
Structure and Glass Transition Temperature of Amorphous Dispersions of Model Pharmaceuticals with Nucleobases from Molecular Dynamics


Abstract
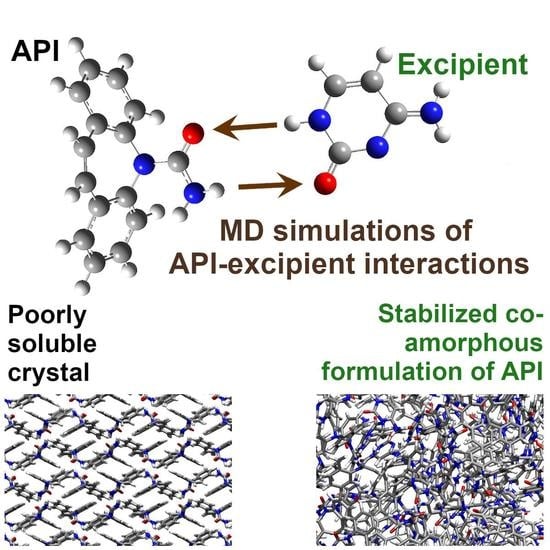
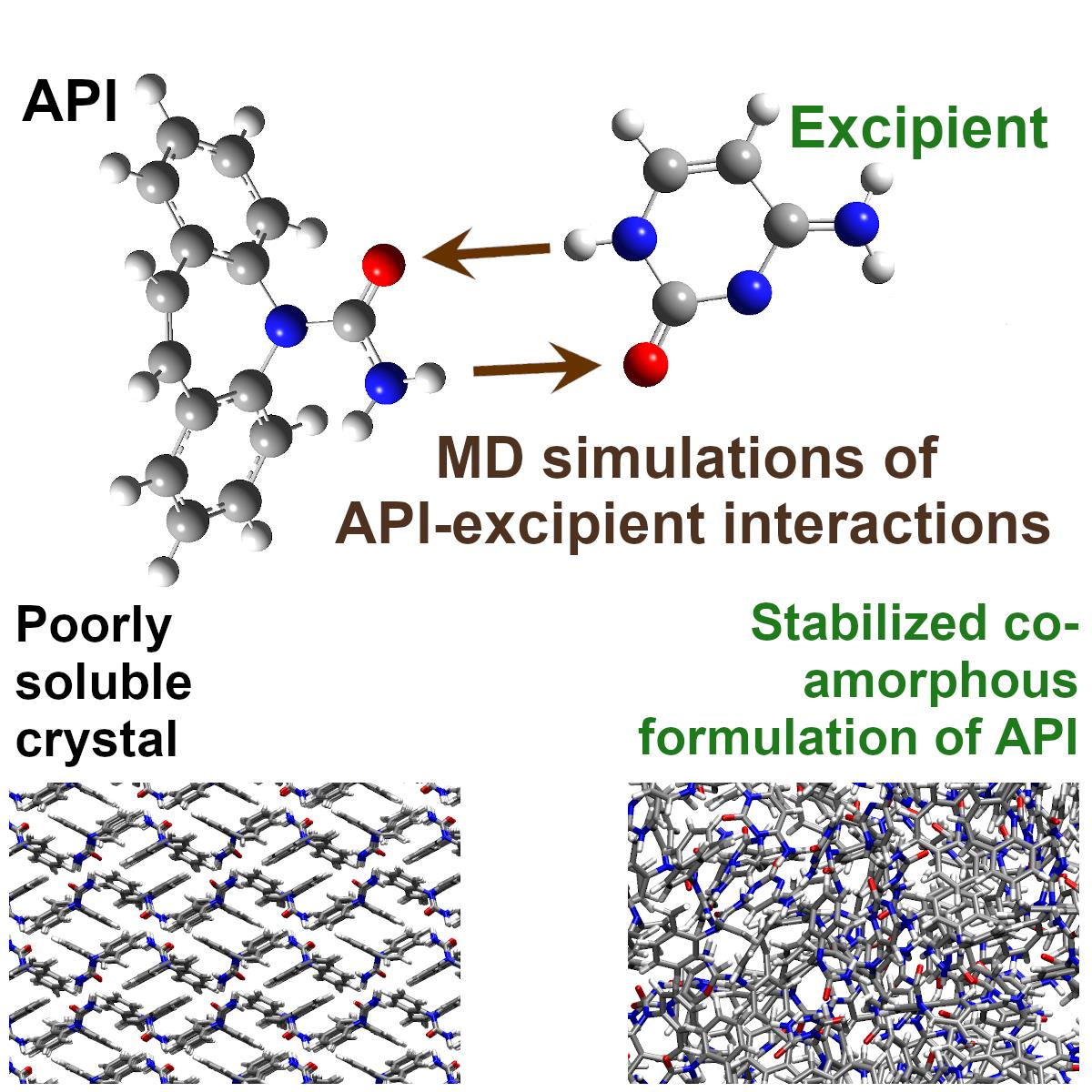
:
1. Introduction
2. Computational Methods
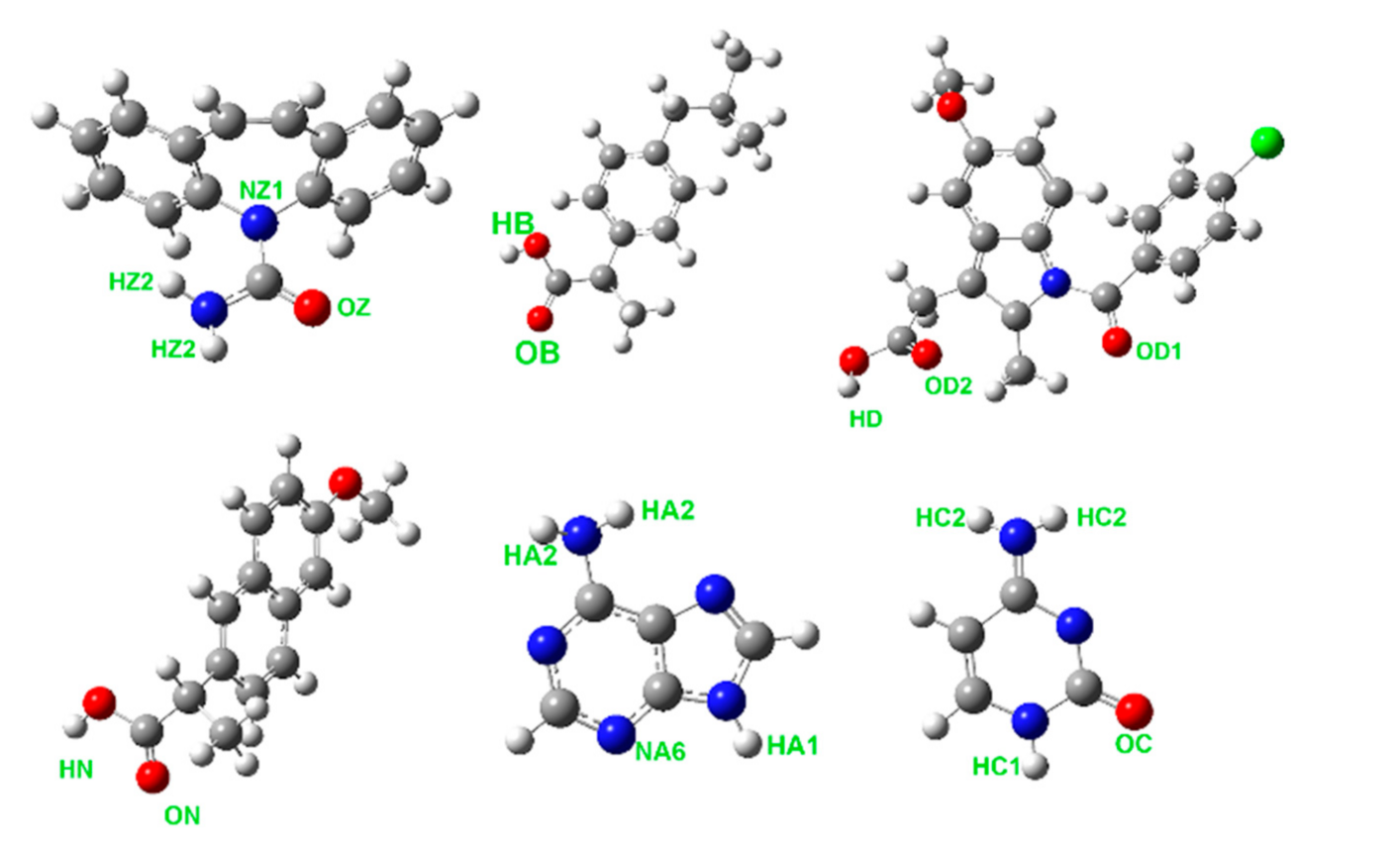
2.1. Force-Field Models
2.2. Molecular Dynamics
3. Results and Discussion
3.1. Force-Field Validation
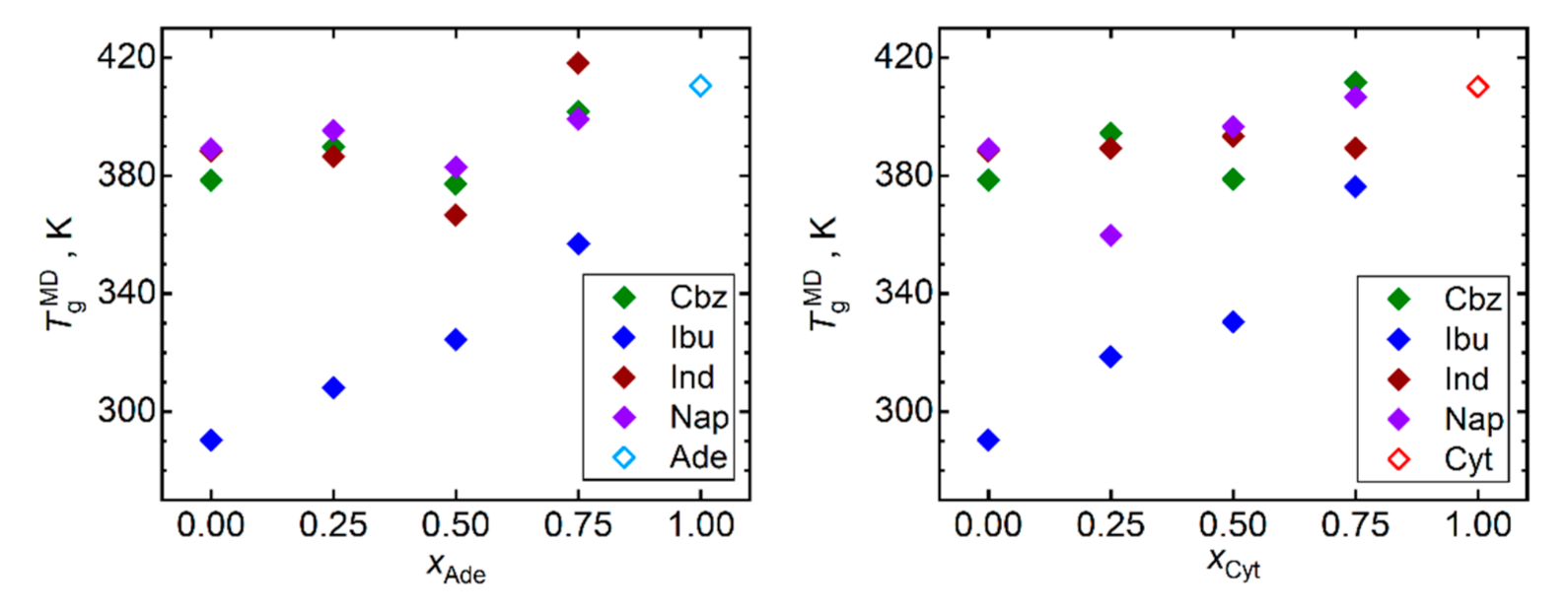
3.2. Vitrification Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Craig, D.Q.M.; Royall, P.G.; Kett, V.L.; Hopton, M.L. The relevance of the amorphous state to pharmaceutical dosage forms: Glassy drugs and freeze dried systems. Int. J. Pharm. 1999, 179, 179–207. [Google Scholar] [CrossRef] [Green Version]
- Hancock, B.C.; Zograf, G. Characteristics and significance of the amorphous state in pharmaceutical systems. J. Pharm. Sci. 1997, 86, 1–12. [Google Scholar] [CrossRef]
- Rams-Baron, M.; Jachowicz, R.; Boldyreva, E.; Zhou, D.; Jamroz, W.; Paluch, M. Amorphous Drugs, Benefits and Challenges; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Barmpalexis, P.; Karagianni, A.; Katopodis, K.; Vardaka, E.; Kachrimanis, K. Molecular modelling and simulation of fusion-based amorphous drug dispersions in polymer/plasticizer blends. Eur. J. Pharm. Sci. 2019, 130, 260–268. [Google Scholar] [CrossRef]
- Medarevic, D.; Djuris, J.; Barmpalexis, P.; Kachrimanis, K.; Ibric, S. Analytical and Computational Methods for the Estimation of Drug-Polymer Solubility and Miscibility in Solid Dispersions Development. Pharmaceutics 2019, 11, 372. [Google Scholar] [CrossRef] [Green Version]
