
DoE-Based Design of a Simple but Efficient Preparation Method for a Non-Effervescent Gastro-Retentive Floating Tablet Containing Metformin HCl


Abstract
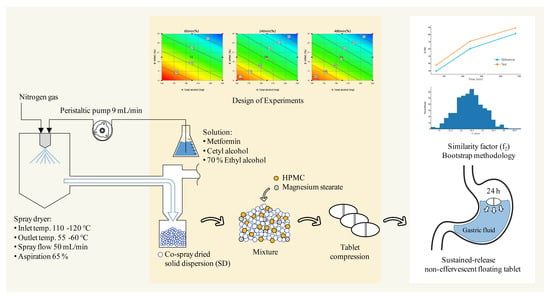
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of a Solid Dispersion of Met HCl with Excipients
2.2.2. Gas Chromatography (GC)
2.2.3. Scanning Electron Microscopy (SEM)
2.2.4. Powder X-ray Diffraction
2.2.5. Bulk Density
2.2.6. Particle Size Distribution
2.2.7. Tablet Preparation
2.2.8. Floating Tests
2.2.9. In Vitro Dissolution Analysis
2.2.10. Release Kinetics Model
2.2.11. High-Performance Liquid Chromatography (HPLC)
2.2.12. Design of Experiments (DoE)
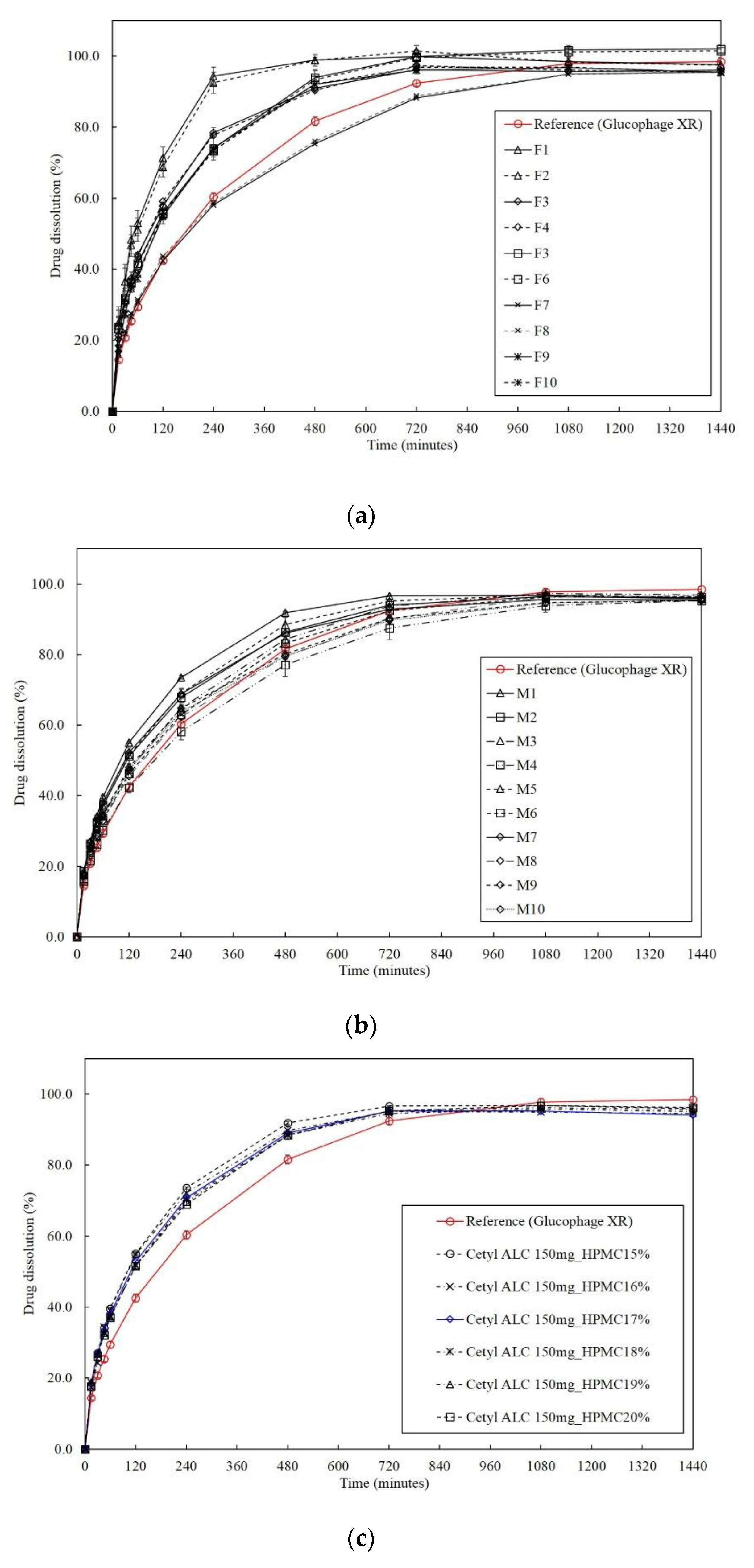
2.2.13. Dissolution Profile Comparison
2.2.14. Swelling and Matrix Erosion
3. Results and Discussion
3.1. Screening of Excipients for Co-Spray-Dried Solid Dispersion of Met HCl
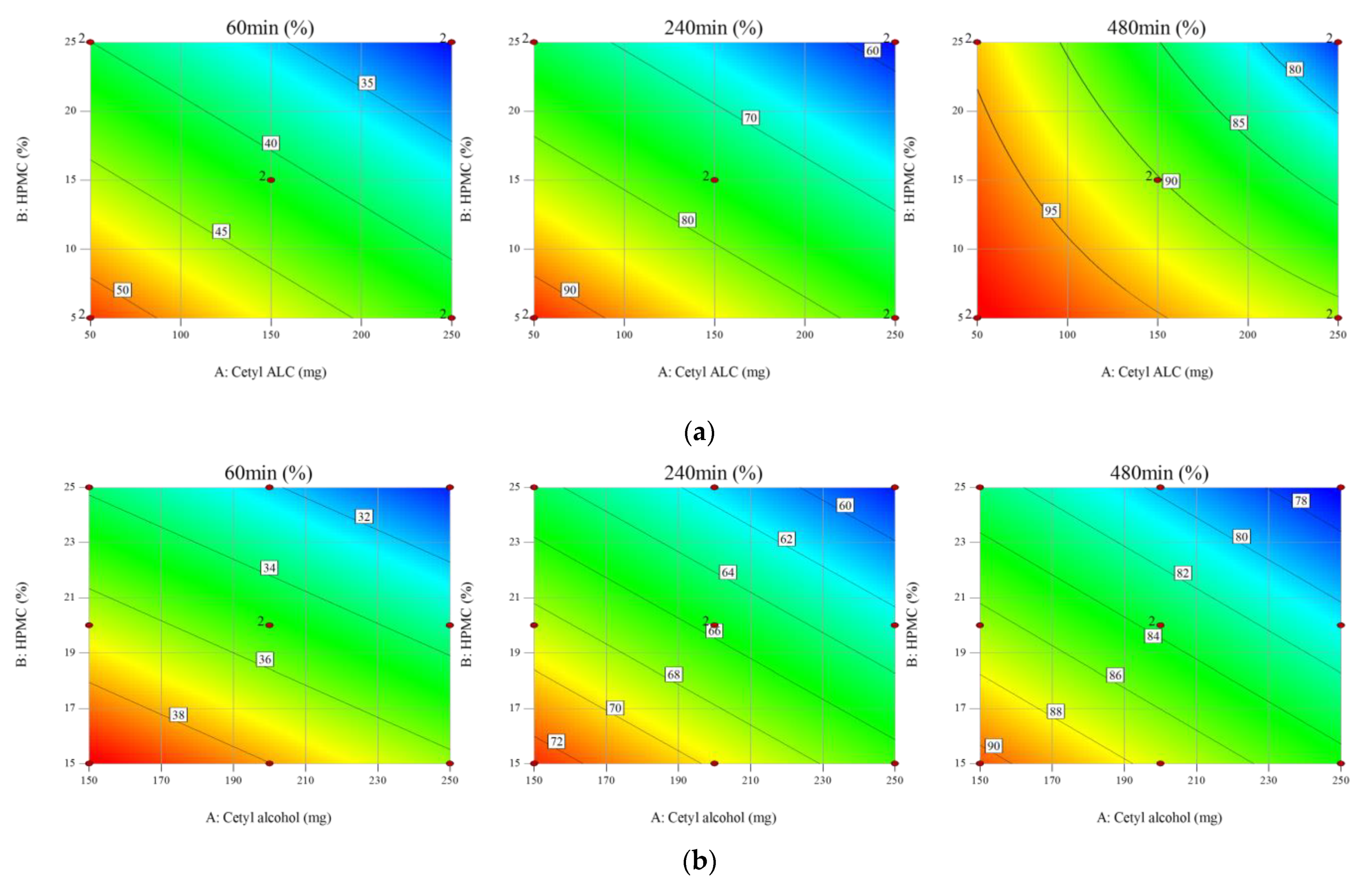
3.2. Design of Experiments (DoE) for the Non-Effervescent Floating Sustained-Release Formulation
3.3. Prediction Using an External Validation Set
3.4. Optimization of the Formulation Using the RSM
3.5. Release Kinetic Models
3.6. Evaluation of Dissolution Profile Equivalence via Bootstrap Analysis
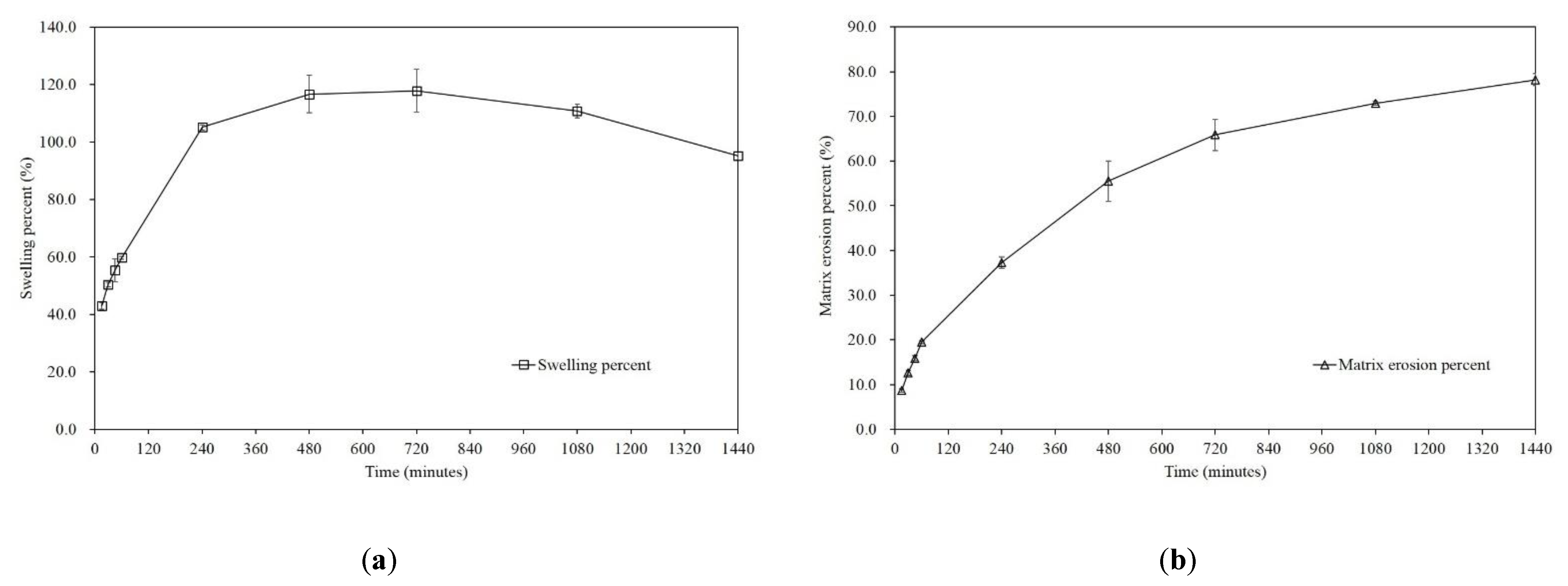
3.7. Floating, Swelling, and Erosion Testing of the Sustained-Release Non-Effervescent Floating Tablet
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rojas, L.B.A.; Gomes, M.B. Metformin: An old but still the best treatment for type 2 diabetes. Diabetol. Metab. Syndr. 2013, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setter, S.M.; Iltz, J.L.; Thams, J.; Campbell, R.K. Metformin hydrochloride in the treatment of type 2 diabetes mellitus: A clinical review with a focus on dual therapy. Clin. Ther. 2003, 25, 2991–3026. [Google Scholar] [CrossRef]
- Robert, F.; Fendri, S.; Hary, L.; Lacroix, C.; Andréjak, M.; Lalau, J.D. Kinetics of plasma and erythrocyte metformin after acute administration in healthy subjects. Diabetes Metab. 2003, 29, 279–283. [Google Scholar] [CrossRef]
- Stepensky, D.; Friedman, M.; Srour, W.; Raz, I.; Hoffman, A. Preclinical evaluation of pharmacokinetic–pharmacodynamic rationale for oral CR metformin formulation. J. Control. Release 2001, 71, 107–115. [Google Scholar] [CrossRef]
- Christofides, E.A. Practical insights into improving adherence to Metformin therapy in patients with type 2 diabetes. ADA 2019, 37, 234–241. [Google Scholar] [CrossRef]
- Lopes, C.M.; Bettencourt, C.; Rossi, A.; Buttini, F.; Barata, P. Overview on gastroretentive drug delivery systems for improving drug bioavailability. Int. J. Pharm. 2016, 510, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Wu, M.; Xu, S.; Wang, X.; Shi, W.; Dong, Y.; Yang, L.; He, W.; Han, X.; Yin, L. Design and optimization of gastro-floating sustained-release tablet of pregabalin: In vitro and in vivo evaluation. Int. J. Pharm. 2018, 545, 37–44. [Google Scholar] [CrossRef]
- Jain, S.K.; Jangdey, M.S. Lectin conjugated gastroretentive multiparticulate delivery system of clarithromycin for the effective treatment of Helicobacter pylori. Mol. Pharm. 2008, 6, 295–304. [Google Scholar] [CrossRef]
