
Polymer Coated Oncolytic Adenovirus to Selectively Target Hepatocellular Carcinoma Cells

 ,
,  , ,
, ,
Abstract
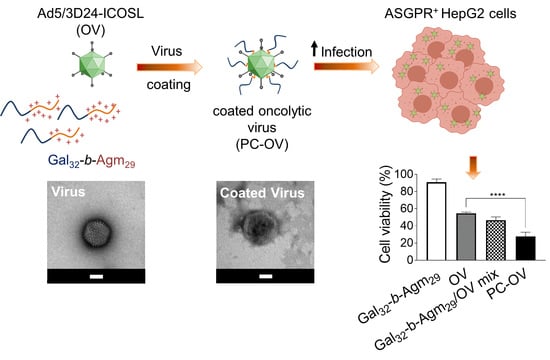
:
{kind=link}
{kind=link}
{kind=link}
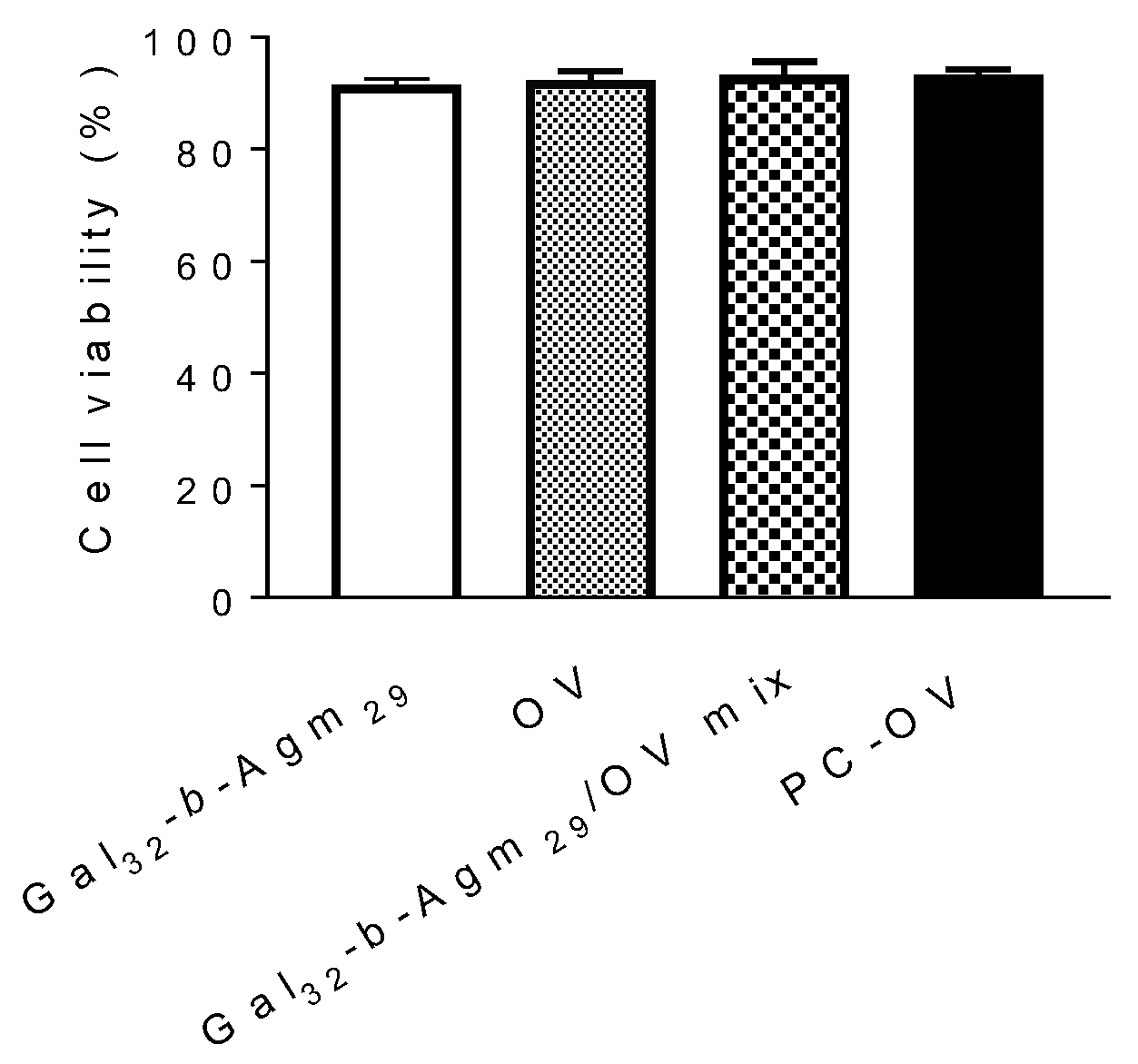{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Nuclear Magnetic Resonance
2.3. Gel Permeation Chromatography (GPC)
2.4. Cell Culture
2.5. Synthesis of 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium Chloride (DMTMM) (2)
2.5.1. Synthesis of 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT) (1)
2.5.2. Synthesis of 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium Chloride (DMTMM) (2)
2.6. Synthesis of 4-cyano-4-(ethylsulfanylthiocarbonylsulfanyl)pentanoic Acid RAFT Agent (4)
2.6.1. Synthesis of bis(ethylsulfanylthiocarbonyl)disulfide (3)
2.6.2. Synthesis of 4-cyano-4-(ethylsulfanylthiocarbonylsulfanyl)pentanoic Acid (4)
2.7. Synthesis of O-β-D-galactopyranosyloxyethyl Acrylamide (6)
2.7.1. Synthesis of 2,3,4,6-O-tetraacetyl-D-galactopyranosyloxyethyl Acrylamide (5)
2.7.2. Deprotection of 2,3,4,6-O-D-galactopyranosyloxyethyl Acrylamide to Give D-galactopyrano- Syloxyethyl Acrylamide (6)
2.8. Synthesis of Agmatine Acrylamide (Agm) (7)
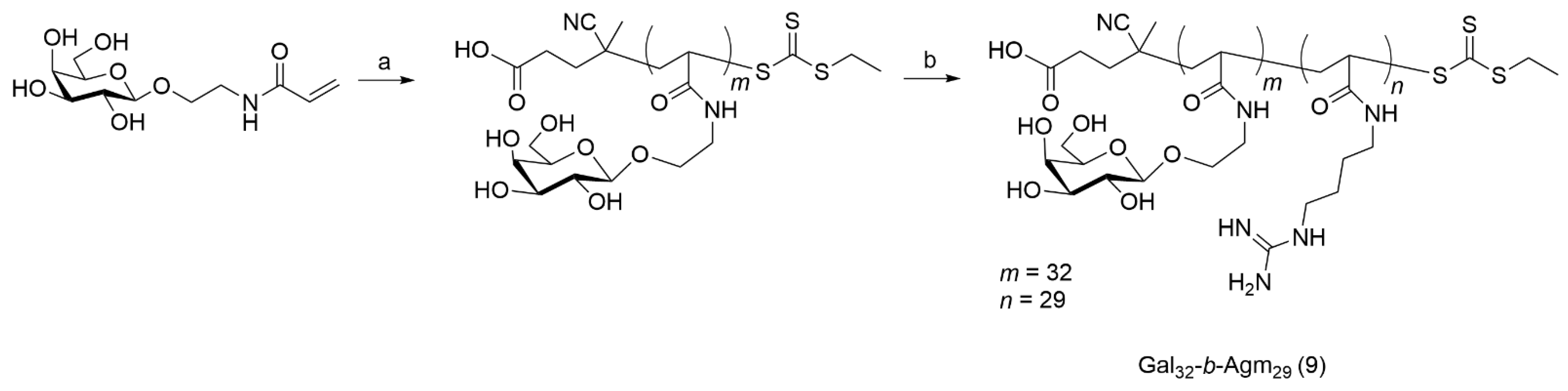
2.9. Synthesis of Gal32-b-Agm29 Cationic Diblock Copolymer (9)
2.10. Synthesis of Cy5 labeled Gal32-b-Agm29 (Cy5-Gal32-b-Agm29) (11)
2.11. Ad5/3-D24-ICOSL Oncolytic Adenovirus Production and Characterization
2.12. Formulation of Unlabeled and Fluorescently Labeled Gal32-b-Agm29 Coated Ad5/3-D24-ICOSL Adenovirus
2.13. Size and Zeta Potential Analyses
2.14. Electron-Microscope Characterizations
2.15. Evaluation of Expression Level of the Coxsackievirus and Adenovirus Receptor (CAR) and Desmoglein-2 (DSG2) in Hepatocellular Carcinoma and Lung Cancer Cells
2.16. Cell Cytotoxicity Studies
2.17. Cell Competition Studies
2.18. Determination of the Infectious Titer
2.19. ATP and HMGB1 Release
2.19.1. ATP Release
2.19.2. HMGB-1 Release
2.19.3. Analysis of Apoptotic and Necrotic Cells
2.20. Statistical Analysis
3. Results and Discussion
3.1. Synthesis of Gal32-b-Agm29 Cationic Block Copolymer
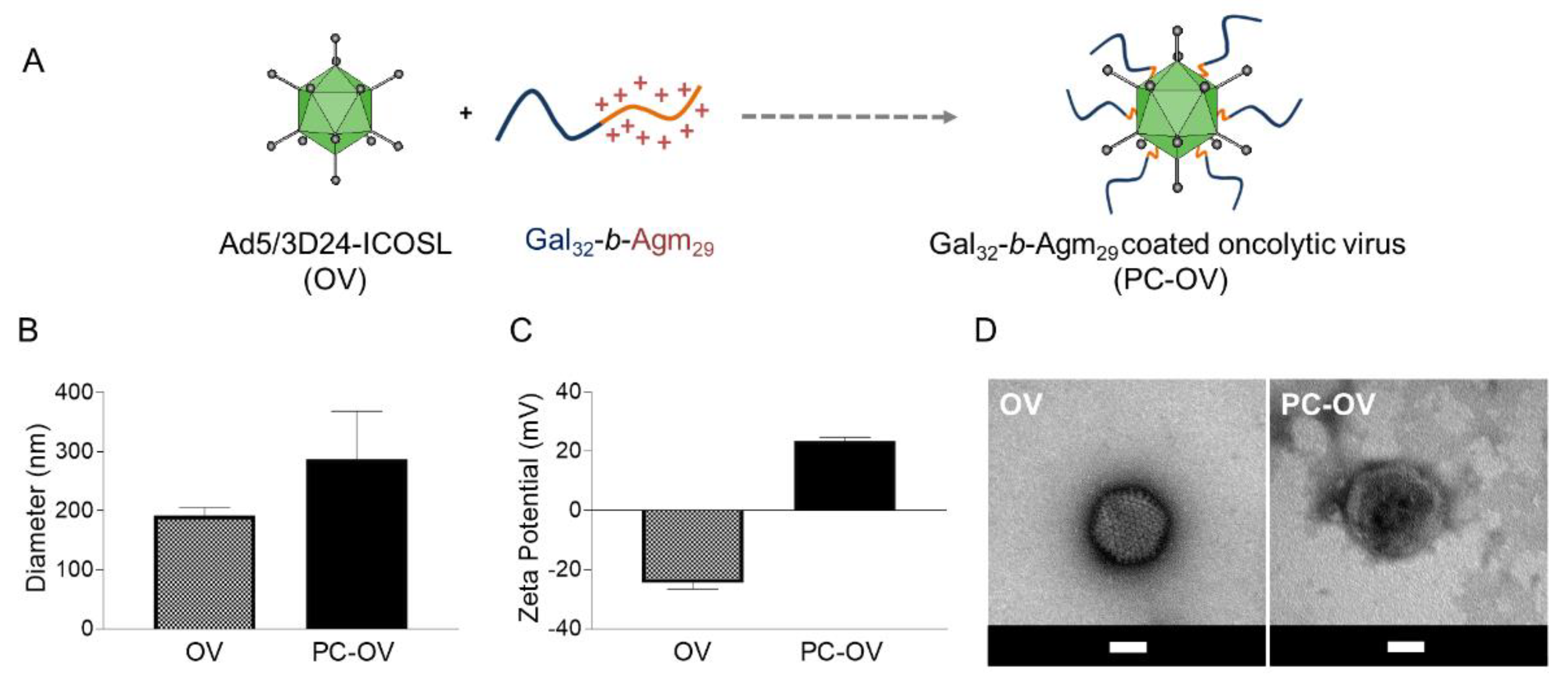
3.2. Oncolytic Adenoviruses Coating with Gal32-b-Agm29
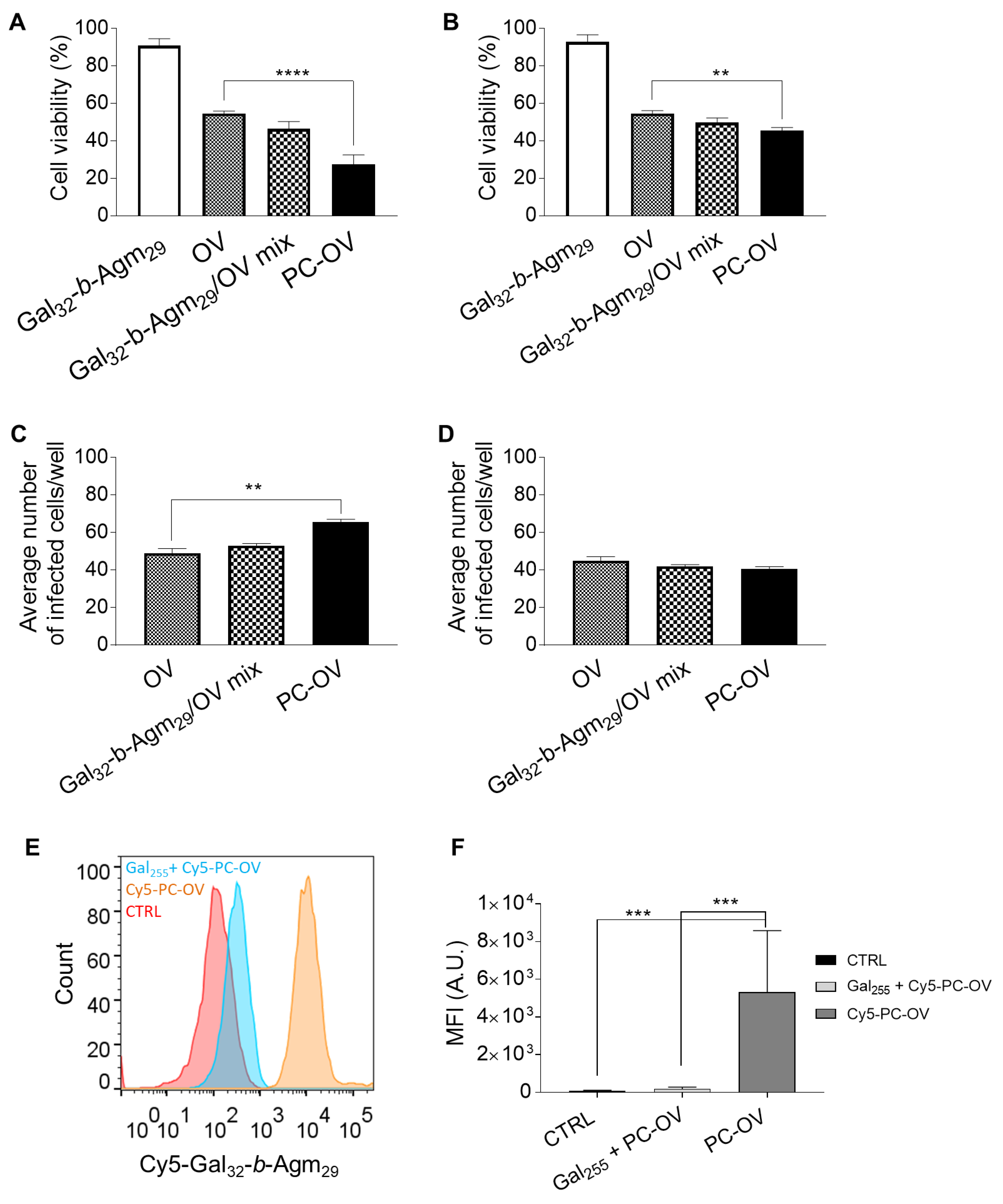
