
Mathematical Modeling of Remdesivir to Treat COVID-19: Can Dosing Be Optimized?

Abstract
:1. Introduction
2. Materials and Methods
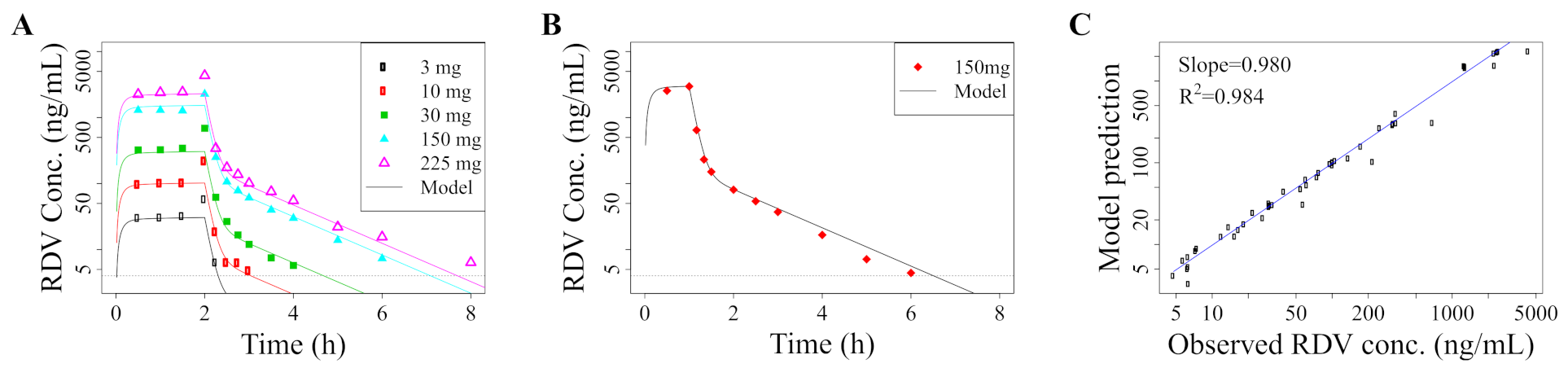
2.1. Pharmacokinetic Model
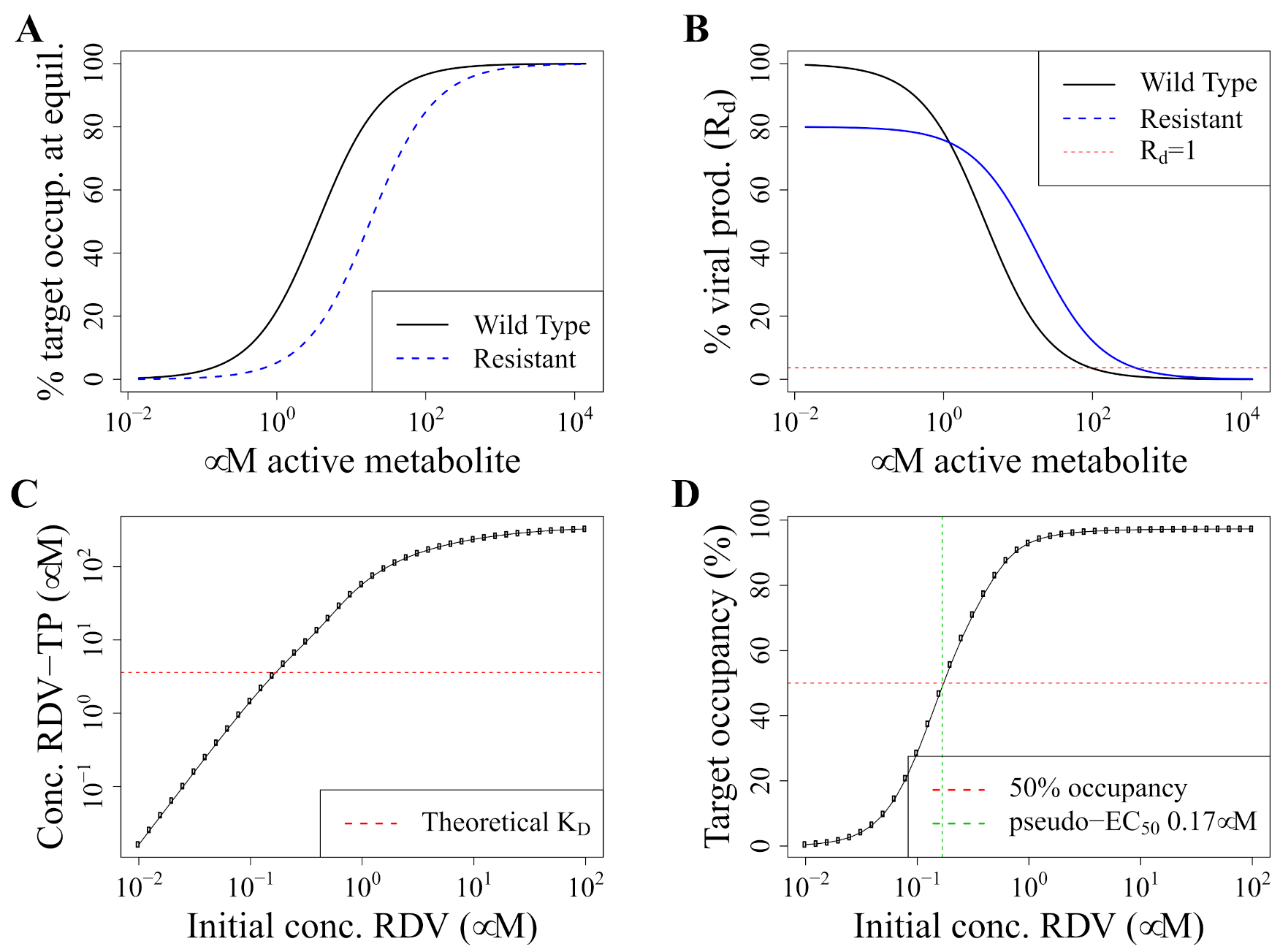
2.2. Pharmacodynamics Model
2.3. Concentrations Selecting for Resistance
2.4. Model Validation: Recapitulation of In Vitro Experiments
3. Results
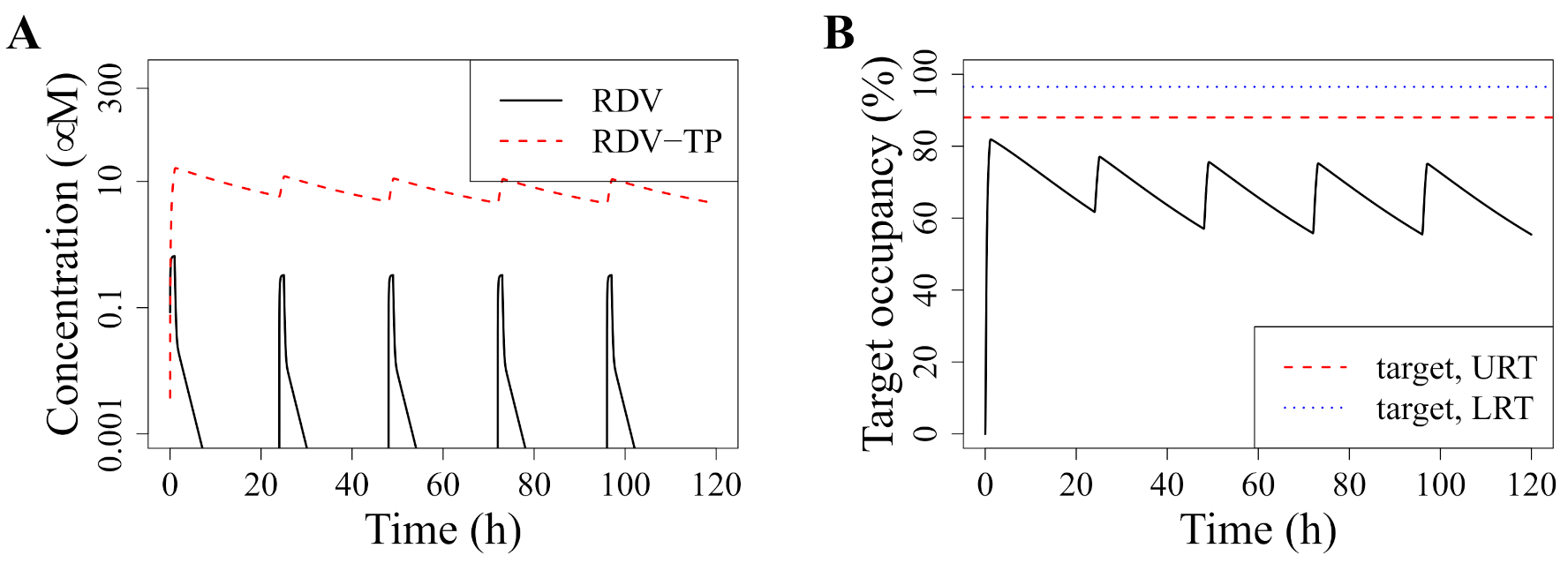
