
Tunable Polyglycerol-Based Redox-Responsive Nanogels for Efficient Cytochrome C Delivery


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Instrumentation and Methods
2.2.1. Characterization Methods
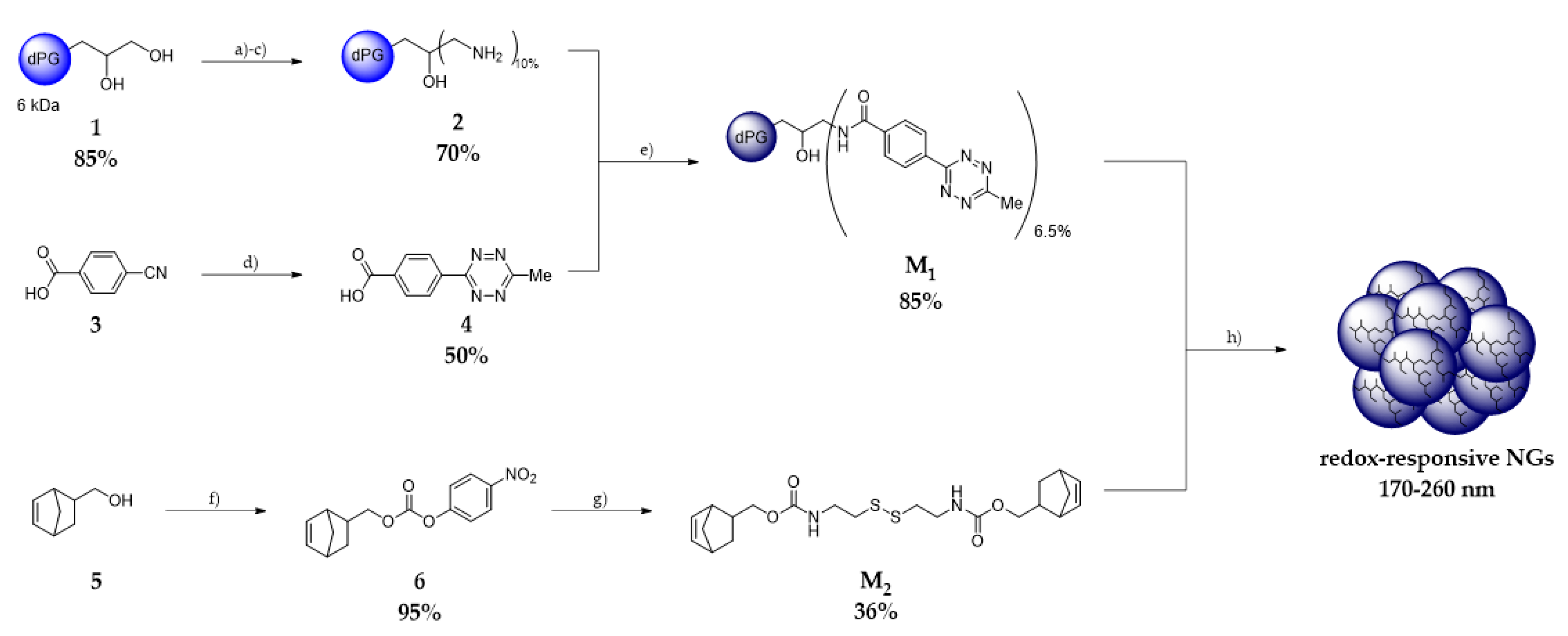
2.2.2. Synthesis of Starting Materials
Synthesis of dPG (1) and dPG-NH2 (2)
4-(6-Methyl-1,2,4,5-tetrazin-3-yl)benzoic Acid (4)
dPG-metTet M1
Bicyclo[2.2.1]hept-5-en-2-ylmethyl (4-nitrophenyl) Carbonate (6)
Norb-Cys-Norb M2
2.2.3. Nanogel Synthesis
2.2.4. Fluorescein Isothiocyanate (FITC) Labeling of M1
2.2.5. Coprecipitation of CC
2.2.6. Degradation of Unloaded NGs
2.2.7. Release of CC
2.2.8. Determining Enzymatic Activity of CC
2.2.9. Cell Viability Studies
2.2.10. Cell Uptake Assay
3. Results and Discussion
3.1. Synthesis of Precursors: dPG metTet and Norb-Cys-Norb
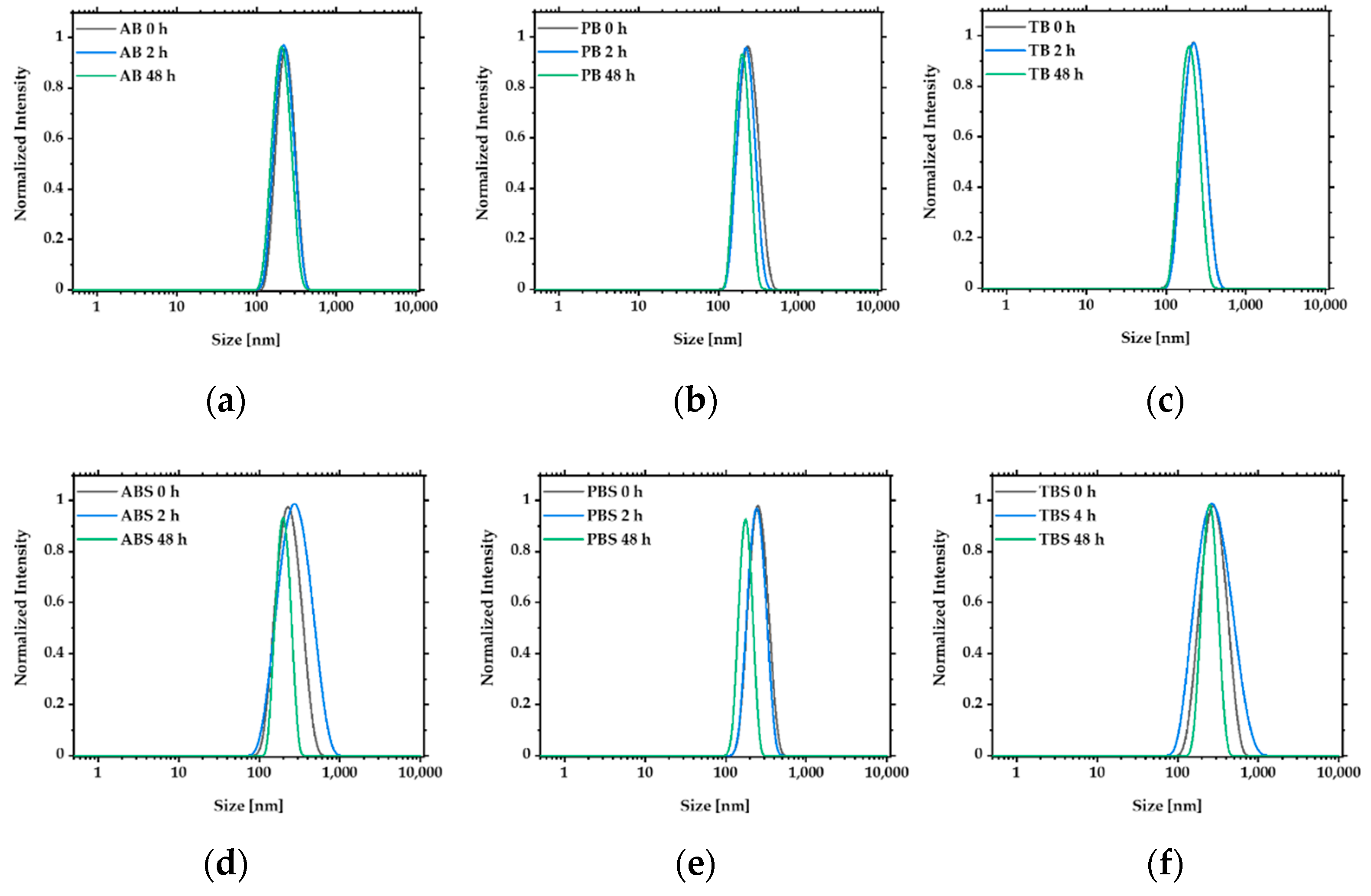
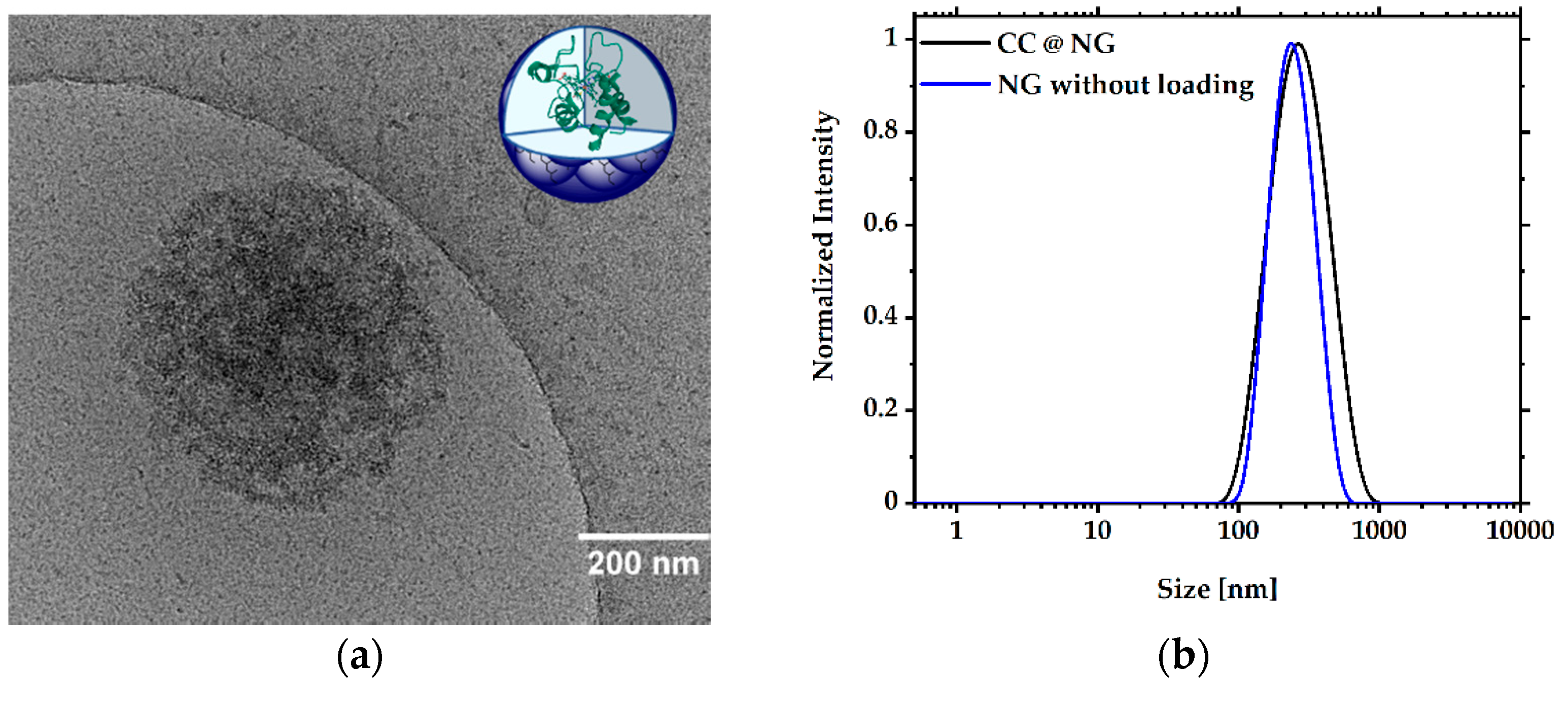
3.2. Formation and Characterization of NGs
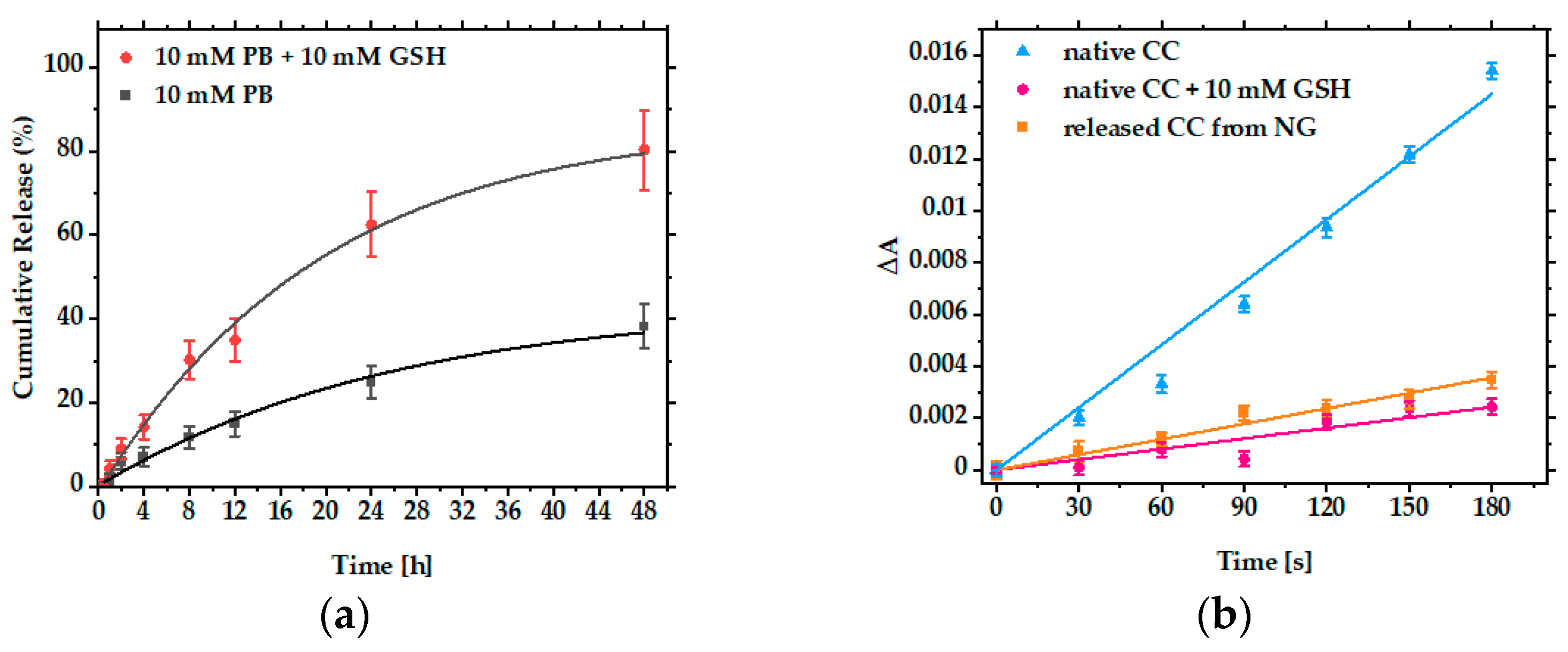
3.3. Encapsulation and Release of CC
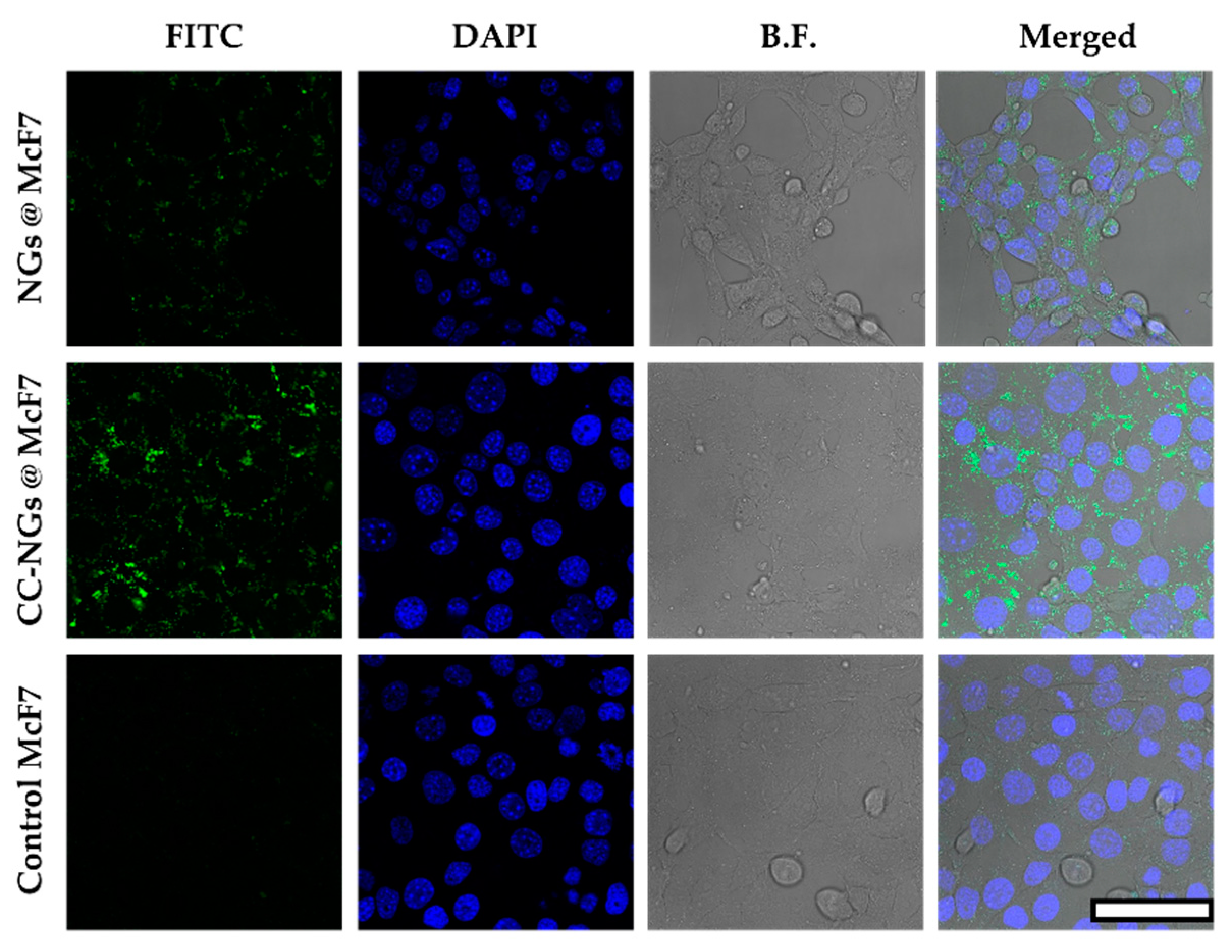
3.4. Cell Viability Studies and Cellular Uptake
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manning, M.C.; Chou, D.K.; Murphy, B.M.; Payne, R.W.; Katayama, D.S. Stability of Protein Pharmaceuticals: An Update. Pharm. Res. 2010, 27, 544–575. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Instability, stabilization, and formulation of liquid protein pharmaceuticals. Int. J. Pharm. 1999, 185, 129–188. [Google Scholar] [CrossRef]