- Hardikar, S.; Bhosale, A. Formulation and evaluation of gastro retentive tablets of clarithromycin prepared by using novel polymer blend. Bull. Fac. Pharm. Cairo Univ. 2018, 56, 147–157. [Google Scholar] [CrossRef]
- Inukai, K.; Takiyama, K.; Noguchi, S.; Iwao, Y.; Itai, S. Effect of gel formation on the dissolution behavior of clarithromycin tablets. Int. J. Pharm. 2017, 521, 33–39. [Google Scholar] [CrossRef]
- Mostafavi, A.; Emami, J.; Varshosaz, J.; Davies, N.M.; Rezazadeh, M. Development of a prolonged-release gastroretentive tablet formulation of ciprofloxacin hydrochloride: Pharmacokinetic characterization in healthy human volunteers. Int. J. Pharm. 2011, 409, 128–136. [Google Scholar] [CrossRef]
- Chavanpatil, M.D.; Jain, P.; Chaudhari, S.; Shear, R.; Vavia, P.R. Novel sustained release, swellable and bioadhesive gastroretentive drug delivery system for ofloxacin. Int. J. Pharm. 2006, 316, 86–92. [Google Scholar] [CrossRef]
- Fu, J.; Yin, H.; Yu, X.; Xie, C.; Jiang, H.; Jin, Y.; Sheng, F. Combination of 3D printing technologies and compressed tablets for preparation of riboflavin floating tablet-in-device (TiD) systems. Int. J. Pharm. 2018, 549, 370–379. [Google Scholar] [CrossRef]
- Kagan, L.; Lapidot, N.; Afargan, M.; Kirmayer, D.; Moor, E.; Mardor, Y.; Friedman, M.; Hoffman, A. Gastroretentive Accordion Pill: Enhancement of riboflavin bioavailability in humans. J. Control. Release. 2006, 113, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.-M.; Cho, C.-H.; Tung, N.-T.; Kim, J.-Y.; Rhee, Y.-S.; Park, E.-S. Release kinetics of highly porous floating tablets containing cilostazol. Eur. J. Pharm. Biopharm. 2017, 115, 39–51. [Google Scholar] [CrossRef]
- Kim, S.; Hwang, K.-M.; Park, Y.S.; Nguyen, T.-T.; Park, E.-S. Preparation and evaluation of non-effervescent gastroretentive tablets containing pregabalin for once-daily administration and dose proportional pharmacokinetics. Int. J. Pharm. 2018, 550, 160–169. [Google Scholar] [CrossRef]
- Ngwuluka, N.C.; Choonara, Y.E.; Kumar, P.; du Toit, L.C.; Modi, G.; Pillay, V. An optimized gastroretentive nanosystem for the delivery of levodopa. Int. J. Pharm. 2015, 494, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Nandi, G.; Changder, A.; Hudati, P.; Sarkar, S.; Ghosh, L.K. Sustained release gastroretentive tablet of metformin hydrochloride based on poly (acrylic acid)-grafted-gellan. Int. J. Biol. Macromol. 2017, 96, 137–148. [Google Scholar] [CrossRef]
- Patil, S.; Talele, G.S. Gastroretentive mucoadhesive tablet of lafutidine for controlled release and enhanced bioavailability. Durg Deliv. 2015, 22, 312–319. [Google Scholar] [CrossRef] [Green Version]
- Sawicki, W. Pharmacokinetics of verapamil and norverapamil from controlled release floating pellets in humans. Eur. J. Pharm. Biopharm. 2002, 53, 29–35. [Google Scholar] [CrossRef]
- Jiménez-Martínez, I.; Quirino-Barreda, T.; Villafuerte-Robles, L. Sustained delivery of captopril from floating matrix tablets. Int. J. Pharm. 2008, 362, 37–43. [Google Scholar] [CrossRef]
- Baumgartner, S.; Kristl, J.; Vrečer, F.; Vodopivec, P.; Zorko, B. Optimisation of floating matrix tablets and evaluation of their gastric residence time. Int. J. Pharm. 2000, 195, 125–135. [Google Scholar] [CrossRef]
- Whitehead, L.; Fell, J.T.; Collett, J.H.; Sharma, H.L.; Smith, A.-M. Floating dosage forms: An in vivo study demonstrating prolonged gastric retention. J. Control. Release 1998, 55, 3–12. [Google Scholar] [CrossRef]