3.1. Standard Dosing Does Not Achieve Effective Concentrations
3.2. Model-Predicted Optimal Dosing Rate
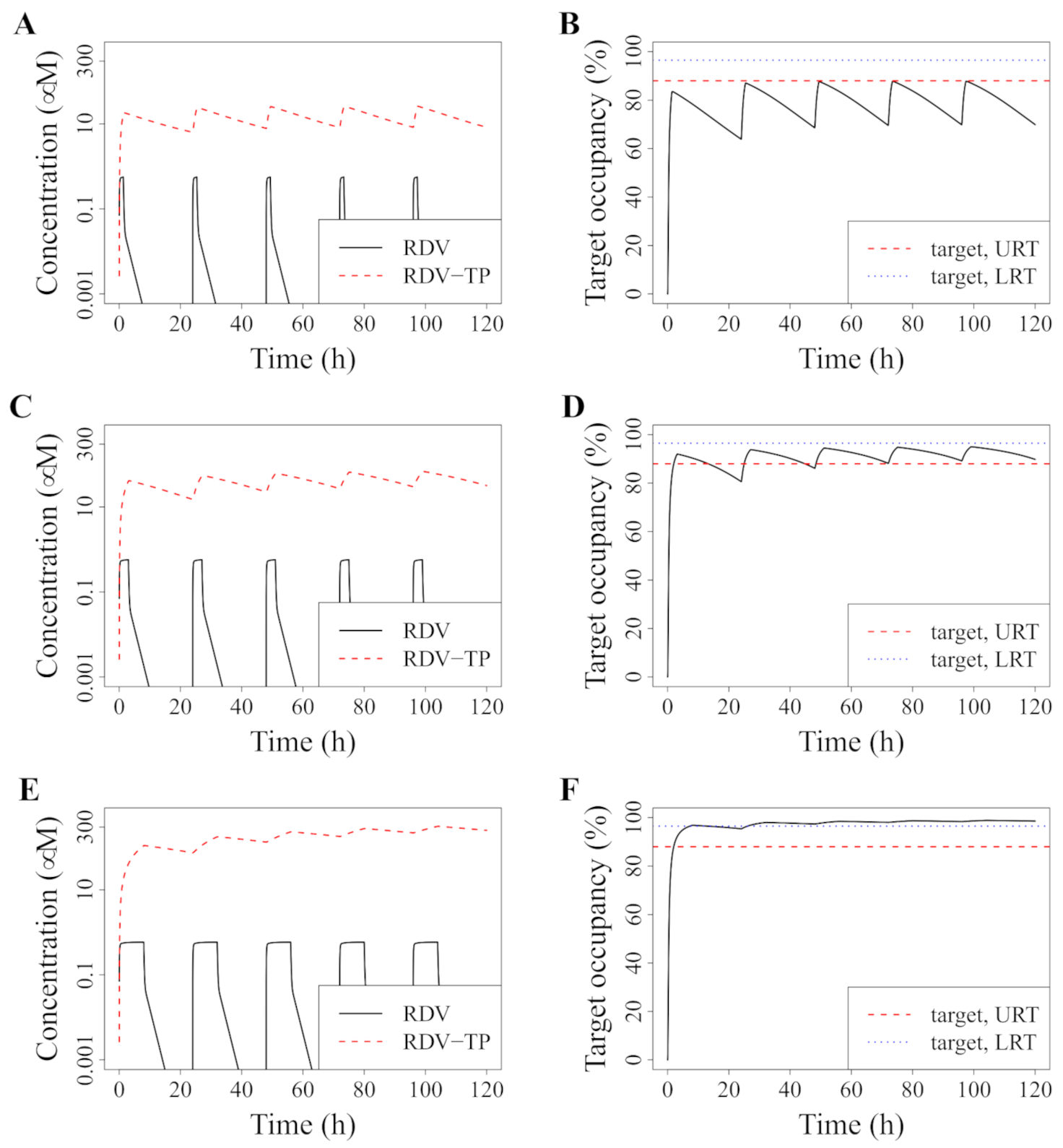
3.3. Improved Dosing with RDV Doses That Were Tested in Phase I Trials
3.4. Alternative: Longer Infusion
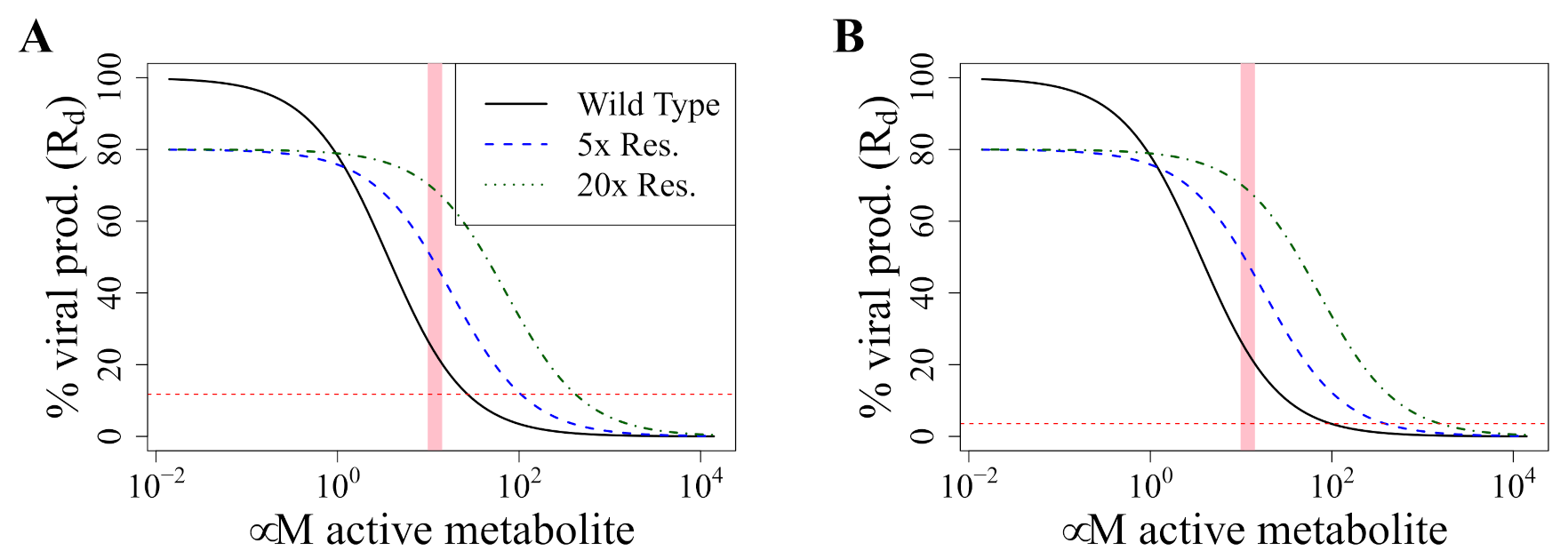
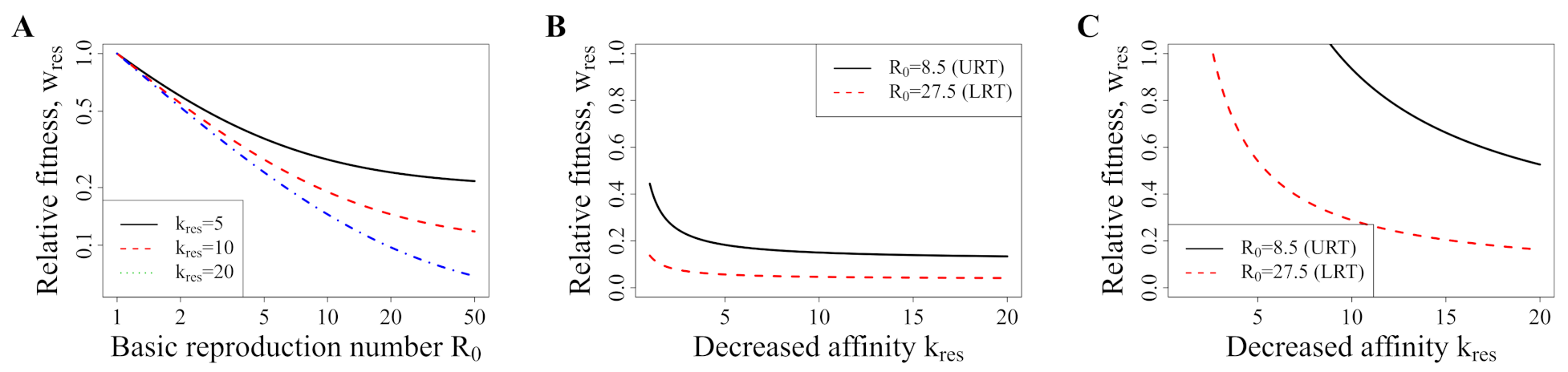
3.5. Avoiding a Potential DR Mutant
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EC | Half-maximal effective concentration |
| EMA | European Medecines Agency |
| FDA | US Food and Drug Administration |
| IC | Half-maximal inhibitory concentration |
| LRT | Lower respiratory tract |
| MERS-CoV | Middle East respiratory syndrome coronavirus |
| PBMCs | Peripheral blood mononuclear cells |
| PK | Pharmacokinetic |
| PD | Pharmacodynamic |
| TE | Target engagement |
| RdRp | RNA-dependent RNA polymerase |
| RDV | Remdesivir |
| RDV-TP | Remdesivir’s active triphosphate metabolite, GS-443902 |
| URT | Upper respiratory tract |
References
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- First COVID-19 Treatment Recommended for EU Authorisation | European Medicines Agency. Available online: https://www.ema.europa.eu/en/news/first-covid-19-treatment-recommended-eu-authorisation (accessed on 11 July 2021).
