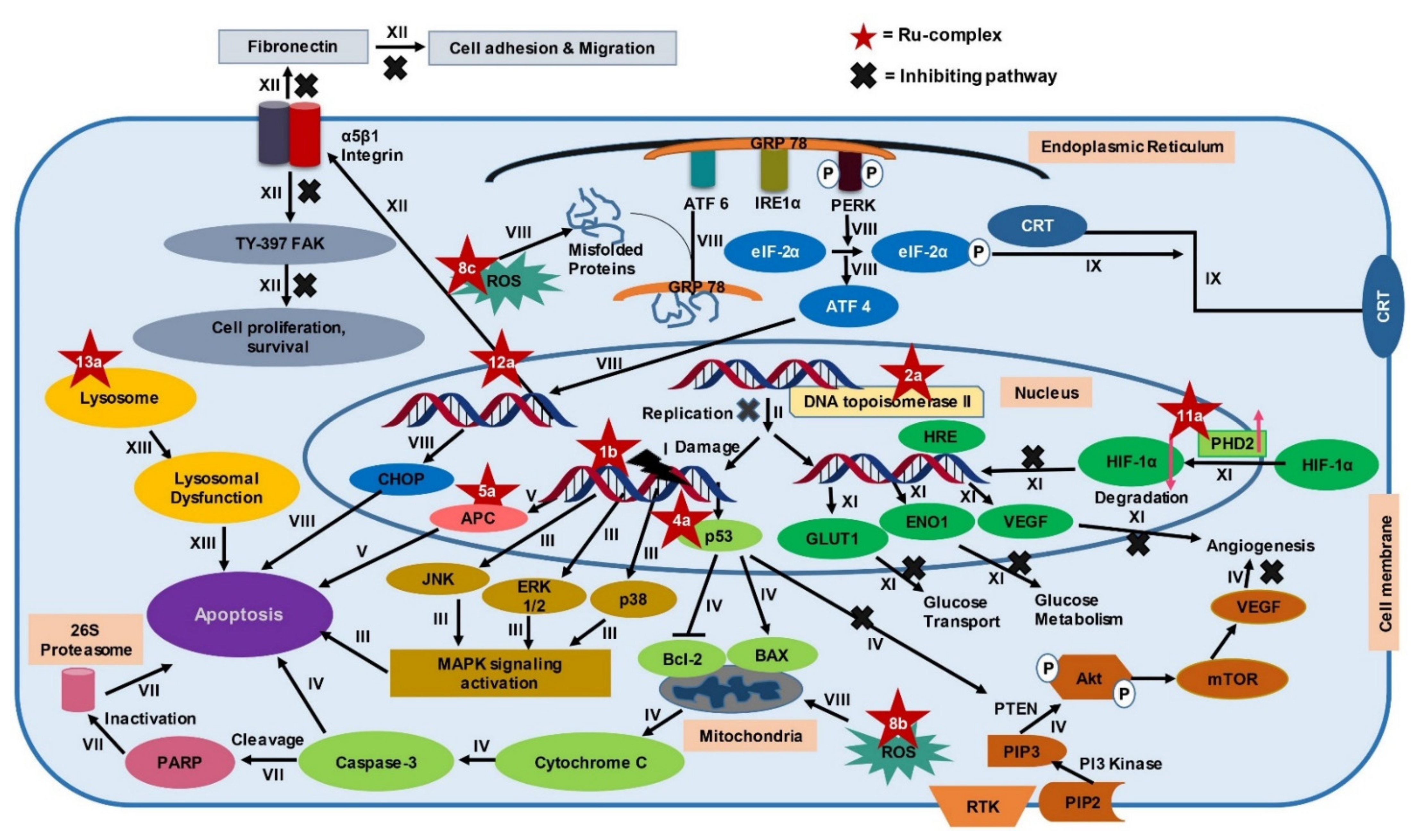{kind=link}