- Hilton, A.K.; Deasy, P.B. In vitro and in vivo evaluation of an oral sustained-release floating dosage form of amoxycillin trihydrate. Int. J. Pharm. 1992, 86, 79–88. [Google Scholar] [CrossRef]
- He, W.; Li, Y.; Zhang, R.; Wu, Z.; Yin, L. Gastro-floating bilayer tablets for the sustained release of metformin and immediate release of pioglitazone: Preparation and in vitro/in vivo evaluation. Int. J. Pharm. 2014, 476, 223–231. [Google Scholar] [CrossRef]
- Hajare, A.A.; Patil, V.A. Formulation and characterization of metformin hydrochloride floating tablets. Asian J. Pharm. Sci. 2012, 2, 111–117. [Google Scholar]
- Raju, D.B.; Sreenivas, R.; Varma, M.M. Formulation and evaluation of floating drug delivery system of metformin hydrochloride. J. Chem. Pharm. 2010, 2, 274–278. [Google Scholar]
- Rajab, M.; Jouma, M.; Neubert, R.H.H.; Dittgen, M. Influence of water-soluble polymers on the in vitro performance of floating mucoadhesive tablets containing metformin. Drug Dev. Ind. Pharm. 2014, 40, 879–885. [Google Scholar] [CrossRef]
- Senjoti, F.G.; Mahmood, S.; Jaffri, J.M.; Mandal, U.K. Design and in-vitro evaluation of sustained release floating tablets of metformin HCl based on effervescence and swelling. Iran. J. Pharm. Sci. 2016, 15, 53–70. [Google Scholar]
- Kumar, R. Development and in vitro evaluation of sustained release floating matrix tablets of metformin hydrochloride. Int. J. Pharm. Sci. 2010, 1, 96–101. [Google Scholar]
- Rajab, M.; Jouma, M.; Neubert, R.H.; Dittgen, M. Optimization of a metformin effervescent floating tablet containing hydroxypropylmethylcellulose and stearic acid. Pharmazie 2010, 65, 97–101. [Google Scholar]
- Djebbar, M.; Chaffai, N.; Bouchal, F.; Aouf, N. Effervescent floating tablets of metformin HCl developed by melt granulation. Part I: Effect of hydrophilic polymer on biopharmaceutical properties. GSCBPS 2019, 6, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Djebbar, M.; Chaffai, N.; Bouchal, F. Development of floating tablets of metformin HCl by thermoplastic granulation. Part II: In vitro evaluation of the combined effect of acacia gum/HPMC on Biopharmaceutical performances. Adv. Pharm. Bull. 2020, 10, 399–407. [Google Scholar] [CrossRef]
- Chandira, R.M.; Arafath, A.A.M.Y.; Bhowmik, D.; Jayakar1, B.; Kumar, K.P.S. Formulation and evaluation of bilayered floating tablets of metformin hydrochloride. Pharma Innovation 2012, 1, 26–39. [Google Scholar]
- Boldhane, S.P.; Kuchekar, B.S. Gastroretentive drug delivery of metformin hydrochloride: Formulation and in vitro evaluation using 32 full factorial design. Curr. Drug Deliv. 2009, 6, 477–485. [Google Scholar] [CrossRef]
- Patel, M.B.; Shaikh, F.; Patel, V.; Surti, N.I. Application of simplex centroid design in formulation and optimization of floating matrix tablets of metformin. J. Appl. Pharm. Sci. 2017, 7, 23–30. [Google Scholar]
- Thapa, P.; Jeong, S.H. Effects of formulation and process variables on gastroretentive floating tablets with a high-dose soluble drug and experimental design approach. Pharmaceutics 2018, 10, 161. [Google Scholar] [CrossRef] [Green Version]
