
Ruthenium Complexes: An Alternative to Platinum Drugs in Colorectal Cancer Treatment





Abstract
:1. Introduction
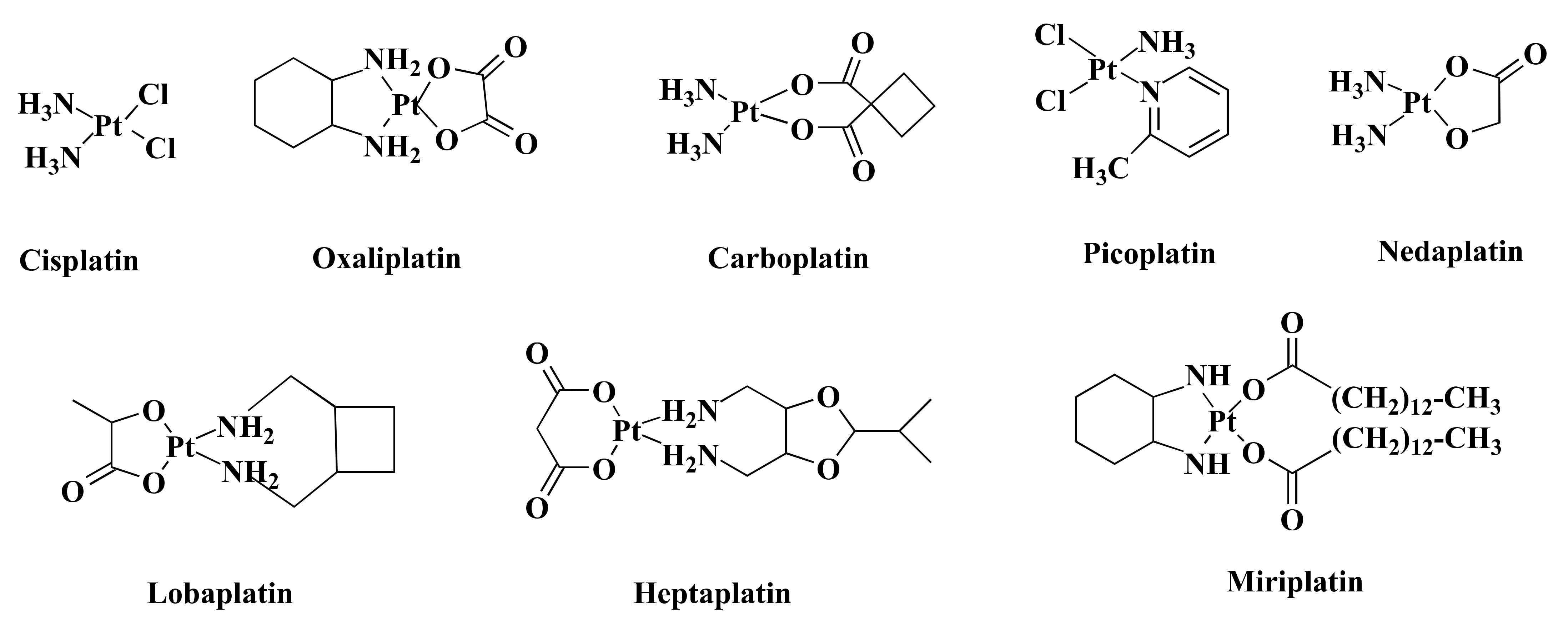
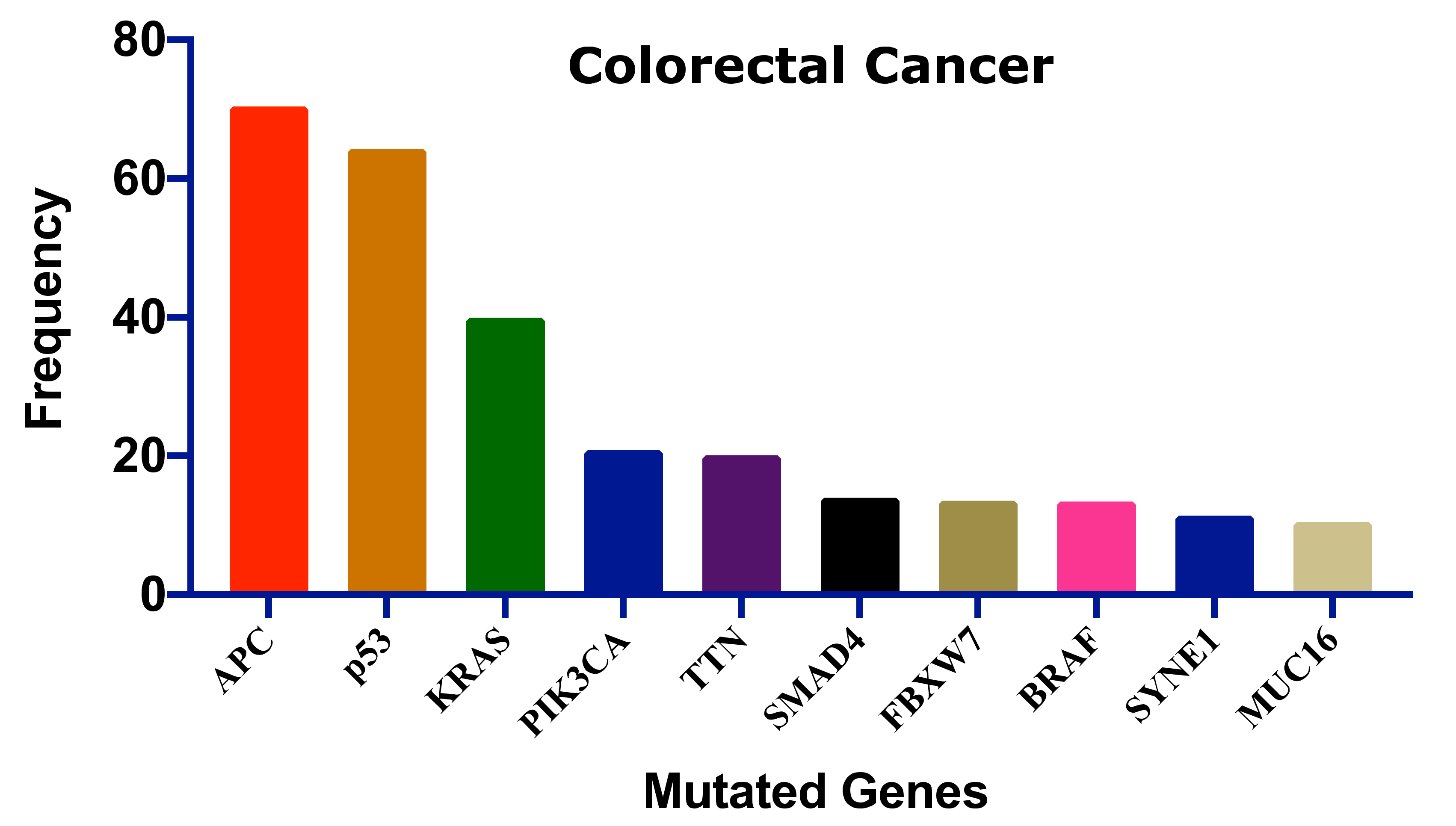
2. Colorectal Cancer and Pt-Based Drugs
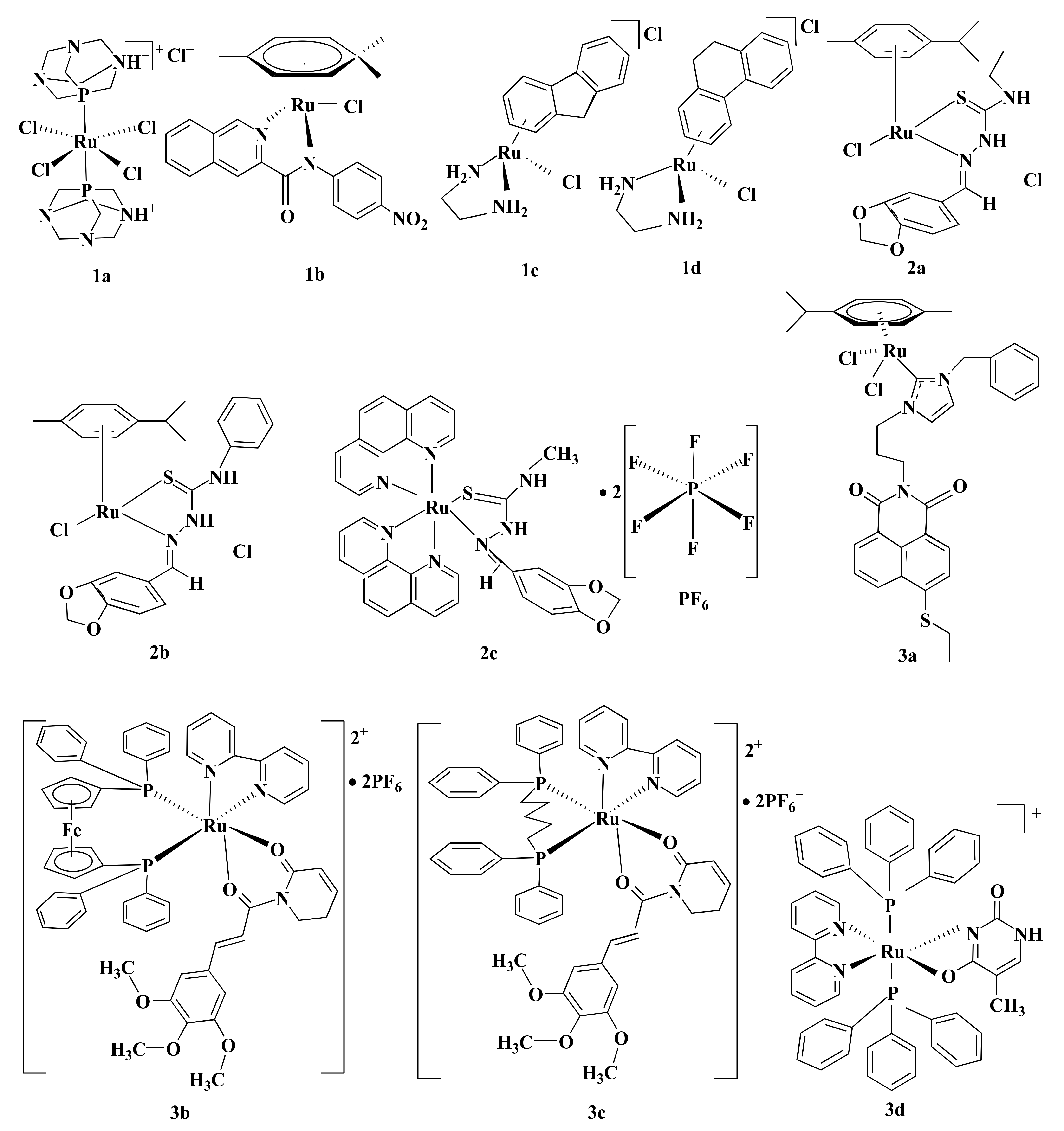
3. Features of Ru-Complexes
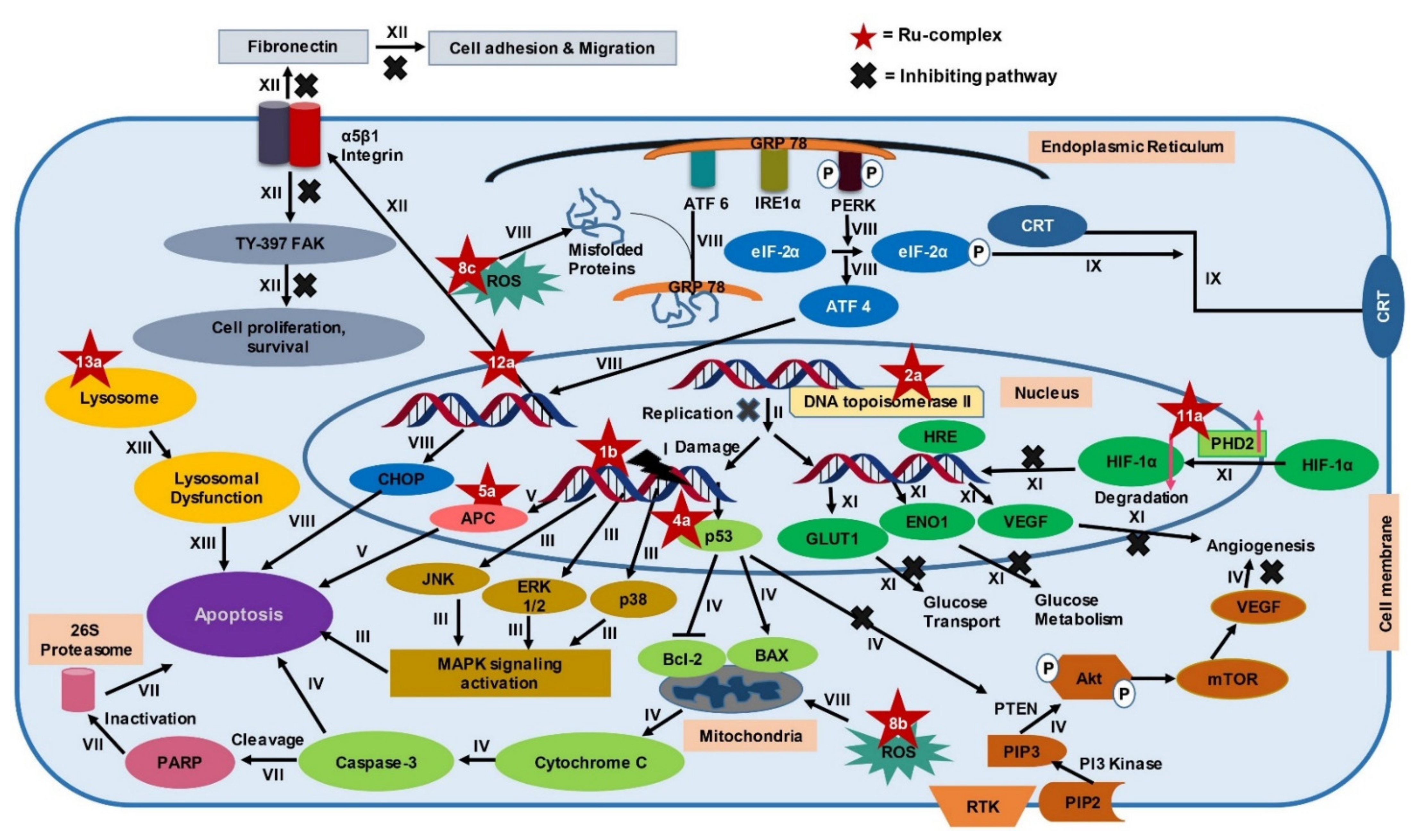
4. Underlying Mechanisms of Ru-Complexes in Targeting CRC
4.1. DNA Damage Mediated Apoptosis
4.2. Inhibition of Topoisomerase II Enzyme
4.3. MAPK Signaling Pathway

4.4. p53 Dependent Caspase-3 Mediated Signaling
4.5. Upregulation of APC and p53 Gene
4.6. p53 Independent Activity
4.7. Inhibition of Proteasome
4.8. ROS-Mediated Apoptosis

4.9. Immunogenic Cell Death
4.10. Inhibition of Thioredoxin Reductase Activity
4.11. Inhibition of HIF-1 Pathway
4.12. Anti-Metastasis Activity
4.13. Lysosomal Dysfunction
4.14. Photodynamic Therapy
5. Ru-Nanocomplexes in CRC Theranostics
5.1. CRC Diagnosis
5.2. CRC Treatment
6. Phase I Dose-Escalation Studies (Phase Ib Clinical Trials)
7. Toxicity of Ru-Drug Candidates
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Centelles, J.J. General aspects of colorectal cancer. Int. Sch. Res. Not. 2012, 2012, 139268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manne, U.; Shanmugam, C.; Katkoori, V.R.; Bumpers, H.L.; Grizzle, W.E. Development and progression of colorectal neoplasia. Cancer Biomark. 2011, 9, 235–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeriouh, W.; Nani, A.; Belarbi, M.; Dumont, A.; de Rosny, C.; Aboura, I.; Ghanemi, F.Z.; Murtaza, B.; Patoli, D.; Thomas, C.; et al. Phenolic extract from oleaster (Olea europaea var. Sylvestris) leaves reduces colon cancer growth and induces caspase-dependent apoptosis in colon cancer cells via the mitochondrial apoptotic pathway. PLoS ONE 2017, 12, e0170823. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.; Oosterling, S.J.; van Egmond, M. Surgery for Colorectal Cancer: A Trigger for Liver Metastases Development? New Insights into the Underlying Mechanisms. Biomedicines 2021, 9, 177. [Google Scholar] [CrossRef]
- Colin, D.J.; Limagne, E.; Ragot, K.; Lizard, G.; Ghiringhelli, F.; Solary, É.; Chauffert, B.; Latruffe, N.; Delmas, D. The role of reactive oxygen species and subsequent DNA-damage response in the emergence of resistance towards resveratrol in colon cancer models. Cell Death Dis. 2014, 5, e1533. [Google Scholar] [CrossRef] [Green Version]
- Denlinger, C.S.; Barsevick, A.M. The challenges of colorectal cancer survivorship. J. Natl. Compr. Canc. Netw. 2009, 7, 883–894. [Google Scholar] [CrossRef]
- Knowles, G.; Haigh, R.; McLean, C.; Phillips, H.A.; Dunlop, M.G.; Din, F.V.N. Long term effect of surgery and radiotherapy for colorectal cancer on defecatory function and quality of life. Eur. J. Oncol. Nurs. 2013, 17, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Benjamin Garbutcheon-Singh, K.; P Grant, M.; W Harper, B.; M Krause-Heuer, A.; Manohar, M.; Orkey, N.R.; Aldrich-Wright, J. Transition Metal Based Anticancer Drugs. Curr. Top. Med. Chem. 2011, 11, 521–542. [Google Scholar] [CrossRef]
- Ndagi, U.; Mhlongo, N.; Soliman, M.E. Metal complexes in cancer therapy—An update from drug design perspective. Drug Des. Dev. Ther. 2017, 11, 599–616. [Google Scholar] [CrossRef] [Green Version]
- Parveen, S. Recent advances in anticancer ruthenium Schiff base complexes. Appl. Organomet. Chem. 2020, 34, e5687. [Google Scholar] [CrossRef]
- Raymond, E.; Chaney, S.; Taamma, A.; Cvitkovic, E. Oxaliplatin: A review of preclinical and clinical studies. Ann. Oncol. 1998, 9, 1053–1071. [Google Scholar] [CrossRef] [PubMed]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef] [PubMed]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Lippard, S.J. Direct cellular responses to platinum-induced DNA damage. Chem. Rev. 2007, 107, 1387–1407. [Google Scholar] [CrossRef]
- Hsu, H.H.; Chen, M.C.; Baskaran, R.; Lin, Y.M.; Day, C.H.; Lin, Y.J.; Tu, C.C.; Vijaya Padma, V.; Kuo, W.W.; Huang, C.Y. Oxaliplatin resistance in colorectal cancer cells is mediated via activation of ABCG2 to alleviate ER stress induced apoptosis. J. Cell. Physiol. 2018, 233, 5458–5467. [Google Scholar] [CrossRef] [PubMed]
- Drott, J.; Fomichov, V.; Starkhammar, H.; Börjeson, S.; Kjellgren, K.; Berterö, C. Oxaliplatin-Induced Neurotoxic Side Effects and Their Impact on Daily Activities: A Longitudinal Study among Patients with Colorectal Cancer. Cancer Nurs. 2019, 42, E40–E48. [Google Scholar] [CrossRef] [Green Version]