3.3. Oncolytic Viruses Complexed with Galactosylated Polymers Exhibit Enhanced In Vitro Efficacy in HepG2 Hepatocarcinoma Cell Line
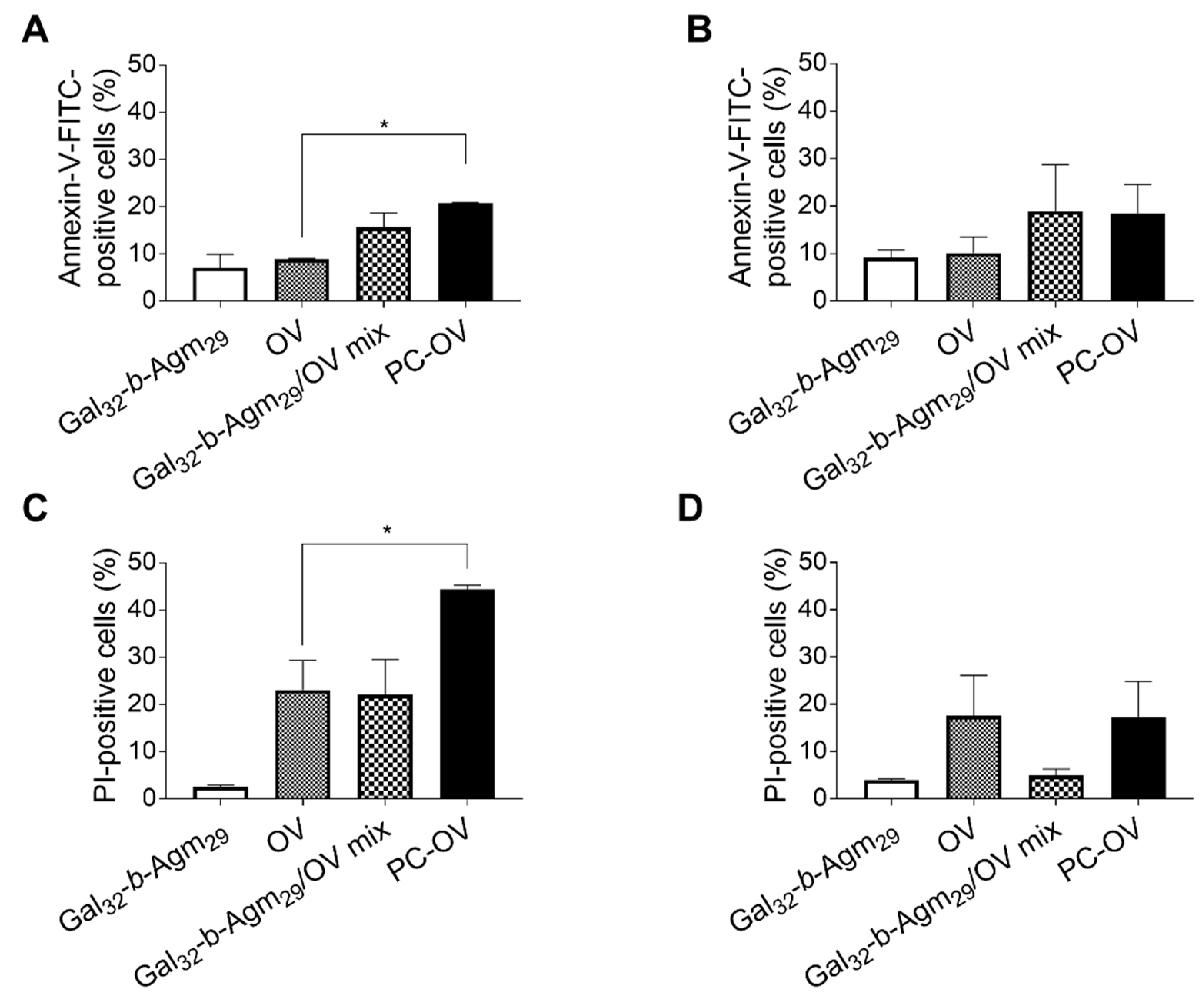
3.4. Oncolytic Viruses Coated with Galactosylated Polymers Induce Immunogenic Cell Death (ICD)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y. Cancer Statistics. CA Cancer J. Clin. 2008, 58, 71–96. [Google Scholar] [CrossRef]
- Goh, G.B.-B.; Chang, P.-E.; Tan, C.-K. Changing epidemiology of hepatocellular carcinoma in Asia. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 919–928. [Google Scholar] [CrossRef]
- Donne, R.; Saroul-Aïnama, M.; Cordier, P.; Celton-Morizur, S.; Desdouets, C. Polyploidy in liver development, homeostasis and disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Fung, J.; Lai, C.L.; Yuen, M.F. Hepatitis B and C virus-related carcinogenesis. Clin. Microbiol. Infect. 2009, 15, 964–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blonski, W.; Kotlyar, D.S.; Forde, K.A. Non-viral causes of hepatocellular carcinoma. World J. Gastroenterol. 2010, 16, 3603–3615. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.L.; Chung, R.T. Viral hepatocarcinogenesis. Oncogene 2010, 29, 2309–2324. [Google Scholar] [CrossRef] [Green Version]
- Daher, S.; Massarwa, M.; Benson, A.A.; Khoury, T. Current and Future Treatment of Hepatocellular Carcinoma: An Updated Comprehensive Review. J. Clin. Transl. Hepatol. 2018, 6, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Balasso, A.; Salmaso, S.; Pontisso, P.; Rosato, A.; Quarta, S.; Malfanti, A.; Mastrotto, F.; Caliceti, P. Re-programming pullulan for targeting and controlled release of doxorubicin to the hepatocellular carcinoma cells. Eur. J. Pharm. Sci. 2017, 103, 104–115. [Google Scholar] [CrossRef]
- Baig, B.; Halim, S.A.; Farrukh, A.; Greish, Y.; Amin, A. Current status of nanomaterial-based treatment for hepatocellular carcinoma. Biomed. Pharm. 2019, 116, 108852. [Google Scholar] [CrossRef]
- Capasso, C.; Hirvinen, M.; Garofalo, M.; Romaniuk, D.; Kuryk, L.; Sarvela, T.; Vitale, A.; Antopolsky, M.; Magarkar, A.; Viitala, T.; et al. Oncolytic adenoviruses coated with MHC-I tumor epitopes increase the antitumor immunity and efficacy against melanoma. Oncoimmunology 2016, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, M.; Villa, A.; Rizzi, N.; Kuryk, L.; Mazzaferro, V.; Ciana, P. Systemic Administration and Targeted Delivery of Immunogenic Oncolytic Adenovirus Encapsulated in Extracellular Vesicles for Cancer Therapies. Viruses 2018, 10, 558. [Google Scholar] [CrossRef] [Green Version]