- Eisenächer, F.; Garbacz, G.; Mäder, K. Physiological relevant in vitro evaluation of polymer coats for gastroretentive floating tablets. Eur. J. Pharm. Biopharm. 2014, 88, 778–786. [Google Scholar] [CrossRef]
- Huh, H.W.; Na, Y.-G.; Kang, H.C.; Kim, M.; Han, M.; Pharm, T.M.A.; Lee, H.; Baek, J.-S.; Lee, H.-K.; Cho, C.W. Novel self-floating tablet for enhanced oral bioavailability of metformin based on cellulose. Int. J. Pharm. 2021, 592, 120113. [Google Scholar]
- Oh, T.-O.; Kim, J.-Y.; Ha, J.-M.; Chi, S.-C.; Rhee, Y.-S.; Park, C.-W.; Park, E.-S. Preparation of highly porous gastroretentive metformin tablets using a sublimation method. Eur. J. Pharm. Sci. 2013, 83, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Pandit, V.; Pai, R.S.; Yadav, V.; Devi, K.; Surekha, B.B.; Inamdar, M.N.; Suresh, S. Pharmacokinetic and pharmacodynamics evaluation of floating microspheres of metformin hydrochloride. Drug Dev. Ind. Pharm. 2013, 39, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.K.; Gupta, A. Development of Gelucire 43/01 beads of metformin hydrochloride for floating delivery. AAPS PharmSciTech 2009, 10, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Groves, M.J. Effect of FITC-dextran molecular weight on its release from floating cetyl alcohol and HPMC tablets. J. Pharm. Pharmacol. 2001, 53, 49–56. [Google Scholar] [CrossRef]
- Ziaee, A.; Albadarin, A.B.; Padrela, L.; Femmer, T.; O’Reilly, E.; Walker, G. Spray drying of pharmaceuticals and biopharmaceutical s: Critical parameters and experimental process optimization approaches. Eur. J. Pharm. Sci. 2019, 127, 300–318. [Google Scholar] [CrossRef] [PubMed]
- Vehring, R. Pharmaceutical particle engineering via spray drying. Pharm. Res. 2008, 25, 999–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacyna, J.; Kordalewska, M.; Markuszewski, M.J. Design of Experiments in metabolomics-related studies: An overview. J. Pharm. Biomed. Anal. 2019, 164, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Maniyar, M.G.; Kokare, C.R. Formulation and evaluation of spray dried liposomes of lopinavir for topical application. J. Pharm. Investig. 2019, 49, 259–270. [Google Scholar] [CrossRef]
- Sander, C.; Madsen, K.D.; Hyrup, B.; Nielsen, H.M.; Rantanen, J.; Jacobsen, J. Characterization of spray dried bioadhesive metformin microparticles for oromucosal administration. Eur. J. Pharm. Biopharam. 2013, 85, 682–688. [Google Scholar] [CrossRef]
- Pandey, K.U.; Joshi, A.; Dalvi, S.V. Evaluating the efcacy of diferent curcumin polymorphs in transdermal drug delivery. J. Pharm. Investig. 2021, 51, 75–84. [Google Scholar] [CrossRef]
- Lee, E.H.; Boerrigter, S.X.M.; Byrn, S.R. Epitaxy of a structurally related compound on the (100) faces of flufenamic acid form I and III single crystals. Cryst. Growth Des. 2010, 10, 518–527. [Google Scholar] [CrossRef]
- JimCnez-Castellanos, M.R.; Zia, H.; Rhodes, C.T. Design and testing in vitro of a bioadhesive and floating drug delivery system for oral application. Int. J. Pharm. 1994, 105, 65–70. [Google Scholar] [CrossRef]
- Zhang, L.; Mao, S. Application of quality by design in the current drug development. Asian J. Pharm. Sci. 2017, 12, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenço, V.; Lochmann, D.; Reich, G.; Menezes, J.C.; Herdling, T.; Schewitz, J. A quality by design study applied to an industrial pharmaceutical fluid bed granulation. Eur. J. Pharm. Biopharam. 2012, 81, 438–447. [Google Scholar] [CrossRef]
