
ChoK-Full of Potential: Choline Kinase in B Cell and T Cell Malignancies


{kind=link}
{kind=link}
Abstract
:1. Introduction
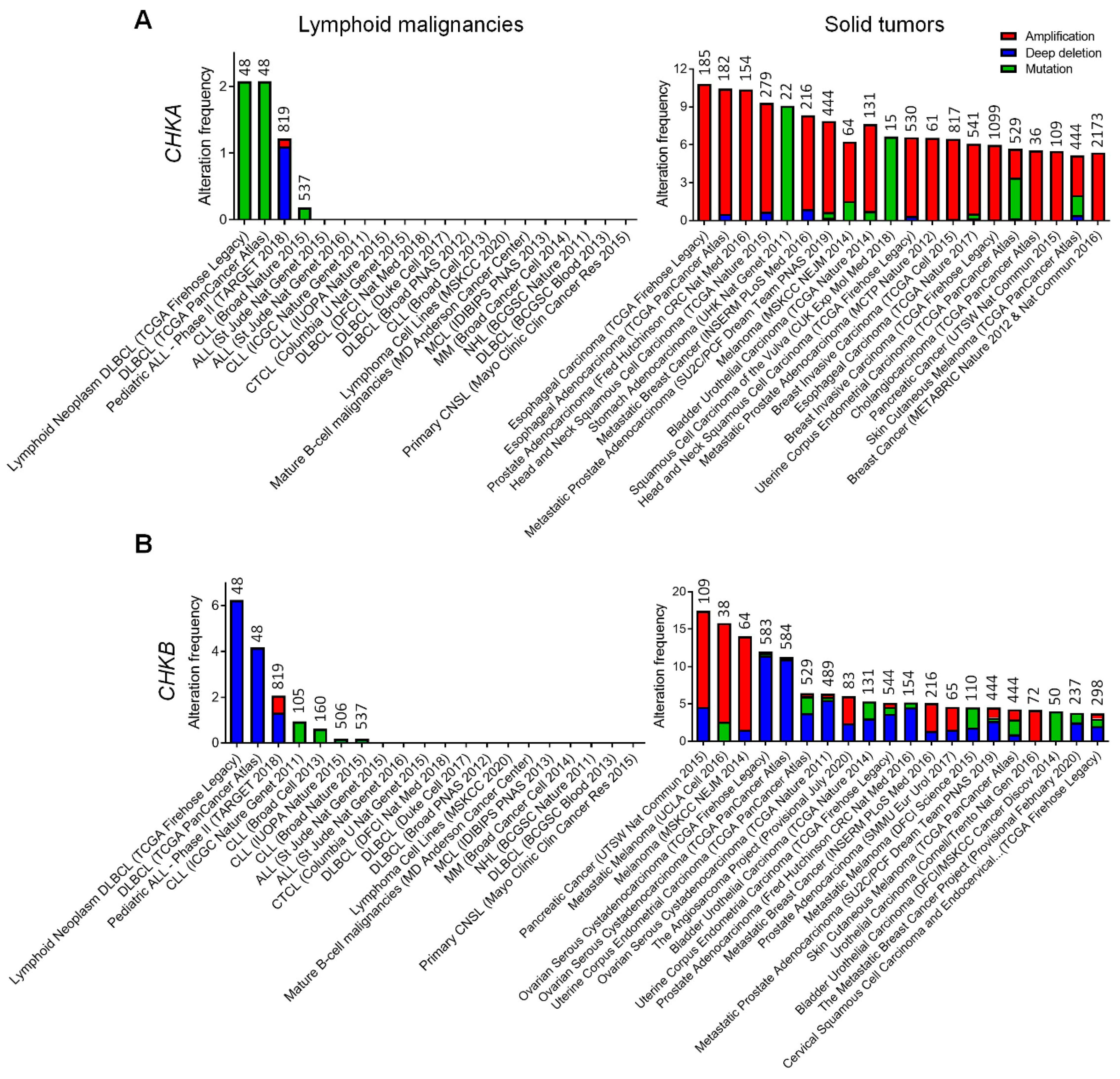
2. Genetic Alterations of Choline Kinase in Human Lymphoid Malignancies
3. Elevated CHKα Expression and Choline Metabolism in B Cell Malignancies
4. Up-Regulated Expression of CHKα and Choline Metabolism in T Cell Malignancies
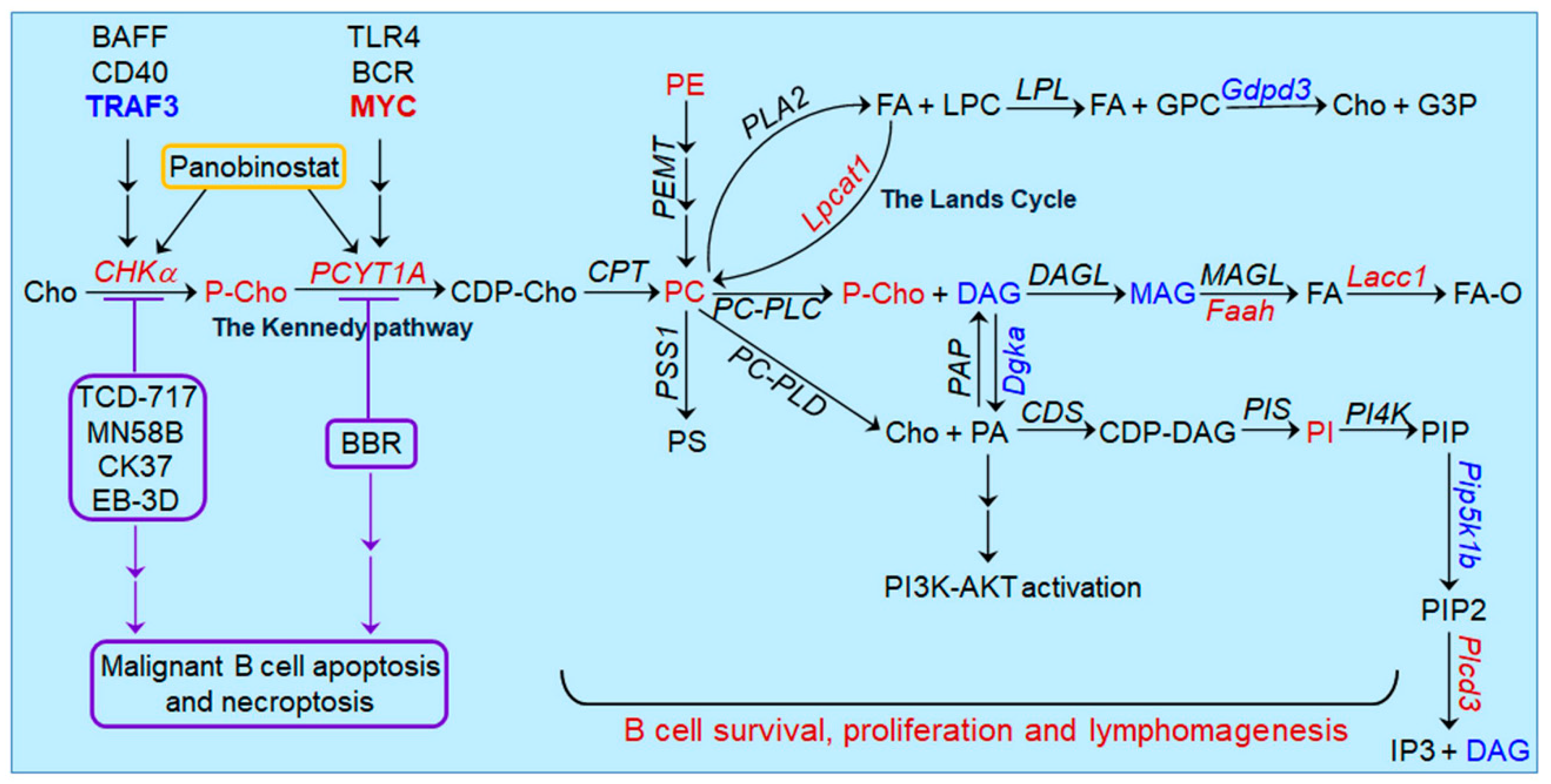
5. Oncogenic Pathways Downstream of CHKα Overexpression in Lymphoid Malignancies
6. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer 2011, 11, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Glunde, K.; Penet, M.F.; Jiang, L.; Jacobs, M.A.; Bhujwalla, Z.M. Choline metabolism-based molecular diagnosis of cancer: An update. Expert Rev. Mol. Diagn. 2015, 15, 735–747. [Google Scholar] [CrossRef]
- Arlauckas, S.P.; Popov, A.V.; Delikatny, E.J. Choline kinase alpha-Putting the ChoK-hold on tumor metabolism. Prog. Lipid Res. 2016, 63, 28–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.; Bhujwalla, Z.M.; Glunde, K. Targeting Phospholipid Metabolism in Cancer. Front. Oncol. 2016, 6, 266. [Google Scholar] [CrossRef] [Green Version]
- Cuccurullo, V.; Di Stasio, G.D.; Evangelista, L.; Castoria, G.; Mansi, L. Biochemical and Pathophysiological Premises to Positron Emission Tomography With Choline Radiotracers. J. Cell Physiol. 2017, 232, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Ruiz, B.; Serran-Aguilera, L.; Hurtado-Guerrero, R.; Conejo-Garcia, A. Recent advances in the design of choline kinase alpha inhibitors and the molecular basis of their inhibition. Med. Res. Rev. 2021, 41, 902–927. [Google Scholar] [CrossRef] [PubMed]
- Van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- McMaster, C.R. From yeast to humans-roles of the Kennedy pathway for phosphatidylcholine synthesis. FEBS Lett. 2018, 592, 1256–1272. [Google Scholar] [CrossRef] [Green Version]