- FDA Approves First Treatment for COVID-19. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-covid-19 (accessed on 11 July 2021).
- de Wit, E.; Feldmann, F.; Cronin, J.; Jordan, R.; Okumura, A.; Thomas, T.; Scott, D.; Cihlar, T.; Feldmann, H. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc. Natl. Acad. Sci. USA 2020, 117, 6771–6776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NIH Clinical Trial Shows Remdesivir Accelerates Recovery from Advanced COVID-19. Available online: https://www.nih.gov/news-events/news-releases/nih-clinical-trial-shows-remdesivir-accelerates-recovery-advanced-covid-19 (accessed on 11 July 2021).
- WHO Solidarity Trial Consortium. Repurposed Antiviral Drugs for Covid-19—Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2020, 384, 497–511. [Google Scholar] [CrossRef]
- Kaka, A.S.; MacDonald, R.; Greer, N.; Vela, K.; Duan-Porter, W.; Obley, A.; Wilt, T.J. Major Update: Remdesivir for Adults with COVID-19. Ann. Intern. Med. 2021, 174, 663–672. [Google Scholar] [CrossRef]
- Garibaldi, B.T.; Fiksel, J.; Muschelli, J.; Robinson, M.L.; Rouhizadeh, M.; Perin, J.; Schumock, G.; Nagy, P.; Gray, J.H.; Malapati, H.; et al. Patient Trajectories Among Persons Hospitalized for COVID-19. Ann. Intern. Med. 2020, 174, 33–41. [Google Scholar] [CrossRef]
- Néant, N.; Lingas, G.; Hingrat, Q.L.; Ghosn, J.; Engelmann, I.; Lepiller, Q.; Gaymard, A.; Ferré, V.; Hartard, C.; Plantier, J.C.; et al. Modeling SARS-CoV-2 viral kinetics and association with mortality in hospitalized patients from the French COVID cohort. Proc. Natl. Acad. Sci. USA 2021, 118, e2017962118. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Duke, E.R.; Cardozo-Ojeda, E.F.; Schiffer, J.T. Mathematical modeling explains differential SARS CoV-2 kinetics in lung and nasal passages in remdesivir treated rhesus macaques. bioRxiv 2020. [Google Scholar] [CrossRef]
- Regan, J.; Flynn, J.P.; Rosenthal, A.; Jordan, H.; Li, Y.; Chishti, R.; Giguel, F.; Corry, H.; Coxen, K.; Fajnzylber, J.; et al. Viral Load Kinetics of SARS-CoV-2 In Hospitalized Individuals with COVID-19. Open Forum Infect. Dis. 2021, ofab153. [Google Scholar] [CrossRef]
- Adaptive COVID-19 Treatment Trial 4 (ACTT-4)—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04640168 (accessed on 11 July 2021).
