
Intracellular Routing and Recognition of Lipid-Based mRNA Nanoparticles

 and
and
Abstract
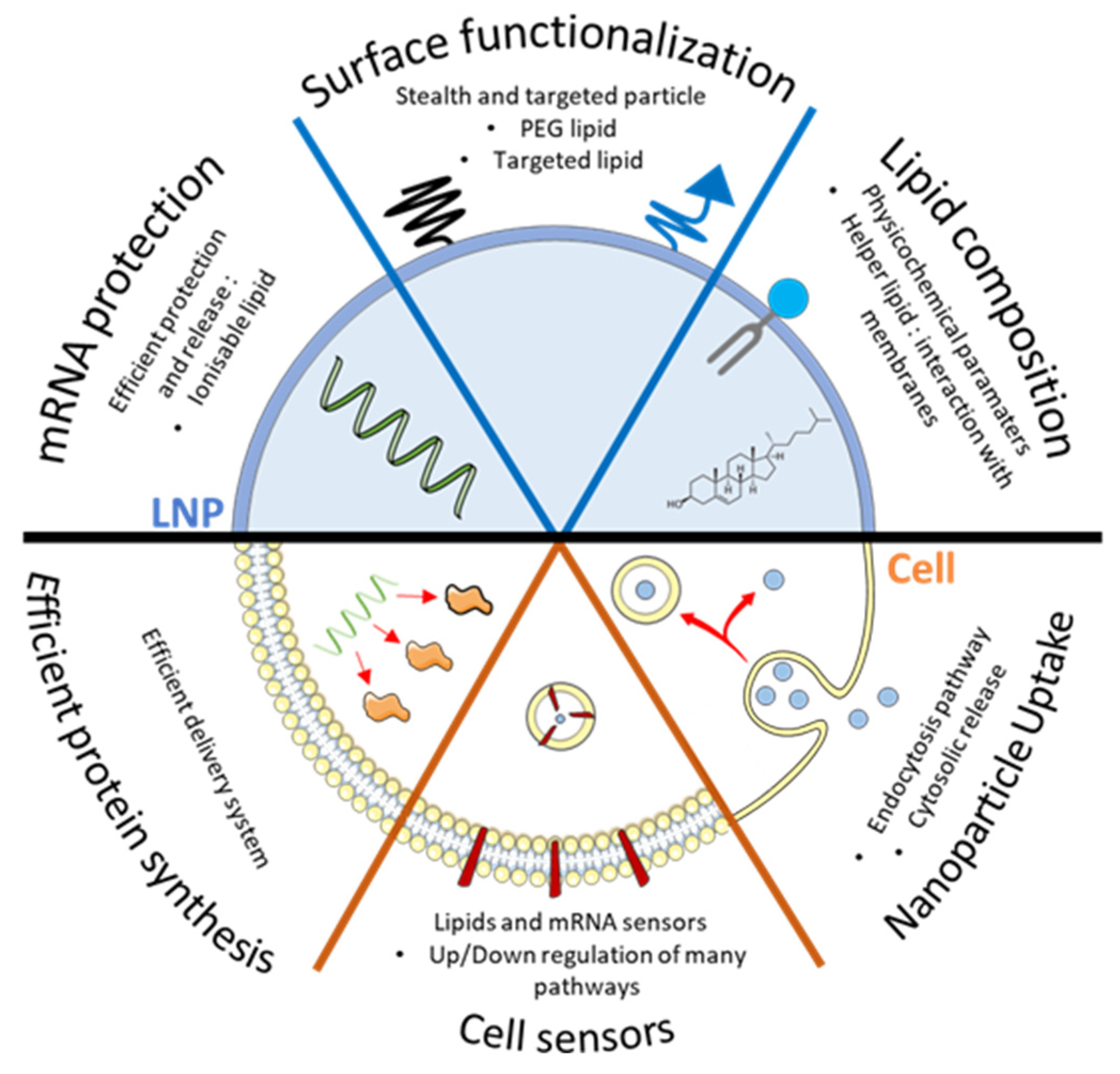
:1. Introduction
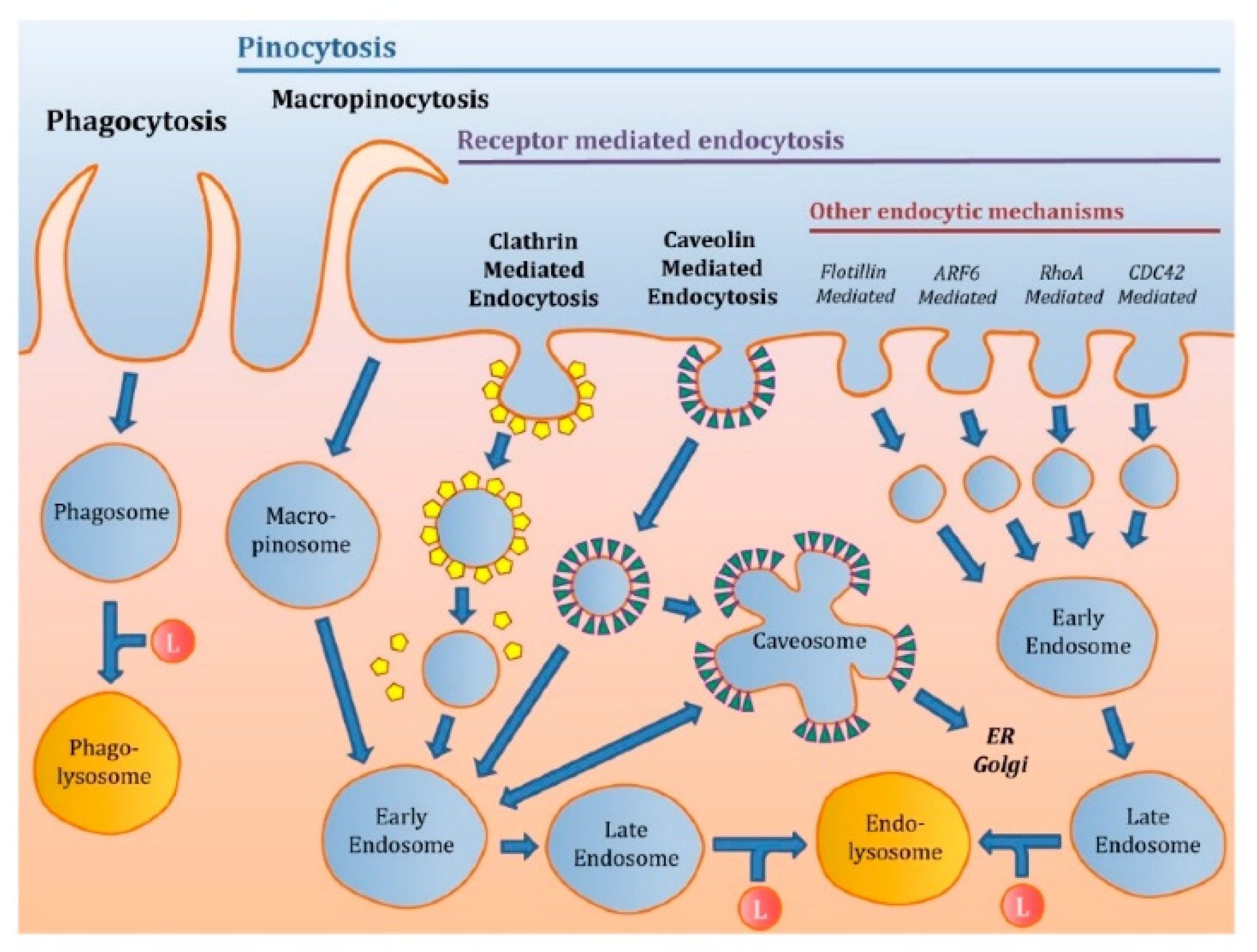
2. Intracellular Trafficking of mRNA
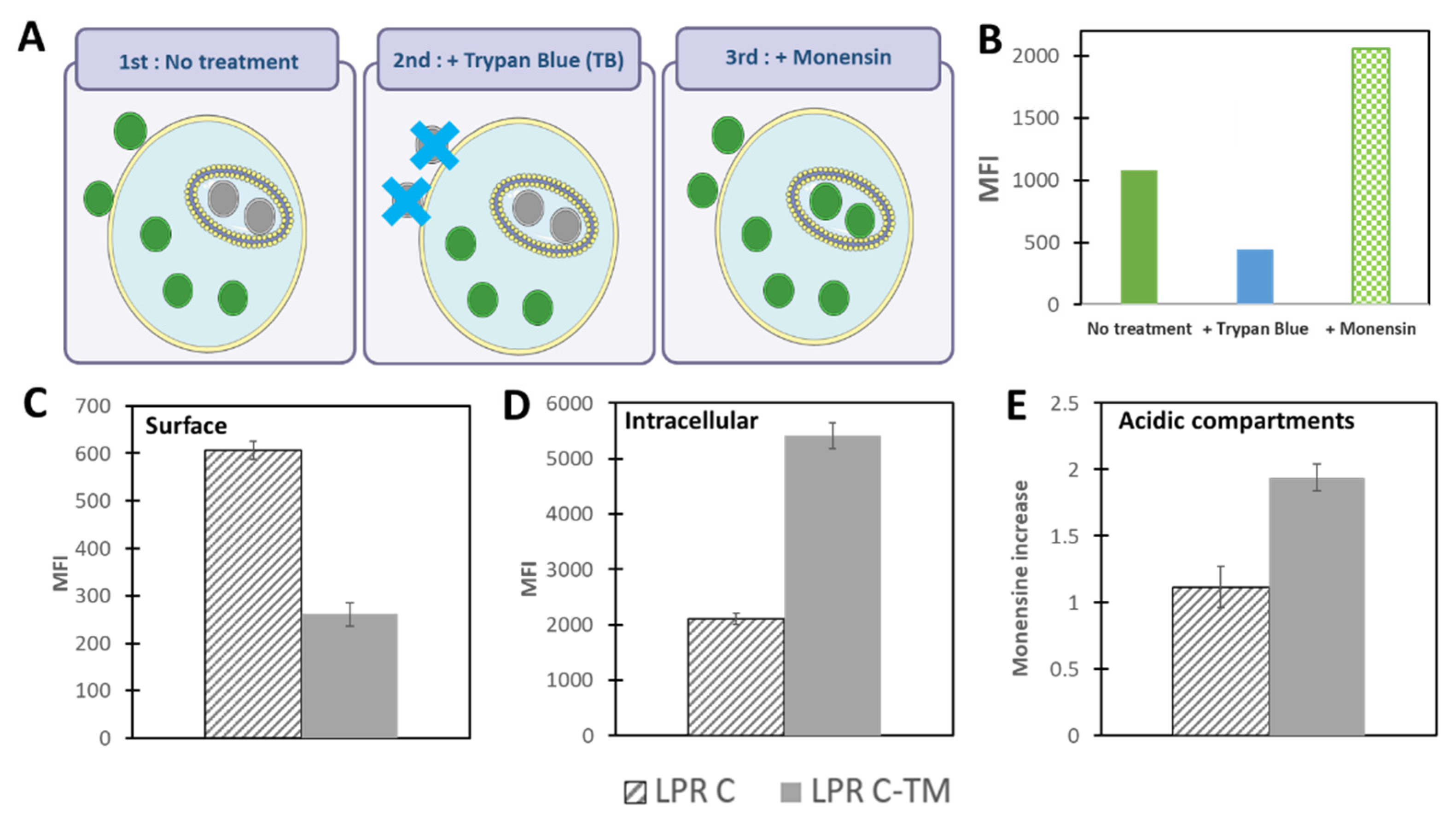
2.1. Endosomal Escape of Nanoparticles
2.2. Cell Sensors: Inflammatory Effect of mRNA NP
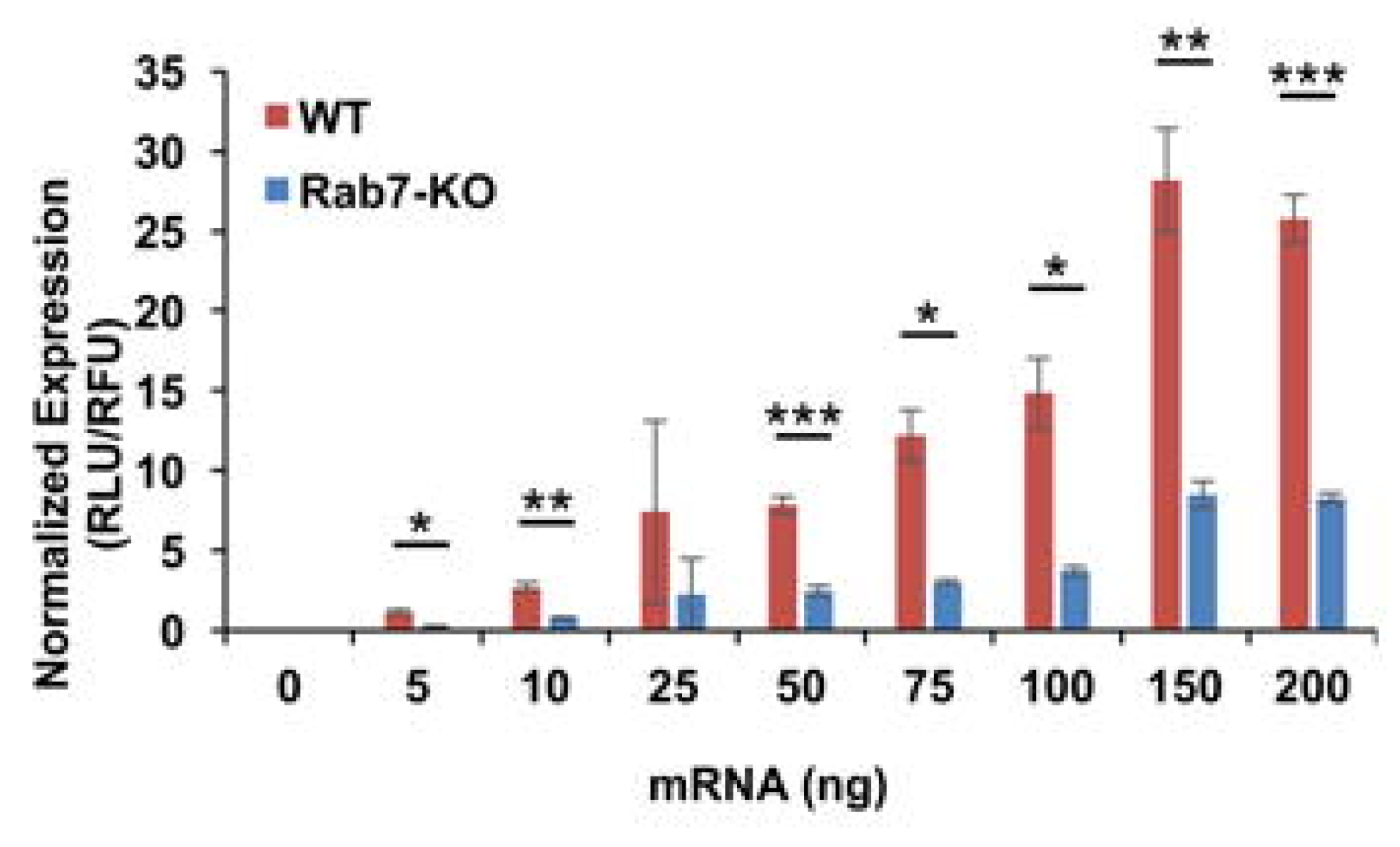
2.3. Other Mechanism to Take in Consideration Regarding LNPs Distribution
3. Quick Look to Other mRNA Loaded Formulations
4. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Uchida, S.; Perche, F.; Pichon, C.; Cabral, H. Nanomedicine-Based Approaches for mRNA Delivery. Mol. Pharm. 2020, 17, 3654–3684. [Google Scholar] [CrossRef]
- Andrée, L.; Yang, F.; Brock, R.; Leeuwenburgh, S.C. Designing biomaterials for the delivery of RNA therapeutics to stimulate bone healing. Mater. Today Bio 2021, 10, 100105. [Google Scholar] [CrossRef]
- Perche, F.; Benvegnu, T.; Berchel, M.; Lebegue, L.; Pichon, C.; Jaffrès, P.-A.; Midoux, P. Enhancement of dendritic cells transfection in vivo and of vaccination against B16F10 melanoma with mannosylated histidylated lipopolyplexes loaded with tumor antigen messenger RNA. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Perche, F.; Clemençon, R.; Schulze, K.; Ebensen, T.; Guzmán, C.A.; Pichon, C. Neutral Lipopolyplexes for In Vivo Delivery of Conventional and Replicative RNA Vaccine. Mol. Ther. Nucleic Acids 2019, 17, 767–775. [Google Scholar] [CrossRef]
- Uchida, S.; Itaka, K.; Uchida, H.; Hayakawa, K.; Ogata, T.; Ishii, T.; Fukushima, S.; Osada, K.; Kataoka, K. In Vivo Messenger RNA Introduction into the Central Nervous System Using Polyplex Nanomicelle. PLoS ONE 2013, 8, e56220. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Perche, F.; Ikegami, M.; Uchida, S.; Kataoka, K.; Itaka, K. Messenger RNA-based therapeutics for brain diseases: An animal study for augmenting clearance of beta-amyloid by intracerebral administration of neprilysin mRNA loaded in polyplex nanomicelles. J. Control. Release 2016, 235, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Perche, F.; Uchida, S.; Akiba, H.; Lin, C.-Y.; Ikegami, M.; Dirisala, A.; Nakashima, T.; Itaka, K.; Tsumoto, K.; Kataoka, K. Improved brain expression of anti-amyloid β scfv by complexation of mRNA including a secretion sequence with PEG-based block catiomer. Curr. Alzheimer Res. 2017, 14, 295–302. [Google Scholar] [CrossRef]
- Kis, Z.; Kontoravdi, C.; Shattok, R.; Shah, N. Resources, Production Scales and Time Required for Producing RNA Vaccines for the Global Pandemic Demand. Vaccines 2021, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Eygeris, Y.; Gupta, M.; Sahay, G. Self-assembled mRNA vaccines. Adv. Drug Deliv. Rev. 2021, 170, 83–112. [Google Scholar] [CrossRef]
- Zhang, H.; Rombouts, K.; Raes, L.; Xiong, R.; De Smedt, S.C.; Braeckmans, K.; Remaut, K. Fluorescence-Based Quantification of Messenger RNA and Plasmid DNA Decay Kinetics in Extracellular Biological Fluids and Cell Extracts. Adv. Biosyst. 2020, 4, e2000057. [Google Scholar] [CrossRef]
- Tsui, N.B.; Ng, E.K.-O.; Lo, Y.D. Stability of Endogenous and Added RNA in Blood Specimens, Serum, and Plasma. Clin. Chem. 2002, 48, 1647–1653. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalski, P.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midoux, P.; Pichon, C. Lipid-based mRNA vaccine delivery systems. Expert Rev. Vaccines 2014, 14, 221–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engin, A.B.; Nikitovic, D.; Neagu, M.; Henrich-Noack, P.; Docea, A.O.; Shtilman, M.I.; Golokhvast, K.; Tsatsakis, A.M. Mechanistic understanding of nanoparticles’ interactions with extracellular matrix: the cell and immune system. Part. Fibre Toxicol. 2017, 14, 22. [Google Scholar] [CrossRef]
- Gómez-Aguado, I.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Rodríguez-Gascón, A.; Aspiazu, M.; Ángeles, S.; Del Pozo-Rodríguez, A. Nanomedicines to Deliver mRNA: State of the Art and Future Perspectives. Nanomaterials 2020, 10, 364. [Google Scholar] [CrossRef] [Green Version]