- Capasso, C.; Magarkar, A.; Cervera-Carrascon, V.; Fusciello, M.; Feola, S.; Muller, M.; Garofalo, M.; Kuryk, L.; Tahtinen, S.; Pastore, L.; et al. A novel in silico framework to improve MHC-I epitopes and break the tolerance to melanoma. Oncoimmunology 2017, 6, e1319028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, M.; Villa, A.; Brunialti, E.; Crescenti, D.; Dell’Omo, G.; Kuryk, L.; Vingiani, A.; Mazzaferro, V.; Ciana, P. Cancer-derived EVs show tropism for tissues at early stage of neoplastic transformation. Nanotheranostics 2021, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kuryk, L.; Bertinato, L.; Staniszewska, M.; Pancer, K.; Wieczorek, M.; Salmaso, S.; Caliceti, P.; Garofalo, M. From Conventional Therapies to Immunotherapy: Melanoma Treatment in Review. Cancers 2020, 12, 3057. [Google Scholar] [CrossRef]
- Yamamoto, M.; Curiel, D.T. Current Issues and Future Directions of Oncolytic Adenoviruses. Mol. Ther. 2010, 18, 243–250. [Google Scholar] [CrossRef]
- Kuryk, L.; Møller, A.-S.W.; Garofalo, M.; Cerullo, V.; Pesonen, S.; Alemany, R.; Jaderberg, M. Anti-tumor specific T-cell responses induced by oncolytic adenovirus ONCOS-102 in peritoneal mesothelioma mouse model. J. Med. Virol. 2018, 90, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Kuryk, L.; Møller, A.-S.W.; Jaderberg, M. Abscopal effect when combining oncolytic adenovirus and checkpoint inhibitor in a humanized NOG mouse model of melanoma. J. Med. Virol. 2019, 91, 1702–1706. [Google Scholar] [CrossRef] [Green Version]
- Kuryk, L.; Møller, A.-S.; Vuolanto, A.; Pesonen, S.; Garofalo, M.; Cerullo, V.; Jaderberg, M. Optimization of Early Steps in Oncolytic Adenovirus ONCOS-401 Production in T-175 and HYPERFlasks. Int. J. Mol. Sci. 2019, 20, 621. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.; Chavez, V.; Khatwani, N.; Ban, Y.; Espejo, A.P.; Chen, X.; Merchan, J.R. In vivo antitumor activity by dual stromal and tumor-targeted oncolytic measles viruses. Cancer Gene Ther. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuryk, L.; Vassilev, L.; Ranki, T.; Hemminki, A.; Karioja-Kallio, A.; Levalampi, O.; Vuolanto, A.; Cerullo, V.; Pesonen, S. Toxicological and bio-distribution profile of a GM-CSF-expressing, double-targeted, chimeric oncolytic adenovirus ONCOS-102—Support for clinical studies on advanced cancer treatment. PLoS ONE 2017, 12, e0182715. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, H.L. Two roads for oncolytic immunotherapy development. J. Immunother. Cancer 2019, 2, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Dermatol. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Abudoureyimu, M.; Lai, Y.; Tian, C.; Wang, T.; Wang, R.; Chu, X. Oncolytic Adenovirus-A Nova for Gene-Targeted Oncolytic Viral Therapy in HCC. Front. Oncol. 2019, 9, 1182. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Zheng, Y.-F.; Ge, W.; Wang, S.-B.; Mou, X.-Z. Synergistic Anti-tumour Effects of Quercetin and Oncolytic Adenovirus expressing TRAIL in Human Hepatocellular Carcinoma. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, A.R.; Hong, J.; Kim, M.; Yun, C.-O. Hepatocellular carcinoma-targeting oncolytic adenovirus overcomes hypoxic tumor microenvironment and effectively disperses through both central and peripheral tumor regions. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Krimmel, J.; Zhang, Z.; Hu, Z.; Seth, P. Systemic Delivery of a Novel Liver-Detargeted Oncolytic Adenovirus Causes Reduced Liver Toxicity but Maintains the Antitumor Response in a Breast Cancer Bone Metastasis Model. Hum. Gene Ther. 2011, 22, 1137–1142. [Google Scholar] [CrossRef]
- Hong, J.; Yun, C.-O. Overcoming the limitations of locally administered oncolytic virotherapy. BMC Biomed. Eng. 2019, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, A.A.; Devarajan, P.V. Asialoglycoprotein receptor mediated hepatocyte targeting—Strategies and applications. J. Control. Release 2015, 203, 126–139. [Google Scholar] [CrossRef]
- Thapa, B.; Kumar, P.; Zeng, H.; Narain, R. Asialoglycoprotein Receptor-Mediated Gene Delivery to Hepatocytes Using Galactosylated Polymers. Biomacromolecules 2015, 16, 3008–3020. [Google Scholar] [CrossRef]
- Cronin, J.S.; Ginah, F.O.; Murray, A.R.; Copp, J.D. An Improved Procedure for the Large Scale Preparation of 2-Chloro-4,6-dimethoxy-1,3,5-triazine. Synth. Commun. 1996, 26, 3491–3494. [Google Scholar] [CrossRef]