- Gupta, J.; Nunes, C.; Jonnalagadda, S. A Molecular Dynamics Approach for Predicting the Glass Transition Temperature and Plasticization Effect in Amorphous Pharmaceuticals. Mol. Pharm. 2013, 10, 4136–4145. [Google Scholar] [CrossRef]
- Russo, M.G.; Baldoni, H.A.; Davila, Y.A.; Brusau, E.V.; Ellena, J.A.; Narda, G.E. Rational Design of a Famotidine-Ibuprofen Coamorphous System: An Experimental and Theoretical Study. J. Phys. Chem. B 2018, 122, 8772–8782. [Google Scholar] [CrossRef]
- Knapik-Kowalczuk, J.; Tu, W.; Chmiel, K.; Rams-Baron, M.; Paluch, M. Co-Stabilization of Amorphous Pharmaceuticals—The Case of Nifedipine and Nimodipine. Mol. Pharm. 2018, 15, 2455–2465. [Google Scholar] [CrossRef]
- Weng, L.D.; Stott, S.L.; Toner, M. Exploring Dynamics and Structure of Biomolecules, Cryoprotectants, and Water Using Molecular Dynamics Simulations: Implications for Biostabilization and Biopreservation. Annu. Rev. Biomed. Eng. 2019, 15, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.D.; Knapik-Kowalczuk, J.; Paluch, M.; Hoang, T.X.; Wakabayashi, K. Theoretical Model for the Structural Relaxation Time in Coamorphous Drugs. Mol. Pharm. 2019, 16, 2992–2998. [Google Scholar] [CrossRef]
- Alzghoul, A.; Alhalaweh, A.; Mahlin, D.; Bergström, C.A.S. Experimental and Computational Prediction of Glass Transition Temperature of Drugs. J. Chem. Inf. Modeling 2014, 54, 3396–3403. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.; Taylor, J.S. Ideal Copolymers and the 2nd-order Transitions of Synthetic Rubbers. 1. Non-crystalline Copolymers. J. Appl. Chem. 1952, 2, 493–500. [Google Scholar] [CrossRef]
- Fox, T.G.; Flory, P.J. 2nd-order Transition Temperatures and Related Properties of Polystyrene. 1. Influence of Molecular Weight. J. Appl. Phys. 1950, 21, 581–591. [Google Scholar] [CrossRef]