- Virag, P.; Fischer-Fodor, E.; Perde-Schrepler, M.; Brie, I.; Tatomir, C.; Balacescu, L.; Berindan-Neagoe, I.; Victor, B.; Balacescu, O. Oxaliplatin induces different cellular and molecular chemoresistance patterns in colorectal cancer cell lines of identical origins. BMC Genomics 2013, 14, 480. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Kim, C.Y.; Nam, T.-G. Ruthenium Complexes as Anticancer Agents: A Brief History and Perspectives. Drug Des. Dev. Ther. 2020, 14, 5375–5392. [Google Scholar] [CrossRef]
- Kostova, I. Ruthenium Complexes as Anticancer Agents. Curr. Med. Chem. 2006, 13, 1085–1107. [Google Scholar] [CrossRef] [PubMed]
- Dougan, S.J.; Habtemariam, A.; McHale, S.E.; Parsons, S.; Sadler, P.J. Catalytic organometallic anticancer complexes. Proc. Natl. Acad. Sci. USA 2008, 105, 11628–11633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandioller, W.; Balsano, E.; Meier, S.M.; Jungwirth, U.; Göschl, S.; Roller, A.; Jakupec, M.A.; Berger, W.; Keppler, B.K.; Hartinger, C.G. Organometallic anticancer complexes of lapachol: Metal centre-dependent formation of reactive oxygen species and correlation with cytotoxicity. Chem. Commun. 2013, 49, 3348–3350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Sadler, P.J. Advances in the design of organometallic anticancer complexes. J. Organomet. Chem. 2017, 839, 5–14. [Google Scholar] [CrossRef]
- Silva, V.R.; Corrêa, R.S.; Santos, L.D.S.; Soares, M.B.P.; Batista, A.A.; Bezerra, D.P. A ruthenium-based 5-fluorouracil complex with enhanced cytotoxicity and apoptosis induction action in HCT116 cells. Sci. Rep. 2018, 8, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shum, J.; Leung, P.K.-K.; Lo, K.K.-W. Luminescent Ruthenium(II) Polypyridine Complexes for a Wide Variety of Biomolecular and Cellular Applications. Inorg. Chem. 2019, 58, 2231–2247. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.-P.; Zhong, Y.-M.; Ji, L.-N.; Mao, Z.-W. Phosphorescent metal complexes as theranostic anticancer agents: Combining imaging and therapy in a single molecule. Chem. Sci. 2021, 12, 2357–2367. [Google Scholar] [CrossRef]
- Xu, M.; Wen, Y.; Liu, Y.; Tan, X.; Chen, X.; Zhu, X.; Wei, C.; Chen, L.; Wang, Z.; Liu, J. Hollow mesoporous ruthenium nanoparticles conjugated bispecific antibody for targeted anti-colorectal cancer response of combination therapy. Nanoscale 2019, 11, 9661–9678. [Google Scholar] [CrossRef]
- Zhu, X.; Gong, Y.; Liu, Y.; Yang, C.; Wu, S.; Yuan, G.; Guo, X.; Liu, J.; Qin, X. Ru@CeO2 yolk shell nanozymes: Oxygen supply in situ enhanced dual chemotherapy combined with photothermal therapy for orthotopic/subcutaneous colorectal cancer. Biomaterials 2020, 242, 119923. [Google Scholar] [CrossRef]
- Thangavel, P.; Viswanath, B.; Kim, S. Recent developments in the nanostructured materials functionalized with ruthenium complexes for targeted drug delivery to tumors. Int. J. Nanomed. 2017, 12, 2749–2758. [Google Scholar] [CrossRef] [Green Version]
- Malki, A.; ElRuz, R.A.; Gupta, I.; Allouch, A.; Vranic, S.; Al Moustafa, A.-E. Molecular Mechanisms of Colon Cancer Progression and Metastasis: Recent Insights and Advancements. Int. J. Mol. Sci. 2021, 22, 130. [Google Scholar] [CrossRef]
- Tariq, K.; Ghias, K. Colorectal cancer carcinogenesis: A review of mechanisms. Cancer Biol.Med. 2016, 13, 120–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Guda, K.; Veigl, M.L.; Varadan, V.; Nosrati, A.; Ravi, L.; Lutterbaugh, J.; Beard, L.; Willson, J.K.V.; Sedwick, W.D.; Wang, Z.J.; et al. Novel recurrently mutated genes in African American colon cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 1149–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasaikar, S.; Huang, C.; Wang, X.; Petyuk, V.A.; Savage, S.R.; Wen, B.; Dou, Y.; Zhang, Y.; Shi, Z.; Arshad, O.A.; et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177, 1035–1049. [Google Scholar] [CrossRef] [Green Version]
- Giannakis, M.; Mu, J.X.; Shukla, A.S.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Brannon, A.R.; Vakiani, E.; Sylvester, B.E.; Scott, S.N.; McDermott, G.; Shah, R.H.; Kania, K.; Viale, A.; Oschwald, D.M.; Vacic, V.; et al. Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol. 2014, 15, 454. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- cBioPortal Colon Cancer. Available online: https://bit.ly/2Od7xhB (accessed on 15 July 2021).