- Strohl, W. Fusion Proteins for Half-Life Extension of Biologics as a Strategy to Make Biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef] [Green Version]
- Kontermann, R.E. Half-life extended biotherapeutics. Expert Opin. Biol. Ther. 2016, 16, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Caliceti, P. Pharmacokinetic and biodistribution properties of poly(ethylene glycol)–protein conjugates. Adv. Drug Deliv. Rev. 2003, 55, 1261–1277. [Google Scholar] [CrossRef]
- Turecek, P.L.; Bossard, M.J.; Schoetens, F.; Ivens, I.A. PEGylation of Biopharmaceuticals: A Review of Chemistry and Nonclinical Safety Information of Approved Drugs. J. Pharm. Sci. 2016, 105, 460–475. [Google Scholar] [CrossRef] [Green Version]
- Ezban, M.; Hansen, M.; Kjalke, M. An overview of turoctocog alfa pegol (N8-GP; ESPEROCT ®) assay performance: Implications for postadministration monitoring. Haemoph. 2019, 26, 156–163. [Google Scholar] [CrossRef]
- Lah, M.; McPheron, M. Palynziq clinic: One year and 43 patients later. Mol. Genet. Metab. 2021, 133, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Radadiya, A.; Zhu, W.; Coricello, A.; Alcaro, S.; Richards, N.G.J. Improving the Treatment of Acute Lymphoblastic Leukemia. Biochem. 2020, 59, 3193–3200. [Google Scholar] [CrossRef] [PubMed]