- Efron, B. Nonparametric estimates of standard error: The jackknife, the bootstrap and other methods. Biometrika 1981, 68, 589–599. [Google Scholar] [CrossRef]
- Efentakis, M.; Vlachou, M. Evaluation of high molecular weight poly (oxyethylene) (polyox) polymer: Studies of flow properties and release rates of furosemide and captopril from controlled-release hard gelatin capsules. Pharm. Dev. Technol. 2000, 5, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Li, H.; Guo, Z.; Wu, L.; Chen, F.; Matas, M.; Shao, Q.; Xiao, T.; York, P.; He, Y.; et al. Quantification of swelling and erosion in the controlled release of a poorly water-soluble drug using synchrotron x-ray computed microtomography. AAPS J. 2013, 15, 1025–1034. [Google Scholar] [CrossRef] [Green Version]
- Al-Zoubi, N.; Odeh, F.; Nikolakakis, I. Co-spray drying of metformin hydrochloride with polymers to improve compaction behavior. Powder Technol. 2017, 307, 163–174. [Google Scholar] [CrossRef]
- Cesur, S.; Cam, M.E.; Sayın, F.S.; Su, S.; Gunduz, O. Controlled Release of Metformin Loaded Polyvinyl Alcohol (Pva) Microbubble/Nanoparticles Using Microfluidic Device for the Treatment of Type 2 Diabetes Mellitus; Bioinformatics and Biomedical Engineering: Granada, Spain, 2020; pp. 185–193. [Google Scholar]
- Tiwari, R.; Gupta, A.; Joshi, M.; Tiwari, G. Bilayer tablet formulation of metformin HCl and acarbose: A novel approach to control diabetes. PDA J. Pharm. Sci. Technol. 2014, 68, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Benmessaoud, I.; Koutchoukali, O.; Bouhelassa, M.; Nouar, A.; Veesler, S. Solvent screening and crystal habit of metformin hydrochloride. J. Cryst. Growth 2016, 451, 42–51. [Google Scholar] [CrossRef]
- Eriksson, L.; Johansson, E.; Kettaneh-Wood, N.; Wikström, C.; Wold, S. Design of Experiments: Principles and Applications; MKS Umetrics AB: Umea, Sweden, 2008; pp. 149–154. [Google Scholar]
- van der Voet, H. Comparing the predictive accuracy of models using a simple randomization test. Chemom. Intell. Lab. Syst. 1994, 25, 313–323. [Google Scholar] [CrossRef]
- DiFoggio, R. Examination of some misconceptions about near-infrared analysis. Appl. Spectrosc. 1995, 49, 67–75. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Shah, V.P.; Tsong, Y.; Sathe, P.; Liu, J.-P. In vitro dissolution profile comparison-Statistics and analysis of the similarity factor, f2. Pharm. Res. 1998, 15, 889–896. [Google Scholar] [CrossRef]
- Mendyk, A.; Pacławski, A.; Szlek, J.; Jachowicz, R. PhEq_bootstrap: Open-source software for the simulation of f2 distribution in cases of large variability in dissolution profiles. Dissolution Technol. 2013, 20, 13–17. [Google Scholar] [CrossRef]
- Noce, L.; Gwaza, L.; Mangas-Sanjuan, V.; Garcia-Arieta, A. Comparison of free software platforms for the calculation of the 90% confidence interval of f2 similarity factor by bootstrap analysis. Eur. J. Pharm. Sci. 2020, 146, 105259. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Peppas, N.A. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv. Drug Deliv. Rev. 2012, 64, 163–174. [Google Scholar] [CrossRef]