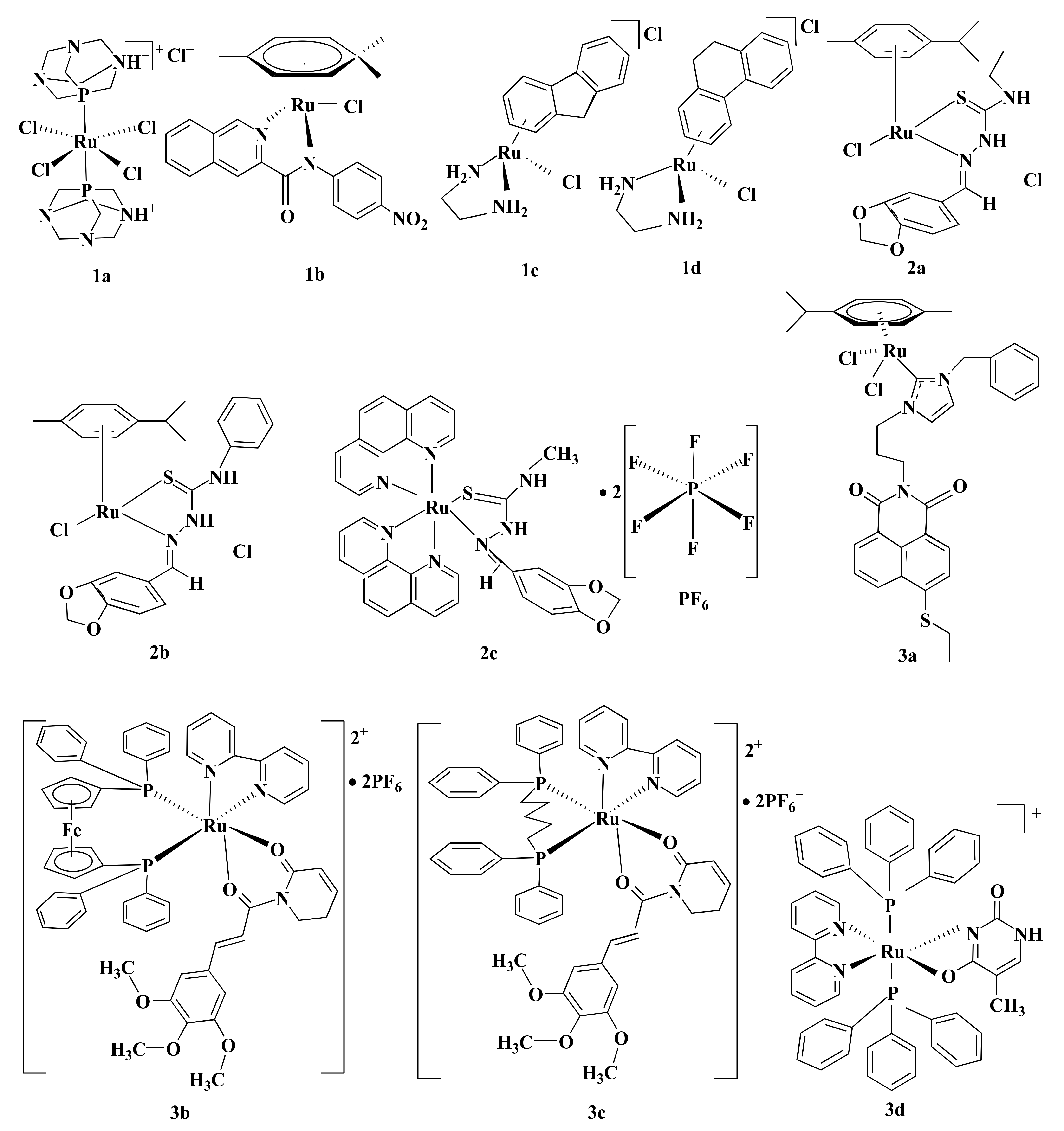{kind=link}
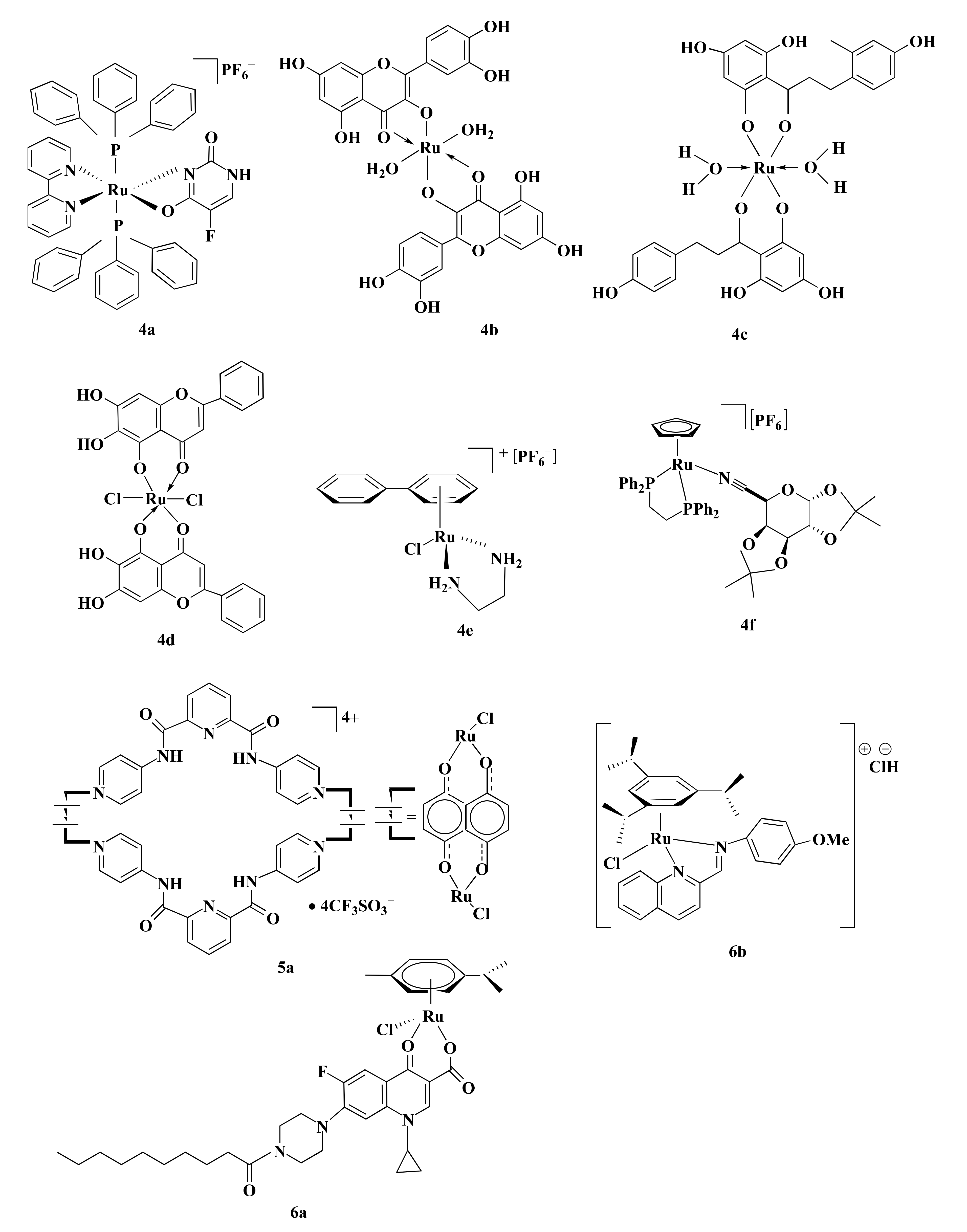