- Cox, F.; Khalib, K.; Conlon, N. PEG That Reaction: A Case Series of Allergy to Polyethylene Glycol. J. Clin. Pharmacol. 2021, 61, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Sellaturay, P.; Nasser, S.; Islam, S.; Gurugama, P.; Ewan, P.W. Polyethylene glycol (PEG) is a cause of anaphylaxis to the Pfizer/BioNTech mRNA COVID-19 vaccine. Clin. Exp. Allergy 2021, 51, 861–863. [Google Scholar] [CrossRef]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew. Chem. Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef] [PubMed]
- Soni, K.S.; Desale, S.S.; Bronich, T.K. Nanogels: An overview of properties, biomedical applications and obstacles to clinical translation. J. Control. Release 2016, 240, 109–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karg, M.; Pich, A.; Hellweg, T.; Hoare, T.; Lyon, L.A.; Crassous, J.J.; Suzuki, D.; Gumerov, R.; Schneider, S.; Potemkin, I.I.; et al. Nanogels and Microgels: From Model Colloids to Applications, Recent Developments, and Future Trends. Langmuir 2019, 35, 6231–6255. [Google Scholar] [CrossRef]
- Chacko, R.T.; Ventura, J.; Zhuang, J.; Thayumanavan, S. Polymer nanogels: A versatile nanoscopic drug delivery platform. Adv. Drug Deliv. Rev. 2012, 64, 836–851. [Google Scholar] [CrossRef] [Green Version]
- Pamfil, D.; Vasile, C. Nanogels of Natural Polymers. Gels Horiz. Sci. Smart Mater. 2018, 71–110. [Google Scholar] [CrossRef]