- Gallego-Ortega, D.; Gomez del Pulgar, T.; Valdes-Mora, F.; Cebrian, A.; Lacal, J.C. Involvement of human choline kinase alpha and beta in carcinogenesis: A different role in lipid metabolism and biological functions. Adv. Enzym. Regul. 2011, 51, 183–194. [Google Scholar] [CrossRef]
- Aoyama, C.; Liao, H.; Ishidate, K. Structure and function of choline kinase isoforms in mammalian cells. Prog. Lipid Res. 2004, 43, 266–281. [Google Scholar] [CrossRef]
- Janardhan, S.; Srivani, P.; Sastry, G.N. Choline kinase: An important target for cancer. Curr. Med. Chem. 2006, 13, 1169–1186. [Google Scholar] [CrossRef]
- Wu, G.; Vance, D.E. Choline kinase and its function. Biochem. Cell Biol. 2010, 88, 559–564. [Google Scholar] [CrossRef]
- Chen, X.; Qiu, H.; Wang, C.; Yuan, Y.; Tickner, J.; Xu, J.; Zou, J. Molecular structure and differential function of choline kinases CHKalpha and CHKbeta in musculoskeletal system and cancer. Cytokine Growth Factor Rev. 2017, 33, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.Z.; Jiang, Y.Y.; Hao, J.J.; Zhang, Y.; Zhang, T.T.; Shang, L.; Liu, S.G.; Shi, F.; Wang, M.R. Identification of putative target genes for amplification within 11q13.2 and 3q27.1 in esophageal squamous cell carcinoma. Clin. Transl. Oncol. 2014, 16, 606–615. [Google Scholar] [CrossRef]
- Lin, X.M.; Hu, L.; Gu, J.; Wang, R.Y.; Li, L.; Tang, J.; Zhang, B.H.; Yan, X.Z.; Zhu, Y.J.; Hu, C.L.; et al. Choline Kinase alpha Mediates Interactions Between the Epidermal Growth Factor Receptor and Mechanistic Target of Rapamycin Complex 2 in Hepatocellular Carcinoma Cells to Promote Drug Resistance and Xenograft Tumor Progression. Gastroenterology 2017, 152, 1187–1202. [Google Scholar] [CrossRef]
- Glunde, K.; Serkova, N.J. Therapeutic targets and biomarkers identified in cancer choline phospholipid metabolism. Pharmacogenomics 2006, 7, 1109–1123. [Google Scholar] [CrossRef]