{kind=link}
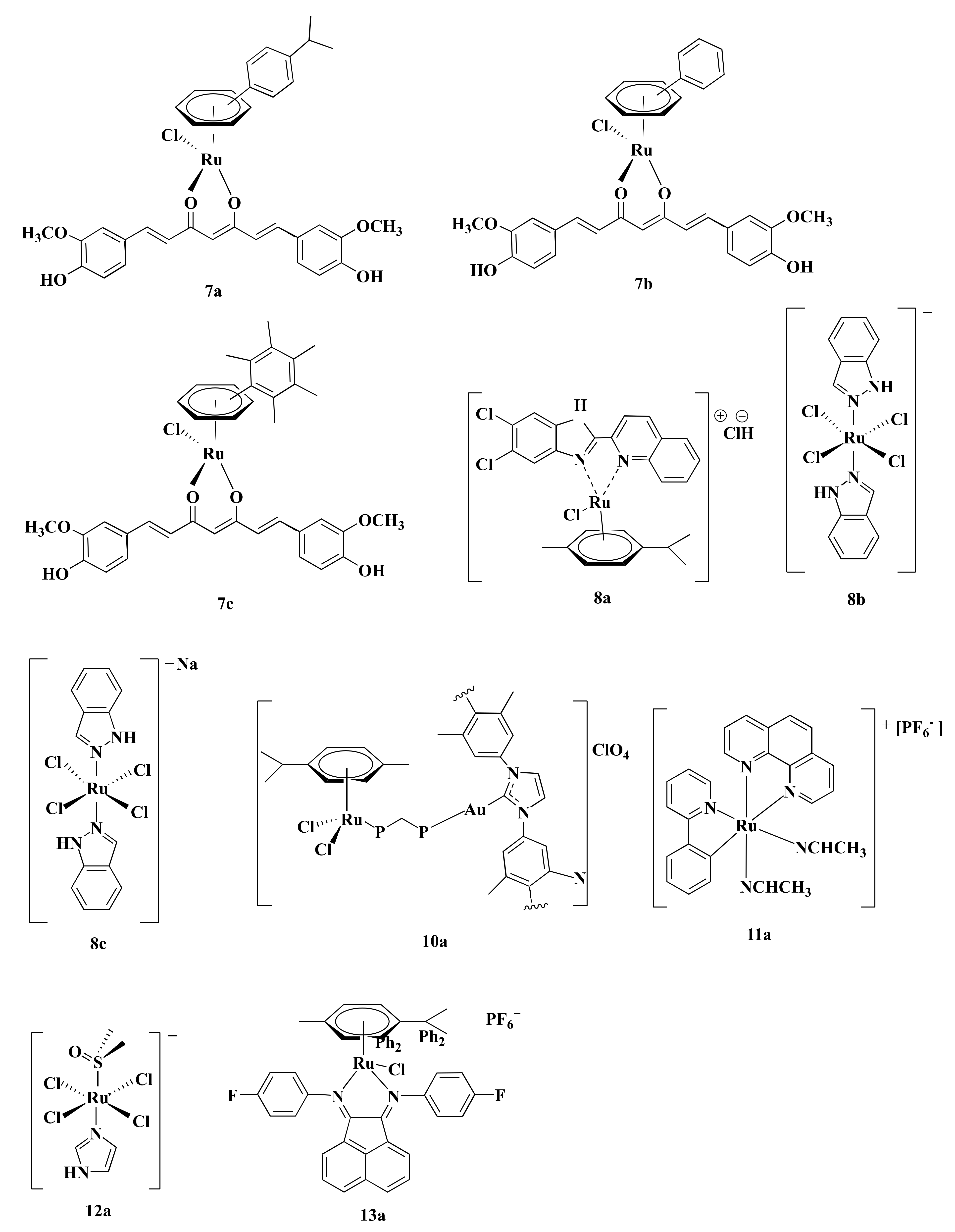{kind=link}