- Duncan, R.; Richardson, S.C.W. Endocytosis and Intracellular Trafficking as Gateways for Nanomedicine Delivery: Opportunities and Challenges. Mol. Pharm. 2012, 9, 2380–2402. [Google Scholar] [CrossRef]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, L.; Li, L.; Huang, Y.; Delcassian, D.; Chahal, J.; Han, J.; Shi, Y.; Sadtler, K.; Gao, W.; Lin, J.; et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol. 2019, 37, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. The dawn of mRNA vaccines: The COVID-19 case. J. Control. Release 2021, 333, 511–520. [Google Scholar] [CrossRef]
- Buschmann, M.; Carrasco, M.; Alishetty, S.; Paige, M.; Alameh, M.; Weissman, D. Nanomaterial Delivery Systems for mRNA Vaccines. Vaccines 2021, 9, 65. [Google Scholar] [CrossRef]
- Evers, M.J.W.; Kulkarni, J.A.; Van Der Meel, R.; Cullis, P.R.; Vader, P.; Schiffelers, R.M. State-of-the-Art Design and Rapid-Mixing Production Techniques of Lipid Nanoparticles for Nucleic Acid Delivery. Small Methods 2018, 2, 1700375. [Google Scholar] [CrossRef]
- Hajj, K.A.; Ball, R.L.; Deluty, S.B.; Singh, S.R.; Strelkova, D.; Knapp, C.M.; Whitehead, K.A. Branched-Tail Lipid Nanoparticles Potently Deliver mRNA In Vivo due to Enhanced Ionization at Endosomal pH. Small 2019, 15, e1805097. [Google Scholar] [CrossRef]
- Paunovska, K.; Sago, C.D.; Monaco, C.M.; Hudson, W.; Castro, M.G.; Rudoltz, T.G.; Kalathoor, S.; Vanover, D.A.; Santangelo, P.J.; Ahmed, R.; et al. A Direct Comparison of in Vitro and in Vivo Nucleic Acid Delivery Mediated by Hundreds of Nanoparticles Reveals a Weak Correlation. Nano Lett. 2018, 18, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Sakurai, Y.; Anindita, J.; Akita, H. Development of lipid-like materials for RNA delivery based on intracellular environment-responsive membrane destabilization and spontaneous collapse. Adv. Drug Deliv. Rev. 2020, 154–155, 210–226. [Google Scholar] [CrossRef] [PubMed]
- Kirschman, J.L.; Bhosle, S.; Vanover, D.; Blanchard, E.L.; Loomis, K.H.; Zurla, C.; Murray, K.; Lam, B.C.; Santangelo, P.J. Characterizing exogenous mRNA delivery, trafficking, cytoplasmic release and RNA-protein correlations at the level of single cells. Nucleic Acids Res. 2017, 45, e113. [Google Scholar] [CrossRef] [Green Version]
- Florence, A.T. Reductionism and complexity in nanoparticle-vectored drug targeting. J. Control. Release 2012, 161, 399–402. [Google Scholar] [CrossRef]
- Torchilin, V.; Amiji, M.M. Handbook of Materials for Nanomedicine; CRC Press: Boca Raton, FL, USA, 2011; Volume 1. [Google Scholar]
- Zukancic, D.; Suys, E.J.A.; Pilkington, E.H.; Algarni, A.; Al-Wassiti, H.; Truong, N.P. The Importance of Poly(ethylene glycol) and Lipid Structure in Targeted Gene Delivery to Lymph Nodes by Lipid Nanoparticles. Pharmaceutics 2020, 12, 1068. [Google Scholar] [CrossRef]
- Thi, T.; Suys, E.; Lee, J.; Nguyen, D.; Park, K.; Truong, N. Lipid-Based Nanoparticles in the Clinic and Clinical Trials: From Cancer Nanomedicine to COVID-19 Vaccines. Vaccines 2021, 9, 359. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Akita, H.; Harashima, H. The Polyethyleneglycol Dilemma: Advantage and Disadvantage of PEGylation of Liposomes for Systemic Genes and Nucleic Acids Delivery to Tumors. Biol. Pharm. Bull. 2013, 36, 892–899. [Google Scholar] [CrossRef] [Green Version]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.F.A.; Jiskoot, W.; Crommelin, D.J.A. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef] [PubMed]
- Perche, F.; Biswas, S.; Torchilin, V.P. Stimuli-Sensitive Polymeric Nanomedicines for Cancer Imaging and Therapy. In Handbook of Polymers for Pharmaceutical Technologies; John Wiley & Sons: Hoboken, NJ, USA, 2015; Volume 2, pp. 311–344. [Google Scholar]
- Perche, F.; Torchilin, V.P. Recent Trends in Multifunctional Liposomal Nanocarriers for Enhanced Tumor Targeting. J. Drug Deliv. 2013, 2013, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Brader, M.L.; Williams, S.J.; Banks, J.M.; Hui, W.H.; Zhou, Z.H.; Jin, L. Encapsulation state of messenger RNA inside lipid nanoparticles. Biophys. J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Manzanares, D.; Ceña, V. Endocytosis: The Nanoparticle and Submicron Nanocompounds Gateway into the Cell. Pharmaceutics 2020, 12, 371. [Google Scholar] [CrossRef] [Green Version]
- Kou, L.; Sun, J.; Zhai, Y.; He, Z. The endocytosis and intracellular fate of nanomedicines: Implication for rational design. Asian J. Pharm. Sci. 2013, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Doherty, G.J.; McMahon, H.T. Mechanisms of Endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Kim, J.; Herrera, M.; Mukherjee, A.; Kabanov, A.V.; Sahay, G. Brief update on endocytosis of nanomedicines. Adv. Drug Deliv. Rev. 2019, 144, 90–111. [Google Scholar] [CrossRef]