- Beloueche-Babari, M.; Arunan, V.; Troy, H.; te Poele, R.H.; te Fong, A.C.; Jackson, L.E.; Payne, G.S.; Griffiths, J.R.; Judson, I.R.; Workman, P.; et al. Histone deacetylase inhibition increases levels of choline kinase alpha and phosphocholine facilitating noninvasive imaging in human cancers. Cancer Res. 2012, 72, 990–1000. [Google Scholar] [CrossRef] [Green Version]
- Castro-Navas, F.F.; Schiaffino-Ortega, S.; Carrasco-Jimenez, M.P.; Rios-Marco, P.; Marco, C.; Espinosa, A.; Gallo, M.A.; Mariotto, E.; Basso, G.; Viola, G.; et al. New more polar symmetrical bipyridinic compounds: New strategy for the inhibition of choline kinase alpha. Future Med. Chem. 2015, 7, 417–436. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino-Ortega, S.; Baglioni, E.; Mariotto, E.; Bortolozzi, R.; Serran-Aguilera, L.; Rios-Marco, P.; Carrasco-Jimenez, M.P.; Gallo, M.A.; Hurtado-Guerrero, R.; Marco, C.; et al. Design, synthesis, crystallization and biological evaluation of new symmetrical biscationic compounds as selective inhibitors of human Choline Kinase alpha1 (ChoKalpha1). Sci. Rep. 2016, 6, 23793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sola-Leyva, A.; Lopez-Cara, L.C.; Rios-Marco, P.; Rios, A.; Marco, C.; Carrasco-Jimenez, M.P. Choline kinase inhibitors EB-3D and EB-3P interferes with lipid homeostasis in HepG2 cells. Sci. Rep. 2019, 9, 5109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bizzi, A.; Movsas, B.; Tedeschi, G.; Phillips, C.L.; Okunieff, P.; Alger, J.R.; Di Chiro, G. Response of non-Hodgkin lymphoma to radiation therapy: Early and long-term assessment with H-1 MR spectroscopic imaging. Radiology 1995, 194, 271–276. [Google Scholar] [CrossRef] [PubMed]
- King, A.D.; Yeung, D.K.; Ahuja, A.T.; Yuen, E.H.; Ho, S.F.; Tse, G.M.; van Hasselt, A.C. Human cervical lymphadenopathy: Evaluation with in vivo 1H-MRS at 1.5 T. Clin. Radiol. 2005, 60, 592–598. [Google Scholar] [CrossRef]
- Bisdas, S.; Fetscher, S.; Feller, A.C.; Baghi, M.; Knecht, R.; Gstoettner, W.; Vogl, T.J.; Balzer, J.O. Primary B cell lymphoma of the sphenoid sinus: CT and MRI characteristics with correlation to perfusion and spectroscopic imaging features. Eur. Arch. Otorhinolaryngol. 2007, 264, 1207–1213. [Google Scholar] [CrossRef]