- McQuade, R.M.; Stojanovska, V.; Bornstein, J.C.; Nurgali, K. PARP inhibition in platinum-based chemotherapy: Chemopotentiation and neuroprotection. Pharmacol. Res. 2018, 137, 104–113. [Google Scholar] [CrossRef]
- Apps, M.G.; Choi, E.H.Y.; Wheate, N.J. The state-of-play and future of platinum drugs Endocr. Relat. Cancer 2015, 22, R219–R233. [Google Scholar] [CrossRef] [Green Version]
- Mehmood, R.K. Review of Cisplatin and oxaliplatin in current immunogenic and monoclonal antibody treatments. Oncol. Rev. 2014, 8, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coverdale, J.P.C.; Laroiya-McCarron, T.; Romero-Canelón, I. Designing Ruthenium Anticancer Drugs: What Have We Learnt from the Key Drug Candidates? Inorganics 2019, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Chiorazzi, A.; Semperboni, S.; Marmiroli, P. Current View in Platinum Drug Mechanisms of Peripheral Neurotoxicity. Toxics 2015, 3, 304–321. [Google Scholar] [CrossRef] [Green Version]
- André, T.; Boni, C.; Mounedji-Boudiaf, L.; Navarro, M.; Tabernero, J.; Hickish, T.; Topham, C.; Zaninelli, M.; Clingan, P.; Bridgewater, J.; et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N. Engl. J. Med. 2004, 350, 2343–2351. [Google Scholar] [CrossRef] [Green Version]
- Rosati, G.; Cordio, S.; Reggiardo, G.; Aprile, G.; Butera, A.; Avallone, A.; Tucci, A.; Novello, G.; Blanco, G.; Caputo, G.J.C. Oxaliplatin-based chemotherapy in patients with metastatic colorectal cancer aged at least 75 Years: A post-hoc subgroup analysis of three phase II trials. Cancers 2019, 11, 578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forcello, N.P.; Khubchandani, S.; Patel, S.J.; Brahaj, D. Oxaliplatin-induced immune-mediated cytopenias: A case report and literature review. J. Oncol. Pharm. Pract. 2015, 21, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Haller, D.G. Safety of oxaliplatin in the treatment of colorectal cancer. Oncology 2000, 14, 15–20. [Google Scholar]
- Rubbia-Brandt, L.; Audard, V.; Sartoretti, P.; Roth, A.; Brezault, C.; Le Charpentier, M.; Dousset, B.; Morel, P.; Soubrane, O.; Chaussade, S.J. Severe hepatic sinusoidal obstruction associated with oxaliplatin-based chemotherapy in patients with metastatic colorectal cancer. Ann. Oncol. 2004, 15, 460–466. [Google Scholar] [CrossRef]
- Overman, M.J.; Maru, D.M.; Charnsangavej, C.; Loyer, E.M.; Wang, H.; Pathak, P.; Eng, C.; Hoff, P.M.; Vauthey, J.-N.; Wolff, R.; et al. Oxaliplatin-mediated increase in spleen size as a biomarker for the development of hepatic sinusoidal injury. J. Clin. Oncol. 2010, 28, 2549–2555. [Google Scholar] [CrossRef] [PubMed]
- Süss-Fink, G. Arene ruthenium complexes as anticancer agents. Dalton Trans. 2010, 39, 1673–1688. [Google Scholar] [CrossRef] [PubMed]
- Jakupec, M.A.; Galanski, M.; Arion, V.B.; Hartinger, C.G.; Keppler, B.K. Antitumour metal compounds: More than theme and variations. Dalton Trans. 2008, 14, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Ang, W.H.; Dyson, P.J. Classical and non-classical ruthenium-based anticancer drugs: Towards targeted chemotherapy. Eur. J. Inorg. Chem. 2006, 2006, 4003–4018. [Google Scholar] [CrossRef]
- Motswainyana, W.M.; Ajibade, P.A. Anticancer Activities of Mononuclear Ruthenium(II) Coordination Complexes. Adv. Chem. 2015, 2015, 859730. [Google Scholar] [CrossRef] [Green Version]
- Savic, M.; Arsenijevic, A.; Milovanovic, J.; Stojanovic, B.; Stankovic, V.; Rilak Simovic, A.; Lazic, D.; Arsenijevic, N.; Milovanovic, M. Antitumor Activity of Ruthenium(II) Terpyridine Complexes towards Colon Cancer Cells In Vitro and In Vivo. Molecules 2020, 25, 4699. [Google Scholar] [CrossRef]
- Fukushi, S.; Yoshino, H.; Yoshizawa, A.; Kashiwakura, I. p53-independent structure-activity relationships of 3-ring mesogenic compounds’ activity as cytotoxic effects against human non-small cell lung cancer lines. BMC Cancer 2016, 16, 521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, E.; Zhu, M.; Liu, L.; Huang, Y.; Wang, L.; Shi, C.; Zhang, W.; Sun, Y. Impact of the carbon chain length of novel palladium (II) complexes on interaction with DNA and cytotoxic activity. Inorg. Chem. 2010, 49, 3261–3270. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Fischer, A.; Xu, Y.; Sun, L. Isolated Seven-Coordinate Ru(IV) Dimer Complex with [HOHOH]—Bridging Ligand as an Intermediate for Catalytic Water Oxidation. J. Am. Chem. Soc. 2009, 131, 10397–10399. [Google Scholar] [CrossRef] [PubMed]
- Jabłońska-Wawrzycka, A.; Rogala, P.; Michałkiewicz, S.; Hodorowicz, M.; Barszcz, B. Ruthenium complexes in different oxidation states: Synthesis, crystal structure, spectra and redox properties. Dalton Trans. 2013, 42, 6092–6101. [Google Scholar] [CrossRef]
- Lin, K.; Zhao, Z.-Z.; Bo, H.-B.; Hao, X.-J.; Wang, J.-Q. Applications of Ruthenium Complex in Tumor Diagnosis and Therapy. Front. Pharmacol. 2018, 9, 1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allardyce, C.S.; Dyson, P.J. Ruthenium in medicine: Current clinical uses and future prospects. Platin. Metals Rev. 2001, 45, 62. [Google Scholar]
- Riccardi, C.; Musumeci, D.; Trifuoggi, M.; Irace, C.; Paduano, L.; Montesarchio, D. Anticancer Ruthenium(III) Complexes and Ru(III)-Containing Nanoformulations: An Update on the Mechanism of Action and Biological Activity. Pharmaceuticals 2019, 12, 146. [Google Scholar] [CrossRef] [Green Version]
- Schluga, P.; Hartinger, C.G.; Egger, A.; Reisner, E.; Galanski, M.; Jakupec, M.A.; Keppler, B.K. Redox behavior of tumor-inhibiting ruthenium(III) complexes and effects of physiological reductants on their binding to GMP. Dalton Trans. 2006, 14, 1796–1802. [Google Scholar] [CrossRef]
- Wiśniewska, J.; Fandzloch, M.; Łakomska, I. The reduction of ruthenium(III) complexes with triazolopyrimidine ligands by ascorbic acid and mechanistic insight into their action in anticancer therapy. Inorg. Chim. Acta 2019, 484, 305–310. [Google Scholar] [CrossRef]
- Blazevic, A.; Hummer, A.A.; Heffeter, P.; Berger, W.; Filipits, M.; Cibin, G.; Keppler, B.K.; Rompel, A. Electronic State of Sodium trans-[Tetrachloridobis (1 H-indazole) ruthenate (III)](NKP-1339) in Tumor, Liver and Kidney Tissue of a SW480-bearing Mouse. Sci. Rep. 2017, 7, 40966. [Google Scholar] [CrossRef] [PubMed]
- Beckford, F.; Dourth, D.; Shaloski, M.; Didion, J.; Thessing, J.; Woods, J.; Crowell, V.; Gerasimchuk, N.; Gonzalez-Sarrías, A.; Seeram, N.P. Half-sandwich ruthenium–arene complexes with thiosemicarbazones: Synthesis and biological evaluation of [(η6-p-cymene)Ru(piperonal thiosemicarbazones)Cl]Cl complexes. J. Inorg. Biochem. 2011, 105, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Beckford, F.A.; Thessing, J.; Shaloski, M.; Mbarushimana, P.C.; Brock, A.; Didion, J.; Woods, J.; Gonzalez-Sarrías, A.; Seeram, N.P. Synthesis and characterization of mixed-ligand diimine-piperonal thiosemicarbazone complexes of ruthenium(II): Biophysical investigations and biological evaluation as anticancer and antibacterial agents. J. Mol. Struct. 2011, 992, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battistin, F.; Scaletti, F.; Balducci, G.; Pillozzi, S.; Arcangeli, A.; Messori, L.; Alessio, E. Water-soluble Ru(II)- and Ru(III)-halide-PTA complexes (PTA=1,3,5-triaza-7-phosphaadamantane): Chemical and biological properties. J. Inorg. Biochem. 2016, 160, 180–188. [Google Scholar] [CrossRef]
- Camm, K.D.; El-Sokkary, A.; Gott, A.L.; Stockley, P.G.; Belyaeva, T.; McGowan, P.C. Synthesis, molecular structure and evaluation of new organometallic ruthenium anticancer agents. Dalton Trans. 2009, 48, 10914–10925. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.; Westhorpe, A.; Romero, M.J.; Habtemariam, A.; Gallevo, C.R.; Bark, Y.; Menezes, N.; Sadler, P.J.; Sharma, R.A. Radiosensitisation of human colorectal cancer cells by ruthenium(II) arene anticancer complexes. Sci. Rep. 2016, 6, 20596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Gallardo, J.; Elie, B.T.; Sanaú, M.; Contel, M. Versatile synthesis of cationic N-heterocyclic carbene–Gold(I) complexes containing a second ancillary ligand. Design of heterobimetallic ruthenium–gold anticancer agents. Chem. Commun. 2016, 52, 3155–3158. [Google Scholar] [CrossRef] [Green Version]