- Iglesias, N.; Galbis, E.; Romero-Azogil, L.; Benito, E.; Lucas, R.; García-Martín, M.G.; de-Paz, M.-V. In-depth study into polymeric materials in low-density gastroretentive formulations. Pharmaceutics 2020, 12, 636. [Google Scholar] [CrossRef] [PubMed]
- Lamoudi, L.; Chaumeil, J.C.; Daoud, K. Swelling, erosion and drug release characteristics of sodium diclofenac from heterogeneous matrix tablets. J. Drug Deliv. Sci. Technol. 2016, 31, 93–100. [Google Scholar] [CrossRef]
- Prinderre, P.; Sauzet, C.; Fuxen, C. Advances in gastro retentive drug-delivery systems. Expert Opin. Drug Deliv. 2011, 8, 1189–1203. [Google Scholar] [CrossRef] [PubMed]
- Mandal, U.K.; Chatterjee, B.; Senjoti, F.G. Gastro-retentive drug delivery systems and their in vivo success: A recent update. Asian J. Pharm. Sci. 2016, 11, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Li, C.L.; Martini, L.G.; Ford, J.L.; Roberts, M. The use of hypromellose in oral drug. J. Pharm. Pharmacol. 2005, 57, 533–546. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NEFT/EFT | Preparation Methods | Polymers for Extended-Release Property | Excipients for Floating Property | Title |
|---|---|---|---|---|
| Effervescent | Wet granulation | HPMC K4M, HPMC K15M, HPMC K100M | SB | Gastro-floating bilayer tablets for the sustained release of metformin and immediate release of pioglitazone: preparation and in vitro/in vivo evaluation [25] |
| Guar gum, κ-carrageenan, HPMC K100 | SB, CA | Formulation and characterization of metformin hydrochloride floating tablets [26] | ||
| HPMC K4M, carbopol 934P | SB | Formulation and evaluation of floating drug delivery system of metformin hydrochloride [27] | ||
| PVP, TSG, HPMC | SB, CA | Influence of water-soluble polymers on the in vitro performance of floating mucoadhesive tablets containing metformin [28] | ||
| HPMC, PEO, SSG | SB | Design and in-vitro evaluation of sustained release floating tablets of metformin HCl based on effervescence and swelling [29] | ||
| HPMC | PB | Development and in vitro evaluation of sustained release floating matrix tablets of metformin hydrochloride [30] | ||
| Melt-granulation | HPMC | SA, SB, CA | Optimization of a metformin effervescent floating tablet containing hydroxypropylmethylcellulose and stearic acid [31] | |
| HPMC K4M, HPMC K15M, HPMC K100M, AG | SB | Effervescent floating tablets of metformin HCl developed by melt granulation. Part I: effect of hydrophilic polymer on biopharmaceutical properties [32] | ||
| HPMC K15M, HPMC K100M, AG | SB | Development of floating tablets of metformin HCl by thermoplastic granulation. Part II: In vitro evaluation of the combined effect of acacia gum/HPMC on Biopharmaceutical performances [33] | ||
| Direct compression | HPMC K4M, HPMC K100M, SSG, PVP-K-30, MCC | SB, CA, | Formulation and evaluation of bilayered floating tablets of metformin hydrochloride [34] | |
| Sodium alginate, sodium CMC | SB | Gastroretentive drug delivery of metformin hydrochloride: formulation and in vitro evaluation using 32 full factorial design [35] | ||
| HPMC K15M, κ-carrageenan | SB | Application of simplex centroid design in formulation and optimization of floating matrix tablets of metformin [36] | ||