- Trombetta, E.S.; Mellman, I. Cell Biology of Antigen Processing In Vitro and In Vivo. Annu. Rev. Immunol. 2005, 23, 975–1028. [Google Scholar] [CrossRef]
- Cheng, J.P.; Nichols, B.J. Caveolae: One function or many? Trends Cell Biol. 2016, 26, 177–189. [Google Scholar] [CrossRef]
- Kiss, A.L.; Botos, E. Endocytosis via caveolae: Alternative pathway with distinct cellular compartments to avoid lysosomal degradation? J. Cell. Mol. Med. 2009, 13, 1228–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrielson, N.P.; Pack, D.W. Efficient polyethylenimine-mediated gene delivery proceeds via a caveolar pathway in HeLa cells. J. Control. Release 2009, 136, 54–61. [Google Scholar] [CrossRef] [Green Version]
- El-Sayed, A.; Harashima, H. Endocytosis of Gene Delivery Vectors: From Clathrin-Dependent to Lipid Raft-Mediated Endocytosis. Mol. Ther. 2013, 21, 1118–1130. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, G.M.; Burwell, T.J.; Peterson, N.C.; Cann, J.A.; Hanna, R.N.; Li, Q.; Ongstad, E.L.; Boyd, J.T.; Kennedy, M.A.; Zhao, W.; et al. Targeted drug delivery via caveolae-associated protein PV1 improves lung fibrosis. Commun. Biol. 2019, 2, 92. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Chan, C.; Peterson, N.; Hanna, R.N.; Alfaro, A.; Allen, K.L.; Wu, H.; Dall’Acqua, W.F.; Borrok, M.J.; Santos, J.L. Engineering Caveolae-Targeted Lipid Nanoparticles to Deliver mRNA to the Lungs. ACS Chem. Biol. 2020, 15, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Liu, D.; Subramanyam, K.; Wang, B.; Yang, J.; Huang, J.; Auguste, D.T.; Moses, M.A. Nanoparticle elasticity directs tumor uptake. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, I.; Kogure, K.; Akita, H.; Harashima, H. Uptake Pathways and Subsequent Intracellular Trafficking in Nonviral Gene Delivery. Pharmacol. Rev. 2006, 58, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Midoux, P.; Breuzard, G.; Gomez, J.P.; Pichon, C. Polymer-Based Gene Delivery: A Current Review on the Uptake and Intracellular Trafficking of Polyplexes. Curr. Gene Ther. 2008, 8, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Midoux, P.; Roche, A.-C.; Monsigny, M. Quantitation of the binding, uptake, and degradation of fluoresceinylated neoglycoproteins by flow cytometry. Cytom. J. Int. Soc. Anal. Cytol. 1987, 8, 327–334. [Google Scholar] [CrossRef]
- Murphy, R.F.; Powers, S.; Cantor, C.R. Endosome pH measured in single cells by dual fluorescence flow cytometry: Rapid acidification of insulin to pH 6. J. Cell Biol. 1984, 98, 1757–1762. [Google Scholar] [CrossRef]
- Perche, F.; Gosset, D.; Mével, M.; Miramon, M.-L.; Yaouanc, J.-J.; Pichon, C.; Benvegnu, T.; Jaffrès, P.-A.; Midoux, P. Selective gene delivery in dendritic cells with mannosylated and histidylated lipopolyplexes. J. Drug Target. 2010, 19, 315–325. [Google Scholar] [CrossRef]
- Nuutila, J.; Lilius, E.-M. Flow cytometric quantitative determination of ingestion by phagocytes needs the distinguishing of overlapping populations of binding and ingesting cells. Cytom. Part A 2005, 65, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.M.; Lindqvist, L. The pH dependence of fluorescein fluorescence. J. Lumin. 1975, 10, 381–390. [Google Scholar] [CrossRef]
- Avraméas, A.; McIlroy, D.; Hosmalin, A.; Autran, B.; Debré, P.; Monsigny, M.; Roche, A.C.; Midoux, P. Expression of a mannose/fucose membrane lectin on human dendritic cells. Eur. J. Immunol. 1996, 26, 394–400. [Google Scholar] [CrossRef]
- Barbeau, J.; Lemiègre, L.; Quelen, A.; Malard, V.; Gao, H.; Gonçalves, C.; Berchel, M.; Jaffrès, P.-A.; Pichon, C.; Midoux, P.; et al. Synthesis of a trimannosylated-equipped archaeal diether lipid for the development of novel glycoliposomes. Carbohydr. Res. 2016, 435, 142–148. [Google Scholar] [CrossRef]
- Vigerust, D.J.; Vick, S.; Shepherd, V.L. Characterization of functional mannose receptor in a continuous hybridoma cell line. BMC Immunol. 2012, 13, 51. [Google Scholar] [CrossRef] [Green Version]
- Mislick, K.A.; Baldeschwieler, J.D. Evidence for the role of proteoglycans in cation-mediated gene transfer. Proc. Natl. Acad. Sci. USA 1996, 93, 12349–12354. [Google Scholar] [CrossRef] [Green Version]
