
Imaging-Based Characterization of a Slco2b1(-/-) Mouse Model Using [11C]Erlotinib and [99mTc]Mebrofenin as Probe Substrates

 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Radiotracers
2.2. Animals
2.3. PET and Planar Scintigraphic Imaging
2.4. Analysis of PET Data
2.5. Analysis of Planar Scintigraphy Data
2.6. Pharmacokinetic Modeling
2.7. Statistical Analysis
3. Results
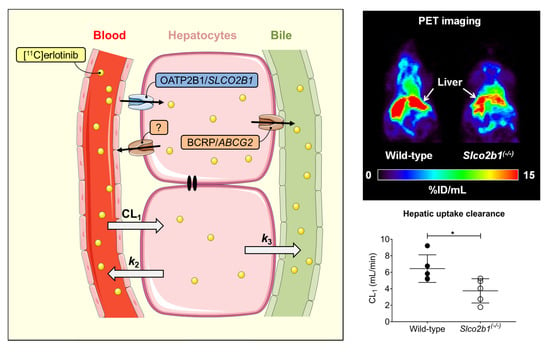
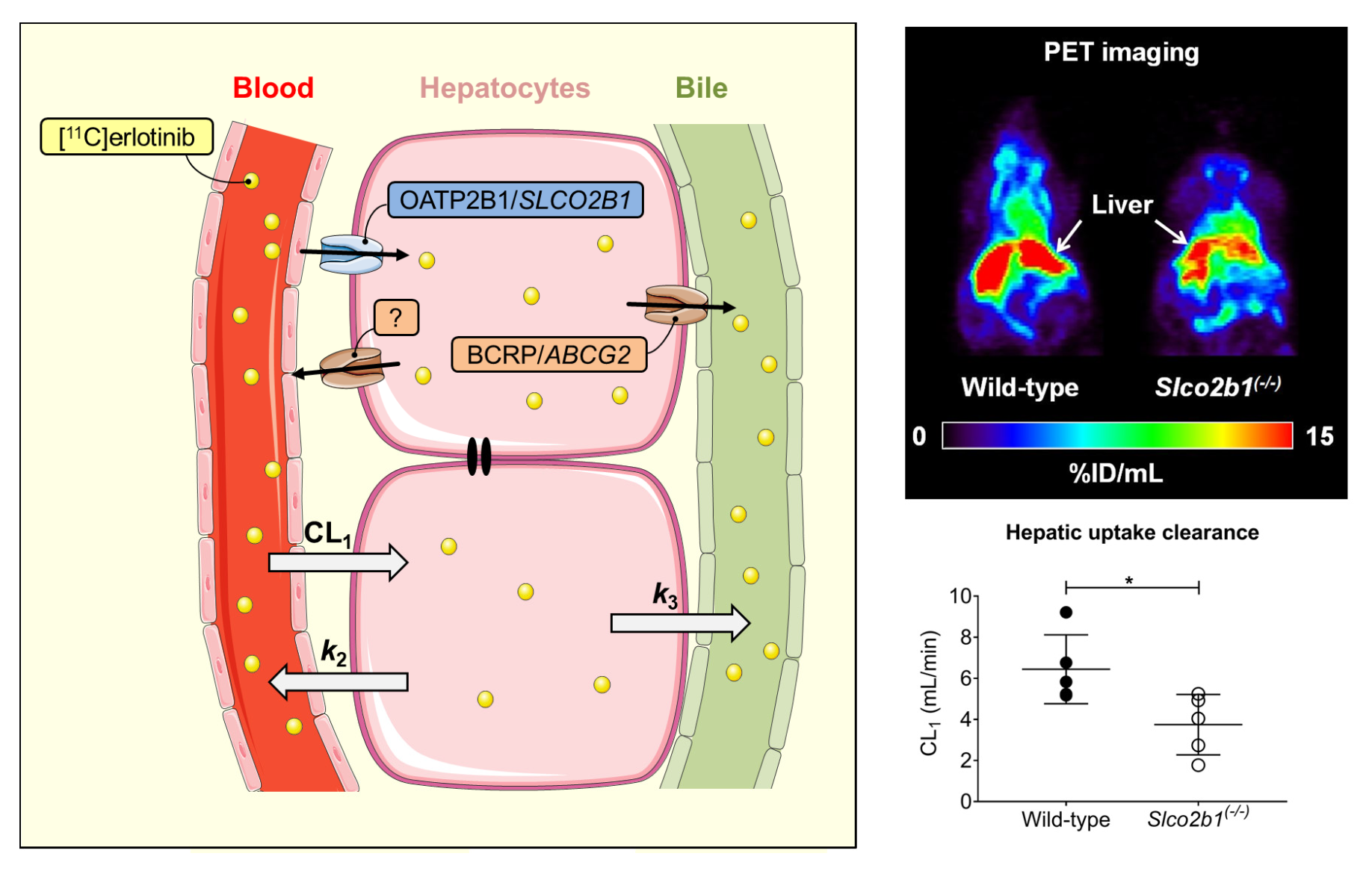
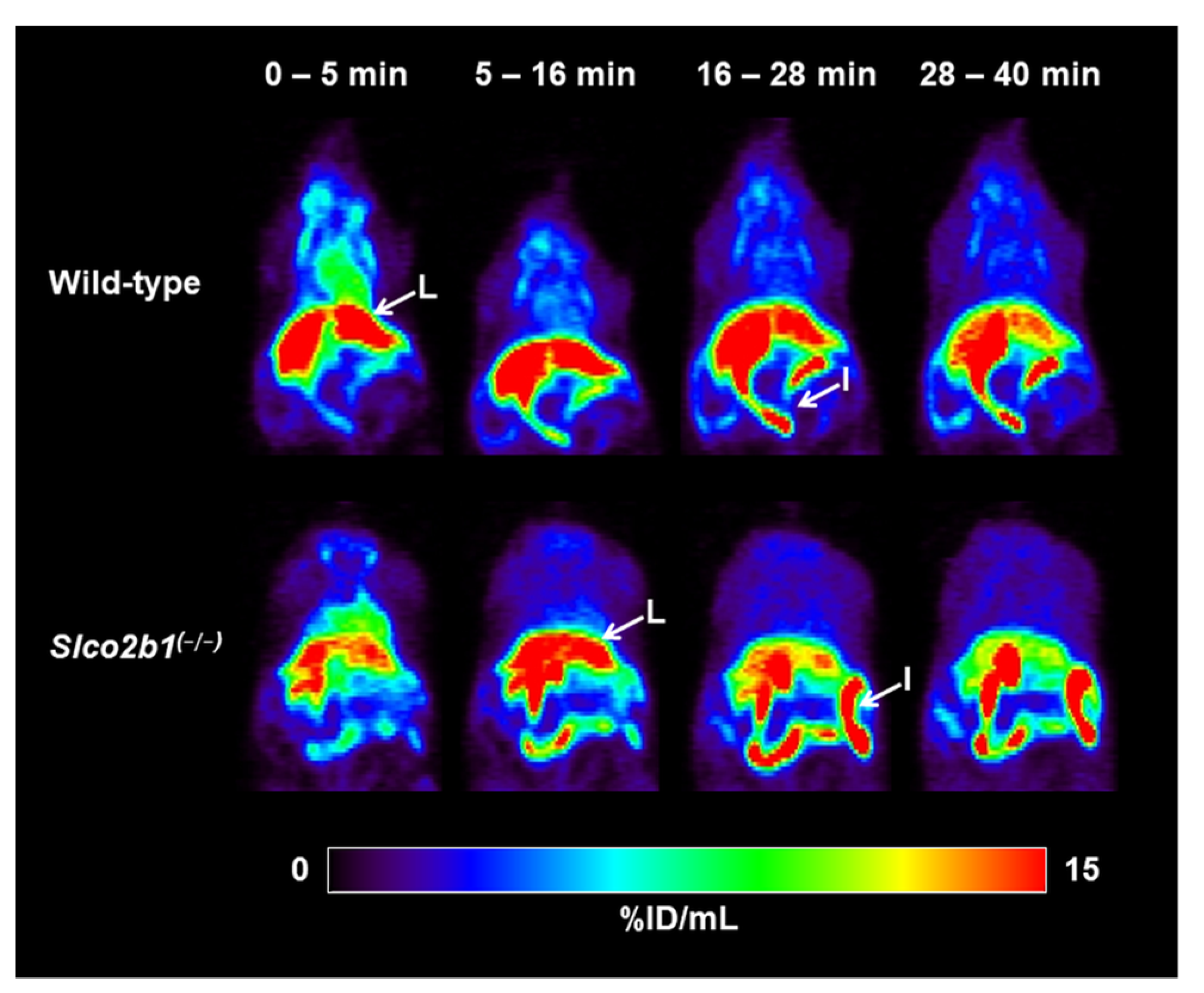
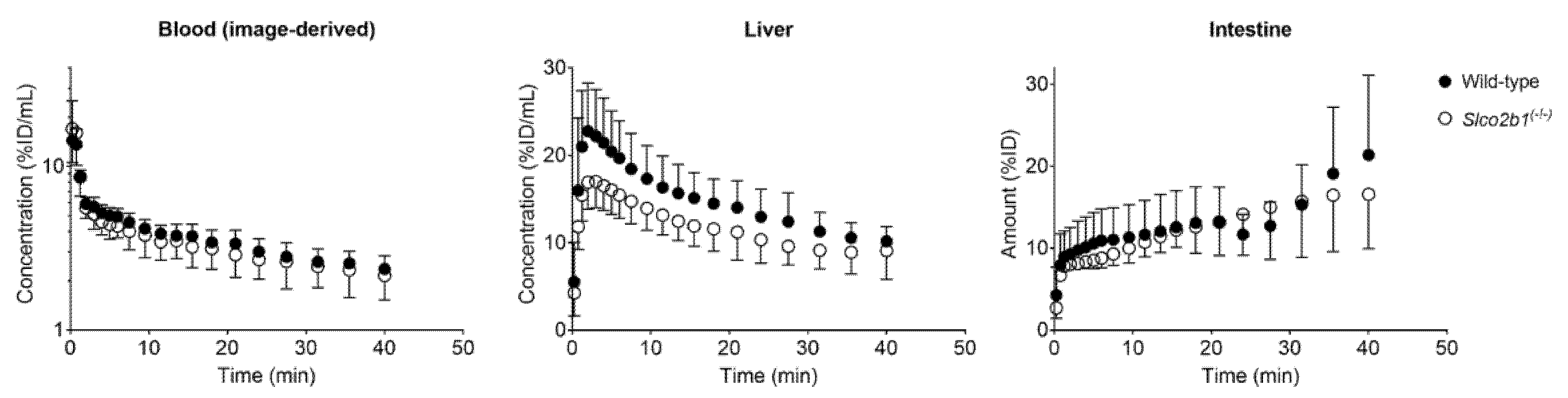
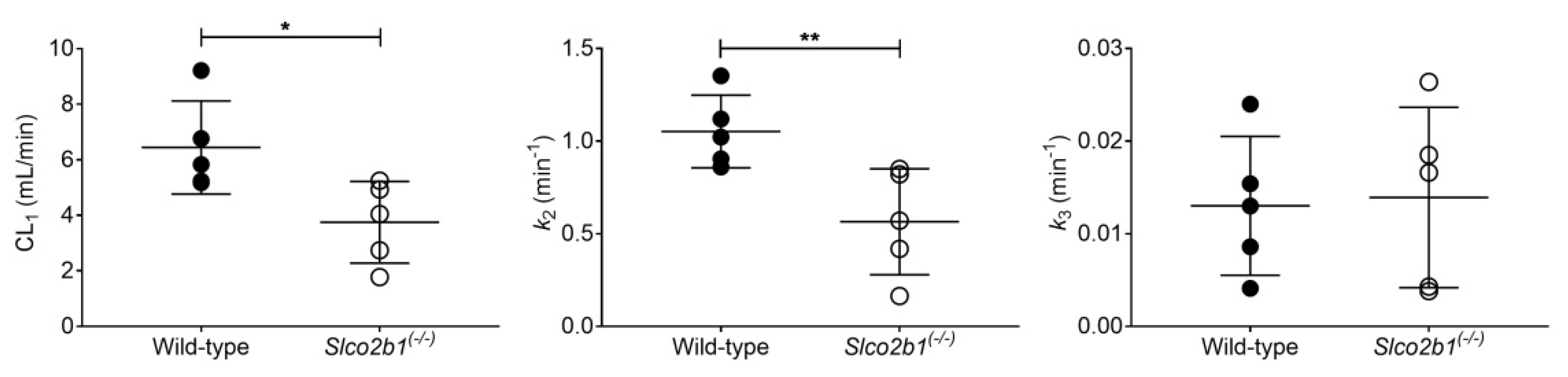
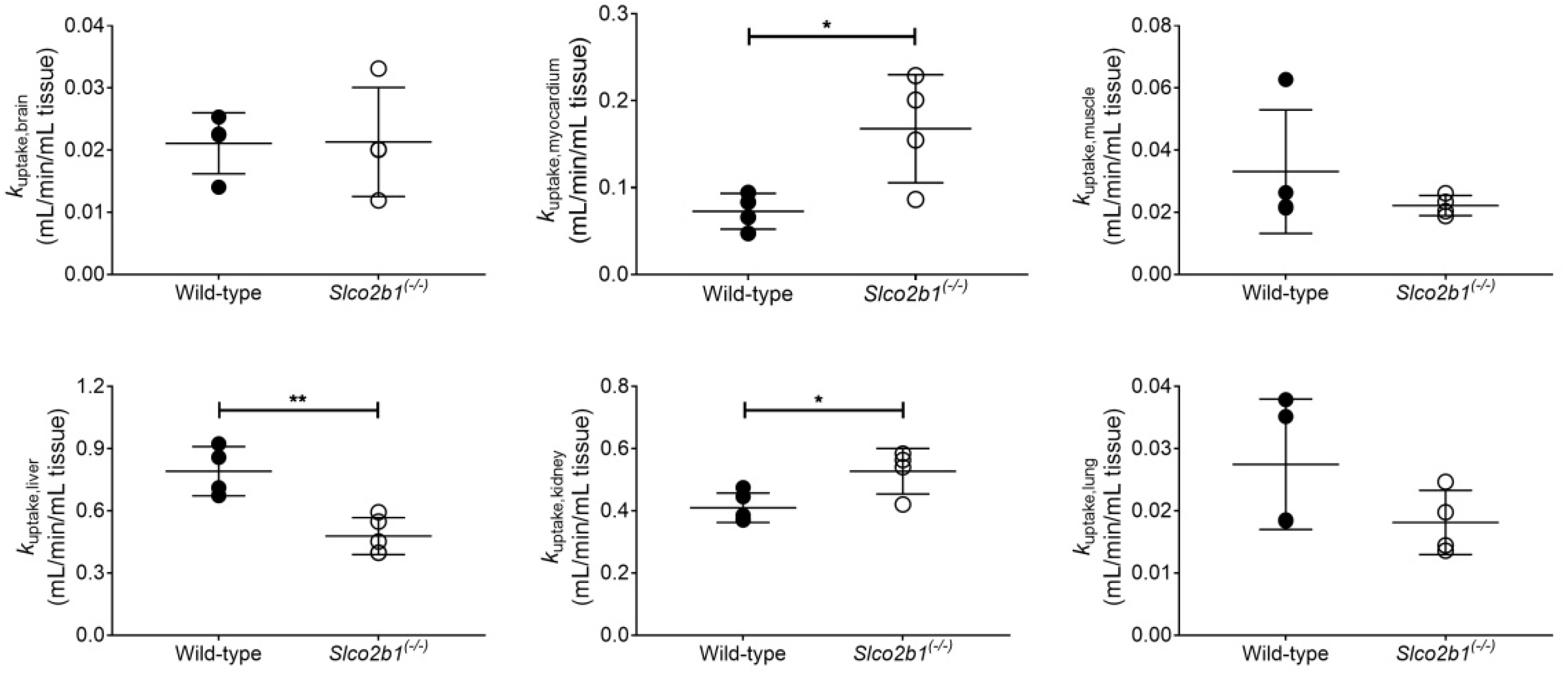
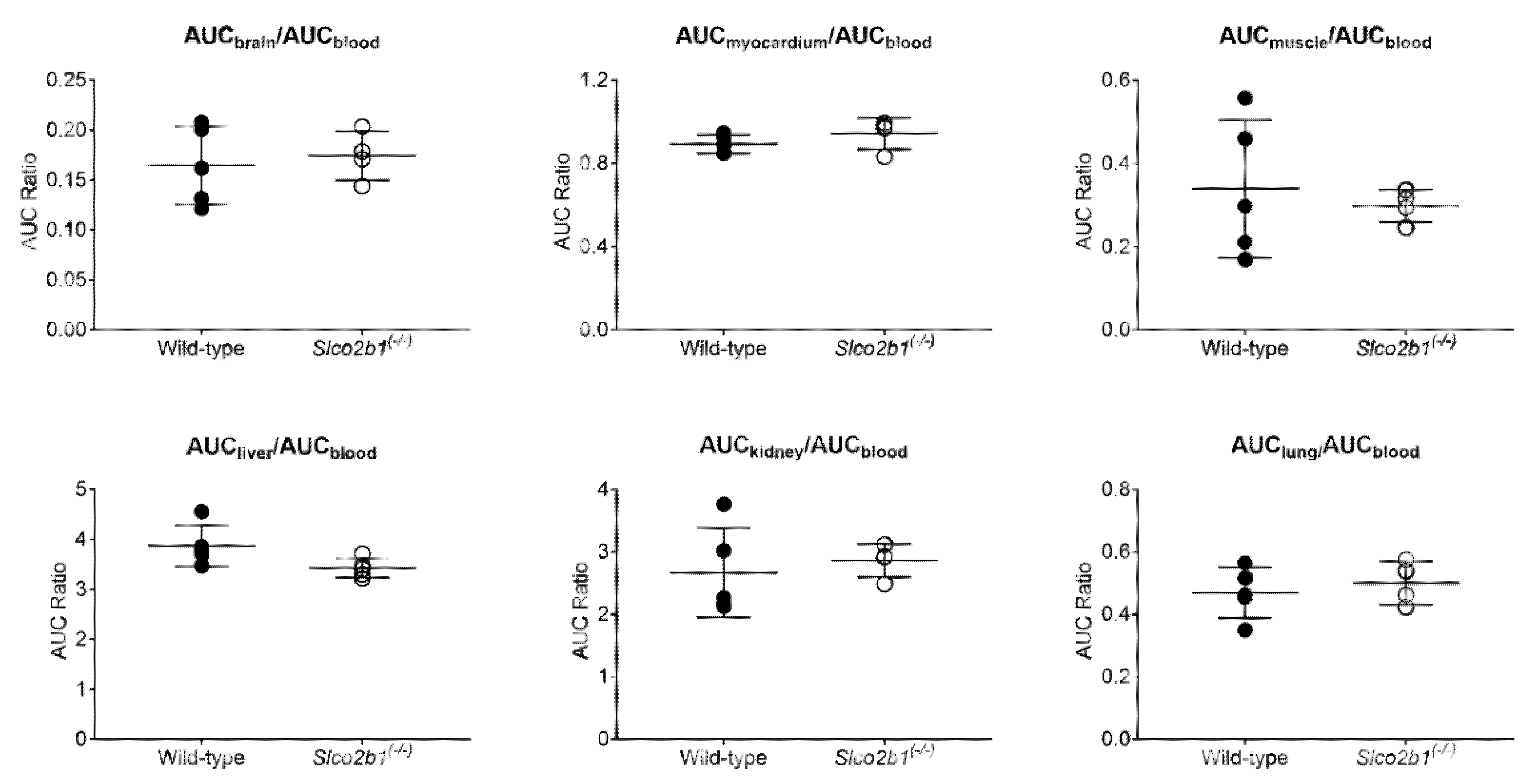
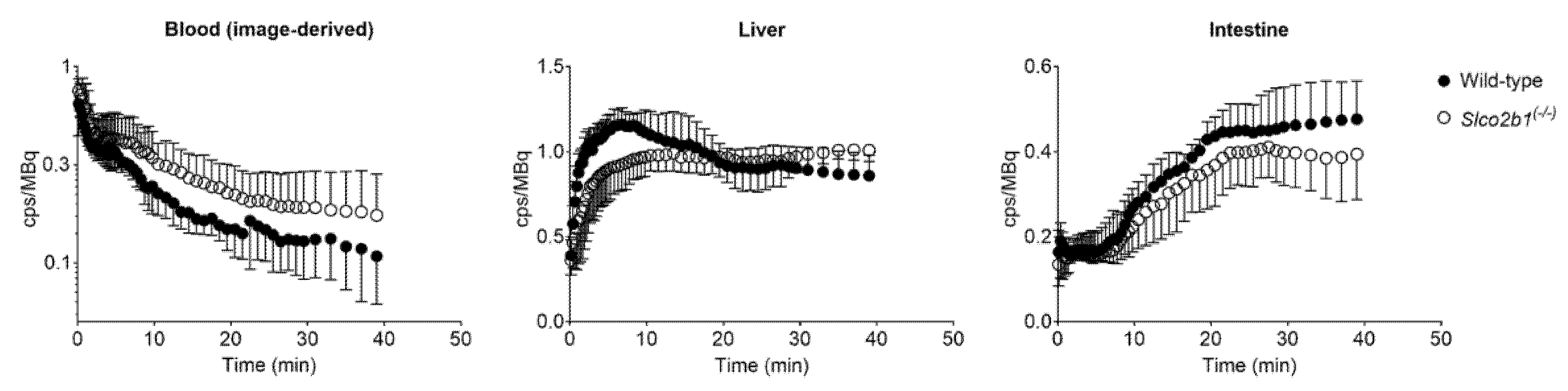
3.1. Influence of OATP2B1 on the Hepatobiliary Disposition of [11C]erlotinib
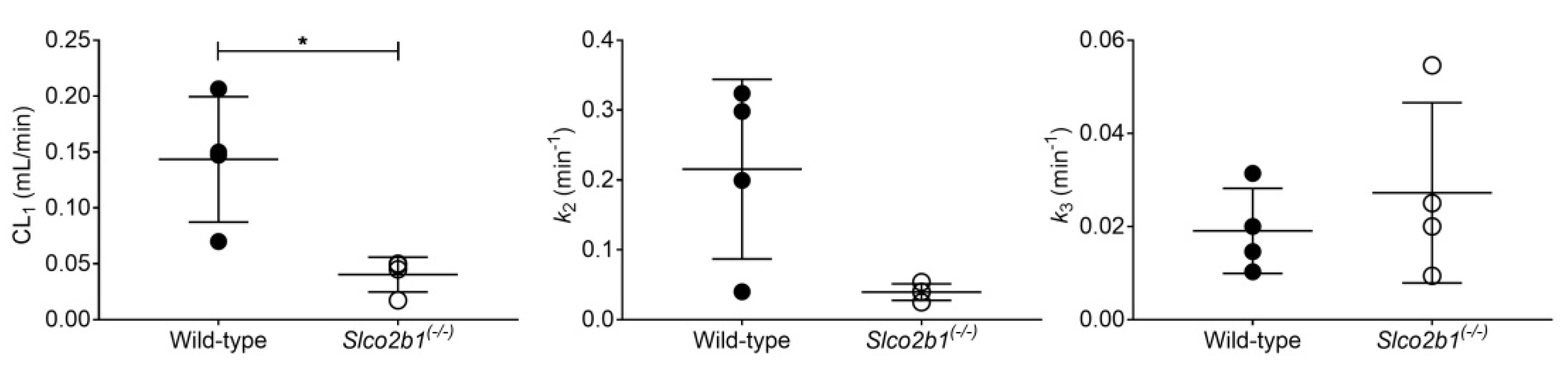
3.2. Influence of OATP2B1 on the Hepatobiliary Disposition of [99mTc]mebrofenin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Roth, M.; Obaidat, A.; Hagenbuch, B. OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br. J. Pharmacol. 2012, 165, 1260–1287. [Google Scholar] [CrossRef] [Green Version]
- Tamai, I.; Nezu, J.; Uchino, H.; Sai, Y.; Oku, A.; Shimane, M.; Tsuji, A. Molecular identification and characterization of novel members of the human organic anion transporter (OATP) family. Biochem. Biophys. Res. Commun. 2000, 273, 251–260. [Google Scholar] [CrossRef]
- Kullak-Ublick, G.A.; Ismair, M.G.; Stieger, B.; Landmann, L.; Huber, R.; Pizzagalli, F.; Fattinger, K.; Meier, P.J.; Hagenbuch, B. Organic anion-transporting polypeptide B (OATP-B) and its functional comparison with three other OATPs of human liver. Gastroenterology 2001, 120, 525–533. [Google Scholar] [CrossRef]