- Dabiri, Y.; Schmid, A.; Theobald, J.; Blagojevic, B.; Streciwilk, W.; Ott, I.; Wölfl, S.; Cheng, X. A Ruthenium(II) N-Heterocyclic Carbene (NHC) Complex with Naphthalimide Ligand Triggers Apoptosis in Colorectal Cancer Cells via Activating the ROS-p38 MAPK Pathway. Int. J. Mol. Sci. 2018, 19, 3964. [Google Scholar] [CrossRef] [Green Version]
- Baliza, I.R.S.; Silva, S.L.R.; Santos, L.D.S.; Neto, J.H.A.; Dias, R.B.; Sales, C.B.S.; Rocha, C.A.G.; Soares, M.B.P.; Batista, A.A.; Bezerra, D.P. Ruthenium Complexes With Piplartine Cause Apoptosis Through MAPK Signaling by a p53-Dependent Pathway in Human Colon Carcinoma Cells and Inhibit Tumor Development in a Xenograft Model. Front. Oncol. 2019, 9, 582. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.L.R.; Baliza, I.R.S.; Dias, R.B.; Sales, C.B.S.; Rocha, C.A.G.; Soares, M.B.P.; Correa, R.S.; Batista, A.A.; Bezerra, D.P. Ru(II)-thymine complex causes DNA damage and apoptotic cell death in human colon carcinoma HCT116 cells mediated by JNK/p38/ERK1/2 via a p53-independent signaling. Sci. Rep. 2019, 9, 11094. [Google Scholar] [CrossRef] [Green Version]
- Jin, G.; Zhao, Z.; Chakraborty, T.; Mandal, A.; Roy, A.; Roy, S.; Guo, Z. Decrypting the Molecular Mechanistic Pathways Delineating the Chemotherapeutic Potential of Ruthenium-Phloretin Complex in Colon Carcinoma Correlated with the Oxidative Status and Increased Apoptotic Events. Oxid. Med. Cell. Longev. 2020, 2020, 7690845. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Das, R.; Ghosh, B.; Chakraborty, T. Deciphering the biochemical and molecular mechanism underlying the in vitro and in vivo chemotherapeutic efficacy of ruthenium quercetin complex in colon cancer. Mol. Carcinog. 2018, 57, 700–721. [Google Scholar] [CrossRef]
- Wang, Y.; Bian, L.; Chakraborty, T.; Ghosh, T.; Chanda, P.; Roy, S. Construing the Biochemical and Molecular Mechanism Underlying the in Vivo and in Vitro Chemotherapeutic Efficacy of Ruthenium-Baicalein Complex in Colon Cancer. Int. J. Biol. Sci. 2019, 15, 1052–1071. [Google Scholar] [CrossRef] [Green Version]
- Hayward, R.L.; Schornagel, Q.C.; Tente, R.; Macpherson, J.S.; Aird, R.E.; Guichard, S.; Habtemariam, A.; Sadler, P.; Jodrell, D.I. Investigation of the role of Bax, p21/Waf1 and p53 as determinants of cellular responses in HCT116 colorectal cancer cells exposed to the novel cytotoxic ruthenium(II) organometallic agent, RM175. Cancer Chemother. Pharmacol. 2005, 55, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Jeong, Y.J.; Jo, J.-H.; Kang, S.C.; Lah, M.S.; Chi, K.-W. Anticancer Potency Studies of Coordination Driven Self-Assembled Arene–Ru-Based Metalla-Bowls. ChemBioChem 2014, 15, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Vidimar, V.; Licona, C.; Cerón-Camacho, R.; Guerin, E.; Coliat, P.; Venkatasamy, A.; Ali, M.; Guenot, D.; Le Lagadec, R.; Jung, A.C.; et al. A redox ruthenium compound directly targets PHD2 and inhibits the HIF1 pathway to reduce tumor angiogenesis independently of p53. Cancer Lett. 2019, 440–441, 145–155. [Google Scholar] [CrossRef]
- Pelillo, C.; Mollica, H.; Eble, J.A.; Grosche, J.; Herzog, L.; Codan, B.; Sava, G.; Bergamo, A. Inhibition of adhesion, migration and of α5β1 integrin in the HCT-116 colorectal cancer cells treated with the ruthenium drug NAMI-A. J. Inorg. Biochem. 2016, 160, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Flocke, L.S.; Trondl, R.; Jakupec, M.A.; Keppler, B.K. Molecular mode of action of NKP-1339—A clinically investigated ruthenium-based drug—Involves ER-and ROS-related effects in colon carcinoma cell lines. Investig. New Drugs 2016, 34, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonfili, L.; Pettinari, R.; Cuccioloni, M.; Cecarini, V.; Mozzicafreddo, M.; Angeletti, M.; Lupidi, G.; Marchetti, F.; Pettinari, C.; Eleuteri, A.M. Arene—RuII Complexes of Curcumin Exert Antitumor Activity via Proteasome Inhibition and Apoptosis Induction. ChemMedChem 2012, 7, 2010–2020. [Google Scholar] [CrossRef]
- Xu, Z.; Huang, J.; Kong, D.; Yang, Y.; Guo, L.; Jia, X.; Zhong, G.; Liu, Z. Potent half-sandwich Ru(Ⅱ) N^N (aryl-BIAN) complexes: Lysosome-mediated apoptosis, in vitro and in vivo anticancer activities. Eur. J. Med. Chem. 2020, 207, 112763. [Google Scholar] [CrossRef]
- Fong, J.; Kasimova, K.; Arenas, Y.; Kaspler, P.; Lazic, S.; Mandel, A.; Lilge, L. A novel class of ruthenium-based photosensitizers effectively kills in vitro cancer cells and in vivo tumors. Photochem. Photobiol. Sci. 2015, 14, 2014–2023. [Google Scholar] [CrossRef]
- Cho, Y.-H.; Ro, E.J.; Yoon, J.-S.; Mizutani, T.; Kang, D.-W.; Park, J.-C.; Il Kim, T.; Clevers, H.; Choi, K.-Y. 5-FU promotes stemness of colorectal cancer via p53-mediated WNT/β-catenin pathway activation. Nat. Commun. 2020, 11, 5321. [Google Scholar] [CrossRef]
- Su, W.; Qian, Q.; Li, P.; Lei, X.; Xiao, Q.; Huang, S.; Huang, C.; Cui, J. Synthesis, Characterization, and Anticancer Activity of a Series of Ketone-N4-Substituted Thiosemicarbazones and Their Ruthenium(II) Arene Complexes. Inorg. Chem. 2013, 52, 12440–12449. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, P.; Yu, B.; Chen, Y.; Wang, J.; Ji, L.; Chao, H. Targeting Nucleus DNA with a Cyclometalated Dipyridophenazineruthenium(II) Complex. J. Med. Chem. 2014, 57, 8971–8983. [Google Scholar] [CrossRef] [PubMed]
- Coury, J.E.; McFail-Isom, L.; Williams, L.D.; Bottomley, L.A. A novel assay for drug-DNA binding mode, affinity, and exclusion number: Scanning force microscopy. Proc. Natl. Acad. Sci. USA 1996, 93, 12283–12286. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Liang, Y.; Huang, C.; Su, W.; Lei, X.; Liu, Y.; Xiao, Q. Systematical investigation of binding interaction between novel ruthenium(II) arene complex with curcumin analogs and ctDNA. Luminescence 2016, 31, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-K.; Parkinson, J.A.; Bella, J.; Wang, F.; Sadler, P.J. Penetrative DNA intercalation and G-base selectivity of an organometallic tetrahydroanthracene RuII anticancer complex. Chem. Sci. 2010, 1, 258–270. [Google Scholar] [CrossRef] [Green Version]
- Martinez, R.; Chacon-Garcia, L. The Search of DNA-Intercalators as Antitumoral Drugs: What it Worked and What did not Work. Curr. Med. Chem. 2005, 12, 127–151. [Google Scholar] [CrossRef]
- Liu, S.; Liang, A.; Wu, K.; Zeng, W.; Luo, Q.; Wang, F. Binding of Organometallic Ruthenium Anticancer Complexes to DNA: Thermodynamic Base and Sequence Selectivity. Int. J. Mol. Sci. 2018, 19, 2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bešker, N.; Coletti, C.; Marrone, A.; Re, N. Binding of antitumor ruthenium complexes to DNA and proteins: A theoretical approach. J. Phys. Chem. B 2007, 111, 9955–9964. [Google Scholar] [CrossRef]
- Das, D.; Dutta, A.; Mondal, P. Interaction of aquated form of ruthenium (III) anticancer complexes with normal and mismatch base pairs: A density functional theoretical study. Comput. Theor. Chem. 2015, 1072, 28–36. [Google Scholar] [CrossRef]
- Zeng, W.; Zhang, Y.; Zheng, W.; Luo, Q.; Han, J.; Liu, J.a.; Zhao, Y.; Jia, F.; Wu, K.; Wang, F. Discovery of cisplatin binding to thymine and cytosine on a single-stranded oligodeoxynucleotide by high resolution FT-ICR mass spectrometry. Molecules 2019, 24, 1852. [Google Scholar] [CrossRef] [Green Version]
- Faivre, S.; Chan, D.; Salinas, R.; Woynarowska, B.; Woynarowski, J.M. DNA strand breaks and apoptosis induced by oxaliplatin in cancer cells. Biochem. Pharmacol. 2003, 66, 225–237. [Google Scholar] [CrossRef]