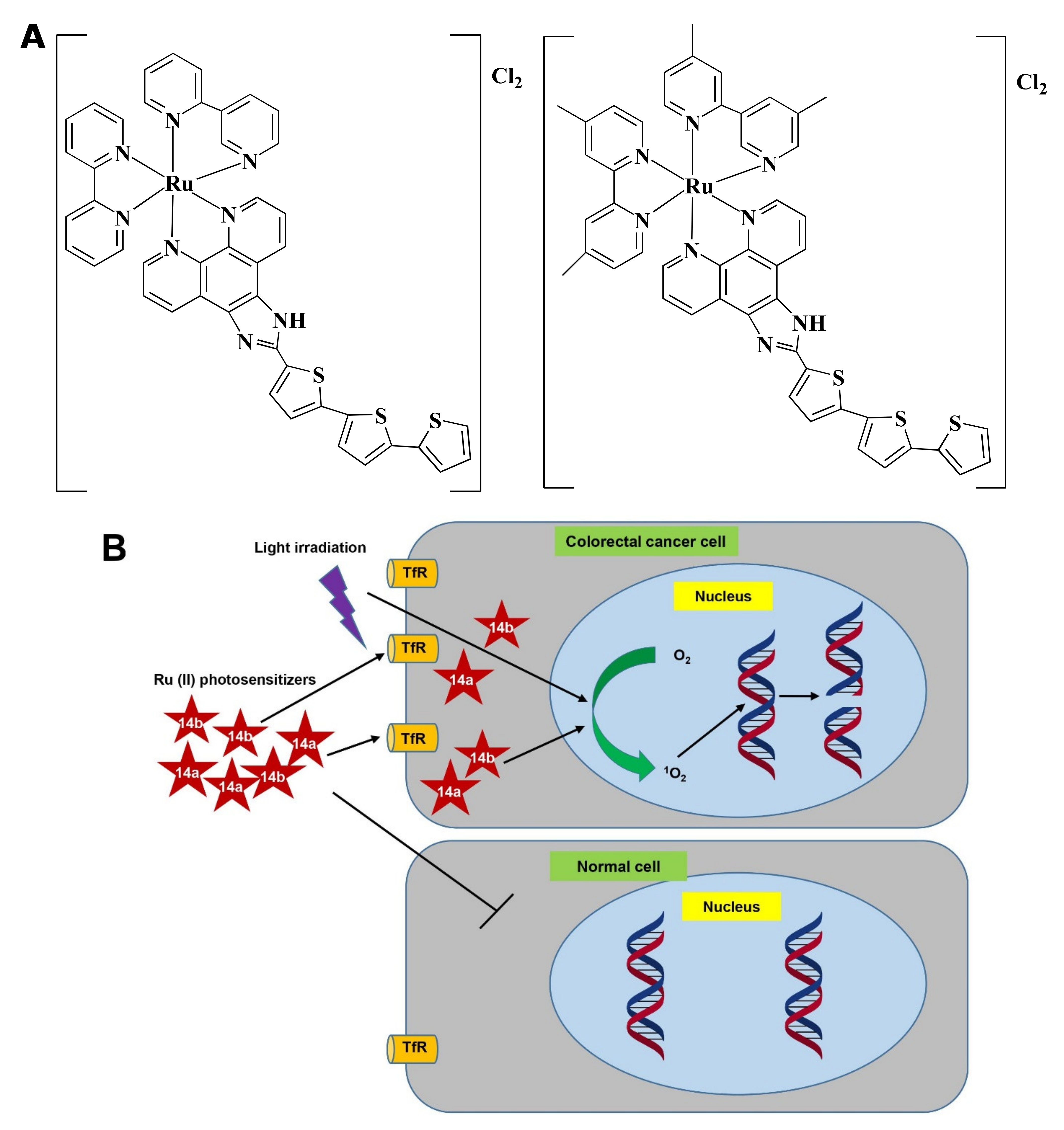{kind=link}
Table 1.
Potency of Ru-based drug candidate(s) compared to conventional anticancer drugs.