- Grube, M.; Kock, K.; Oswald, S.; Draber, K.; Meissner, K.; Eckel, L.; Bohm, M.; Felix, S.B.; Vogelgesang, S.; Jedlitschky, G.; et al. Organic anion transporting polypeptide 2B1 is a high-affinity transporter for atorvastatin and is expressed in the human heart. Clin. Pharmacol. Ther. 2006, 80, 607–620. [Google Scholar] [CrossRef]
- Seki, S.; Kobayashi, M.; Itagaki, S.; Hirano, T.; Iseki, K. Contribution of organic anion transporting polypeptide OATP2B1 to amiodarone accumulation in lung epithelial cells. Biochim. Biophys. Acta 2009, 1788, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Knauer, M.J.; Urquhart, B.L.; Meyer zu Schwabedissen, H.E.; Schwarz, U.I.; Lemke, C.J.; Leake, B.F.; Kim, R.B.; Tirona, R.G. Human skeletal muscle drug transporters determine local exposure and toxicity of statins. Circ. Res. 2010, 106, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Tapaninen, T.; Neuvonen, P.J.; Niemi, M. Orange and apple juice greatly reduce the plasma concentrations of the OATP2B1 substrate aliskiren. Br. J. Clin. Pharmacol. 2011, 71, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Akamine, Y.; Miura, M.; Komori, H.; Tamai, I.; Ieiri, I.; Yasui-Furukori, N.; Uno, T. The change of pharmacokinetics of fexofenadine enantiomers through the single and simultaneous grapefruit juice ingestion. Drug. Metab. Pharmacokinet. 2015, 30, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Ieiri, I.; Doi, Y.; Maeda, K.; Sasaki, T.; Kimura, M.; Hirota, T.; Chiyoda, T.; Miyagawa, M.; Irie, S.; Iwasaki, K.; et al. Microdosing clinical study: Pharmacokinetic, pharmacogenomic (SLCO2B1), and interaction (grapefruit juice) profiles of celiprolol following the oral microdose and therapeutic dose J. Clin. Pharmacol. 