- Mannisto, M.; Reinisalo, M.; Ruponen, M.; Honkakoski, P.; Tammi, M.; Urtti, A. Polyplex-mediated gene transfer and cell cycle: Effect of carrier on cellular uptake and intracellular kinetics, and significance of glycosaminoglycans. J. Gene Med. Cross-Discip. J. Res. Sci. Gene Transf. Its Clin. Appl. 2007, 9, 479–487. [Google Scholar] [CrossRef]
- Rejman, J.; Bragonzi, A.; Conese, M. Role of clathrin- and caveolae-mediated endocytosis in gene transfer mediated by lipo- and polyplexes. Mol. Ther. 2005, 12, 468–474. [Google Scholar] [CrossRef]
- Fujimoto, T.; Kogo, H.; Nomura, R.; Une, T. Isoforms of caveolin-1 and caveolar structure. J. Cell Sci. 2000, 113, 3509–3517. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.B.; Rovere, P.; Kleijmeer, M.J.; Rescigno, M.; Assmann, C.U.; Oorschot, V.M.; Geuze, H.J.; Trucy, J.; Demandolx, D.; Davoust, J.; et al. Intracellular routes and selective retention of antigens in mildly acidic cathepsin D/lysosome-associated membrane protein-1/MHC class II-positive vesicles in immature dendritic cells. J. Immunol. 1997, 159, 3707–3716. [Google Scholar] [PubMed]
- Murray, D.H.; Jahnel, M.; Lauer, J.; Avellaneda, M.; Brouilly, N.; Cezanne, A.; Morales-Navarrete, A.; Perini, E.D.; Ferguson, C.; Lupas, A.N.; et al. An endosomal tether undergoes an entropic collapse to bring vesicles together. Nat. Cell Biol. 2016, 537, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Casey, J.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 2009, 11, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Martens, T.F.; Remaut, K.; Deemester, J.; De Smedt, S.C.; Braeckmans, K. Intracellular delivery of nanomaterials: How to catch endosomal escape in the act. Nano Today 2014, 9, 344–364. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, M.; Nawaz, M.; Papadimitriou, A.; Angerfors, A.; Camponeschi, A.; Na, M.; Hölttä, M.; Skantze, P.; Johansson, S.; Sundqvist, M.; et al. Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Sabnis, S.; Kumarasinghe, E.S.; Salerno, T.; Mihai, C.; Ketova, T.; Senn, J.J.; Lynn, A.; Bulychev, A.; McFadyen, I.; Chan, J.; et al. A Novel Amino Lipid Series for mRNA Delivery: Improved Endosomal Escape and Sustained Pharmacology and Safety in Non-Human Primates. Mol. Ther. 2018, 26, 1509–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
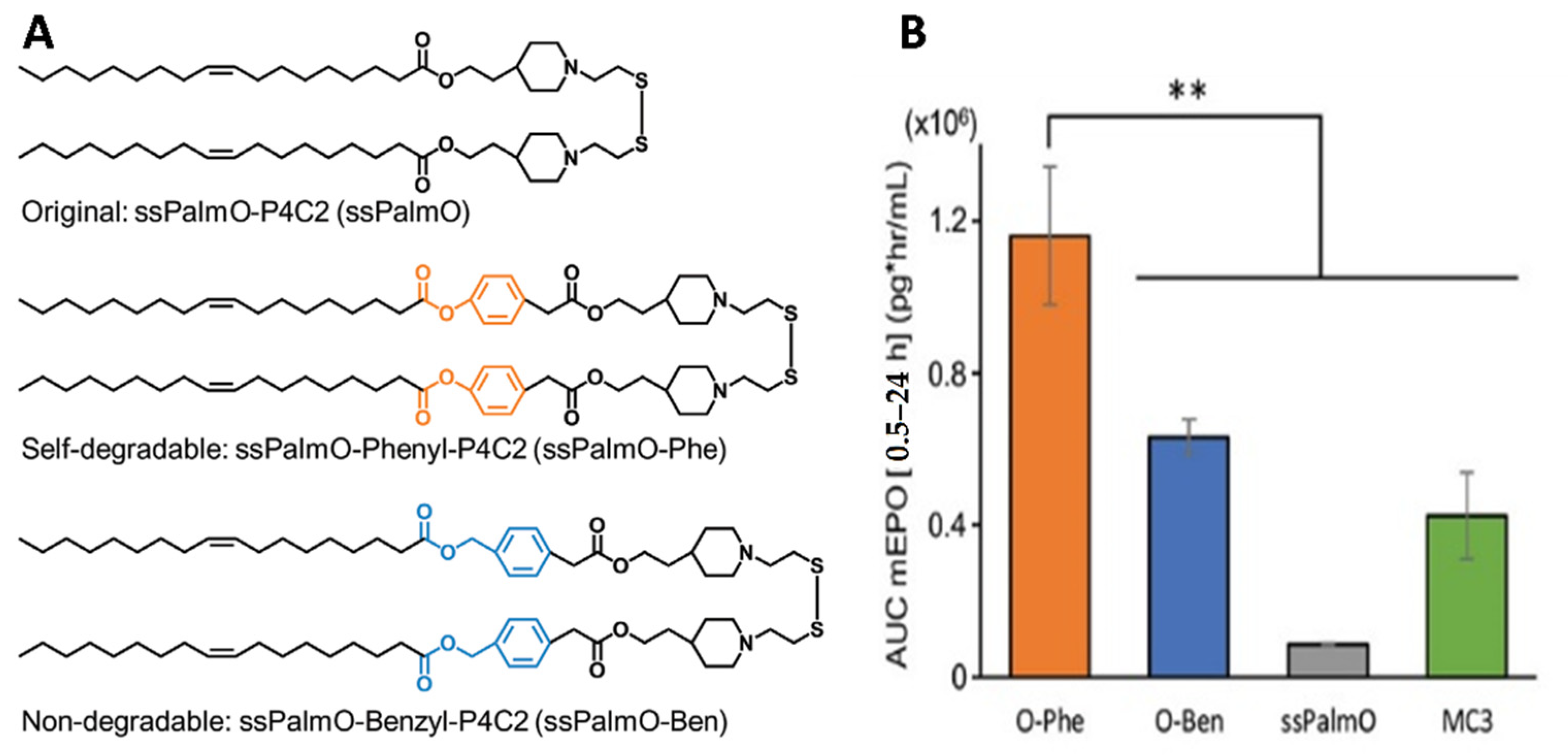
- Akita, H. Development of an SS-Cleavable pH-Activated Lipid-Like Material (ssPalm) as a Nucleic Acid Delivery Device. Biol. Pharm. Bull. 2020, 43, 1617–1625. [Google Scholar] [CrossRef]
- Tanaka, H.; Takahashi, T.; Konishi, M.; Takata, N.; Gomi, M.; Shirane, D.; Miyama, R.; Hagiwara, S.; Yamasaki, Y.; Sakurai, Y.; et al. Self-Degradable Lipid-Like Materials Based on “Hydrolysis accelerated by the intra-Particle Enrichment of Reactant (HyPER)” for Messenger RNA Delivery. Adv. Funct. Mater. 2020, 30, 1910575. [Google Scholar] [CrossRef]
- Lee, S.; Cheng, Q.; Yu, X.; Liu, S.; Johnson, L.T.; Siegwart, D.J. A Systematic Study of Unsaturation in Lipid Nanoparticles Leads to Improved mRNA Transfection In Vivo. Angew. Chem. Int. Ed. 2020, 60, 5848–5853. [Google Scholar] [CrossRef]
- Patel, S.; Ryals, R.C.; Weller, K.K.; Pennesi, M.E.; Sahay, G. Lipid nanoparticles for delivery of messenger RNA to the back of the eye. J. Control. Release 2019, 303, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, J.; Qiu, M.; Li, Y.; Glass, Z.; Xu, Q. Imidazole-Based Synthetic Lipidoids for In Vivo mRNA Delivery into Primary T Lymphocytes. Angew. Chem. 2020, 132, 20258–20264. [Google Scholar] [CrossRef]
- Patel, S.; Ashwanikumar, N.; Robinson, E.; Xia, Y.; Mihai, C.; Griffith, J.P., III; Hou, S.; Esposito, A.A.; Ketova, T.; Welsher, K.; et al. Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ohto, T.; Konishi, M.; Tanaka, H.; Onomoto, K.; Yoneyama, M.; Nakai, Y.; Tange, K.; Yoshioka, H.; Akita, H. Inhibition of the Inflammatory Pathway Enhances Both the in Vitro and in Vivo Transfection Activity of Exogenous in Vitro-Transcribed mRNAs Delivered by Lipid Nanoparticles. Biol. Pharm. Bull. 2019, 42, 299–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; You, X.; Wang, X.; Cui, L.; Wang, Z.; Xu, F.; Li, M.; Yang, Z.; Liu, J.; Huang, P.; et al. Delivery of mRNA vaccine with a lipid-like material potentiates antitumor efficacy through Toll-like receptor 4 signaling. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Madisen, L.; Zwingman, T.A.; Sunkin, S.M.; Oh, S.W.; Zariwala, H.A.; Gu, H.; Ng, L.L.; Palmiter, R.D.; Hawrylycz, M.J.; Jones, A.R.; et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2009, 13, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sago, C.D.; Krupczak, B.; Lokugamage, M.; Gan, Z.; Dahlman, J.E. Cell Subtypes Within the Liver Microenvironment Differentially Interact with Lipid Nanoparticles. Cell. Mol. Bioeng. 2019, 12, 389–397. [Google Scholar] [CrossRef]