- Bergstrom, C.A.S.; Charman, W.N.; Porter, C.J.H. Computational prediction of formulation strategies for beyond-rule-of-5 compounds. Adv. Drug Deliv. Rev. 2016, 101, 6–21. [Google Scholar] [CrossRef]
- Grzybowska, K.; Grzybowski, A.; Knapik-Kowalczuk, J.; Chmiel, K.; Woyna-Orlewicz, K.; Szafraniec-Szczęsny, J.; Antosik-Rogóż, A.; Jachowicz, R.; Kowalska-Szojda, K.; Lodowski, P.; et al. Molecular Dynamics and Physical Stability of Ibuprofen in Binary Mixtures with an Acetylated Derivative of Maltose. Mol. Pharm. 2020, 17, 3087–3105. [Google Scholar] [CrossRef] [PubMed]
- Koperwas, K.; Adrjanowicz, K.; Wojnarowska, Z.; Jedrzejowska, A.; Knapik, J.; Paluch, M. Glass-Forming Tendency of Molecular Liquids and the Strength of the Intermolecular Attractions. Sci. Rep. 2016, 6, 36934. [Google Scholar] [CrossRef] [Green Version]
- Glotzer, S.C.; Paul, W. Molecular and mesoscale simulation methods for polymer materials. Annu. Rev. Mater. Res. 2002, 32, 401–436. [Google Scholar] [CrossRef] [Green Version]
- Paul, W.; Smith, G.D. Structure and dynamics of amorphous polymers: Computer simulations compared to experiment and theory. Rep. Prog. Phys. 2004, 67, 1117–1185. [Google Scholar] [CrossRef]
- Roe, R.J. Short-time Dynamics of Polymer Liquid and Glass Studied by Molecular-Dynamics Simulation. J. Chem. Phys. 1994, 100, 1610–1619. [Google Scholar] [CrossRef]
- Han, J.; Gee, R.H.; Boyd, R.H. Glass-transition Temperatures of Polymers from Molecular-Dynamics Simulations. Macromolecules 1994, 27, 7781–7784. [Google Scholar] [CrossRef]
- Greiner, M.; Elts, E.; Schneider, J.; Reuter, K.; Briesen, H. Dissolution study of active pharmaceutical ingredients using molecular dynamics simulations with classical force fields. J. Cryst. Growth 2014, 405, 122–130. [Google Scholar] [CrossRef]