2012, 52, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- McFeely, S.J.; Wu, L.; Ritchie, T.K.; Unadkat, J. Organic anion transporting polypeptide 2B1 - More than a glass-full of drug interactions. Pharmacol. Ther. 2019, 196, 204–215. [Google Scholar] [CrossRef]
- Zamek-Gliszczynski, M.J.; Taub, M.E.; Chothe, P.P.; Chu, X.; Giacomini, K.M.; Kim, R.B.; Ray, A.S.; Stocker, S.L.; Unadkat, J.D.; Wittwer, M.B.; et al. Transporters in drug development: 2018 ITC recommendations for transporters of emerging clinical importance. Clin. Pharmacol. Ther. 2018, 104, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Medwid, S.; Li, M.M.J.; Knauer, M.J.; Lin, K.; Mansell, S.E.; Schmerk, C.L.; Zhu, C.; Griffin, K.E.; Yousif, M.D.; Dresser, G.K.; et al. Fexofenadine and rosuvastatin pharmacokinetics in mice with targeted disruption of organic anion transporting polypeptide 2B1. Drug. Metab. Dispos. 2019, 47, 832–842. [Google Scholar] [CrossRef]
- Chen, M.; Hu, S.; Li, Y.; Gibson, A.A.; Fu, Q.; Baker, S.D.; Sparreboom, A. Role of Oatp2b1 in drug absorption and drug-drug interactions. Drug. Metab. Dispos. 2020, 48, 419–425. [Google Scholar] [CrossRef]
- Tournier, N.; Stieger, B.; Langer, O. Imaging techniques to study drug transporter function in vivo. Pharmacol. Ther. 2018, 189, 104–122. [Google Scholar] [CrossRef] [Green Version]
- Hernández Lozano, I.; Karch, R.; Bauer, M.; Blaickner, M.; Matsuda, A.; Wulkersdorfer, B.; Hacker, M.; Zeitlinger, M.; Langer, O. Towards improved pharmacokinetic models for the analysis of transporter-mediated hepatic disposition of drug molecules with positron emission tomography. AAPS J. 2019, 21, 61. [Google Scholar] [CrossRef]