- Jiang, Y.; Lu, Q.; Wang, Y.; Xu, E.; Ho, A.; Singh, P.; Wang, Y.; Jiang, Z.; Yang, F.; Tietjen, G.T.; et al. Quantitating Endosomal Escape of a Library of Polymers for mRNA Delivery. Nano Lett. 2020, 20, 1117–1123. [Google Scholar] [CrossRef]
- Lu, Q.; Grotzke, J.E.; Cresswell, P. A novel probe to assess cytosolic entry of exogenous proteins. Nat. Commun. 2018, 9, 3104. [Google Scholar] [CrossRef]
- Meng, F.; Hennink, W.E.; Zhong, Z. Reduction-sensitive polymers and bioconjugates for biomedical applications. Biomaterials 2009, 30, 2180–2198. [Google Scholar] [CrossRef]
- Guigas, G.; Kalla, C.; Weiss, M. The degree of macromolecular crowding in the cytoplasm and nucleoplasm of mammalian cells is conserved. FEBS Lett. 2007, 581, 5094–5098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, S.B.; Minton, A. Macromolecular Crowding: Biochemical, Biophysical, and Physiological Consequences. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 27–65. [Google Scholar] [CrossRef]
- Sayers, E.; Peel, S.E.; Schantz, A.; England, R.M.; Beano, M.; Bates, S.M.; Desai, A.S.; Puri, S.; Ashford, M.B.; Jones, A.T. Endocytic Profiling of Cancer Cell Models Reveals Critical Factors Influencing LNP-Mediated mRNA Delivery and Protein Expression. Mol. Ther. 2019, 27, 1950–1962. [Google Scholar] [CrossRef]
- Patel, S.; Ashwanikumar, N.; Robinson, E.; DuRoss, A.; Sun, C.; Murphy-Benenato, K.E.; Mihai, C.; Almarsson, Ö.; Sahay, G. Boosting Intracellular Delivery of Lipid Nanoparticle-Encapsulated mRNA. Nano Lett. 2017, 17, 5711–5718. [Google Scholar] [CrossRef]
- Perera, R.M.; Zoncu, R. The Lysosome as a Regulatory Hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.-Y.; Zoncu, R. The lysosome as a command-and-control center for cellular metabolism. J. Cell Biol. 2016, 214, 653–664. [Google Scholar] [CrossRef]
- Heyes, J.; Palmer, L.; Bremner, K.; MacLachlan, I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J. Control. Release 2005, 107, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stöter, M.; et al. Image-based analysis of lipid nanoparticle–mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eygeris, Y.; Patel, S.; Jozic, A.; Sahay, G. Deconvoluting Lipid Nanoparticle Structure for Messenger RNA Delivery. Nano Lett. 2020, 20, 4543–4549. [Google Scholar] [CrossRef] [PubMed]
- Jahanafrooz, Z.; Baradaran, B.; Mosafer, J.; Hashemzaei, M.; Rezaei, T.; Mokhtarzadeh, A.; Hamblin, M.R. Comparison of DNA and mRNA vaccines against cancer. Drug Discov. Today 2020, 25, 552–560. [Google Scholar] [CrossRef]
- Liang, X.; Li, D.; Leng, S.; Zhu, X. RNA-based pharmacotherapy for tumors: From bench to clinic and back. Biomed. Pharmacother. 2020, 125, 109997. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzaro, S.; Giovani, C.; Mangiavacchi, S.; Magini, D.; Maione, D.; Baudner, B.; Geall, A.J.; De Gregorio, E.; D’Oro, U.; Buonsanti, C. CD8 T-cell priming upon mRNA vaccination is restricted to bone-marrow-derived antigen-presenting cells and may involve antigen transfer from myocytes. Immunology 2015, 146, 312–326. [Google Scholar] [CrossRef]
- Dan, M.; Zheng, D.; Field, L.L.; Bonnevie-Nielsen, V. Induction and activation of antiviral enzyme 2′,5′-oligoadenylate synthetase by in vitro transcribed insulin mRNA and other cellular RNAs. Mol. Biol. Rep. 2012, 39, 7813–7822. [Google Scholar] [CrossRef] [PubMed]
- Freund, I.; Eigenbrod, T.; Helm, M.; Dalpke, A.H. RNA Modifications Modulate Activation of Innate Toll-Like Receptors. Genes 2019, 10, 92. [Google Scholar] [CrossRef] [Green Version]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Anderson, B.R.; Muramatsu, H.; Jha, B.K.; Silverman, R.H.; Weissman, D.; Karikó, K. Nucleoside modifications in RNA limit activation of 2′-5′-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res. 2011, 39, 9329–9338. [Google Scholar] [CrossRef] [Green Version]