| Compounds or Drugs | Oxidation State | Assay Name | CRC Cell Lines and IC50 (µM) | Up-Regulated Protein | Down Regulated Protein | Cell Cycle Arrest | Corresponding Conventional Drugs IC50 (µM) | References |
|---|---|---|---|---|---|---|---|---|
| 1a | III | TBE | HCT116 (>100) * | NR | NR | NR | HCT116 (CIS = 7.65 µM) * | [75] |
| 1b | II | SRB | LS174T (7.7 µmol/L), LoVo (8.1 µmol/L) ***** | NR | NR | NR | LS174T (CIS = 4.6), LoVo (CIS = 0.7) ***** | [76] |
| 1c, 1d | II | CS | DLD1(1c = 10.2), (1d = 7.5) * | p53, p21, GAPDH | PARP | G2/M | DLD1 (OXA = 11.3) * | [77] |
| 2a, 2b | II | MTT | HCT116 (2a = 50.5, 2b = 153), Caco-2 (2a = 26.3, 2b = 121) *** | NR | NR | NR | HCT116 (CIS = 41.7, ETP = 18.3), Caco-2 (CIS = 14.9, ETP = 16.5) *** | [73] |
| 2c | II | MTT | HCT116 (8.6), Caco-2 (6.6) *** | NR | NR | NR | HCT116 (ETP = 18.3), Caco-2 (ETP = 16.5) *** | [74] |
| 4a | II | AB | HCT116 (1.5) *** | caspase-3 | NR | ND | HCT116 (DOX = 0.5, OXA = 4.3, 5-FLU = 4.1) | [27] |
| 4f | II | MTS | HCT116 (0.45) *** | Caspase-3, caspase-7 | NR | NR | HCT116 (OXA = 0.45, 5-FLU = 3.80) *** | [115] |
| 5a | II | MTT | HT-15 (6.9) * | p53, APC | NR | NR | HT-15 (CIS = 13.2, DOX = 15.9) * | [86] |
| 6a | II | SRB | HCT116 (1.33) *** | NR | NR | Both S and G2/M phase | HCT116 (CIS = 5.1, OXA = 3.99) *** | [116] |
| 6b | II | MTT | HCT116 (1.04), SW480 (7.3) *** | NR | NR | G0/G1 (2.5 µM) G2/M (10 µM) | HCT116 (OXA = 2.06), SW480 (OXA = 2.65) *** | [117] |
| 8a | II | MTT | Caco-2 (6.16) ** | NR | NR | G0/G1 | Caco-2 (CIS = 17.9) ** | [118] |
| 10a | II | BP | HCT116 (5.22) *** | NR | NR | NR | CIS = No effect | [78] |
| 11a | II | MTT | HCT116 (2.63) ** | NAD+ | HIF1α, VEGF GLUT1 ENO1 | NR | HCT116 (CIS = 6.33) ** | [87] |
| 13a | II | MTT | HT29 (3.2), HCT116 (2.7), CT-26 (2.3) * | Cathepsin B | NF-kB p65, MMP-2, MMP-9, LAMP1 | NR | HT29 (CIS = 18.9), HCT116 (CIS = 42.8), CT-26 (CIS = 25.6) * | [91] |
trans-[RuCl4(1,3,5-triaza-7-phosphaadamantane protonated at one N atom)2]Cl: 1a, Ru-5-chloro-3-((5-(3-(4-methyl-1,4-diazepane-1-carbonyl)phenyl)furan-2-yl)methylene)indolin-2-one: 1b, [(η6-fluorene)RuII(ethylenediamine)Cl]+: 1c, [(η6-9,10-dihydrophenanthrene)RuII(ethylenediamine)Cl]+: 1d, [(η6-p-cymene)Ru(piperonal-N(4)-ethylthiosemicarbazone)Cl]Cl: 2a, [(η6-p-cymene)Ru(piperonal-N(4)-phenylthiosemicarbazone)Cl]Cl: 2b, [(1,10-phenanthroline)2Ru(2-(benzo[d][1,3]dioxol-5-ylmethylene)-N-methylhydrazinecarbothioamide)](hexafluorophosphate)2: 2c, [Ru(piplartine)(1,1 bis(diphenylphosphino)ferrocene)(2,2′-bipyridine)](hexafluorophosphate)2: 3b, [Ru(piplartine)(1,4-bis(diphenylphosphino)butane)(2,2′-bipyridine)](hexafluorophosphate)2: 3c, Ru(II) thymin complex: 3d, [Ru(5-Fluorouracil)(triphenylphosphine)2(2,2′-bipyridine)]hexafluorophosphate: 4a, [(η5-C5H5)Ru(1,2-bis(diphenylphosphino)ethane)]+ bearing the galactose nitrile derivative ligands: 