- Hernández Lozano, I.; Langer, O. Use of imaging to assess the activity of hepatic transporters. Expert. Opin. Drug. Metab. Toxicol. 2020, 16, 149–164. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Lozano, I.; Wanek, T.; Sauberer, M.; Filip, T.; Mairinger, S.; Stanek, J.; Traxl, A.; Karch, R.; Schuetz, J.D.; Langer, O. Influence of ABC transporters on the excretion of ciprofloxacin assessed with PET imaging in mice. Eur. J. Pharm. Sci. 2021, 163, 105854. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Matsuda, A.; Wulkersdorfer, B.; Philippe, C.; Traxl, A.; Özvegy-Laczka, C.; Stanek, J.; Nics, L.; Klebermass, E.M.; Poschner, S.; et al. Influence of OATPs on hepatic disposition of erlotinib measured with positron emission tomography. Clin. Pharmacol. Ther. 2018, 104, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.; Traxl, A.; Matsuda, A.; Karch, R.; Philippe, C.; Nics, L.; Klebermass, E.M.; Wulkersdorfer, B.; Weber, M.; Poschner, S.; et al. Effect of rifampicin on the distribution of [11C]erlotinib to the liver, a translational PET study in humans and in mice. Mol. Pharm. 2018, 15, 4589–4598. [Google Scholar] [CrossRef]
- De Graaf, W.; Hausler, S.; Heger, M.; van Ginhoven, T.M.; van Cappellen, G.; Bennink, R.J.; Kullak-Ublick, G.A.; Hesselmann, R.; van Gulik, T.M.; Stieger, B. Transporters involved in the hepatic uptake of [99mTc]mebrofenin and indocyanine green. J. Hepatol. 