| PEO WSR 303 | SB | Effects of formulation and process variables on gastroretentive floating tablets with a high-dose soluble drug and experimental design approach [37] | ||
| Polymer coating | Polyvinyl acetate, ammonio-methacrylate copolymer type A | SB, CA | Physiological relevant in vitro evaluation of polymer coats for gastroretentive floating tablets [38] | |
| Non-effervescent | Mold–hollow-core floating tablet (HCFT) | HPMC K100M, MCC | n/a | Novel self-floating tablet for enhanced oral bioavailability of metformin based on cellulose [39] |
| Wet-granulation, Sublimation | PEO WSR 301, HPC | D, L-Camphor | Preparation of highly porous gastroretentive metformin tablets using a sublimation method [40] | |
| Emulsion solvent evaporation method | n/a | Liquid paraffin, Span 60, petroleum ether | Pharmacokinetic and pharmacodynamics evaluation of floating microspheres of metformin hydrochloride [41] | |
| Beads | n/a | Gelucire 43/01 | Development of Gelucire 43/01 beads of metformin hydrochloride for floating delivery [42] | |
| AG: acacia gum; CA: citric acid; HPC: hydroxypropyl cellulose; HPMC: hydroxypropylmethylcellulose; MCC: microcrystalline cellulose; PB: potassium bicarbonate; PEO: polyethylene glycol; PVP: polyvinylprrolidone; SA: stearic acid; SB: sodium bicarbonate; sodium CMC: sodium carboxymethylcellulose; SSG: sodium starch glycolate; TSG: tarmarind seed gum | ||||
| Components (mg) | Solvent/mL | SD1 | SD2 | SD3 | SD4 | SD5 | SD6 |
|---|---|---|---|---|---|---|---|
| Metformin HCl | - | 500 | 500 | 500 | 500 | 500 | 500 |
| Sodium alginate | Water/750 | 250 | - | - | - | - | - |
| PVP K-30 | Water/750 | - | 250 | - | - | - | - |
| PEG 6000 | Water/750 | - | - | 250 | - | - | - |
| Polyvinyl alcohol | Water/750 | - | - | - | 250 | - | - |
| HPMC K15M | Water/750 | - | - | - | - | 250 | - |
| Cetyl alcohol | 70% Ethanol/750 | - | - | - | - | - | 250 |
| Magnesium stearate | - | 7.58 | 7.58 | 7.58 | 7.58 | 7.58 | 7.58 |
| Tablet total (mg) | - | 757.58 | 757.58 | 757.58 | 757.58 | 757.58 | 757.58 |
| Test (n = 3) | SD1 | SD2 | SD3 | SD4 | SD5 | SD6 |
|---|---|---|---|---|---|---|
| Bulk density of SD (g/cm3) | 0.41 ± 0.01 | 0.41 ± 0.01 | 0.42 ± 0.01 | 0.40 ± 0.01 | 0.51 ± 0.01 | 0.30 ± 0.01 |
| Particle size distribution of SD (d 0.5, um) | 7.15 ± 0.23 | 5.32 ± 0.13 | 7.62 ± 0.22 | 6.38 ± 0.25 | 5.06 ± 0.15 | 7.15 ± 0.18 |
| Content (%) of metformin in SD | 99.31 ± 0.73 | 98.59 ± 0.68 | 99.30 ± 0.79 | 99.26 ± 0.59 | 99.67 ± 0.50 | 99.75 ± 0.56 |
| Floating of tablet in 0.1 N HCl buffer (pH 1.2) | Not floated | Not floated | Not floated | Not floated | Not floated | Floated |
| Residual solvent of SD6 (Ethanol < 5000 ppm) | n/a | n/a | n/a | n/a | n/a | 134.10 ± 2.08 |
| ANOVA (p-Value < 0.05) | Y Variable (Response) | ||
|---|---|---|---|
| (R1) 60 min | (R2) 240 min | (R3) 480 min | |
| Model | <0.0001 | <0.0001 | 0.0081 |
| A: Cetyl alcohol | 0.0002 | 0.0002 | 0.0280 |
| B: HPMC K15M | <0.0001 | <0.0001 | 0.0086 |
| R-Squared | 96.50% | 95.45% | 74.71% |
| Adj R-Squared | 95.38% | 94.15% | 67.49% |
| Coded equation | R1 = +35.06 − 1.72 × A − 2.95 × B | R2 = +65.61 − 3.05 × A − 4.17 × B | R3 = +83.64 − 2.98 × A − 3.90 × B |
| Actual equation | R1 = +53.73 − 0.03 × A − 0.59 × B | R2 = +94.48 − 0.06 × A − 0.83 × B | R3 = +111.17 − 0.06 × A − 0.78 × B |