- Monteil, V.; Dyczynski, M.; Lauschke, V.M.; Kwon, H.; Wirnsberger, G.; Youhanna, S.; Zhang, H.; Slutsky, A.S.; del Pozo, C.H.; Horn, M.; et al. Human soluble ACE2 improves the effect of remdesivir in SARS-CoV-2 infection. EMBO Mol. Med. 2021, 13, e13426. [Google Scholar] [CrossRef]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with Covid-19. N. Engl. J. Med. 2020, 384, 795–807. [Google Scholar] [CrossRef]
- Humeniuk, R.; Mathias, A.; Cao, H.; Osinusi, A.; Shen, G.; Chng, E.; Ling, J.; Vu, A.; German, P. Safety, Tolerability, and Pharmacokinetics of Remdesivir, An Antiviral for Treatment of COVID-19, in Healthy Subjects. Clin. Transl. Sci. 2020, 13, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef]
- Humeniuk, R.; Mathias, A.; Kirby, B.J.; Lutz, J.D.; Cao, H.; Osinusi, A.; Babusis, D.; Porter, D.; Wei, X.; Ling, J.; et al. Pharmacokinetic, Pharmacodynamic, and Drug-Interaction Profile of Remdesivir, a SARS-CoV-2 Replication Inhibitor. Clin. Pharmacokinet. 2021, 60, 569–583. [Google Scholar] [CrossRef]
- Mager, D.E.; Jusko, W.J. General Pharmacokinetic Model for Drugs Exhibiting Target-Mediated Drug Disposition. J. Pharmacokinet. Pharmacodyn. 2001, 28, 507–532. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Rabi, S.A.; Sedaghat, A.R.; Shan, L.; Lai, J.; Xing, S.; Siliciano, R.F. A Critical Subset Model Provides a Conceptual Basis for the High Antiviral Activity of Major HIV Drugs. Sci. Transl. Med. 2011, 3, 91ra63. [Google Scholar] [CrossRef] [Green Version]
- Walkup, G.K.; You, Z.; Ross, P.L.; Allen, E.K.H.; Daryaee, F.; Hale, M.R.; O’Donnell, J.; Ehmann, D.E.; Schuck, V.J.A.; Buurman, E.T.; et al. Translating Slow-Binding Inhibition Kinetics into Cellular and in Vivo Effects. Nat. Chem. Biol. 2015, 11, 416–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel zur Wiesch, P.; Abel, S.; Gkotzis, S.; Ocampo, P.; Engelstädter, J.; Hinkley, T.; Magnus, C.; Waldor, M.K.; Udekwu, K.; Cohen, T. Classic reaction kinetics can explain complex patterns of antibiotic action. Sci. Transl. Med. 2015, 7, 287ra73. [Google Scholar] [CrossRef] [Green Version]
- Baeder, D.Y.; Yu, G.; Hozé, N.; Rolff, J.; Regoes, R.R. Antimicrobial combinations: Bliss independence and Loewe additivity derived from mechanistic multi-hit models. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150294. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, X.; Doddareddy, R.; Fink, D.; McIntosh, T.; Davis, H.M.; Zhou, H. Mechanistic Pharmacokinetic/Target Engagement/Pharmacodynamic (PK/TE/PD) Modeling in Deciphering Interplay Between a Monoclonal Antibody and Its Soluble Target in Cynomolgus Monkeys. AAPS J. 2014, 16, 129–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampejo, T. Influenza and antiviral resistance: An overview. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1201–1208. [Google Scholar] [CrossRef]
- Martinot, M.; Jary, A.; Fafi-Kremer, S.; Leducq, V.; Delagreverie, H.; Garnier, M.; Pacanowski, J.; Mékinian, A.; Pirenne, F.; Tiberghien, P.; et al. Remdesivir failure with SARS-CoV-2 RNA-dependent RNA-polymerase mutation in a B-cell immunodeficient patient with protracted Covid-19. Clin. Infect. Dis. 2020, ciaa1474. [Google Scholar] [CrossRef] [PubMed]
- Szemiel, A.M.; Merits, A.; Orton, R.J.; MacLean, O.; Pinto, R.M.; Wickenhagen, A.; Lieber, G.; Turnbull, M.L.; Wang, S.; Mair, D.; et al. In vitro evolution of Remdesivir resistance reveals genome plasticity of SARS-CoV-2. bioRxiv 2021. [Google Scholar] [CrossRef]
- Padhi, A.K.; Shukla, R.; Saudagar, P.; Tripathi, T. High-throughput rational design of the remdesivir binding site in the RdRp of SARS-CoV-2: Implications for potential resistance. iScience 2021, 24, 101992. [Google Scholar] [CrossRef] [PubMed]