2011, 54, 738–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghibellini, G.; Leslie, E.M.; Pollack, G.M.; Brouwer, K.L. Use of tc-99m mebrofenin as a clinical probe to assess altered hepatobiliary transport: Integration of in vitro, pharmacokinetic modeling, and simulation studies. Pharm. Res. 2008, 25, 1851–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marie, S.; Hernández-Lozano, I.; Langer, O.; Tournier, N. Repurposing 99mTc-mebrofenin as a probe for molecular imaging of hepatocyte transporters. J. Nucl. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Bahce, I.; Smit, E.F.; Lubberink, M.; van der Veldt, A.A.; Yaqub, M.; Windhorst, A.D.; Schuit, R.C.; Thunnissen, E.; Heideman, D.A.; Postmus, P.E.; et al. Development of [11C]erlotinib positron emission tomography for in vivo evaluation of EGF receptor mutational status. Clin. Cancer. Res. 2013, 19, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loening, A.M.; Gambhir, S.S. AMIDE: A free software tool for multimodality medical image analysis. Mol. Imaging 2003, 2, 131–137. [Google Scholar] [CrossRef]
- Traxl, A.; Mairinger, S.; Filip, T.; Sauberer, M.; Stanek, J.; Poschner, S.; Jäger, W.; Zoufal, V.; Novarino, G.; Tournier, N.; et al. Inhibition of ABCB1 and ABCG2 at the mouse blood-brain barrier with marketed drugs to improve brain delivery of the model ABCB1/ABCG2 substrate [11C]erlotinib. Mol. Pharm. 2019, 16, 1282–1293. [Google Scholar] [CrossRef]
- Shingaki, T.; Hume, W.E.; Takashima, T.; Katayama, Y.; Okauchi, T.; Hayashinaka, E.; Wada, Y.; Cui, Y.; Kusuhara, H.; Sugiyama, Y.; et al. Quantitative evaluation of mMate1 function based on minimally invasive measurement of tissue concentration using PET with [11C]metformin in mouse. Pharm. Res. 2015, 32, 2538–2547. [Google Scholar] [CrossRef]