- Ni, G.; Ma, Z.; Damania, B. cGAS and STING: At the intersection of DNA and RNA virus-sensing networks. PLoS Pathog. 2018, 14, e1007148. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nat. Cell Biol. 2009, 461, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Hopfner, K.-P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Korbel, D.S.; Schneider, B.E.; Schaible, U.E. Innate immunity in tuberculosis: myths and truth. Microbes Infect. 2008, 10, 995–1004. [Google Scholar] [CrossRef]
- Marinho, F.V.; Benmerzoug, S.; Rose, S.; Campos, P.C.; Marques, J.T.; Báfica, A.; Barber, G.; Ryffel, B.; Oliveira, S.C.; Quesniaux, V.F. The cGAS/STING Pathway Is Important for Dendritic Cell Activation but Is Not Essential to Induce Protective Immunity against Mycobacterium tuberculosis Infection. J. Innate Immun. 2018, 10, 239–252. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef]
- Islam, M.A.; Xu, Y.; Tao, W.; Ubellacker, J.M.; Lim, M.; Aum, D.; Lee, G.Y.; Zhou, K.; Zope, H.; Yu, M.; et al. Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat. Biomed. Eng. 2018, 2, 850–864. [Google Scholar] [CrossRef]
- Zhu, X.; Tao, W.; Liu, D.; Wu, J.; Guo, Z.; Ji, X.; Bharwani, Z.; Zhao, L.; Zhao, X.; Farokhzad, O.C.; et al. Surface De-PEGylation Controls Nanoparticle-Mediated siRNA Delivery In Vitro and In Vivo. Theranostics 2017, 7, 1990–2002. [Google Scholar] [CrossRef] [Green Version]
- Mui, B.L.; Tam, Y.K.; Jayaraman, M.; Ansell, S.M.; Du, X.; Tam, Y.Y.C.; Lin, P.J.; Chen, S.; Narayanannair, J.K.; Rajeev, K.G.; et al. Influence of Polyethylene Glycol Lipid Desorption Rates on Pharmacokinetics and Pharmacodynamics of siRNA Lipid Nanoparticles. Mol. Ther. Nucleic Acids 2013, 2, e139. [Google Scholar] [CrossRef]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [Green Version]
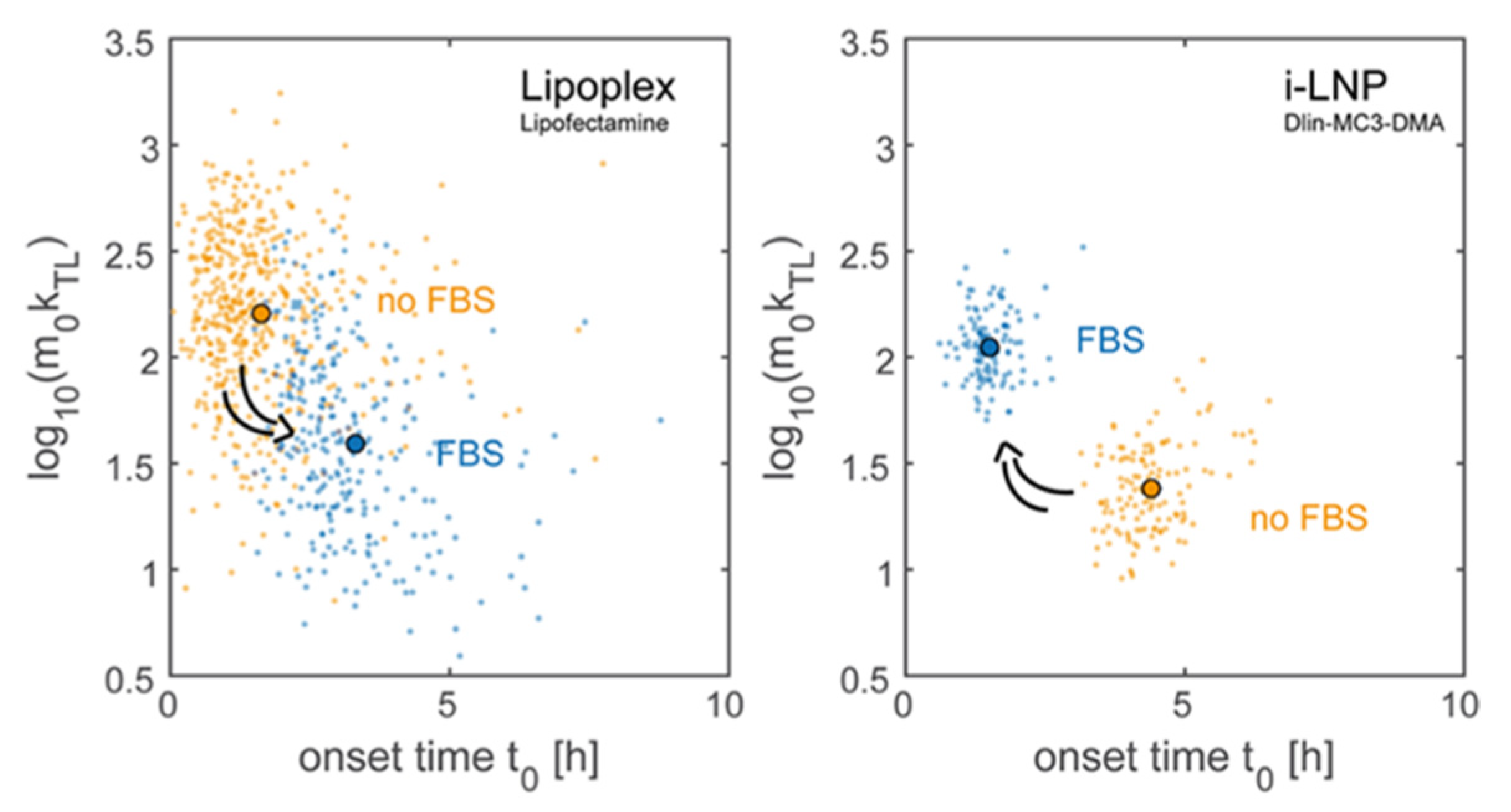
- Reiser, A.; Woschée, D.; Mehrotra, N.; Krzysztoń, R.S.; Strey, H.; Rädler, J.O. Correlation of mRNA delivery timing and protein expression in lipid-based transfection. Integr. Biol. 2019, 11, 362–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caracciolo, G.; Callipo, L.; De Sanctis, M.C.; Cavaliere, C.; Pozzi, D.; Laganà, A. Surface adsorption of protein corona controls the cell internalization mechanism of DC-Chol–DOPE/DNA lipoplexes in serum. Biochim. Biophys. Acta (BBA) Biomembr. 2010, 1798, 536–543. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Yu, L.; Monopoli, M.P.; Sandin, P.; Mahon, E.; Salvati, A.; Dawson, K.A. The biomolecular corona is retained during nanoparticle uptake and protects the cells from the damage induced by cationic nanoparticles until degraded in the lysosomes. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted Delivery of RNAi Therapeutics with Endogenous and Exogenous Ligand-Based Mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef]
- Sebastiani, F.; Yanez Arteta, M.; Lerche, M.; Porcar, L.; Lang, C.; Bragg, R.A.; Elmore, C.S.; Krishnamurthy, V.R.; Russell, R.A.; Darwish, T.; et al. Apolipoprotein E binding drives structural and compositional rearrangement of mRNA-containing lipid nanoparticles. ACS Nano 2021, 15, 6709–6722. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felgner, P.L.; Ringold, G.M. Cationic liposome-mediated transfection. Nat. Cell Biol. 1989, 337, 387–388. [Google Scholar] [CrossRef]
- Rädler, J.O.; Koltover, I.; Salditt, T.; Safinya, C.R. Structure of DNA-Cationic Liposome Complexes: DNA Intercalation in Multilamellar Membranes in Distinct Interhelical Packing Regimes. Science 1997, 275, 810–814. [Google Scholar] [CrossRef] [Green Version]
- Guevara, M.L.; Persano, S.; Persano, F. Lipid-based vectors for therapeutic mRNA-based anti-cancer vaccines. Curr. Pharm. Des. 2019, 25, 1443–1454. [Google Scholar] [CrossRef]
- Wahane, A.; Waghmode, A.; Kapphahn, A.; Dhuri, K.; Gupta, A.; Bahal, R. Role of Lipid-Based and Polymer-Based Non-Viral Vectors in Nucleic Acid Delivery for Next-Generation Gene Therapy. Molecules 2020, 25, 2866. [Google Scholar] [CrossRef]
- Franco, M.S.; Gomes, E.R.; Roque, M.C.; Oliveira, M.C. Triggered Drug Release from Liposomes: Exploiting the Outer and Inner Tumor Environment. Front. Oncol. 2021, 11, 470. [Google Scholar] [CrossRef]
- Chen, W.; Deng, W.; Xu, X.; Zhao, X.; Vo, J.N.; Anwer, A.G.; Williams, T.; Cui, H.; Goldys, E.M. Photoresponsive endosomal escape enhances gene delivery using liposome–polycation–DNA (LPD) nanovectors. J. Mater. Chem. B 2018, 6, 5269–5281. [Google Scholar] [CrossRef] [PubMed]