- Moshe, H.; Davizon, Y.; Raskin, M.M.; Sosnik, A. Novel poly(vinyl alcohol)-based amphiphilic nanogels by non-covalent boric acid crosslinking of polymeric micelles. Biomater. Sci. 2017, 5, 2295–2309. [Google Scholar] [CrossRef]
- Birjand, M.A.; Bergueiro, J.; Rancan, F.; Cuggino, J.C.; Mutihac, R.-C.; Achazi, K.; Dernedde, J.; Blume-Peytayi, U.; Vogt, A.; Calderón, M. Engineering thermoresponsive polyether-based nanogels for temperature dependent skin penetration. Polym. Chem. 2015, 6, 5827–5831. [Google Scholar] [CrossRef] [Green Version]
- Kabanov, A.V.; Vinogradov, S.V. Nanogels as Pharmaceutical Carriers: Finite Networks of Infinite Capabilities. Angew. Chem. Int. Ed. 2009, 48, 5418–5429. [Google Scholar] [CrossRef] [Green Version]
- Mauri, E.; Perale, G.; Rossi, F. Nanogel Functionalization: A Versatile Approach To Meet the Challenges of Drug and Gene Delivery. ACS Appl. Nano Mater. 2018, 1, 6525–6541. [Google Scholar] [CrossRef]
- Takeuchi, T.; Kitayama, Y.; Sasao, R.; Yamada, T.; Toh, K.; Matsumoto, Y.; Kataoka, K. Molecularly Imprinted Nanogels Acquire Stealth In Situ by Cloaking Themselves with Native Dysopsonic Proteins. Angew. Chem. Int. Ed. 2017, 56, 7088–7092. [Google Scholar] [CrossRef]
- Basak, S. The Age of Multistimuli-responsive Nanogels: The Finest Evolved Nano Delivery System in Biomedical Sciences. Biotechnol. Bioprocess Eng. 2020, 25, 655–669. [Google Scholar] [CrossRef]