- Römer, F.; Kraska, T. A force field for naproxen. Mol. Simul. 2012, 38, 152–160. [Google Scholar] [CrossRef]
- Xiang, T.-X.; Anderson, B.D. Molecular Dynamics Simulation of Amorphous Indomethacin. Mol. Pharm. 2013, 10, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Pranata, J.; Wierschke, S.G.; Jorgensen, W.L. OPLS potential functions for nucleotide bases. Relative association constants of hydrogen-bonded base pairs in chloroform. J. Am. Chem. Soc. 1991, 113, 2810–2819. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hanus, M.; Kabeláč, M.; Rejnek, J.; Ryjáček, F.; Hobza, P. Correlated ab Initio Study of Nucleic Acid Bases and Their Tautomers in the Gas Phase, in a Microhydrated Environment, and in Aqueous Solution. Part 3. Adenine. J. Phys. Chem. B 2004, 108, 2087–2097. [Google Scholar] [CrossRef]
- Trygubenko, S.A.; Bogdan, T.V.; Rueda, M.; Orozco, M.; Luque, F.J.; Šponer, J.; Slavíček, P.; Hobza, P. Correlated ab initio study of nucleic acid bases and their tautomers in the gas phase, in a microhydrated environment and in aqueous solution Part 1. Cytosine. Phys. Chem. Chem. Phys. 2002, 4, 4192–4203. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Martinez, L.; Andrade, R.; Birgin, E.G.; Martinez, J.M. PACKMOL: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Alejandre, J.; Lopez-Rendon, R.; Jochim, A.L.; Martyna, G.J. A Liouville-operator derived. measure-preserving integrator for molecular dynamics simulations in the isothermal-isobaric ensemble. J. Phys. A Math. Gen. 2006, 39, 5629–5651. [Google Scholar] [CrossRef]
- Shinoda, W.; Shiga, M.; Mikami, M. Rapid estimation of elastic constants by molecular dynamics simulation under constant stress. Phys. Rev. B 2004, 69, 134103. [Google Scholar] [CrossRef]