- Ray, B.; Gupta, B.; Mehrotra, R. Binding of platinum derivative, oxaliplatin to deoxyribonucleic acid: Structural insight into antitumor action. J. Biomol. Struct. Dyn. 2019, 37, 3838–3847. [Google Scholar] [CrossRef]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anti-Cancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Zaniboni, A.; Labianca, R.; Pancera, G.; Barni, S.; Frontini, L.; Marini, G.; Luporini, G. Oral Etoposide as Second-Line Chemotherapy for Colorectal Cancer: A GISCAD Study. J. Chemother. 1995, 7, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Seminara, P.; Pastore, C.; Iascone, C.; Cicconetti, F.; Nigita, G.; Ielapi, T.; Franchi, F. Mitomycin C and Etoposide in Advanced Colorectal Carcinoma. Chemotheraphy 2007, 53, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Passalacqua, R.; Bisagni, G.; Cocconi, G.; Boni, C.; Di Blasio, B.; Ceci, G. Cisplatin and etoposide in advanced colorectal carcinoma. Ann. Oncol. 1991, 2, 687–688. [Google Scholar] [CrossRef]
- Zhou, G.; Yang, J.; Song, P. Correlation of ERK/MAPK signaling pathway with proliferation and apoptosis of colon cancer cells. Oncol. Lett. 2019, 17, 2266–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhou, L.; Bao, Y.L.; Wu, Y.; Yu, C.L.; Huang, Y.X.; Sun, Y.; Zheng, L.H.; Li, Y.X. Butyrate induces cell apoptosis through activation of JNK MAP kinase pathway in human colon cancer RKO cells. Chem.-Biol. Interact. 2010, 185, 174–181. [Google Scholar] [CrossRef]
- Shin, D.Y.; Lee, W.S.; Lu, J.N.; Kang, M.H.; Ryu, C.H.; Kim, G.Y.; Kang, H.S.; Shin, S.C.; Choi, Y.H. Induction of apoptosis in human colon cancer HCT-116 cells by anthocyanins through suppression of Akt and activation of p38-MAPK. Int. J. Oncol. 2009, 35, 1499–1504. [Google Scholar]
- Zilfou, J.T.; Lowe, S.W. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Med. 2009, 1, a001883. [Google Scholar] [CrossRef] [PubMed]
- Florindo, P.R.; Pereira, D.M.; Borralho, P.M.; Rodrigues, C.M.P.; Piedade, M.F.M.; Fernandes, A.C. Cyclopentadienyl–Ruthenium(II) and Iron(II) Organometallic Compounds with Carbohydrate Derivative Ligands as Good Colorectal Anticancer Agents. J. Med. Chem. 2015, 58, 4339–4347. [Google Scholar] [CrossRef] [PubMed]
- Ude, Z.; Romero-Canelón, I.; Twamley, B.; Fitzgerald Hughes, D.; Sadler, P.J.; Marmion, C.J. A novel dual-functioning ruthenium(II)–arene complex of an anti-microbial ciprofloxacin derivative—Anti-proliferative and anti-microbial activity. J. Inorg. Biochem. 2016, 160, 210–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, M.J.; Licona, C.; Yuan Qiang Wong, D.; Pastorin, G.; Gaiddon, C.; Ang, W.H. Discovery and Investigation of Anticancer Ruthenium–Arene Schiff-Base Complexes via Water-Promoted Combinatorial Three-Component Assembly. J. Med. Chem. 2014, 57, 6043–6059. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Sen, U.; Roy, N.; Muthukumar, V.; Sahoo, S.K.; Bose, B.; Paira, P. DNA targeting half sandwich Ru(II)-p-cymene-N^N complexes as cancer cell imaging and terminating agents: Influence of regioisomers in cytotoxicity. Dalton Trans. 2021, 50, 979–997. [Google Scholar] [CrossRef] [PubMed]
- Kapitza, S.; Jakupec, M.A.; Uhl, M.; Keppler, B.K.; Marian, B. The heterocyclic ruthenium(III) complex KP1019 (FFC14A) causes DNA damage and oxidative stress in colorectal tumor cells. Cancer Lett. 2005, 226, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Wernitznig, D.; Kiakos, K.; Del Favero, G.; Harrer, N.; Machat, H.; Osswald, A.; Jakupec, M.A.; Wernitznig, A.; Sommergruber, W.; Keppler, B.K. First-in-class ruthenium anticancer drug (KP1339/IT-139) induces an immunogenic cell death signature in colorectal spheroids in vitro. Metallomics 2019, 11, 1044–1048. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, J.-S.; Lin, S.-R.; Chang, M.-Y.; Chen, F.-M.; Lu, C.-Y.; Huang, T.-J.; Huang, Y.-S.; Huang, C.-J.; Wang, J.-Y. APC, K-ras, and p53 Gene Mutations in Colorectal Cancer Patients: Correlation to Clinicopathologic Features and Postoperative Surveillance. Am. Surg. 2005, 71, 336–343. [Google Scholar] [CrossRef]
- Nakayama, M.; Oshima, M. Mutant p53 in colon cancer. J. Mol. Cell Biol. 2018, 11, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Fodde, R. The APC gene in colorectal cancer. Eur. J. Cancer 2002, 38, 867–871. [Google Scholar] [CrossRef]
- Kwong, L.N.; Dove, W.F. APC and its modifiers in colon cancer. Adv. Exp. Med. Biol. 2009, 656, 85–106. [Google Scholar] [PubMed] [Green Version]
- Kaeser, M.D.; Pebernard, S.; Iggo, R.D. Regulation of p53 Stability and Function in HCT116 Colon Cancer Cells. J. Biol. Chem. 2004, 279, 7598–7605. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Lovejoy, K.S.; Shima, J.E.; Lagpacan, L.L.; Shu, Y.; Lapuk, A.; Chen, Y.; Komori, T.; Gray, J.W.; Chen, X.; et al. Organic Cation Transporters Are Determinants of Oxaliplatin Cytotoxicity. Cancer Res. 2006, 66, 8847–8857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landis-Piwowar, K.R.; Milacic, V.; Chen, D.; Yang, H.; Zhao, Y.; Chan, T.H.; Yan, B.; Dou, Q.P. The proteasome as a potential target for novel anticancer drugs and chemosensitizers. Drug Resist. Updates 2006, 9, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Mani, A.; Gelmann, E.P. The ubiquitin-proteasome pathway and its role in cancer. J. Clin. Oncol. 2005, 23, 4776–4789. [Google Scholar] [CrossRef]
- Richardson, P.G.; Mitsiades, C.; Hideshima, T.; Anderson, K.C. Proteasome Inhibition in the Treatment of Cancer. Cell Cycle 2005, 4, 289–295. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Richardson, P.G.; Hideshima, T.; Anderson, K.C. Proteasome Inhibition As a Novel Therapeutic Target in Human Cancer. J. Clin. Oncol. 2005, 23, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Milacic, V.; Banerjee, S.; Landis-Piwowar, K.R.; Sarkar, F.H.; Majumdar, A.P.N.; Dou, Q.P. Milacic, V.; Banerjee, S.; Landis-Piwowar, K.R.; Sarkar, F.H.; et al. Curcumin inhibits the proteasome activity in human colon cancer cells in vitro and in vivo. Cancer Res. 2008, 68, 7283–7292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.B.; Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [Green Version]
- Arnold, J.; Grune, T. PARP-mediated proteasome activation: A co-ordination of DNA repair and protein degradation? BioEssays 2002, 24, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Kotsafti, A.; Scarpa, M.; Castagliuolo, I.; Scarpa, M. Reactive Oxygen Species and Antitumor Immunity—From Surveillance to Evasion. Cancers 2020, 12, 1748. [Google Scholar] [CrossRef]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updates 2004, 7, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Sreevalsan, S.; Safe, S. Reactive Oxygen Species and Colorectal Cancer. Curr. Colorectal Cancer Rep. 2013, 9, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Hartinger, C.G.; Jakupec, M.A.; Zorbas-Seifried, S.; Groessl, M.; Egger, A.; Berger, W.; Zorbas, H.; Dyson, P.J.; Keppler, B.K. KP1019, A New Redox-Active Anticancer Agent—Preclinical Development and Results of a Clinical Phase I Study in Tumor Patients. Chem. Biodivers. 2008, 5, 2140–2155. [Google Scholar] [CrossRef]
- Hartinger, C.G.; Zorbas-Seifried, S.; Jakupec, M.A.; Kynast, B.; Zorbas, H.; Keppler, B.K. From bench to bedside—Preclinical and early clinical development of the anticancer agent indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019 or FFC14A). J. Inorg. Biochem. 2006, 100, 891–904. [Google Scholar] [CrossRef]