- Chander, N.; Morstein, J.; Bolten, J.S.; Shemet, A.; Cullis, P.R.; Trauner, D.; Witzigmann, D. Optimized Photoactivatable Lipid Nanoparticles Enable Red Light Triggered Drug Release. Small 2021, 2008198. [Google Scholar] [CrossRef]
- Chen, W.; Deng, W.; Goldys, E.M. Light-Triggerable Liposomes for Enhanced Endolysosomal Escape and Gene Silencing in PC12 Cells. Mol. Ther. Nucleic Acids 2017, 7, 366–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksoy, Y.A.; Yang, B.; Chen, W.; Hung, T.; Kuchel, R.P.; Zammit, N.W.; Grey, S.T.; Goldys, E.M.; Deng, W. Spatial and Temporal Control of CRISPR-Cas9-Mediated Gene Editing Delivered via a Light-Triggered Liposome System. ACS Appl. Mater. Interfaces 2020, 12, 52433–52444. [Google Scholar] [CrossRef]
- Kontturi, L.-S.; Dikkenberg, J.V.D.; Urtti, A.; Hennink, W.E.; Mastrobattista, E. Light-Triggered Cellular Delivery of Oligonucleotides. Pharmaceutics 2019, 11, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias-Alpizar, G.; Kong, L.; Vlieg, R.C.; Rabe, A.; Papadopoulou, P.; Meijer, M.S.; Bonnet, S.; Vogel, S.; Van Noort, J.; Kros, A.; et al. Light-triggered switching of liposome surface charge directs delivery of membrane impermeable payloads in vivo. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
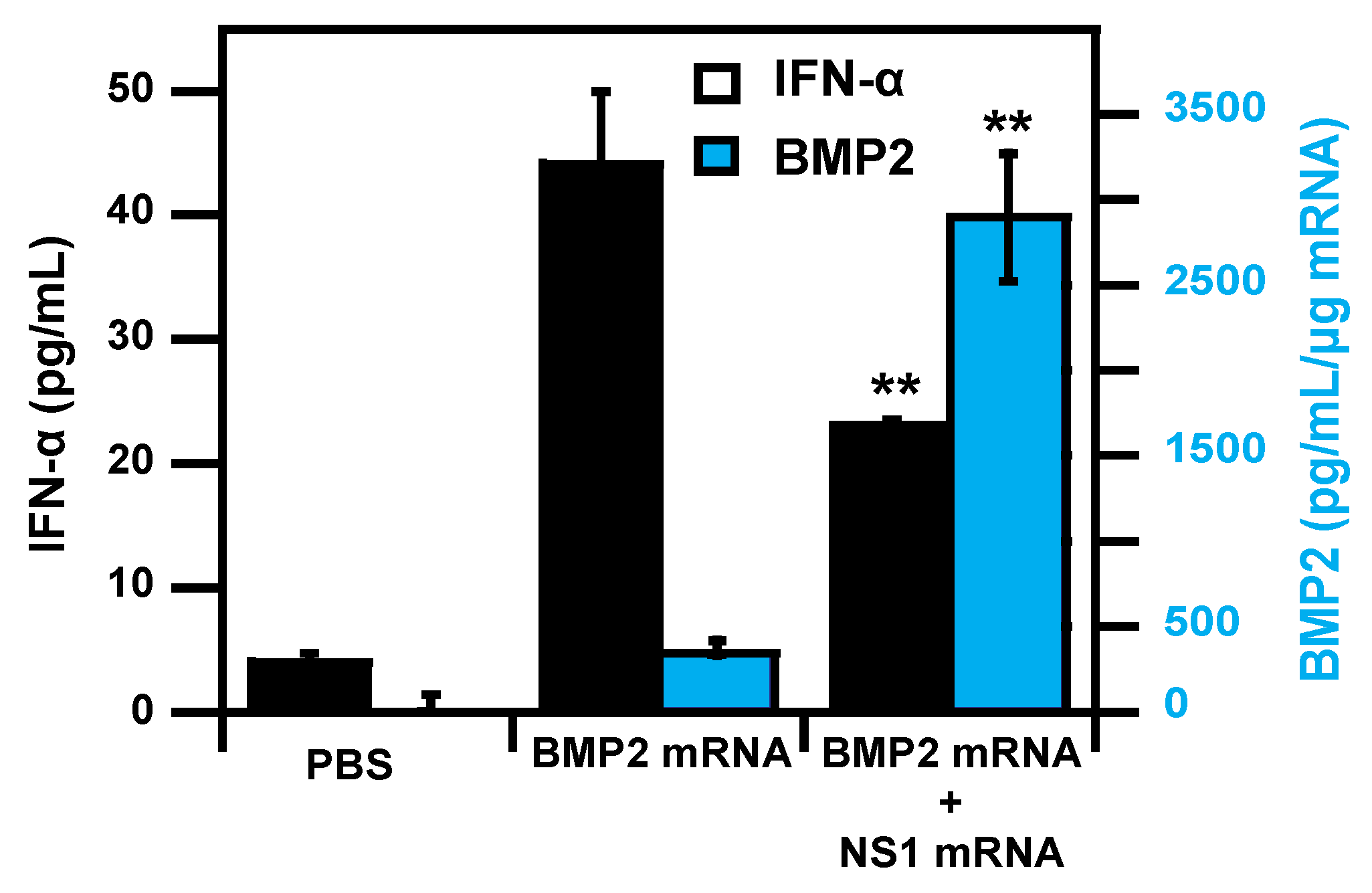
- Wang, P.; Logeart-Avramoglou, D.; Petite, H.; Goncalves, C.; Midoux, P.; Perche, F.; Pichon, C. Co-delivery of NS1 and BMP2 mRNAs to murine pluripotent stem cells leads to enhanced BMP-2 expression and osteogenic differentiation. Acta Biomater. 2020, 108, 337–346. [Google Scholar] [CrossRef]
- Wang, P.; Perche, F.; Midoux, P.; Cabral, S.; Malard, V.; Correia, I.J.; Ei-Hafci, H.; Petite, H.; Logeart-Avramoglou, D.; Pichon, C. In Vivo bone tissue induction by freeze-dried collagen-nanohydroxyapatite matrix loaded with BMP2/NS1 mRNAs lipopolyplexes. J. Control. Release 2021, 334, 188–200. [Google Scholar] [CrossRef]
- Gao, X.; Huang, L. Potentiation of Cationic Liposome-Mediated Gene Delivery by Polycations. Biochemistry 1996, 35, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Simion, V.; Henriet, E.; Juric, V.; Aquino, R.; Loussouarn, C.; Laurent, Y.; Martin, F.; Midoux, P.; Garcion, E.; Pichon, C.; et al. Intracellular trafficking and functional monitoring of miRNA delivery in glioblastoma using lipopolyplexes and the miRNA-ON RILES reporter system. J. Control. Release 2020, 327, 429–443. [Google Scholar] [CrossRef]
- Gonçalves, C.; Berchel, M.; Gosselin, M.-P.; Malard, V.; Cheradame, H.; Jaffrès, P.-A.; Guégan, P.; Pichon, C.; Midoux, P. Lipopolyplexes comprising imidazole/imidazolium lipophosphoramidate, histidinylated polyethyleneimine and siRNA as efficient formulation for siRNA transfection. Int. J. Pharm. 2014, 460, 264–272. [Google Scholar] [CrossRef]
- Van der Jeught, K.; De Koker, S.; Bialkowski, L.; Heirman, C.; Tjok Joe, P.; Perche, F.; Maenhout, S.; Bevers, S.; Broos, K.; Deswarte, K.; et al. Dendritic cell targeting mRNA lipopolyplexes combine strong antitumor T-cell immunity with improved inflammatory safety. ACS Nano 2018, 12, 9815–9829. [Google Scholar] [CrossRef]
- Yen, H.-C.; Cabral, H.; Mi, P.; Toh, K.; Matsumoto, Y.; Liu, X.; Koori, H.; Kim, A.; Miyazaki, K.; Miura, Y.; et al. Light-Induced Cytosolic Activation of Reduction-Sensitive Camptothecin-Loaded Polymeric Micelles for Spatiotemporally Controlled in Vivo Chemotherapy. ACS Nano 2014, 8, 11591–11602. [Google Scholar] [CrossRef]
- Zhu, K.; Liu, G.; Hu, J.; Liu, S. Near-Infrared Light-Activated Photochemical Internalization of Reduction-Responsive Polyprodrug Vesicles for Synergistic Photodynamic Therapy and Chemotherapy. Biomacromolecules 2017, 18, 2571–2582. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhang, C.-J.; Liu, B. A Photoactivatable AIE Polymer for Light-Controlled Gene Delivery: Concurrent Endo/Lysosomal Escape and DNA Unpacking. Angew. Chem. Int. Ed. 2015, 54, 11419–11423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; De Smedt, S.C.; Remaut, K. Fluorescence Correlation Spectroscopy to find the critical balance between extracellular association and intracellular dissociation of mRNA complexes. Acta Biomater. 2018, 75, 358–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setten, R.; Rossi, J.J.; Han, S.-P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Main Subject of Study | Enhancement Strategy | Model | Results | Ref |