- Hockney, R.W.; Eastwood, J.W. Computer Simulation Using Particles; Taylor&Francis: New York, NY, USA, 1988; p. 540. [Google Scholar]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical-integration of cartesian equations of motion of a system with constraints -molecular-dynamics of n-alkanes. J. Comp. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Mahapatra, S.; Nayak, S.K.; Prathapa, S.J.; Guru Row, T.N. Anhydrous Adenine: Crystallization, Structure, and Correlation with Other Nucleobases. Cryst. Growth Des. 2008, 8, 1223–1225. [Google Scholar] [CrossRef]
- McClure, R.J.; Craven, B.M. New investigations of cytosine and its monohydrate. Acta Crystallogr. Sect. B 1973, 29, 1234–1238. [Google Scholar] [CrossRef]
- Tang, G.-M.; Wang, J.-H.; Zhao, C.; Wang, Y.-T.; Cui, Y.-Z.; Cheng, F.-Y.; Ng, S.W. Multi odd–even effects on cell parameters, melting points, and optical properties of chiral crystal solids based on S-naproxen. CrystEngComm 2015, 17, 7258–7261. [Google Scholar] [CrossRef]
- Cox, P.J.; Manson, P.L. [gamma]-Indomethacin at 120 K. Acta Crystallogr. Sect. E 2003, 59, o986–o988. [Google Scholar] [CrossRef]
- Chen, X.; Morris, K.R.; Griesser, U.J.; Byrn, S.R.; Stowell, J.G. Reactivity Differences of Indomethacin Solid Forms with Ammonia Gas. J. Am. Chem. Soc. 2002, 124, 15012–15019. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska, K.; Kropidłowska, M.; Katrusiak, A. High-Pressure Crystallization and Structural Transformations in Compressed R,S-Ibuprofen. Cryst. Growth Des. 2015, 15, 1512–1517. [Google Scholar] [CrossRef]
- Eccles, K.S.; Stokes, S.P.; Daly, C.A.; Barry, N.M.; McSweeney, S.P.; O’Neill, D.J.; Kelly, D.M.; Jennings, W.B.; Ni Dhubhghaill, O.M.; Moynihan, H.A.; et al. Evaluation of the Bruker SMART X2S: Crystallography for the nonspecialist? J. Appl. Crystallogr. 2011, 44, 213–215. [Google Scholar] [CrossRef] [Green Version]