- Peti, W.; Pieper, T.; Sommer, M.; Keppler, B.K.; Giester, G. Synthesis of Tumor-Inhibiting Complex Salts Containing the Anion trans-Tetrachlorobis(indazole)ruthenate(III) and Crystal Structure of the Tetraphenylphosphonium Salt. Eur. J. Inorg. Chem. 1999, 1999, 1551–1555. [Google Scholar] [CrossRef]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Wernitznig, D.; Favero, G.D.; Kiakos, K.; Harrer, N.; Machat, H.; Marko, D.; Jakupec, M.; Sommergruber, W.; Keppler, B.K. KP-1339 (IT-139) induces the hallmarks of immunogenic cell death in a colon cancer 3D model in vitro. Cancer Res. 2018, 78, 4395. [Google Scholar]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Kepp, O.; Menger, L.; Vacchelli, E.; Locher, C.; Adjemian, S.; Yamazaki, T.; Martins, I.; Sukkurwala, A.Q.; Michaud, M.; Senovilla, L.; et al. Crosstalk between ER stress and immunogenic cell death. Cytokine Growth Factor Rev. 2013, 24, 311–318. [Google Scholar] [CrossRef]
- Osman, R.; Tacnet-Delorme, P.; Kleman, J.-P.; Millet, A.; Frachet, P. Calreticulin Release at an Early Stage of Death Modulates the Clearance by Macrophages of Apoptotic Cells. Front. Immunol. 2017, 8, 1034. [Google Scholar] [CrossRef] [Green Version]
- Dudek, A.M.; Garg, A.D.; Krysko, D.V.; De Ruysscher, D.; Agostinis, P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 2013, 24, 319–333. [Google Scholar] [CrossRef]
- Ruan, H.; Leibowitz, B.J.; Zhang, L.; Yu, J. Immunogenic cell death in colon cancer prevention and therapy. Mol. Carcinog. 2020, 59, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhao, L.; Loos, F.; Iribarren, K.; Lachkar, S.; Zhou, H.; Gomes-da-Silva, L.C.; Chen, G.; Bezu, L.; Boncompain, G.; et al. Identification of pharmacological agents that induce HMGB1 release. Sci. Rep. 2017, 7, 14915. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, P.B.; Edes, K.; Nelson, C.C.; Parsawar, K.; Fitzpatrick, F.A.; Moos, P.J. Thioredoxin reductase is required for the inactivation of tumor suppressor p53 and for apoptosis induced by endogenous electrophiles. Carcinogenesis 2006, 27, 2538–2549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, S.; Müller-Ladner, U.; Neumann, E.; Spöttl, T.; Schlottmann, K.; Rüschoff, J.; Schölmerich, J.; Kullmann, F. Thioredoxin Reductase 1 Expression in Colon Cancer: Discrepancy between in Vitro and in Vivo Findings. Lab. Investig. 2003, 83, 1321–1331. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.-J.; Geng, W.-S.; Wang, Z.-Q.; Chen, L.; Zeng, X.-S. The role of thioredoxin system in cancer: Strategy for cancer therapy. Cancer Chemother. Pharmacol. 2019, 84, 453–470. [Google Scholar] [CrossRef]
- Burkitt, K.; Chun, S.Y.; Dang, D.T.; Dang, L.H. Targeting both HIF-1 and HIF-2 in human colon cancer cells improves tumor response to sunitinib treatment. Mol. Cancer Ther. 2009, 8, 1148–1156. [Google Scholar] [CrossRef] [Green Version]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharmacol. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Imamura, T.; Kikuchi, H.; Herraiz, M.-T.; Park, D.-Y.; Mizukami, Y.; Mino-Kenduson, M.; Lynch, M.P.; Rueda, B.R.; Benita, Y.; Xavier, R.J.; et al. HIF-1α and HIF-2α have divergent roles in colon cancer. Int. J. Cancer 2009, 124, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Sadlecki, P.; Bodnar, M.; Grabiec, M.; Marszalek, A.; Walentowicz, P.; Sokup, A.; Zegarska, J.; Walentowicz-Sadlecka, M. The role of Hypoxia-inducible factor-1α, glucose transporter-1,(GLUT-1) and carbon anhydrase IX in endometrial cancer patients. Biomed. Res. Int. 2014, 2014, 616850. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Jang, W.-C.; Fung, F.K.C.; Lo, A.C.Y.; Wong, I.Y.H. Up-Regulation of ENO1 by HIF-1α in Retinal Pigment Epithelial Cells after Hypoxic Challenge Is Not Involved in the Regulation of VEGF Secretion. PLoS ONE 2016, 11, e0147961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, W.; Cui, G.; Tang, C.-W.; Zhang, X.-L.; Dai, C.; Xu, Y.-Q.; Gong, H.; Xue, T.; Guo, H.-H.; Bao, Y. Role of glucose metabolism related gene GLUT1 in the occurrence and prognosis of colorectal cancer. Oncotarget 2017, 8, 56850–56857. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Wang, Y.; Zhao, S.; Liu, C.; Wang, Y.; Wen, M.; Mao, J.-H.; Wei, G.; Zhang, P. FBXW7 negatively regulates ENO1 expression and function in colorectal cancer. Lab. Investig. 2015, 95, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Takahashi, Y.; Liu, W.; Shaheen, R.M. Vascular Endothelial Growth Factor in Human Colon Cancer: Biology and Therapeutic Implications. Oncologist 2000, 5, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-Y.; He, Y.-L.; Zhu, L.-L. Possible Role of PHD Inhibitors as Hypoxia-Mimicking Agents in the Maintenance of Neural Stem Cells’ Self-Renewal Properties. Front. Cell Dev. Biol. 2018, 6, 169. [Google Scholar] [CrossRef]
- Leyva, L.; Sirlin, C.; Rubio, L.; Franco, C.; Le Lagadec, R.; Spencer, J.; Bischoff, P.; Gaiddon, C.; Loeffler, J.-P.; Pfeffer, M. Synthesis of Cycloruthenated Compounds as Potential Anticancer Agents. Eur. J. Inorg. Chem. 2007, 2007, 3055–3066. [Google Scholar] [CrossRef]
- Alessio, E.; Balducci, G.; Calligaris, M.; Costa, G.; Attia, W.M.; Mestroni, G. Synthesis, molecular structure, and chemical behavior of hydrogen trans-bis(dimethyl sulfoxide)tetrachlororuthenate(III) and mer-trichlorotris(dimethyl sulfoxide)ruthenium(III): The first fully characterized chloride-dimethyl sulfoxide-ruthenium(III) complexes. Inorg. Chem. 1991, 30, 609–618. [Google Scholar]
- Sava, G.; Zorzet, S.; Turrin, C.; Vita, F.; Soranzo, M.; Zabucchi, G.; Cocchietto, M.; Bergamo, A.; DiGiovine, S.; Pezzoni, G.; et al. Dual Action of NAMI-A in inhibition of solid tumor metastasis: Selective targeting of metastatic cells and binding to collagen. Clin. Cancer Res. 2003, 9, 1898–1905. [Google Scholar]
- Gava, B.; Zorzet, S.; Spessotto, P.; Cocchietto, M.; Sava, G. Inhibition of B16 melanoma metastases with the ruthenium complex imidazolium trans-imidazoledimethylsulfoxide-tetrachlororuthenate and down-regulation of tumor cell invasion. J. Pharmacol. Exp. Ther. 2006, 317, 284–291. [Google Scholar] [CrossRef]
- Zorzet, S.; Bergamo, A.; Cocchietto, M.; Sorc, A.; Gava, B.; Alessio, E.; Iengo, E.; Sava, G. Lack of In vitro cytotoxicity, associated to increased G(2)-M cell fraction and inhibition of matrigel invasion, may predict IN vivo-selective antimetastasis activity of ruthenium complexes. J. Pharmacol. Exp. Ther. 2000, 295, 927–933. [Google Scholar]
- Alessio, E. Thirty Years of the Drug Candidate NAMI-A and the Myths in the Field of Ruthenium Anticancer Compounds: A Personal Perspective. Eur. J. Inorg. Chem. 2017, 2017, 1549–1560. [Google Scholar] [CrossRef]
- Janouskova, H.; Ray, A.-M.; Noulet, F.; Lelong-Rebel, I.; Choulier, L.; Schaffner, F.; Lehmann, M.; Martin, S.; Teisinger, J.; Dontenwill, M. Activation of p53 pathway by Nutlin-3a inhibits the expression of the therapeutic target α5 integrin in colon cancer cells. Cancer Lett. 2013, 336, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Pelillo, C.; Chambery, A.; Sava, G. Influence of components of tumour microenvironment on the response of HCT-116 colorectal cancer to the ruthenium-based drug NAMI-A. J. Inorg. Biochem. 2017, 168, 90–97. [Google Scholar] [CrossRef] [Green Version]
- Pelillo, C.; Bergamo, A.; Mollica, H.; Bestagno, M.; Sava, G. Colorectal Cancer Metastases Settle in the Hepatic Microenvironment Through α5β1 Integrin. J. Cell. Biochem. 2015, 116, 2385–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Hochwald, S.N. The role of FAK in tumor metabolism and therapy. Pharmacol. Ther. 2014, 142, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Tian, Z.; Xu, Z.; Zhang, S.; Feng, Y.; Zhang, L.; Liu, Z. Highly potent half-sandwich iridium and ruthenium complexes as lysosome-targeted imaging and anticancer agents. Dalton Trans. 2018, 47, 15772–15782. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Li, J.; Zhang, S.; Xu, Z.; Yang, Y.; Kong, D.; Zhang, H.; Ge, X.; Zhang, J.; Liu, Z. Lysosome-Targeted Chemotherapeutics: Half-Sandwich Ruthenium(II) Complexes That Are Selectively Toxic to Cancer Cells. Inorg. Chem. 2018, 57, 10498–10502. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; Jäättelä, M. Lysosomal involvement in cell death and cancer. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 746–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piao, S.; Amaravadi, R.K. Targeting the lysosome in cancer. Ann. N. Y. Acad. Sci. 2016, 1371, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Dielschneider, R.F.; Henson, E.S.; Gibson, S.B. Lysosomes as Oxidative Targets for Cancer Therapy. Oxid. Med. Cell. Longev. 2017, 2017, 3749157. [Google Scholar] [CrossRef] [Green Version]