- Cai, M.-H.; Chen, X.-Y.; Fu, L.-Q.; Du, W.-L.; Yang, X.; Mou, X.-Z.; Hu, P.-Y. Design and Development of Hybrid Hydrogels for Biomedical Applications: Recent Trends in Anticancer Drug Delivery and Tissue Engineering. Front. Bioeng. Biotechnol. 2021, 9, 630943. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, S.; Torres, F.G.; Arroyo, J.; Gonzales, K.N.; Troncoso, O.P.; López, D. Synthesis of highly stable κ/ι-hybrid carrageenan micro- and nanogels via a sonication-assisted microemulsion route. Polym. Renew. Resour. 2020, 11, 69–82. [Google Scholar] [CrossRef]
- Yu, K.; Yang, X.; He, L.; Zheng, R.; Min, J.; Su, H.; Shan, S.; Jia, Q. Facile preparation of pH/reduction dual-stimuli responsive dextran nanogel as environment-sensitive carrier of doxorubicin. Polymer 2020, 200, 122585. [Google Scholar] [CrossRef]
- Steinhilber, D.; Witting, M.; Zhang, X.; Staegemann, M.; Paulus, F.; Friess, W.; Küchler, S.; Haag, R. Surfactant free preparation of biodegradable dendritic polyglycerol nanogels by inverse nanoprecipitation for encapsulation and release of pharmaceutical biomacromolecules. J. Control. Release 2013, 169, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Chow, S.F.; Zheng, Y. Application of flash nanoprecipitation to fabricate poorly water-soluble drug nanoparticles. Acta Pharm. Sin. B 2019, 9, 4–18. [Google Scholar] [CrossRef]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6998. [Google Scholar] [CrossRef] [Green Version]