- Štejfa, V.; Pokorný, V.; Mathers, A.; Růžička, K.; Fulem, M. Heat capacities of selected active pharmaceutical ingredients. J. Chem. Thermodyn. 2021, 106585, in press. [Google Scholar]
- Klajmon, M. Investigating Various Parametrization Strategies for Pharmaceuticals within the PC-SAFT Equation of State. J. Chem. Eng. Data 2020, 65, 5753–5767. [Google Scholar] [CrossRef]
- Buchholz, H.; Emel’yanenko, V.N.; Lorenz, H.; Verevkin, S.P. An Examination of the Phase Transition Thermodynamics of (S)- and (RS)-Naproxen as a Basis for the Design of Enantioselective Crystallization Processes. J. Pharm. Sci. 2016, 105, 1676–1683. [Google Scholar] [CrossRef] [Green Version]
- Perlovich, G.L.; Kurkov, S.V.; Hansen, L.K.; Bauer-Brandl, A. Thermodynamics of sublimation, crystal lattice energies, and crystal structures of racemates and enantiomers: (+)- and (±)-ibuprofen. J. Pharm. Sci. 2004, 93, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Červinka, C.; Fulem, M.; Růžička, K. Evaluation of Accuracy of Ideal-Gas Heat Capacity and Entropy Calculations by Density Functional Theory (DFT) for Rigid Molecules. J. Chem. Eng. Data 2012, 57, 227–232. [Google Scholar] [CrossRef]
- Červinka, C.; Štejfa, V. Computational assessment of the crystallization tendency of 1-ethyl-3-methylimidazolium ionic liquids. Phys. Chem. Chem. Phys. 2021, 23, 4951–4962. [Google Scholar] [CrossRef]
- Zemánková, A. Screening and Evaluation of Co-Amorphous Drug Delivery Systems. Bachelor’s Thesis, University of Chemistry and Technology, Prague, Czech Republic, 2020. [Google Scholar]