- Kunishima, M.; Kawachi, C.; Monta, J.; Terao, K.; Iwasaki, F.; Tani, S. 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride: An efficient condensing agent leading to the formation of amides and esters. Tetrahedron 1999, 55, 13159–13170. [Google Scholar] [CrossRef]
- Truong, N.P.; Dussert, M.V.; Whittaker, M.R.; Quinn, J.F.; Davis, T.P. Rapid synthesis of ultrahigh molecular weight and low polydispersity polystyrene diblock copolymers by RAFT-mediated emulsion polymerization. Polym. Chem. 2015, 6, 3865–3874. [Google Scholar] [CrossRef]
- Luisa, M.-P.F.M.; Giuseppe, M. Sulfated Glycopolymers. Patent WO/2018/007827, 11 January 2018. [Google Scholar]
- Mattias, A.P.B.; Nicolas, T. Method for Synthesis of Acrylamide Derivatives. U.S. Patent 7,294,743, 13 November 2007. [Google Scholar]
- Gody, G.; Maschmeyer, T.; Zetterlund, P.B.; Perrier, S. Pushing the Limit of the RAFT Process: Multiblock Copolymers by One-Pot Rapid Multiple Chain Extensions at Full Monomer Conversion. Macromolecules 2014, 47, 3451–3460. [Google Scholar] [CrossRef]
- Mastrotto, F.; Breen, A.F.; Sicilia, G.; Murdan, S.; Johnstone, A.D.; Marsh, G.E.; Grainger-Boultby, C.; Russell, N.A.; Alexander, C.; Mantovani, G. One-pot RAFT and fast polymersomes assembly: A ‘beeline’ from monomers to drug-loaded nanovectors. Polym. Chem. 2016, 7, 6714–6724. [Google Scholar] [CrossRef] [Green Version]
- Kanerva, A.; Zinn, K.R.; Chaudhuri, T.R.; Lam, J.T.; Suzuki, K.; Uil, T.G.; Hakkarainen, T.; Bauerschmitz, G.J.; Wang, M.; Liu, B.; et al. Enhanced therapeutic efficacy for ovarian cancer with a serotype 3 receptor-targeted oncolytic adenovirus. Mol. Ther. 2003, 8, 449–458. [Google Scholar] [CrossRef]
- Kanerva, A.; Hemminki, A. Modified adenoviruses for cancer gene therapy. Int. J. Cancer 2004, 110, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Kuryk, L.; Møller, A.-S.W.; Jaderberg, M. Quantification and functional evaluation of CD40L production from the adenovirus vector ONCOS-401. Cancer Gene Ther. 2019, 26, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Bertinato, L.; Staniszewska, M.; Wieczorek, M.; Salmaso, S.; Schrom, S.; Rinner, B.; Pancer, K.W.; Kuryk, L. Combination Therapy of Novel Oncolytic Adenovirus with Anti-PD1 Resulted in Enhanced Anti-Cancer Effect in Syngeneic Immunocompetent Melanoma Mouse Model. Pharmaceutics 2021, 13, 547. [Google Scholar] [CrossRef] [PubMed]