- Guaresti, O.; Maiz-Fernández, S.; Palomares, T.; Alonso-Varona, A.; Eceiza, A.; Pérez-Álvarez, L.; Gabilondo, N. Dual charged folate labelled chitosan nanogels with enhanced mucoadhesion capacity for targeted drug delivery. Eur. Polym. J. 2020, 134, 109847. [Google Scholar] [CrossRef]
- Dimde, M.; Neumann, F.; Reisbeck, F.; Ehrmann, S.; Cuellar-Camacho, J.L.; Steinhilber, D.; Ma, N.; Haag, R. Defined pH-sensitive nanogels as gene delivery platform for siRNA mediated in vitro gene silencing. Biomater. Sci. 2017, 5, 2328–2336. [Google Scholar] [CrossRef] [PubMed]
- Nagel, G.; Sousa-Herves, A.; Wedepohl, S.; Calderón, M. Matrix Metalloproteinase-sensitive Multistage Nanogels Promote Drug Transport in 3D Tumor Model. Theranostics 2020, 10, 91–108. [Google Scholar] [CrossRef]
- Choi, H.; Kwon, M.; Choi, H.; Hahn, S.; Kim, K. Non-Invasive Topical Drug-Delivery System Using Hyaluronate Nanogels Crosslinked via Click Chemistry. Materials 2021, 14, 1504. [Google Scholar] [CrossRef]
- Oehrl, A.; Schötz, S.; Haag, R. Systematic Screening of Different Polyglycerin-Based Dienophile Macromonomers for Efficient Nanogel Formation through IEDDA Inverse Nanoprecipitation. Macromol. Rapid Commun. 2020, 41, e1900510. [Google Scholar] [CrossRef] [Green Version]
- Haag, R.; Kratz, F. Polymer Therapeutics: Concepts and Applications. Angew. Chem. Int. Ed. 2006, 45, 1198–1215. [Google Scholar] [CrossRef]