- Marie, S.; Hernández-Lozano, I.; Breuil, L.; Saba, W.; Novell, A.; Gennisson, J.L.; Langer, O.; Truillet, C.; Tournier, N. Validation of pharmacological protocols for targeted inhibition of canalicular MRP2 activity in hepatocytes using [99mTc]mebrofenin imaging in rats. Pharmaceutics 2020, 12, 486. [Google Scholar] [CrossRef]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef]
- Kinzi, J.; Grube, M.; Meyer Zu Schwabedissen, H.E. OATP2B1—The underrated member of the organic anion transporting polypeptide family of drug transporters? Biochem. Pharmacol. 2021, 188, 114534. [Google Scholar] [CrossRef]
- Munk, O.L.; Keiding, S.; Bass, L. Impulse-response function of splanchnic circulation with model-independent constraints: Theory and experimental validation. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Sørensen, M.; Munk, O.L.; Ørntoft, N.W.; Frisch, K.; Andersen, K.J.; Mortensen, F.V.; Alstrup, A.K.; Ott, P.; Hofmann, A.F.; Keiding, S. Hepatobiliary secretion kinetics of conjugated bile acids measured in pigs by [11C]cholylsarcosine PET. J. Nucl. Med. 2016, 57, 961–966. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, S.; Krishnamurthy, G.T. Technetium-99m-iminodiacetic acid organic anions: Review of biokinetics and clinical application in hepatology. Hepatology 1989, 9, 139–153. [Google Scholar] [CrossRef]
- Bhargava, K.K.; Joseph, B.; Ananthanarayanan, M.; Balasubramaniyan, N.; Tronco, G.G.; Palestro, C.J.; Gupta, S. Adenosine triphosphate-binding cassette subfamily C member 2 is the major transporter of the hepatobiliary imaging agent [99mTc]mebrofenin. J. Nucl. Med. 2009, 50, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- Traxl, A.; Wanek, T.; Mairinger, S.; Stanek, J.; Filip, T.; Sauberer, M.; Müller, M.; Kuntner, C.; Langer, O. Breast cancer resistance protein and P-glycoprotein influence in vivo disposition of [11C]erlotinib. J. Nucl. Med. 2015, 56, 1930–1936. [Google Scholar] [CrossRef] [Green Version]
- Johnston, R.A.; Rawling, T.; Chan, T.; Zhou, F.; Murray, M. Selective inhibition of human solute carrier transporters by multikinase inhibitors. Drug. Metab. Dispos. 2014, 42, 1851–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.X.; Huang, Y.; Frassetto, L.A.; Benet, L.Z. Elucidating rifampin’s inducing and inhibiting effects on glyburide pharmacokinetics and blood glucose in healthy volunteers: Unmasking the differential effects of enzyme induction and transporter inhibition for a drug and its primary metabolite. Clin. Pharmacol. Ther. 2009, 85, 78–85. [Google Scholar] [CrossRef]
- Karlgren, M.; Vildhede, A.; Norinder, U.; Wisniewski, J.R.; Kimoto, E.; Lai, Y.; Haglund, U.; Artursson, P. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): Influence of protein expression on drug-drug interactions. J. Med. Chem. 2012, 55, 4740–4763. [Google Scholar] [CrossRef] [PubMed]
- Traxl, A.; Komposch, K.; Glitzner, E.; Wanek, T.; Mairinger, S.; Langer, O.; Sibilia, M. Hepatocyte-specific deletion of EGFR in mice reduces hepatic Abcg2 transport activity measured by [11C]erlotinib and positron emission tomography. Drug. Metab. Dispos. 2017, 45, 1093–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neyt, S.; Huisman, M.T.; Vanhove, C.; De Man, H.; Vliegen, M.; Moerman, L.; Dumolyn, C.; Mannens, G.; De Vos, F. In vivo visualization and quantification of (disturbed) Oatp-mediated hepatic uptake and Mrp2-mediated biliary excretion of 99mTc-mebrofenin in mice. J. Nucl. Med. 2013, 54, 624–630. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mice | CL1 (mL/min) | k2 (min−1) | k3 (min−1) | |