- Koski, A.; Kangasniemi, L.; Escutenaire, S.; Pesonen, S.; Cerullo, V.; Diaconu, I.; Nokisalmi, P.; Raki, M.; Rajecki, M.; Guse, K.; et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1874–1884. [Google Scholar] [CrossRef] [PubMed]
- Kuryk, L.; Møller, A.-S.W. Chimeric oncolytic Ad5/3 virus replicates and lyses ovarian cancer cells through desmoglein-2 cell entry receptor. J. Med. Virol. 2020, 92, 1309–1315. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, M.; Iovine, B.; Kuryk, L.; Capasso, C.; Hirvinen, M.; Vitale, A.; Yliperttula, M.; Bevilacqua, M.A.; Cerullo, V. Oncolytic Adenovirus Loaded with L-carnosine as Novel Strategy to Enhance the Antitumor Activity. Mol. Cancer Ther. 2016, 15, 651–660. [Google Scholar] [CrossRef] [Green Version]
- Yousef, S.; Alsaab, H.O.; Sau, S.; Iyer, A.K. Development of asialoglycoprotein receptor directed nanoparticles for selective delivery of curcumin derivative to hepatocellular carcinoma. Heliyon 2018, 4, e01071. [Google Scholar] [CrossRef] [Green Version]
- Weigels, P.H.; Oka, J.A. The Large Intracellular Pool of Asialoglycoprotein Receptors Functions during the Endocytosis of Asialoglycoproteins by Isolated Rat Hepatocytes. J. Biol. Chem. 1983, 258, 5095–5102. [Google Scholar] [CrossRef]
- Rice, K.G.; Lee, Y.C. Oligosaccharide Valency and Conformation in Determining Binding to the Asialoglycoprotein Receptor of Rat Hepatocytes. Adv. Enzymol. Relat. Areas Mol. Biol. 1993, 66, 41–83. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; van der Horst, A.; van Steenbergen, M.J.; Akeroyd, N.; van Nostrum, C.F.; Schoenmakers, P.J.; Hennink, W.E. Molar-Mass Characterization of Cationic Polymers for Gene Delivery by Aqueous Size-Exclusion Chromatography. Pharm. Res. 2006, 23, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Lu, M.; Xian, L.; Zhang, J.; Yang, T.; Yang, L.; Ding, P. Molecular weight determination of a newly synthesized guanidinylated disulfide-containing poly(amido amine) by gel permeation chromatography. Asian J. Pharm. Sci. 2017, 12, 292–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasbender, A.; Zabner, J.; Chillo, M.; Moninger, T.O.; Puga, A.P.; Davidson, B.L.; Welsh, M.J. Complexes of Adenovirus with