- Červinka, C.; Pádua, A.A.H.; Fulem, M. Thermodynamic Properties of Selected Homologous Series of Ionic Liquids Calculated Using Molecular Dynamics. J. Phys. Chem. B 2016, 120, 2362–2371. [Google Scholar] [CrossRef] [PubMed]
- Klajmon, M.; Červinka, C. Does explicit polarizability improve simulations of phase behavior of ionic liquids? J. Chem. Theory Comput. 2021. submitted. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
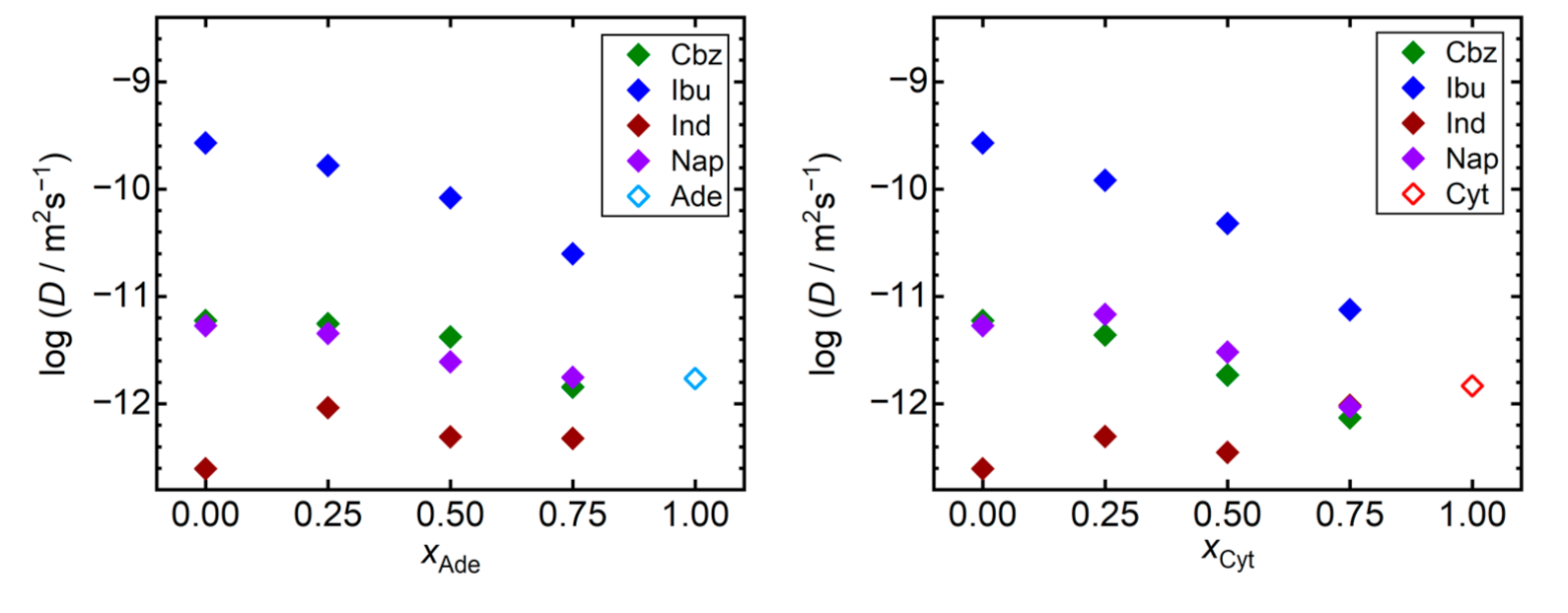{kind=link}
{kind=link}
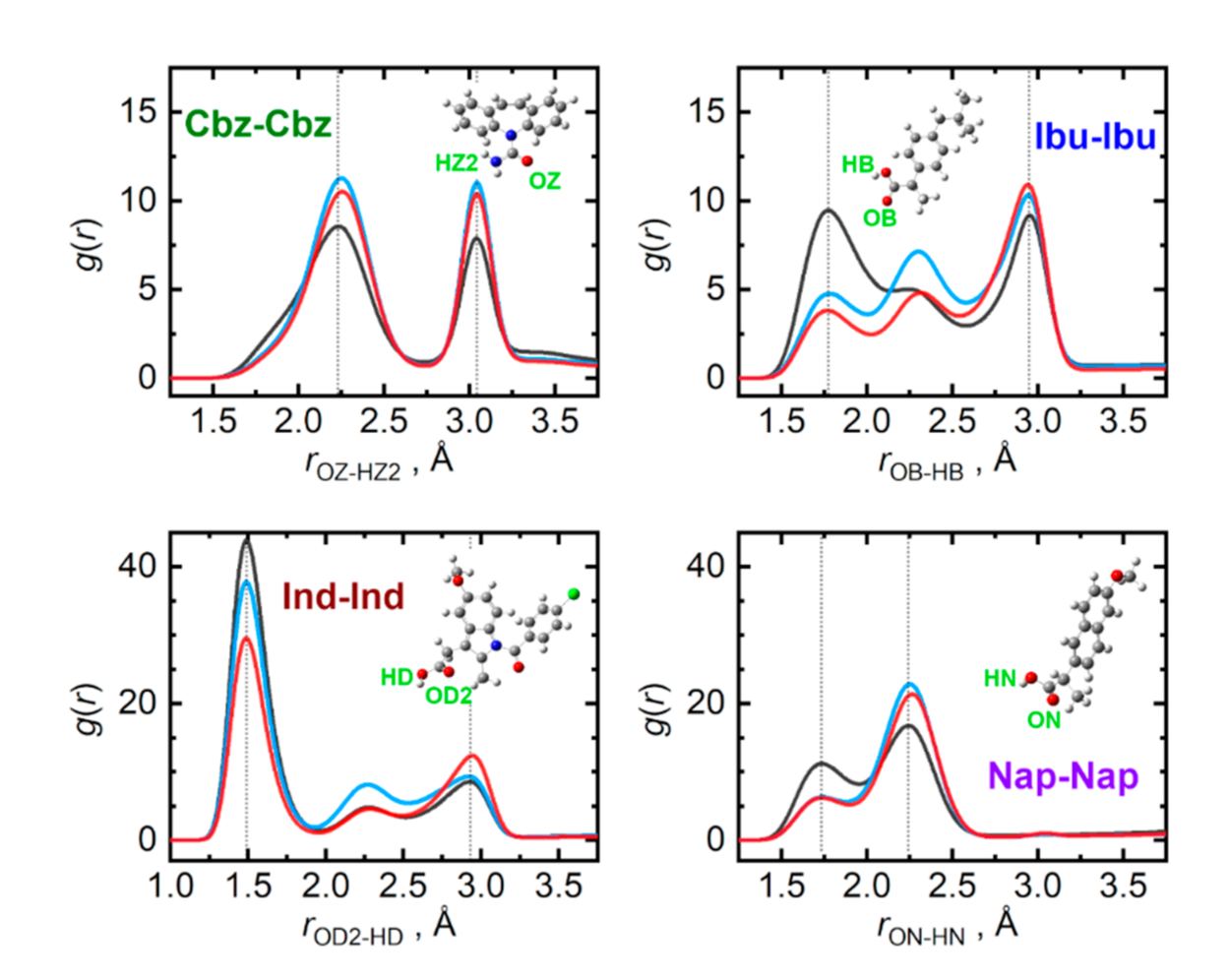{kind=link}
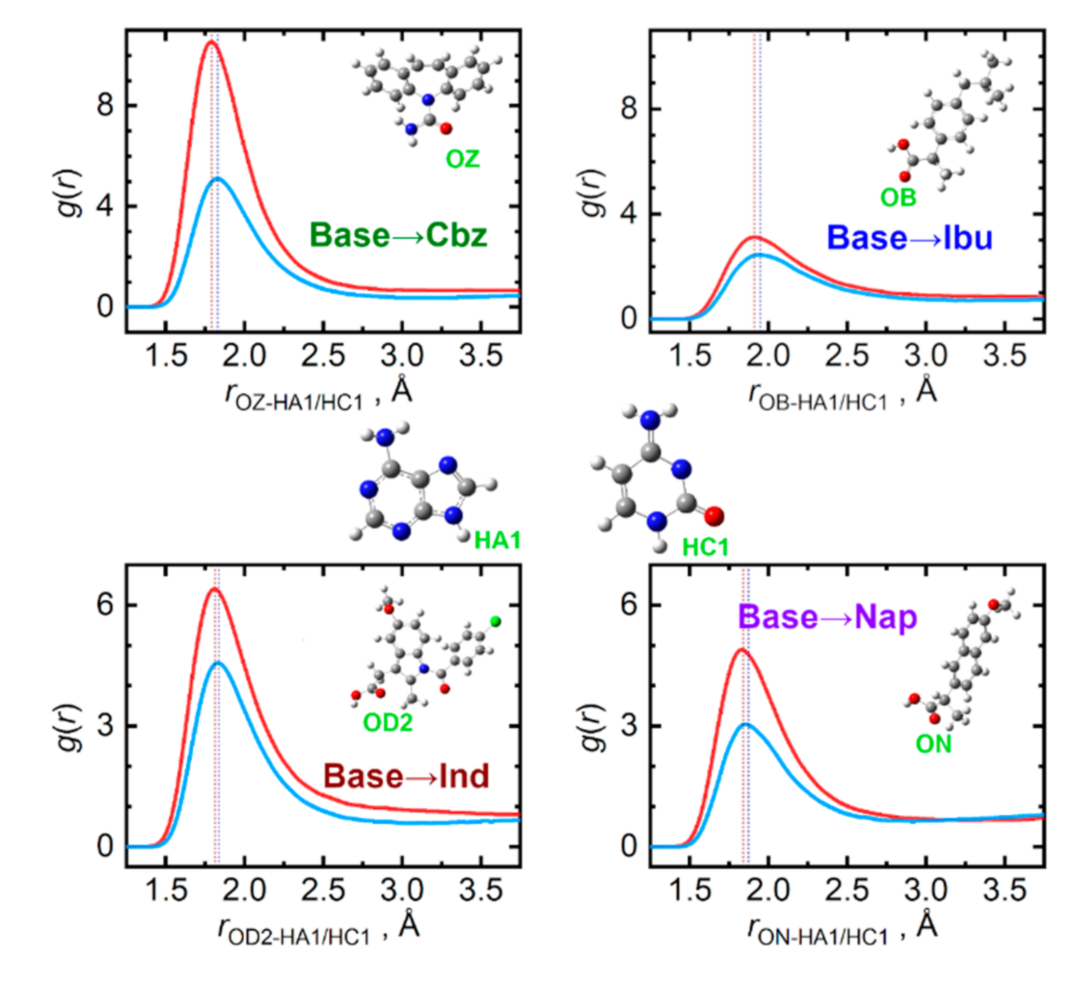{kind=link}
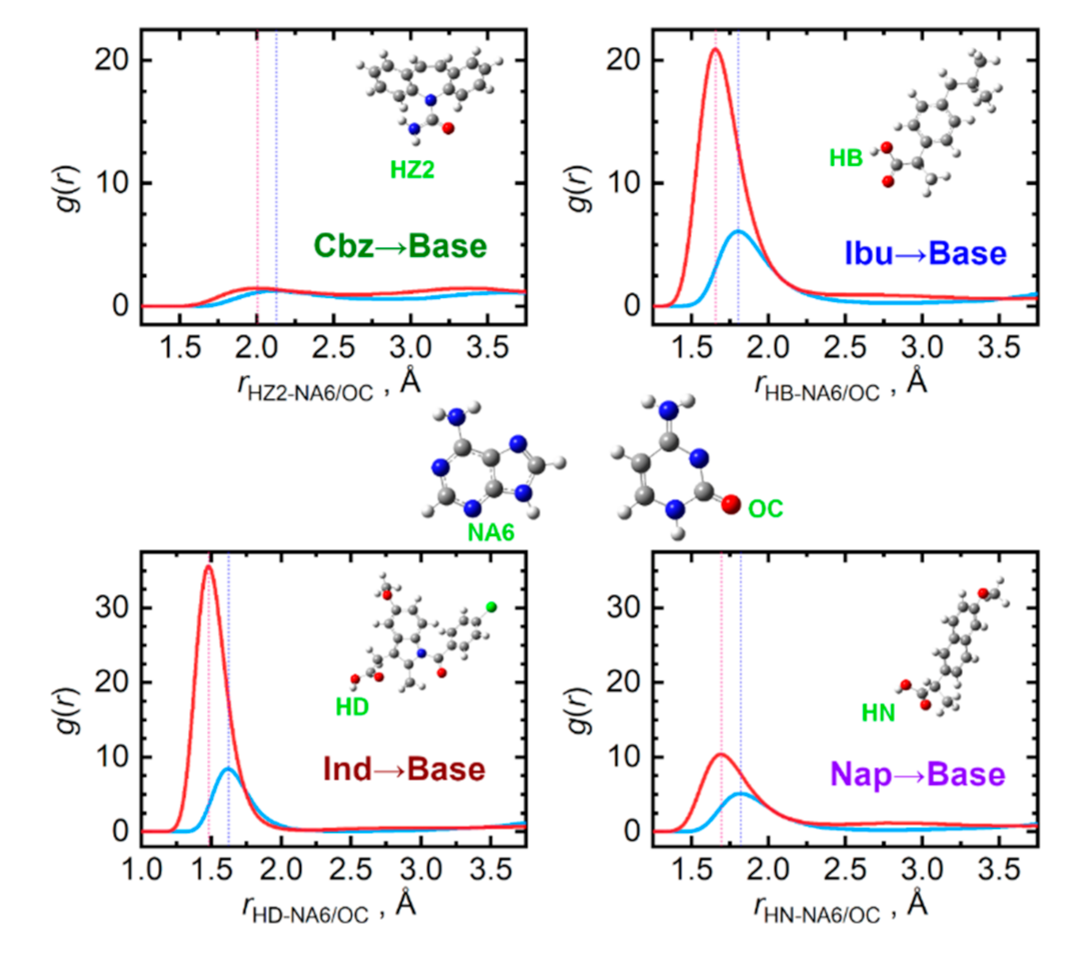{kind=link}
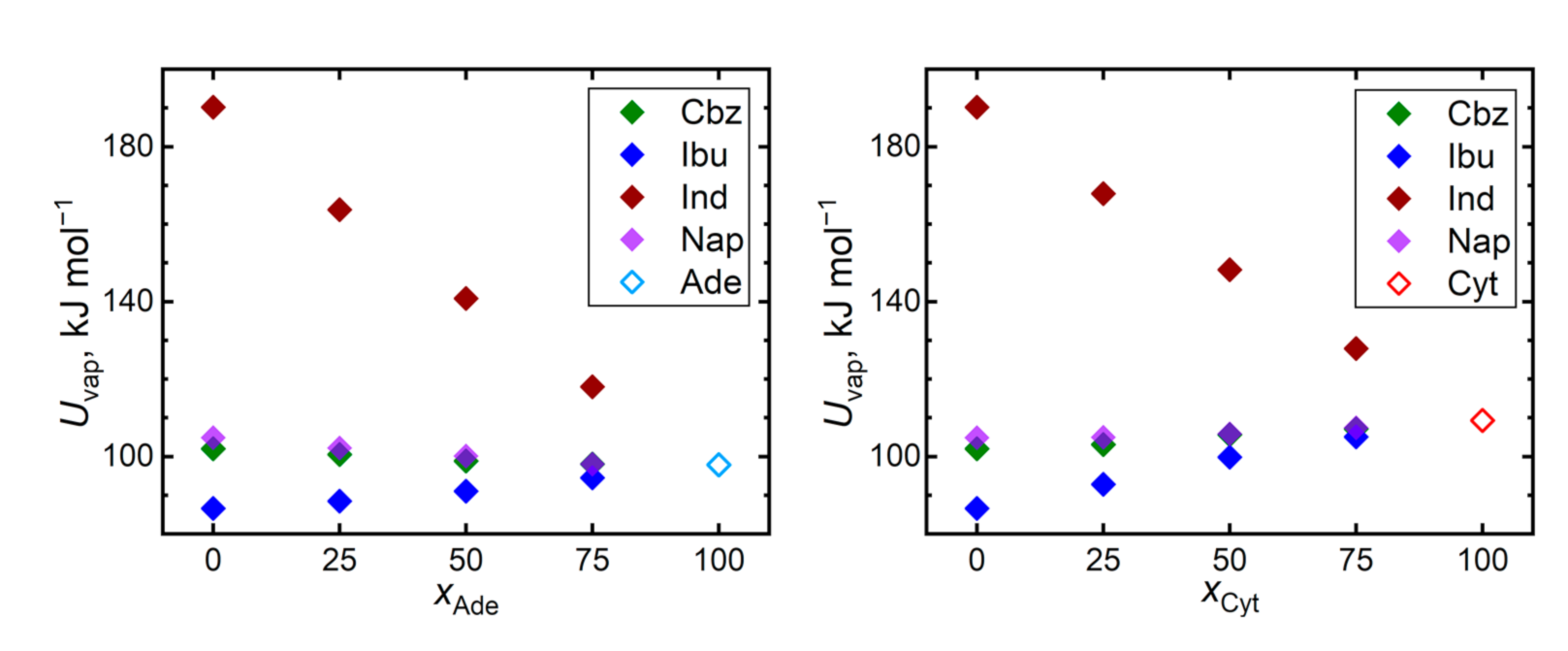{kind=link}
| Term | Carbamazepine | Ibuprofen | Indomethacin | Naproxen | Adenine | Cytosine |
|---|---|---|---|---|---|---|
| Atomic charges | CHELPG, this work a | Greiner et al. [21] | Xiang and Anderson [23] | CHELPG, this work a | CHELPG, this work b | CHELPG, this work b |
| Dispersion | Original OPLS [24] | Greiner et al. [21] | Xiang and Anderson [23] | Römer and Kraska. [22] | Pranata et al. [25] | Pranata et al. [25] |
| Molecular geometry | QM, this worka | Greiner et al. [21] | Xiang and Anderson [23] | Römer and Kraska. [22] | Pranata et al. [25] | Pranata et al. [25] |
| Force constants | Original OPLS [24] | Greiner et al. [21] | Original OPLS [24] | Römer and Kraska. [22] | Pranata et al. [25] | Pranata et al. [25] |
| Compound | Phase | Temperature, K a | ρMD | ρexp | 100(ρMD/ρexp−1) |
|---|---|---|---|---|---|
| Carbamazepine | Crystal III | 293 | 1.335 | 1.333 [41] | 0.1 |
| Ibuprofen | Crystal I | 296 | 1.115 | 1.117 [40] | −0.2 |
| Liquid | 350 | 1.006 | 0.966 [43] | 4.1 | |
| 400 | 0.968 | 0.924 [43] | 4.7 | ||
| Indomethacin | Crystal α | 203 | 1.408 | 1.420 [39] | −0.9 |