- Mari, A.; Roloff, T.; Stange, M.; Søgaard, K.K.; Asllanaj, E.; Tauriello, G.; Alexander, L.T.; Schweitzer, M.; Leuzinger, K.; Gensch, A.; et al. Global Genomic Analysis of SARS-CoV-2 RNA Dependent RNA Polymerase Evolution and Antiviral Drug Resistance. Microorganisms 2021, 9, 1094. [Google Scholar] [CrossRef]
- Ke, R.; Zitzmann, C.; Ribeiro, R.M.; Perelson, A.S. Kinetics of SARS-CoV-2 infection in the human upper and lower respiratory tracts and their relationship with infectiousness. medRxiv 2020. [Google Scholar] [CrossRef]
- Xu, Y.; Barauskas, O.; Kim, C.; Babusis, D.; Murakami, E.; Kornyeyev, D.; Lee, G.; Stepan, G.; Perron, M.; Bannister, R.; et al. Off-target in vitro profiling demonstrates that remdesivir is a highly selective antiviral agent. Antimicrob. Agents Chemother. 2021, 65, e02237-20. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Zhang, L.; Zhou, R. Structural Basis of the Potential Binding Mechanism of Remdesivir to SARS-CoV-2 RNA-Dependent RNA Polymerase. J. Phys. Chem. B 2020, 124, 6955–6962. [Google Scholar] [CrossRef]
- Götte, M. Remdesivir for the treatment of Covid-19: The value of biochemical studies. Curr. Opin. Virol. 2021, 49, 81–85. [Google Scholar] [CrossRef]
- Perelson, A.S. Modelling viral and immune system dynamics. Nat. Rev. Immunol. 2002, 2, 28–36. [Google Scholar] [CrossRef]
- Avataneo, V.; de Nicolò, A.; Cusato, J.; Antonucci, M.; Manca, A.; Palermiti, A.; Waitt, C.; Walimbwa, S.; Lamorde, M.; di Perri, G.; et al. Development and validation of a UHPLC-MS/MS method for quantification of the prodrug remdesivir and its metabolite GS-441524: A tool for clinical pharmacokinetics of SARS-CoV-2/COVID-19 and Ebola virus disease. J. Antimicrob. Chemother. 2020, 75, 1772–1777. [Google Scholar] [CrossRef]
- Pruijssers, A.J.; George, A.S.; Schäfer, A.; Leist, S.R.; Gralinksi, L.E.; Dinnon, K.H.; Yount, B.L.; Agostini, M.L.; Stevens, L.J.; Chappell, J.D.; et al. Remdesivir Inhibits SARS-CoV-2 in Human Lung Cells and Chimeric SARS-CoV Expressing the SARS-CoV-2 RNA Polymerase in Mice. Cell Rep. 2020, 32, 107940. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Zhang, B.; Ma, J.; Zhang, S. Safety profile of the antiviral drug remdesivir: An update. Biomed. Pharmacother. 2020, 130, 110532. [Google Scholar] [CrossRef]
- Mo, H.; Lu, L.; Pilot-Matias, T.; Pithawalla, R.; Mondal, R.; Masse, S.; Dekhtyar, T.; Ng, T.; Koev, G.; Stoll, V.; et al. Mutations conferring resistance to a hepatitis C virus (HCV) RNA-dependent RNA polymerase inhibitor alone or in combination with an HCV serine protease inhibitor in vitro. Antimicrob. Agents Chemother. 2005, 49, 4305–4314. [Google Scholar] [CrossRef] [Green Version]
- Regoes, R.R.; Bonhoeffer, S. Emergence of Drug-Resistant Influenza Virus: Population Dynamical Considerations. Science 2006, 312, 389–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühnert, D.; Kouyos, R.; Shirreff, G.; Pečerska, J.; Scherrer, A.U.; Böni, J.; Yerly, S.; Klimkait, T.; Aubert, V.; Günthard, H.F.; et al. Quantifying the fitness cost of HIV-1 drug resistance mutations through phylodynamics. PLoS Pathog. 2018, 14, e1006895. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; StJohn, S.E.; Osswald, H.L.; O’Brien, A.; Banach, B.S.; Sleeman, K.; Ghosh, A.K.; Mesecar, A.D.; Baker, S.C. Coronaviruses resistant to a 3C-like protease inhibitor are attenuated for replication and pathogenesis, revealing a low genetic barrier but high fitness cost of resistance. J. Virol. 2014, 88, 11886–11898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipsitch, M.; Cohen, T.; Murray, M.; Levin, B.R. Antiviral Resistance and the Control of Pandemic Influenza. PLoS Med. 2007, 4, e15. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Xiao, Y.; Zhang, Y.; Geng, D.; Cong, D.; Shi, K.X.; Knapp, R.J. Challenges of drug development during the COVID-19 pandemic: Key considerations for clinical trial designs. Br. J. Clin. Pharmacol. 2021, 87, 2170–2185. [Google Scholar] [CrossRef]