Polycationic Polymers and Cationic Lipids Increase the Efficiency of Gene Transfer in Vitro and in Vivo. J. Biol. Chem. 1997, 272, 6479–6489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, K.D.; Stallwood, Y.; Green, N.K.; Ulbrich, K.; Mautner, V.; Seymour, L.W. Polymer-coated adenovirus permits efficient retargeting and evades neutralising antibodies. Gene Ther. 2001, 8, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Nikitin, N.; Trifonova, E.; Evtushenko, E.; Kirpichnikov, M.; Atabekov, J.; Karpova, O. Comparative Study of Non-Enveloped Icosahedral Viruses Size. PLoS ONE 2015, 10, e0142415. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.L.; van den Berg, C.W.; Bowen, D.J. ASGR1 and ASGR2, the Genes that Encode the Asialoglycoprotein Receptor (Ashwell Receptor), Are Expressed in Peripheral Blood Monocytes and Show Interindividual Differences in Transcript Profile. Mol. Biol. Int. 2012, 2012, 283974. [Google Scholar] [CrossRef] [Green Version]
- Peng, D.-J.; Sun, J.; Wang, Y.-Z.; Tian, J.; Zhang, Y.-H.; Noteborn, M.H.M.; Qu, S. Inhibition of hepatocarcinoma by systemic delivery of Apoptin gene via the hepatic asialoglycoprotein receptor. Cancer Gene Ther. 2007, 14, 66–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segerman, A.; Atkinson, J.P.; Marttila, M.; Dennerquist, V.; Wadell, G.; Arnberg, N. Adenovirus Type 11 Uses CD46 as a Cellular Receptor. J. Virol. 2003, 77, 9183–9191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stasiak, A.C.; Stehle, T. Human adenovirus binding to host cell receptors: A structural view. Med. Microbiol. Immunol. 2020, 209, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Zhang, W.; Mese, K.; Bunz, O.; Lu, F.; Ehrhardt, A. Transient Chimeric Ad5/37 Fiber Enhances NK-92 Carrier Cell-Mediated Delivery of Oncolytic Adenovirus Type 5 to Tumor Cells. Mol. Ther. Methods Clin. Dev. 2020, 18, 376–389. [Google Scholar] [CrossRef]
- Bersani, S.; Salmaso, S.; Mastrotto, F.; Ravazzolo, E.; Semenzato, A.; Caliceti, P. Star-Like Oligo-Arginyl-Maltotriosyl Derivatives as Novel Cell-Penetrating Enhancers for the Intracellular Delivery of Colloidal Therapeutic Systems. Bioconjugate Chem. 2012, 23, 1415–1425. [Google Scholar] [CrossRef]
- Kepp, O.; Senovilla, L.; Vitale, I. Consensus guidelines for the detection of immunogenic cell death. OncoImmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [Green Version]