4f, [Ru4(p-cymene)4(5,8-dioxydo-1,4-naphthaquinonato-)2(2,6-bis(N-(4-pyridyl)carbamoyl)pyridine)2][4CF3SO3]: 5a, [Ru(η6-p-cymene)( 7-(4-(Decanoyl)piperazin-1-yl)-ciprofloxacin-H)Cl]: 6a, [(η6-1,3,5-triisopropylbenzene)RuCl(4-methoxy-N (2-quinolinylmethylene)aniline)]Cl: 6b, [(η6-p-cymene)RuCl[118]]: 8a, trans-[tetrachloro-bis(1H-indazole)ruthenate(III)]: 8b, Sodium trans-[tetrachloride-bis(1H-indazole)ruthenate(III)]: 8c, [Ru(p-cymene)Cl2(µ-1,1-bis(diphenylphosphino)methane)Au(IMes)]-ClO4: 10a, Ruthenium derived compound 11: 11a, Half-sandwich Ru(II) complexes bearing aryl-BIAN chelating ligands: 13a, Cisplatin: CIS, Oxaliplatin: OXA, Doxorubicin: DOX, 5-Fluorouracil: 5-FLU, Etoposide: ETP; The half maximal inhibitory concentration: IC50, Not reported: NR, Not detected: ND, Trypan blue exclusion: TBE, Sulforhodamine B: SRB, Clonogenic survival: CS, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide: MTT, Alamar blue: AB, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium): MTS, Neutral red uptake: NRU, BluePrestoTM: BP, Tumor protein P53: p53, Cyclin-dependent kinase inhibitor 1: p21, Glyceraldehyde 3 phosphate dehydrogenase: GAPDH, Poly (ADP-ribose) polymerase: PARP, Phosphorylated c-Jun N-terminal kinases 2: p-JNK 2, Extracellular signal-regulated kinases 1: ERK1, Mitogen-activated protein kinases α: p38α, H2A histone family member X: H2AX, Adenomatous polyposis coli: APC, Matrix metalloproteinase-2: MMP-2, B-cell lymphoma 2: Bcl-2, Nicotinamide adenine dinucleotide: NAD+, Hypoxia inducible factor-1 α: HIF-1α, Vascular endothelial growth factor: VEGF, Glucose transporter 1: GLUT1, Enolase 1: ENO1, Nuclear factor of the κ-chain in B-cells p65: NFkB p65, Matrix metalloproteinase-2: MMP-2, Matrix metalloproteinase-9: MMP-9, Lysosomal associated membrane protein 1: LAMP1,. References: Ref, After 24 h: *, After 48 h: **, After 72 h: ***, After 6 days: *****.
Table 2.
Cytotoxic potential of some Ru-complexes towards CRC cells.
| Compounds or Drugs | Oxidation State | Assay Name | CRC Cell Lines and IC50 (µM) | Up-Regulated Protein | Down Regulated Protein | Cell Cycle Arrest | References |
|---|---|---|---|---|---|---|---|
| 3a | II | NR | HCT116 | p21, Bad, p-p38 MAPK, ATF2, Stat1, MMP | Bax | G1 | [79] |
| 4b | II | MTT | HT29 (<100) ** | p53, caspase-3, Bax | Akt1, mTOR, VEGF, Bcl-2, PCNA, WNT, β-catenin | G0/G1 | [83] |
| 4c | II | MTT | HT29 (>100) * | p53, caspase-3, Bax | Akt1, p-Akt, mTOR, p-mTOR, VEGF, Bcl-2, NF-κΒ, MMP-9, PCNA | G0/G1 | [82] |
| 4d | II | MTT | HT29 (~30) ** | p53, caspase-3, Bax | Akt1, mTOR, VEGF, Bcl-2, PCNA, WNT, β-catenin | G0/G1 | [84] |
| 4e | II | SRB | HCT116 (8) HCT116 p53 (16) **** | p53, p21/WAF, Bax | NR | G1 and G2 | [85] |
| 7a, 7b, 7c | II | MTT | HCT116 (NR) | p53, caspase-3 | PARP | NR | [90] |
| 8b | III | NRU | SW480 (30), LT97 (50) * | Caspase-3 | PARP, MMP, Bcl-2 | NR | [119] |
| 8c | III | MTT | HCT116 (20), SW480 (40) * | eIF2α, ATF4, CHOP | NR | NR | [89] |