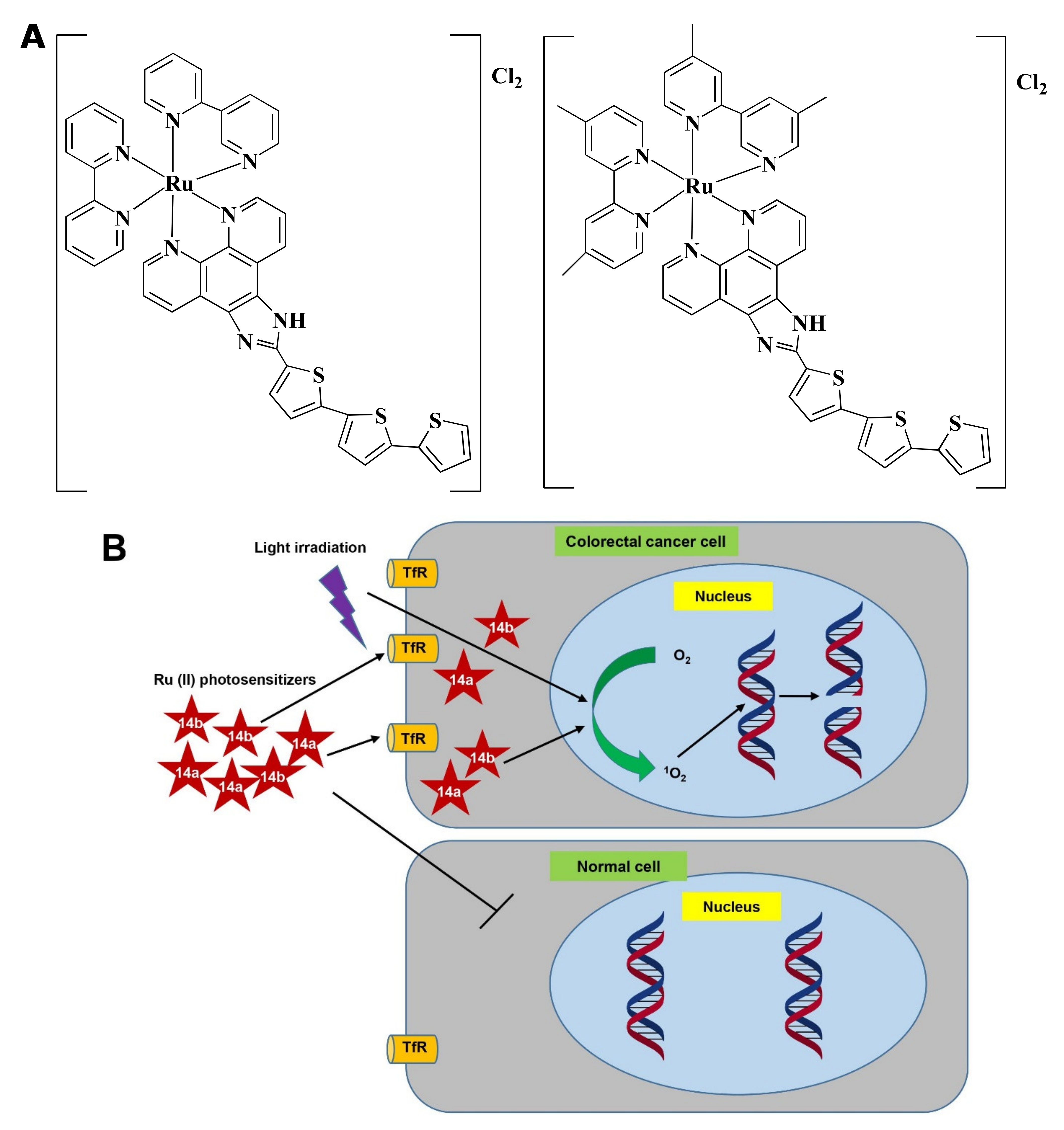
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Niloy, M.S.; Shakil, M.S.; Hossen, M.S.; Alam, M.; Rosengren, R.J. Promise of gold nanomaterials as a lung cancer theranostic agent: A systematic review. Int. Nano Lett. 2021, 11, 93–111. [Google Scholar] [CrossRef]
- Monro, S.; Colón, K.L.; Yin, H.; Roque, J.; Konda, P.; Gujar, S.; Thummel, R.P.; Lilge, L.; Cameron, C.G.; McFarland, S.A. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2019, 119, 797–828. [Google Scholar] [CrossRef]
- Levina, A.; Mitra, A.; Lay, P.A. Recent developments in ruthenium anticancer drugs. Metallomics 2009, 1, 458–470. [Google Scholar] [CrossRef]
- Zhou, Q.-X.; Lei, W.-H.; Sun, Y.; Chen, J.-R.; Li, C.; Hou, Y.-J.; Wang, X.-S.; Zhang, B.-W. [Ru(bpy)3−n(dpb)n]2+: Unusual Photophysical Property and Efficient DNA Photocleavage Activity. Inorg. Chem. 2010, 49, 4729–4731. [Google Scholar] [CrossRef]
- Doherty, R.E.; Sazanovich, I.V.; McKenzie, L.K.; Stasheuski, A.S.; Coyle, R.; Baggaley, E.; Bottomley, S.; Weinstein, J.A.; Bryant, H.E. Photodynamic killing of cancer cells by a Platinum(II) complex with cyclometallating ligand. Sci. Rep. 2016, 6, 22668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, S.A.; Farrell, D.; Grodzinski, P. Nanotechnologies in Cancer Treatment and Diagnosis. J. Natl. Compr. Cancer Netw. 2014, 12, 1727–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampado, R.; Crotti, S.; Caliceti, P.; Pucciarelli, S.; Agostini, M. Nanovectors Design for Theranostic Applications in Colorectal Cancer. J. Oncol. 2019, 2019, 2740923. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Xu, L.; Song, G.; Liu, Z. Emerging nanomedicine approaches fighting tumor metastasis: Animal models, metastasis-targeted drug delivery, phototherapy, and immunotherapy. Chem. Soc. Rev. 2016, 45, 6250–6269. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Song, G.; Liang, C.; Yi, X.; Zhao, Q.; Cheng, L.; Yang, K.; Liu, Z. Perfluorocarbon-loaded hollow Bi2Se3 nanoparticles for timely supply of oxygen under near-infrared light to enhance the radiotherapy of cancer. Adv. Mater. 2016, 28, 2716–2723. [Google Scholar] [CrossRef]
- Heffeter, P.; Riabtseva, A.; Senkiv, Y.; Kowol, C.R.; Körner, W.; Jungwith, U.; Mitina, N.; Keppler, B.K.; Konstantinova, T.; Yanchuk, I.; et al. Nanoformulation Improves Activity of the (pre)Clinical Anticancer Ruthenium Complex KP1019. J. Biomed. Nanotechnol. 2014, 10, 877–884. [Google Scholar] [CrossRef]
- Liu, H.-J.; Luan, X.; Feng, H.-Y.; Dong, X.; Yang, S.-C.; Chen, Z.-J.; Cai, Q.-Y.; Lu, Q.; Zhang, Y.; Sun, P.; et al. Integrated Combination Treatment Using a “Smart” Chemotherapy and MicroRNA Delivery System Improves Outcomes in an Orthotopic Colorectal Cancer Model. Adv. Funct. Mater. 2018, 28, 1801118. [Google Scholar] [CrossRef]
- Tomlinson, J.S.; Jarnagin, W.R.; DeMatteo, R.P.; Fong, Y.; Kornprat, P.; Gonen, M.; Kemeny, N.; Brennan, M.F.; Blumgart, L.H.; D’Angelica, M. Actual 10-year survival after resection of colorectal liver metastases defines cure. J. Clin. Oncol. 2007, 25, 4575–4580. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Cheng, L.; Gong, F.; Dong, Z.; Liang, C.; Wang, M.; Feng, L.; Li, Y.; Liu, Z.; Li, C.; et al. Platinum nanoworms for imaging-guided combined cancer therapy in the second near-infrared window. J. Mater. Chem. B 2018, 6, 5069–5079. [Google Scholar] [CrossRef]
- Zeng, X.; Sun, J.; Li, S.; Shi, J.; Gao, H.; Sun Leong, W.; Wu, Y.; Li, M.; Liu, C.; Li, P.; et al. Blood-triggered generation of platinum nanoparticle functions as an anti-cancer agent. Nat. Commun. 2020, 11, 567. [Google Scholar] [CrossRef] [Green Version]
- Gehrke, H.; Pelka, J.; Hartinger, C.G.; Blank, H.; Bleimund, F.; Schneider, R.; Gerthsen, D.; Bräse, S.; Crone, M.; Türk, M.; et al. Platinum nanoparticles and their cellular uptake and DNA platination at non-cytotoxic concentrations. Arch. Toxicol. 2011, 85, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Porcel, E.; Liehn, S.; Remita, H.; Usami, N.; Kobayashi, K.; Furusawa, Y.; Le Sech, C.; Lacombe, S. Platinum nanoparticles: A promising material for future cancer therapy? Nanotechnology 2010, 21, 85103. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Dang, M.; Tao, J.; Li, Y.; Tang, Y. Mesoporous platinum nanoparticle-based nanoplatforms for combined chemo-photothermal breast cancer therapy. J. Colloid Interface Sci. 2020, 570, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Shieh, M.-J. Platinum(II) Drug-Loaded Gold Nanoshells for Chemo-Photothermal Therapy in Colorectal Cancer. ACS Appl. Mater. Interfaces 2020, 12, 4254–4264. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Lee, J.J.; Siu, L.L. Dose escalation methods in phase I cancer clinical trials. J. Natl. Cancer Inst. 2009, 101, 708–720. [Google Scholar] [CrossRef] [Green Version]
- Rademaker-Lakhai, J.M.; van den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H.M.; Rademaker-Lakhai, J.M.; Van Den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H. A phase I and pharmacological study with imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a novel ruthenium anticancer agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef] [Green Version]
- Leijen, S.; Burgers, S.A.; Baas, P.; Pluim, D.; Tibben, M.; van Werkhoven, E.; Alessio, E.; Sava, G.; Beijnen, J.H.; Schellens, J.H. Phase I/II study with ruthenium compound NAMI-A and gemcitabine in patients with non-small cell lung cancer after first line therapy. Investig. New Drugs 2015, 33, 201–214. [Google Scholar] [CrossRef] [Green Version]
- Lentz, F.; Drescher, A.; Lindauer, A.; Henke, M.; Hilger, R.A.; Hartinger, C.G.; Scheulen, M.E.; Dittrich, C.; Keppler, B.K.; Jaehde, U. Pharmacokinetics of a novel anticancer ruthenium complex (KP1019, FFC14A) in a phase I dose-escalation study. Anticancer Drugs 2009, 20, 97–103. [Google Scholar] [CrossRef]
- Thompson, D.S.; Weiss, G.J.; Jones, S.F.; Burris, H.A.; Ramanathan, R.K.; Infante, J.R.; Bendell, J.C.; Ogden, A.; Von Hoff, D.D. NKP-1339: Maximum tolerated dose defined for first-in-human GRP78 targeted agent. J. Clin. Oncol. 2012, 30, 3033. [Google Scholar] [CrossRef]
- Trondl, R.; Heffeter, P.; Kowol, C.R.; Jakupec, M.A.; Berger, W.; Keppler, B.K. NKP-1339, the first ruthenium-based anticancer drug on the edge to clinical application. Chem. Sci. 2014, 5, 2925–2932. [Google Scholar] [CrossRef] [Green Version]
- clinicaltrial.gov. Available online: https://clinicaltrials.gov (accessed on 17 July 2021).
- Burris, H.A.; Bakewell, S.; Bendell, J.C.; Infante, J.; Jones, S.F.; Spigel, D.R.; Weiss, G.J.; Ramanathan, R.K.; Ogden, A.; Von Hoff, D. Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: A first-in-human, open-label, dose-escalation phase I study with expansion cohort. ESMO Open 2016, 1, e000154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beale, P.; Judson, I.; O’Donnell, A.; Trigo, J.; Rees, C.; Raynaud, F.; Turner, A.; Simmons, L.; Etterley, L. A Phase I clinical and pharmacological study of cis-diamminedichloro(2-methylpyridine) platinum II (AMD473). Br. J. Cancer 2003, 88, 1128–1134. [Google Scholar] [CrossRef]