- Workenhe, S.T.; Mossman, K.L. Oncolytic Virotherapy and Immunogenic Cancer Cell Death: Sharpening the Sword for Improved Cancer Treatment Strategies. Mol. Ther. 2014, 22, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, M.; Staniszewska, M.; Salmaso, S.; Caliceti, P.; Pancer, K.W.; Wieczorek, M.; Kuryk, L. Prospects of Replication-Deficient Adenovirus Based Vaccine Development against SARS-CoV-2. Vaccines 2020, 8, 293. [Google Scholar] [CrossRef]
- Kuryk, L.; Haavisto, E.; Garofalo, M.; Capasso, C.; Hirvinen, M.; Pesonen, S.; Ranki, T.; Vassilev, L.; Cerullo, V. Synergistic anti-tumor efficacy of immunogenic adenovirus ONCOS-102 (Ad5/3-D24-GM-CSF) and standard of care chemotherapy in preclinical mesothelioma model. Int. J. Cancer 2016, 139, 1883–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuryk, L. Combination of immunogenic oncolytic adenovirus ONCOS-102 with anti-PD-1 pembrolizumab exhibits synergistic antitumor effect in humanized A2058 melanoma huNOG mouse model. Oncoimmunology 2019, 8, e1532763. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, T. Intratumoral injection of taxol in vivo suppresses A549 tumor showing cytoplasmic vacuolization. J. Cell. Biochem. 2012, 113, 1397–1406. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garofalo, M.; Bellato, F.; Magliocca, S.; Malfanti, A.; Kuryk, L.; Rinner, B.; Negro, S.; Salmaso, S.; Caliceti, P.; Mastrotto, F. Polymer Coated Oncolytic Adenovirus to Selectively Target Hepatocellular Carcinoma Cells. Pharmaceutics 2021, 13, 949. https://doi.org/10.3390/pharmaceutics13070949
Garofalo M, Bellato F, Magliocca S, Malfanti A, Kuryk L, Rinner B, Negro S, Salmaso S, Caliceti P, Mastrotto F. Polymer Coated Oncolytic Adenovirus to Selectively Target Hepatocellular Carcinoma Cells. Pharmaceutics. 2021; 13(7):949. https://doi.org/10.3390/pharmaceutics13070949
Chicago/Turabian StyleGarofalo, Mariangela, Federica Bellato, Salvatore Magliocca, Alessio Malfanti, Lukasz Kuryk, Beate Rinner, Samuele Negro, Stefano Salmaso, Paolo Caliceti, and Francesca Mastrotto. 2021. "Polymer Coated Oncolytic Adenovirus to Selectively Target Hepatocellular Carcinoma Cells" Pharmaceutics 13, no. 7: 949. https://doi.org/10.3390/pharmaceutics13070949