| Crystal γ | 120 | 1.418 | 1.401 [38] | 1.2 | |
| Liquid | 400 | 1.284 | 1.231 [43] | 4.3 | |
| 450 | 1.264 | 1.183 [43] | 6.9 | ||
| Naproxen | Crystal | 293 | 1.308 | 1.263 [37] | 3.6 |
| Liquid | 430 | 1.154 | 1.088 [43] | 6.1 | |
| 480 | 1.116 | 1.048 [43] | 6.5 | ||
| Adenine | Crystal | 293 | 1.506 | 1.494 [35] | 0.8 |
| Cytosine | Crystal | 293 | 1.537 | 1.502 [36] | −1.6 |
| Compound | Phase | Temperature, K | ΔfusHMD | ΔfusHexp a | 100(ΔfusHMD/ΔfusHexp−1) |
|---|---|---|---|---|---|
| Carbamazepin | Crystal III | 293 | 30.6 | 27.4 | 12 |
| Ibuprofen | Crystal I | 296 | 15.1 | 26.4 | −43 |
| Indomethacin | Crystal γ | 203 | 20.6 | 38.1 | −46 |
| Naproxen | Crystal | 293 | 29.1 | 32.4 | −10 |
| Compound | Tg,expa | 100(Tg,MD/Tg,exp − 1) | |||
|---|---|---|---|---|---|
| Carbamazepin | 384 | 374 | 379 | 315 | 20 |
| Ibuprofen | 295 | 286 | 290 | 228 | 27 |
| Indomethacin | 388 | 388 | 388 | 313 | 24 |
| Naproxen | 343 | 347 | 345 | 278 | 24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Červinka, C.; Fulem, M. Structure and Glass Transition Temperature of Amorphous Dispersions of Model Pharmaceuticals with Nucleobases from Molecular Dynamics. Pharmaceutics 2021, 13, 1253. https://doi.org/10.3390/pharmaceutics13081253
Červinka C, Fulem M. Structure and Glass Transition Temperature of Amorphous Dispersions of Model Pharmaceuticals with Nucleobases from Molecular Dynamics. Pharmaceutics. 2021; 13(8):1253. https://doi.org/10.3390/pharmaceutics13081253
Chicago/Turabian StyleČervinka, Ctirad, and Michal Fulem. 2021. "Structure and Glass Transition Temperature of Amorphous Dispersions of Model Pharmaceuticals with Nucleobases from Molecular Dynamics" Pharmaceutics 13, no. 8: 1253. https://doi.org/10.3390/pharmaceutics13081253