- Li, S.; Zhang, J.; Deng, C.; Meng, F.; Yu, L.; Zhong, Z. Redox-Sensitive and Intrinsically Fluorescent Photoclick Hyaluronic Acid Nanogels for Traceable and Targeted Delivery of Cytochrome c to Breast Tumor in Mice. ACS Appl. Mater. Interfaces 2016, 8, 21155–21162. [Google Scholar] [CrossRef]
- Wallert, M.; Plaschke, J.; Dimde, M.; Ahmadi, V.; Block, S.; Haag, R. Automated Solvent-Free Polymerization of Hyperbranched Polyglycerol with Tailored Molecular Weight by Online Torque Detection. Macromol. Mater. Eng. 2021, 306, 2000688. [Google Scholar] [CrossRef]
- Dimde, M.; Steinhilber, D.; Neumann, F.; Li, Y.; Paulus, F.; Ma, N.; Haag, R. Synthesis of pH-Cleavable dPG-Amines for Gene Delivery Application. Macromol. Biosci. 2016, 17, 1600190. [Google Scholar] [CrossRef]
- Cheng, R.; Feng, F.; Meng, F.; Deng, C.; Feijen, J.; Zhong, Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J. Control. Release 2011, 152, 2–12. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, K.; Haag, R. Multi-stage, charge conversional, stimuli-responsive nanogels for therapeutic protein delivery. Biomater. Sci. 2015, 3, 1487–1496. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | M1:M2 | Size d (nm) 1 | PDI 1 | ζ-Potential (mV) 1 |
|---|---|---|---|---|
| 1 | 1:2.00 | 257 ± 1 | 0.11 ± 0.03 | 2.0 ± 0.7 |
| 2 | 1:1.50 | 263 ± 5 | 0.10 ± 0.03 | 3.0 ± 0.6 |