|---|---|---|---|---|
| [11C]erlotinib | Wild-type | 6.442 ± 1.675 (16.7–62.1) | 1.052 ± 0.196 (15.4–54.2) | 0.013 ± 0.008 (4.9–12.6) |
| Slco2b1(-/-) | 3.748 ± 1.470 * (5.7–57.9) | 0.565 ± 0.287 ** (5.8–54.9) | 0.014 ± 0.010 (3.1–10.7) | |
| [99mTc]mebrofenin | Wild-type | 0.144 ± 0.056 (2.4–3.7) | 0.215 ± 0.129 (2.8–6.7) | 0.019 ± 0.009 (1.3–2.5) |
| Slco2b1(-/-) | 0.040 ± 0.016 * (4.3–43.0) | 0.040 ± 0.012 (7.3–90.3) | 0.027 ± 0.019 (2.3–14.4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marie, S.; Hernández-Lozano, I.; Breuil, L.; Truillet, C.; Hu, S.; Sparreboom, A.; Tournier, N.; Langer, O. Imaging-Based Characterization of a Slco2b1(-/-) Mouse Model Using [11C]Erlotinib and [99mTc]Mebrofenin as Probe Substrates. Pharmaceutics 2021, 13, 918. https://doi.org/10.3390/pharmaceutics13060918
Marie S, Hernández-Lozano I, Breuil L, Truillet C, Hu S, Sparreboom A, Tournier N, Langer O. Imaging-Based Characterization of a Slco2b1(-/-) Mouse Model Using [11C]Erlotinib and [99mTc]Mebrofenin as Probe Substrates. Pharmaceutics. 2021; 13(6):918. https://doi.org/10.3390/pharmaceutics13060918
Chicago/Turabian StyleMarie, Solène, Irene Hernández-Lozano, Louise Breuil, Charles Truillet, Shuiying Hu, Alex Sparreboom, Nicolas Tournier, and Oliver Langer. 2021. "Imaging-Based Characterization of a Slco2b1(-/-) Mouse Model Using [11C]Erlotinib and [99mTc]Mebrofenin as Probe Substrates" Pharmaceutics 13, no. 6: 918. https://doi.org/10.3390/pharmaceutics13060918