| 8c | III | NR | HT15, HCT116, HT29 (NR) | eIF2α, CRT, HMGB-1, ATP, Beclin-1, LC3A/B-II | NR | NR | [120] |
| 12a | III | MTT | HCT116 | RND-1, SIK-1 | α5β1 integrin, VEGF, MCP-1 | NR | [88] |
| 14a, 14b | II | PBCV | CT-26 (NR) | NR | NR | NR | [92] |
Ru(II) naphthalimide N-Heterocyclic Carbene compounds: 3a, Ru-Quercetin: 4b, Ru-Phloretin: 4c, Ru-Baicalein: 4d, [Ru(biphenyl)Cl(1,2-ethylenediamine)]+ with hexafluorophosphate: 4e, [Ru(p-cymene)(curcumin)Cl]: 7a, [(Benzene)Ru(curcumin)Cl]: 7b, [Ru(hexamethylbenzene)(curcumin)Cl]: 7c, trans-[tetrachloro-bis(1H-indazole)ruthenate(III)]: 8b, Sodium trans-[tetrachloride-bis(1H-indazole)ruthenate(III)]: 8c, [Imidazolium-trans-tetrachloro(dimethylsulfoxide)imidazoleruthenium(III)]: 12a, [Ru(2,2′-biyridine)2(2-(2′,2″:5″,2′′′-terthiophene)-imidazo[4,5-f][1,10]phenanthroline)]2+: 14a, [Ru(4,4′-dimethyl-2,2′-bipyridine)2(2-(2′,2″:5″,2′′′-terthiophene)- imidazo[4,5-f][1,10] phenanthroline)]2 +: 14b. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide: MTT, Sulforhodamine B: SRB, Neutral red uptake: NRU, Presto Blue Cell Viability: PBCV, Cyclin-dependent kinase inhibitor 1: p21, Bcl-2 associated agonist of cell death: Bad, BCl-2 associated X: BAX, Tumor protein P53: p53, Phosphorylated p38 MAP Kinase: p-p38 MAPK, Activating transcription factor 2: ATF2, Signal transducer and activator of transcription 1: Stat1, Matrix metallo proteinases: MMP, Serine/threonine kinase 1: Akt1; Mammalian target of rapamycin: mTOR; Vascular endothelial growth factor: VEGF, B-cell lymphoma 2: Bcl-2, Proliferating cell nuclear antigen: PCNA, Wingless-related integration site: WNT, Nuclear factor of the κ-chain in B-cells: NFkB, Matrix metalloproteinase 9: MMP-9, Poly (ADP-ribose) polymerase: PARP, Eukaryotic translation initiation factor 2α: eIF2α, Activating transcription factor 4: ATF4, C/eBP homologous protein: CHOP, Calreticulin: CRT, Adenosine triphosphate: ATP, High mobility group protein B1: HMGB-1, Rho Family GTPase 1: RND1, Salt inducible kinase 1: SIK-1, Monocyte chemotactic protein-1: MCP-1. After 24 h: *, After 48 h: **, After 96 h: ****.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mahmud, K.M.; Niloy, M.S.; Shakil, M.S.; Islam, M.A. Ruthenium Complexes: An Alternative to Platinum Drugs in Colorectal Cancer Treatment. Pharmaceutics 2021, 13, 1295. https://doi.org/10.3390/pharmaceutics13081295
AMA Style
Mahmud KM, Niloy MS, Shakil MS, Islam MA. Ruthenium Complexes: An Alternative to Platinum Drugs in Colorectal Cancer Treatment. Pharmaceutics. 2021; 13(8):1295. https://doi.org/10.3390/pharmaceutics13081295
Chicago/Turabian StyleMahmud, Kazi Mustafa, Mahruba Sultana Niloy, Md Salman Shakil, and Md Asiful Islam. 2021. "Ruthenium Complexes: An Alternative to Platinum Drugs in Colorectal Cancer Treatment" Pharmaceutics 13, no. 8: 1295. https://doi.org/10.3390/pharmaceutics13081295
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.