| 3 | 1:1.00 | 236 ± 7 | 0.08 ± 0.01 | 2.4 ± 0.4 |
| 4 | 1:0.50 | 185 ± 4 | 0.07 ± 0.03 | 2.3 ± 0.4 |
| 5 | 1:0.25 | 171 ± 8 | 0.03 ± 0.04 | 2.5 ± 0.1 |
| Entry | Theory wt.% | Determined wt.% 1 | PLE (%) 1 | Size d (nm) 2 | PDI 2 | ζ-Potential (mV) 2 |
|---|---|---|---|---|---|---|
| 1 | 5 | 4.3 | 85 | 242 ± 1 | 0.2 ± 0.01 | −0.47 ± 0.01 |
| 2 | 10 | 8.7 | 87 | 406 ± 9 | 0.2 ± 0.01 | −0.21 ± 0.06 |
| 3 | 20 | 7.5 | 38 | 450 ± 20 | 0.4 ± 0.01 | −0.30 ± 0.08 |
| Cell Line | IC50 (μg·mL−1) |
|---|---|
| McF7 | 30.20 |
| A549 | 29.93 |
| HeLa | 29.79 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schötz, S.; Reisbeck, F.; Schmitt, A.-C.; Dimde, M.; Quaas, E.; Achazi, K.; Haag, R. Tunable Polyglycerol-Based Redox-Responsive Nanogels for Efficient Cytochrome C Delivery. Pharmaceutics 2021, 13, 1276. https://doi.org/10.3390/pharmaceutics13081276
Schötz S, Reisbeck F, Schmitt A-C, Dimde M, Quaas E, Achazi K, Haag R. Tunable Polyglycerol-Based Redox-Responsive Nanogels for Efficient Cytochrome C Delivery. Pharmaceutics. 2021; 13(8):1276. https://doi.org/10.3390/pharmaceutics13081276
Chicago/Turabian StyleSchötz, Sebastian, Felix Reisbeck, Ann-Cathrin Schmitt, Mathias Dimde, Elisa Quaas, Katharina Achazi, and Rainer Haag. 2021. "Tunable Polyglycerol-Based Redox-Responsive Nanogels for Efficient Cytochrome C Delivery" Pharmaceutics 13, no. 8: 1276. https://doi.org/10.3390/pharmaceutics13081276