- Hanafin, P.O.; Jermain, B.; Hickey, A.J.; Kabanov, A.V.; Kashuba, A.D.; Sheahan, T.P.; Rao, G.G. A mechanism-based pharmacokinetic model of remdesivir leveraging interspecies scaling to simulate COVID-19 treatment in humans. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 89–99. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Leenaars, C.H.C.; Kouwenaar, C.; Stafleu, F.R.; Bleich, A.; Ritskes-Hoitinga, M.; Vries, R.B.M.D.; Meijboom, F.L.B. Animal to human translation: A systematic scoping review of reported concordance rates. J. Transl. Med. 2019, 17, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.C.; Chen, C.H.; Wang, C.Y.; Chen, K.H.; Wang, Y.H.; Hsueh, P.R. Clinical efficacy and safety of remdesivir in patients with COVID-19: A systematic review and network meta-analysis of randomized controlled trials. J. Antimicrob. Chemother. 2021, dkab093. [Google Scholar] [CrossRef]
- Baker, S.; Duy, P.T.; Nga, T.V.T.; Dung, T.T.N.; Phat, V.V.; Chau, T.T.; Turner, A.K.; Farrar, J.; Boni, M.F. Fitness benefits in fluoroquinolone-resistant Salmonella Typhi in the absence of antimicrobial pressure. eLife 2013, 2, e01229. [Google Scholar] [CrossRef]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio 2018, 9, e00221-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleeson, M.P.; Hersey, A.; Montanari, D.; Overington, J. Probing the links between in Vitro Potency, ADMET Physicochemical Parameters. Nat. Rev. Drug Discov. 2011, 10, 197–208. [Google Scholar] [CrossRef]
- Wang, Y.C.; Lipsitch, M. Upgrading antibiotic use within a class: Tradeoff between resistance and treatment success. Proc. Natl. Acad. Sci. USA 2006, 103, 9655–9660. [Google Scholar] [CrossRef] [Green Version]
- Arachchige, C.N.P.G.; Prendergast, L.A.; Staudte, R.G. Robust analogs to the coefficient of variation. J. Appl. Stat. 2020, 1–23. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Description | Units | Value |
|---|---|---|---|
| Remdesivir | |||
| Dose | Total mass of RDV infused | mg | varies |
| Duration of infusion | h | varies | |
| Apparent volume | L | 6.09 | |
| Elimination rate | per hour | 4.16 | |
| k | Periphery-to-plasma RDV concentration | per hour | 0.13 |
| transition rate | |||
| Plasma-to-periphery RDV concentration | per hour | 4.42 | |
| transition rate | |||
| d | Nonlinear plasma RDV to RDV-TP | mg/L/h | 31.57 |
| (intracellular GS-443902) transport and | |||
| activation rate | |||
| Conversion of concentrations from mg/L | mol/mg | 1 | |
| to M | |||
| D | RDV concentration yielding 50% of the | mg/L | 3516.27 |
| maximal transport/activation rate | |||
| Remdesivir concentration in the periphery | |||
| Elimination rate | per hour | 1.55 | |
| RDV-TP (Intracellular GS-433902) | |||
| Linear elimination rate of RDV-TP | per hour | 1.55 | |
| Nonlinear elimination rate of RDV-TP | M/h | ||
| M | RDV-TP concentration at which the | M | 17.19 |
| nonlinear elimination rate is 50% its | |||
| maximum | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conway, J.M.; Abel zur Wiesch, P. Mathematical Modeling of Remdesivir to Treat COVID-19: Can Dosing Be Optimized? Pharmaceutics 2021, 13, 1181. https://doi.org/10.3390/pharmaceutics13081181
Conway JM, Abel zur Wiesch P. Mathematical Modeling of Remdesivir to Treat COVID-19: Can Dosing Be Optimized? Pharmaceutics. 2021; 13(8):1181. https://doi.org/10.3390/pharmaceutics13081181
Chicago/Turabian StyleConway, Jessica M., and Pia Abel zur Wiesch. 2021. "Mathematical Modeling of Remdesivir to Treat COVID-19: Can Dosing Be Optimized?" Pharmaceutics 13, no. 8: 1181. https://doi.org/10.3390/pharmaceutics13081181