| Cetyl alcohol (mg) | HPMC K15M (%) | Code | Mass (mg) | In Vitro Drug-Release Model | Sample Mean (f2) | Bootstrap Analysis (500) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Zero-order | Higuchi | Korsmeyer-Peppas | f2 | E(f2) | PI | Bcα | |||||||
| R2 | R2 | R2 | n | k | |||||||||
| 150 | 5 | E1 | 722.2 | 0.6193 | 0.8050 | 0.9116 | 0.3531 | 0.9749 | 37.68 | 37.69 | 37.67 | (36.88, 38.49) | (36.84, 38.44) |
| 10 | E2 | 764.2 | 0.6621 | 0.8402 | 0.9284 | 0.3729 | 0.9111 | 42.98 | 42.98 | 42.95 | (42.29, 43.62) | (42.35, 43.67) | |
| 15 | M1 | 812.4 | 0.6939 | 0.8648 | 0.9432 | 0.3743 | 0.8992 | 47.36 | 47.36 | 47.35 | (46.61, 48.13) | (46.63, 48.15) | |
| 16 | P1 | 822.8 | 0.6921 | 0.7573 | 0.9399 | 0.3779 | 0.8819 | 49.85 | 49.86 | 49.81 | (48.75, 51.00) | (48.84, 51.12) | |
| 17 | P2 | 833.4 | 0.7089 | 0.7847 | 0.9551 | 0.3664 | 0.9094 | 52.41 | 52.41 | 52.43 | (51.42, 53.60) | (51.45, 53.65) | |
| 20 | M5 | 866.6 | 0.7355 | 0.8950 | 0.9619 | 0.3802 | 0.8716 | 55.41 | 55.44 | 55.43 | (53.69, 57.29) | (53.62, 57.17) | |
| 25 | M3 | 928.5 | 0.7782 | 0.9233 | 0.9761 | 0.3939 | 0.8216 | 69.08 | 69.09 | 68.67 | (65.58, 71.99) | (66.25, 72.91) | |
| 200 | 5 | E3 | 777.8 | 0.6397 | 0.8221 | 0.9223 | 0.3488 | 0.9806 | 40.09 | 40.06 | 40.04 | (39.47, 40.69) | (39.50, 40.74) |
| 10 | E4 | 823.5 | 0.6923 | 0.8635 | 0.9437 | 0.3617 | 0.9305 | 47.68 | 47.73 | 47.64 | (46.79, 48.53) | (46.91, 48.75) | |
| 15 | M7 | 874.9 | 0.7419 | 0.8995 | 0.9611 | 0.3677 | 0.9611 | 56.15 | 56.12 | 56.05 | (54.44, 57.69) | (54.44, 57.69) | |
| 20 | M9 | 933.2 | 0.7892 | 0.9305 | 0.9779 | 0.3752 | 0.8638 | 69.32 | 69.08 | 69.02 | (67.13, 70.80) | (67.24, 70.88) | |
| 25 | M8 | 1000.0 | 0.8109 | 0.9435 | 0.9837 | 0.3925 | 0.8127 | 77.88 | 77.86 | 77.50 | (75.89, 79.16) | (76.54, 80.05) | |
| 250 | 5 | E5 | 833.2 | 0.6594 | 0.8380 | 0.9318 | 0.3306 | 1.0235 | 42.40 | 42.45 | 42.43 | (41.69, 43.18) | (41.70, 43.19) |
| 10 | E6 | 882.3 | 0.7222 | 0.8856 | 0.9559 | 0.3600 | 0.9327 | 50.70 | 50.81 | 50.64 | (49.41, 51.73) | (49.43, 51.79) | |
| 15 | M2 | 937.4 | 0.7496 | 0.9047 | 0.9677 | 0.3704 | 0.8940 | 58.15 | 58.13 | 57.99 | (55.51, 60.81) | (55.48, 60.79) | |
| 20 | M6 | 1000.0 | 0.7856 | 0.9279 | 0.9772 | 0.4016 | 0.7939 | 76.76 | 76.58 | 76.43 | (73.01, 79.94) | (72.82, 79.60) | |
| 25 | M4 | 1071.3 | 0.8249 | 0.9513 | 0.9862 | 0.4126 | 0.7412 | 75.47 | 75.62 | 75.17 | (68.91, 82.46) | (69.42, 83.50) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.; Byun, Y.; Lee, E.H. DoE-Based Design of a Simple but Efficient Preparation Method for a Non-Effervescent Gastro-Retentive Floating Tablet Containing Metformin HCl. Pharmaceutics 2021, 13, 1225. https://doi.org/10.3390/pharmaceutics13081225
Kim B, Byun Y, Lee EH. DoE-Based Design of a Simple but Efficient Preparation Method for a Non-Effervescent Gastro-Retentive Floating Tablet Containing Metformin HCl. Pharmaceutics. 2021; 13(8):1225. https://doi.org/10.3390/pharmaceutics13081225
Chicago/Turabian StyleKim, Byungsuk, Youngjoo Byun, and Eun Hee Lee. 2021. "DoE-Based Design of a Simple but Efficient Preparation Method for a Non-Effervescent Gastro-Retentive Floating Tablet Containing Metformin HCl" Pharmaceutics 13, no. 8: 1225. https://doi.org/10.3390/pharmaceutics13081225