|---|---|---|---|---|
| Nanoparticle uptake | Anti-PV1 conjugation for endothelial cell targeting in combination with 160 nm LNPs. | In cellulo In vivo | Increasing lung accumulation in combination with a 50-fold increase in mRNA expression compared to standard LNPs. | [50] |
| Nanoparticle uptake | Incorporation of tri-mannose lipid for mannose receptor targeting on dendritic cell, with lipopolyplexes. | In cellulo | Increase in the binding on the surface of dendritic cells was observed when liposome incorporate a tri-mannose moiety. | [60] |
| Endosomal escape enhancement | LNPs comprising lipid-like material ssPalmO for mRNA complexation and release after cleavage of disulfide bond by intracellular glutathione (GSH). | In vivo | LNPs made of ssPalmO-Phe (self-degradable ionizable phenyl ester lipid) showed the highest EPO blood level 24 h after transfection with EPO mRNA than LNPs made of non-degradable benzyl ester or with D-Lin-MC3-DMA. | [72,73] |
| Endosomal escape/transfection enhancement | Optimization of the length and saturation of the ionizable lipid tail. | In cellulo In vivo | Ionizable lipid with a length of 10 carbons exhibited highest luciferase expression in mice liver and spleen. Tails with more than 2 unsaturations showed lower luciferase expression. | [26,74] |
| Endosomal escape/transfection enhancement | Optimization of the polar head of the ionizable lipid, tertiary versus quaternary amine group. | In vivo | LNPs made with tertiary amine group lipids exhibited transfection on retinal pigment epithelial cells. LNPs based on quaternary amine group lacks transfection. | [75] |
| Endosomal escape/transfection enhancement | Screening of polar head and lipid tail for T lymphocyte transfection. | In vivo | Imidazole head and heteroatoms (O, S, Se) in tail exhibited highest transfection of primary T cells. | [76] |
| Endosomal escape/transfection enhancement | Screening of amino lipid series of ionizable lipids. | In cellulo In vivo | Lipid-5 exhibited a 6-fold increase in endosomal escape versus classic MC3-LNPs in cells. In vivo study showed a 5-fold enhancement of plasma level EPO in cynomolgus monkey after mRNA transfection versus MC3-LNPs. | [71] |
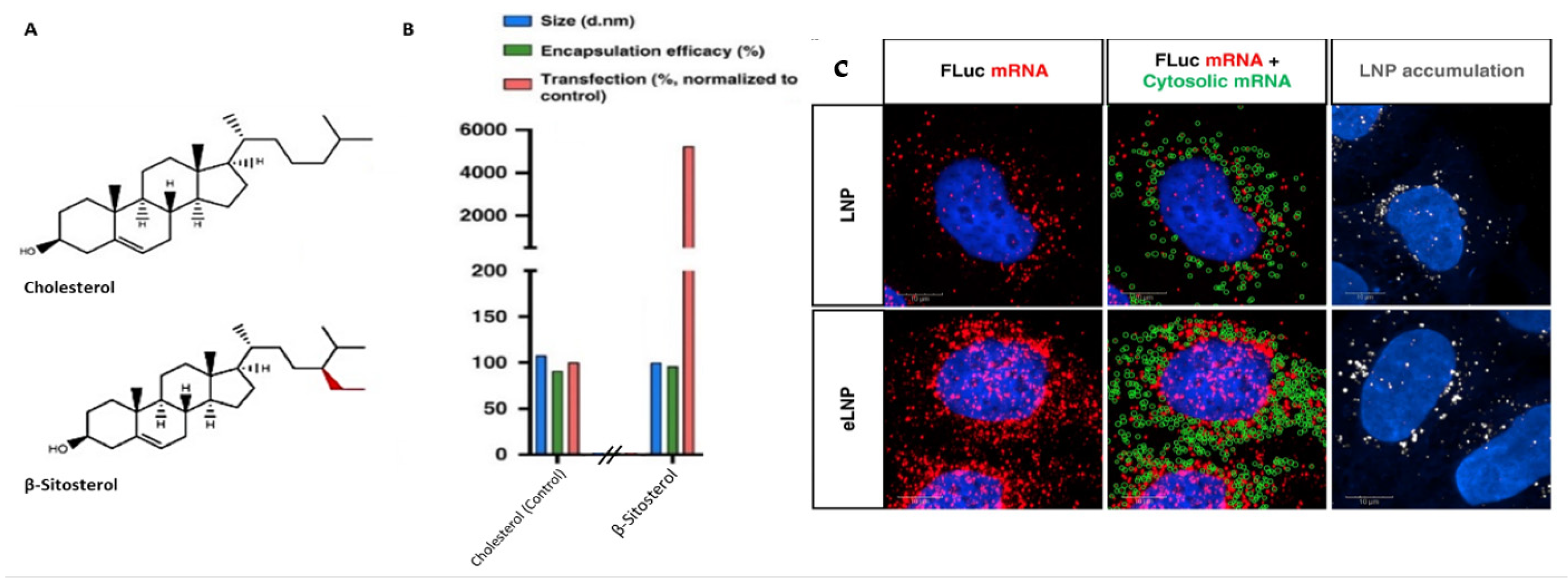
| mRNA translation enhancement | Screening of cholesterol analog. | In cellulo In vivo | LNPs made with cholesterol analog β-sitosterol exhibit equivalent uptake and endosomal escape, but a 48-fold enhancement in transfection versus cholesterol LNPs. | [77] |
| Immune sensors elusion | Combination of unmodified mRNA LNPs with drugs acting as inhibitors, as follows: ISRIB (eIF2a phosphorylation) and DXM (steroidal anti-inflammatory drug). | In-cellulo In vivo | Increase in mRNA translation was observed with both drugs in vitro, with different patterns. Co-delivery of DXM-palmitate and mRNA improve luciferase expression in mice liver. | [78] |
| Adjuvant effect of LNPs composition | Delivery of mRNA to APCs with LNPs made of lipid-like material allowing Toll-like receptor 4 activation for T-cell activation. | In cellulo In vivo | Lipid-like material induced maturation and activation of dendritic cell. Inhibitor of TLRs showed an effect specific to TLR4 signaling. | [79] |
| Adjuvant effect of LNPs composition | Screening of heterocyclic lipid for activation of STING pathway among APCs transfection. | In cellulo In vivo | Lipids with heterocyclic amine head group induced highest antigen-specific cytotoxic T lymphocyte response among OVA mRNA transfection. Empty LNPs made of cyclic group were also capable of DC activation, up to 2–3 fold versus linear LNPs, dependent on STING pathway. | [22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delehedde, C.; Even, L.; Midoux, P.; Pichon, C.; Perche, F. Intracellular Routing and Recognition of Lipid-Based mRNA Nanoparticles. Pharmaceutics 2021, 13, 945. https://doi.org/10.3390/pharmaceutics13070945
Delehedde C, Even L, Midoux P, Pichon C, Perche F. Intracellular Routing and Recognition of Lipid-Based mRNA Nanoparticles. Pharmaceutics. 2021; 13(7):945. https://doi.org/10.3390/pharmaceutics13070945
Chicago/Turabian StyleDelehedde, Christophe, Luc Even, Patrick Midoux, Chantal Pichon, and Federico Perche. 2021. "Intracellular Routing and Recognition of Lipid-Based mRNA Nanoparticles" Pharmaceutics 13, no. 7: 945. https://doi.org/10.3390/pharmaceutics13070945