- Goldberg, R.M.; Sargent, D.J.; Morton, R.F.; Fuchs, C.S.; Ramanathan, R.K.; Williamson, S.K.; Findlay, B.P.; Pitot, H.C.; Alberts, S.R. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J. Clin. Oncol. 2004, 22, 23–30. [Google Scholar] [CrossRef]
- De Gramont, A.; Figer, A.; Seymour, M.; Homerin, M.; Hmissi, A.; Cassidy, J.; Boni, C.; Cortes-Funes, H.; Cervantes, A.; Freyer, G.; et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J. Clin. Oncol. 2000, 18, 2938–2947. [Google Scholar] [CrossRef]
- Cassidy, J.; Clarke, S.; Díaz-Rubio, E.; Scheithauer, W.; Figer, A.; Wong, R.; Koski, S.; Rittweger, K.; Gilberg, F.; Saltz, L. XELOX vs. FOLFOX-4 as first-line therapy for metastatic colorectal cancer: NO16966 updated results. Br. J. Cancer 2011, 105, 58–64. [Google Scholar] [CrossRef]
- Earhart, R.; Cheporov, S.; Gladkov, O.; Biakhov, M.; Breitz, H.; De Jager, R. Randomized phase II study of picoplatin in combination with 5-fluorouracil and leucovorin (FOLPI) as a neuropathy-sparing alternative to modified FOLFOX-6 as first-line therapy for colorectal cancer (CRC). J. Clin. Oncol. 2009, 27, 4026. [Google Scholar] [CrossRef]
- Hartmann, J.T.; Lipp, H.P. Toxicity of platinum compounds. Expert Opin. Pharmacother. 2003, 4, 889–901. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Park, G.Y.; Lippard, S. Understanding and improving platinum anticancer drugs–phenanthriplatin. Anticancer Res. 2014, 34, 471–476. [Google Scholar]
- Dos Santos, E.R.; Graminha, A.E.; Schultz, M.S.; Correia, I.; Selistre-de-Araújo, H.S.; Correa, R.S.; Ellena, J.; Elisângela de Paula, S.L.; Pessoa, J.C.; Batista, A.A. Cytotoxic activity and structural features of Ru(II)/phosphine/amino acid complexes. J. Inorg. Biochem. 2018, 182, 48–60. [Google Scholar] [CrossRef]
- Mello-Andrade, F.; Cardoso, C.G.; e Silva, C.R.; Chen-Chen, L.; de Melo-Reis, P.R.; de Lima, A.P.; Oliveira, R.; Ferraz, I.B.M.; Grisolia, C.K.; Almeida, M.A.P. Acute toxic effects of ruthenium (II)/amino acid/diphosphine complexes on Swiss mice and zebrafish embryos. Biomed. Pharmacother. 2018, 107, 1082–1092. [Google Scholar] [CrossRef]
- Bergamo, A.; Riedel, T.; Dyson, P.J.; Sava, G. Preclinical combination therapy of the investigational drug NAMI-A+ with doxorubicin for mammary cancer. Investig. New Drugs 2015, 33, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Gagliardi, R.; Scarcia, V.; Furlani, A.; Alessio, E.; Mestroni, G.; Sava, G. In vitro cell cycle arrest, in vivo action on solid metastasizing tumors, and host toxicity of the antimetastatic drug NAMI-A and cisplatin. J. Pharmacol. Exp. Ther. 1999, 289, 559–564. [Google Scholar] [PubMed]
- Koch, J.H.; Gyarfas, E.C.; Dwyer, F. Biological Activity of Complex Ions Mechanism of Inhibition of Acetylcholinesterase. Aust. J. Biol. Sci. 1956, 9, 371–381. [Google Scholar] [CrossRef]
- Fiskum, G.; Cockrell, R. Ruthenium red sensitive and insensitive calcium transport in rat liver and Ehrlich ascites tumor cell mitochondria. FEBS Lett. 1978, 92, 125–128. [Google Scholar] [CrossRef] [Green Version]
- Rahamimoff, R.; Alnaes, E. Inhibitory Action of Ruthenium Red on Neuromuscular Transmission. Proc. Natl. Acad. Sci. USA 1973, 70, 3613–3616. [Google Scholar] [CrossRef] [Green Version]
- Kruszyna, H.; Kruszyna, R.; Hurst, J.; Smith, R.P. Toxicology and pharmacology of some ruthenium compounds: Vascular smooth muscle relaxation by nitrosyl derivatives of ruthenium and iridium. J. Toxicol. Environ. Health 1980, 6, 757–773. [Google Scholar] [CrossRef]
- Shakil, M.S.; Parveen, S.; Rana, Z.; Walsh, F.; Movassaghi, S.; Söhnel, T.; Azam, M.; Shaheen, M.A.; Jamieson, S.M.F.; Hanif, M.; et al. High Antiproliferative Activity of Hydroxythiopyridones over Hydroxypyridones and Their Organoruthenium Complexes. Biomedicines 2021, 9, 123. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds or Drugs | Oxidation State | Assay Name | CRC Cell Lines and IC50 (µM) | Up-Regulated Protein | Down Regulated Protein | Cell Cycle Arrest | Corresponding Conventional Drugs IC50 (µM) | References |
|---|---|---|---|---|---|---|---|---|
| 1a | III | TBE | HCT116 (>100) * | NR | NR | NR | HCT116 (CIS = 7.65 µM) * | [75] |
| 1b | II | SRB | LS174T (7.7 µmol/L), LoVo (8.1 µmol/L) ***** | NR | NR | NR | LS174T (CIS = 4.6), LoVo (CIS = 0.7) ***** | [76] |
| 1c, 1d | II | CS | DLD1(1c = 10.2), (1d = 7.5) * | p53, p21, GAPDH | PARP | G2/M | DLD1 (OXA = 11.3) * | [77] |
| 2a, 2b | II | MTT | HCT116 (2a = 50.5, 2b = 153), Caco-2 (2a = 26.3, 2b = 121) *** | NR | NR | NR | HCT116 (CIS = 41.7, ETP = 18.3), Caco-2 (CIS = 14.9, ETP = 16.5) *** | [73] |
| 2c | II | MTT | HCT116 (8.6), Caco-2 (6.6) *** | NR | NR | NR | HCT116 (ETP = 18.3), Caco-2 (ETP = 16.5) *** | [74] |
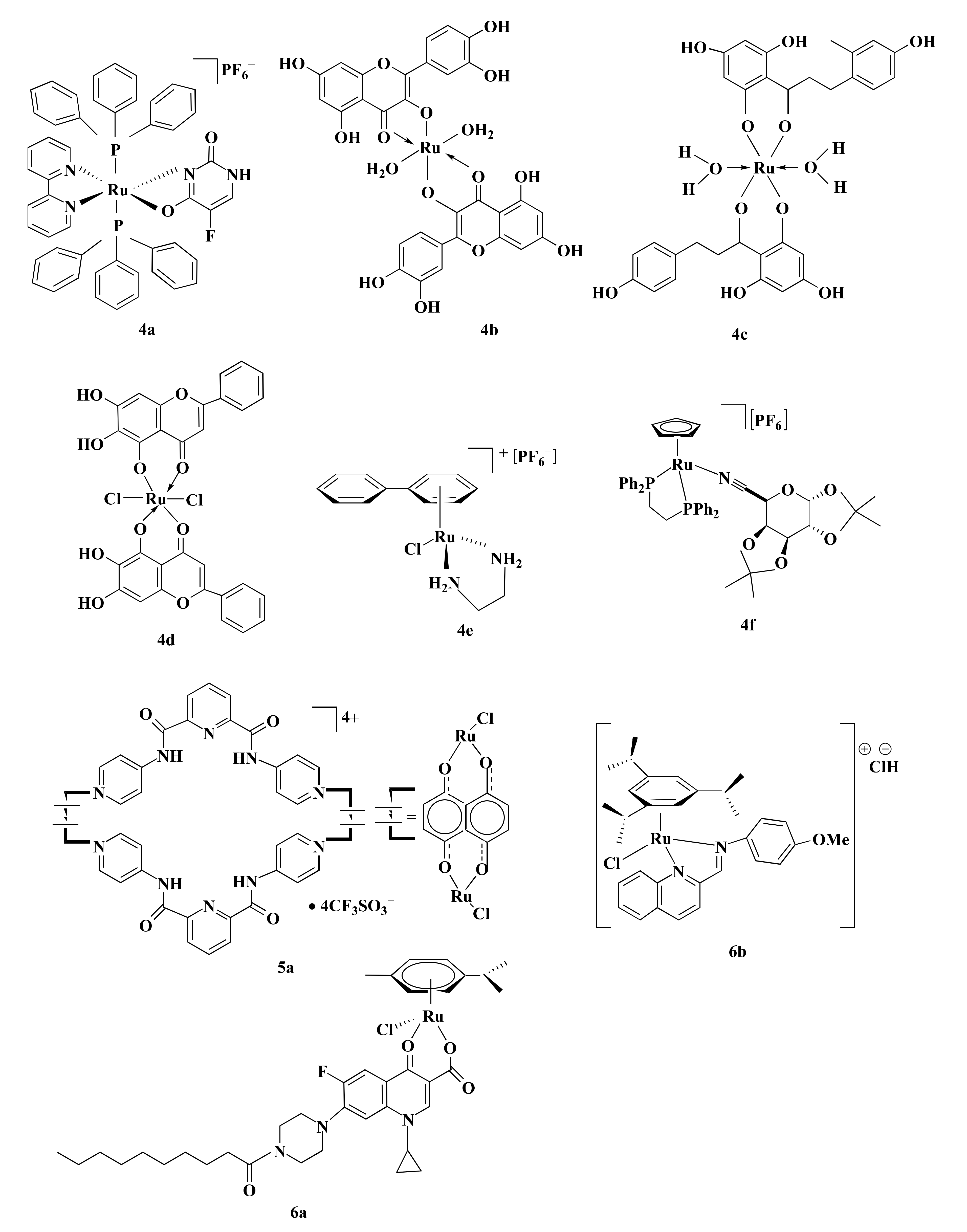
| 4a | II | AB | HCT116 (1.5) *** | caspase-3 | NR | ND | HCT116 (DOX = 0.5, OXA = 4.3, 5-FLU = 4.1) | [27] |
| 4f | II | MTS | HCT116 (0.45) *** | Caspase-3, caspase-7 | NR | NR | HCT116 (OXA = 0.45, 5-FLU = 3.80) *** | [115] |
| 5a | II | MTT | HT-15 (6.9) * | p53, APC | NR | NR | HT-15 (CIS = 13.2, DOX = 15.9) * | [86] |
| 6a | II | SRB | HCT116 (1.33) *** | NR | NR | Both S and G2/M phase | HCT116 (CIS = 5.1, OXA = 3.99) *** | [116] |
| 6b | II | MTT | HCT116 (1.04), SW480 (7.3) *** | NR | NR | G0/G1 (2.5 µM) G2/M (10 µM) | HCT116 (OXA = 2.06), SW480 (OXA = 2.65) *** | [117] |
| 8a | II | MTT | Caco-2 (6.16) ** | NR | NR | G0/G1 | Caco-2 (CIS = 17.9) ** | [118] |
| 10a | II | BP | HCT116 (5.22) *** | NR | NR | NR | CIS = No effect | [78] |
| 11a | II | MTT | HCT116 (2.63) ** | NAD+ | HIF1α, VEGF GLUT1 ENO1 | NR | HCT116 (CIS = 6.33) ** | [87] |
| 13a | II | MTT | HT29 (3.2), HCT116 (2.7), CT-26 (2.3) * | Cathepsin B | NF-kB p65, MMP-2, MMP-9, LAMP1 | NR | HT29 (CIS = 18.9), HCT116 (CIS = 42.8), CT-26 (CIS = 25.6) * | [91] |
| Compounds or Drugs | Oxidation State | Assay Name | CRC Cell Lines and IC50 (µM) | Up-Regulated Protein | Down Regulated Protein | Cell Cycle Arrest | References |
|---|---|---|---|---|---|---|---|
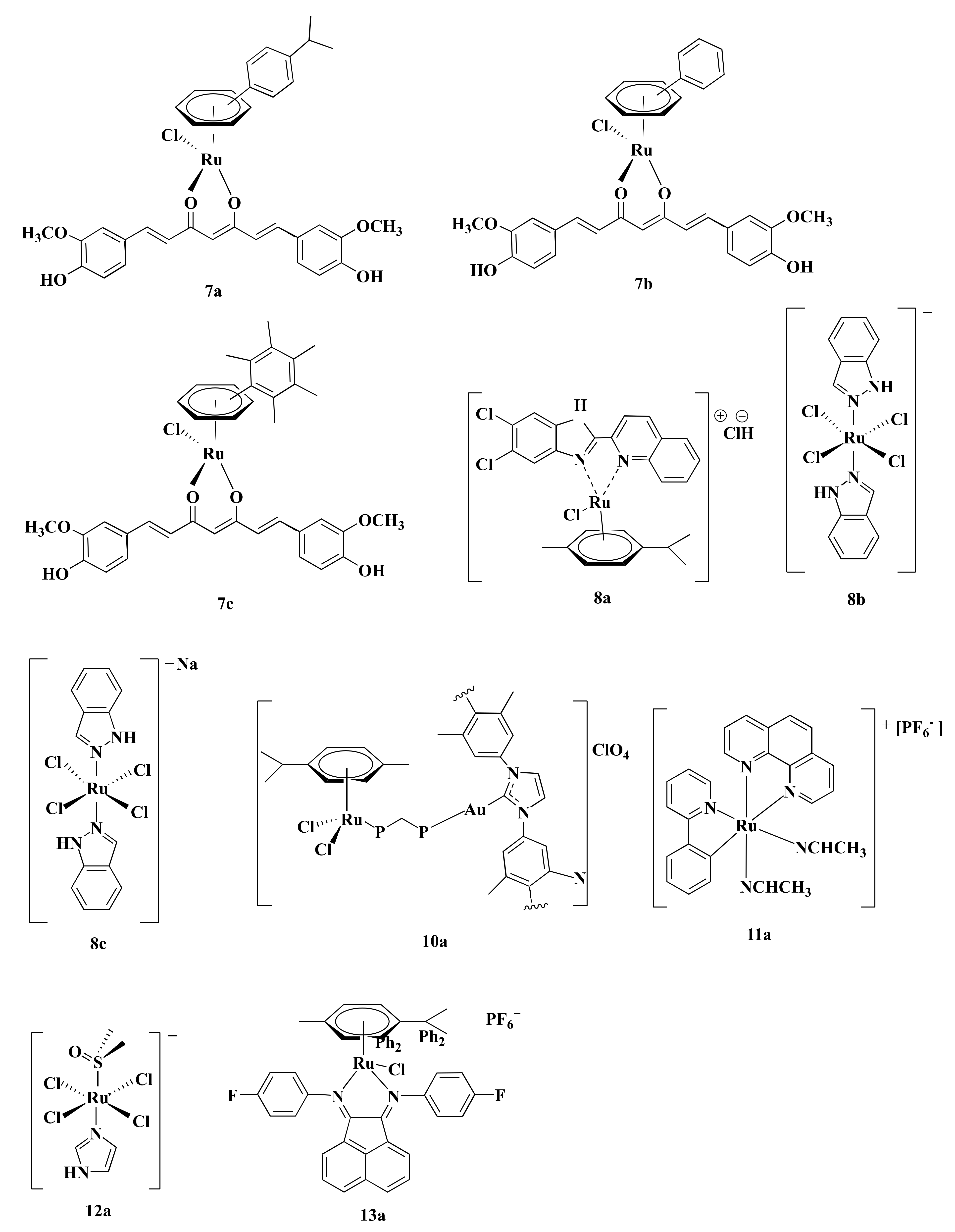
| 3a | II | NR | HCT116 | p21, Bad, p-p38 MAPK, ATF2, Stat1, MMP | Bax | G1 | [79] |
| 4b | II | MTT | HT29 (<100) ** | p53, caspase-3, Bax | Akt1, mTOR, VEGF, Bcl-2, PCNA, WNT, β-catenin | G0/G1 | [83] |
| 4c | II | MTT | HT29 (>100) * | p53, caspase-3, Bax | Akt1, p-Akt, mTOR, p-mTOR, VEGF, Bcl-2, NF-κΒ, MMP-9, PCNA | G0/G1 | [82] |
| 4d | II | MTT | HT29 (~30) ** | p53, caspase-3, Bax | Akt1, mTOR, VEGF, Bcl-2, PCNA, WNT, β-catenin | G0/G1 | [84] |
| 4e | II | SRB | HCT116 (8) HCT116 p53 (16) **** | p53, p21/WAF, Bax | NR | G1 and G2 | [85] |
| 7a, 7b, 7c | II | MTT | HCT116 (NR) | p53, caspase-3 | PARP | NR | [90] |
| 8b | III | NRU | SW480 (30), LT97 (50) * | Caspase-3 | PARP, MMP, Bcl-2 | NR | [119] |
| 8c | III | MTT | HCT116 (20), SW480 (40) * | eIF2α, ATF4, CHOP | NR | NR | [89] |
| 8c | III | NR | HT15, HCT116, HT29 (NR) | eIF2α, CRT, HMGB-1, ATP, Beclin-1, LC3A/B-II | NR | NR | [120] |
| 12a | III | MTT | HCT116 | RND-1, SIK-1 | α5β1 integrin, VEGF, MCP-1 | NR | [88] |
| 14a, 14b | II | PBCV | CT-26 (NR) | NR | NR | NR | [92] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmud, K.M.; Niloy, M.S.; Shakil, M.S.; Islam, M.A. Ruthenium Complexes: An Alternative to Platinum Drugs in Colorectal Cancer Treatment. Pharmaceutics 2021, 13, 1295. https://doi.org/10.3390/pharmaceutics13081295
Mahmud KM, Niloy MS, Shakil MS, Islam MA. Ruthenium Complexes: An Alternative to Platinum Drugs in Colorectal Cancer Treatment. Pharmaceutics. 2021; 13(8):1295. https://doi.org/10.3390/pharmaceutics13081295
Chicago/Turabian StyleMahmud, Kazi Mustafa, Mahruba Sultana Niloy, Md Salman Shakil, and Md Asiful Islam. 2021. "Ruthenium Complexes: An Alternative to Platinum Drugs in Colorectal Cancer Treatment" Pharmaceutics 13, no. 8: 1295. https://doi.org/10.3390/pharmaceutics13081295