- Zacharia, T.T.; Law, M.; Naidich, T.P.; Leeds, N.E. Central nervous system lymphoma characterization by diffusion-weighted imaging and MR spectroscopy. J. Neuroimaging 2008, 18, 411–417. [Google Scholar] [CrossRef]
- Harting, I.; Hartmann, M.; Jost, G.; Sommer, C.; Ahmadi, R.; Heiland, S.; Sartor, K. Differentiating primary central nervous system lymphoma from glioma in humans using localised proton magnetic resonance spectroscopy. Neurosci. Lett. 2003, 342, 163–166. [Google Scholar] [CrossRef]
- Yamasaki, F.; Takayasu, T.; Nosaka, R.; Amatya, V.J.; Doskaliyev, A.; Akiyama, Y.; Tominaga, A.; Takeshima, Y.; Sugiyama, K.; Kurisu, K. Magnetic resonance spectroscopy detection of high lipid levels in intraaxial tumors without central necrosis: A characteristic of malignant lymphoma. J. Neurosurg. 2015, 122, 1370–1379. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, T.H.; Filho, S.R.; Castro, A.C.; Paulino, E., Jr.; Mamede, M. Targeting personalized medicine in a non-Hodgkin lymphoma patient with 18F-FDG and 18F-choline PET/CT. Rev. Assoc. Med. Bras. 2017, 63, 109–111. [Google Scholar] [CrossRef] [Green Version]
- De Leiris, N.; Riou, L.; Leenhardt, J.; Vuillez, J.P.; Djaileb, L. 18F-Choline and 18F-FDG PET/CT in a Patient With Diffuse Large B-Cell Lymphoma and Recurrent Prostate Cancer. Clin. Nucl. Med. 2018, 43, e471–e472. [Google Scholar] [CrossRef] [PubMed]
- Matteucci, F.; Paganelli, G.; Martinelli, G.; Cerchione, C. PET/CT in Multiple Myeloma: Beyond FDG. Front. Oncol. 2020, 10, 622501. [Google Scholar] [CrossRef] [PubMed]
- Mesguich, C.; Hulin, C.; Lascaux, A.; Bordenave, L.; Marit, G.; Hindie, E. Choline PET/CT in Multiple Myeloma. Cancers 2020, 12, 1394. [Google Scholar] [CrossRef]
- Millard, T.; Chau, I.; Iyengar, S.; El-Sharkawi, D.; Cunningham, D.; Sharma, B. (18) F-choline radiotracer positron emission tomography as a new means to monitor central nervous system lymphoma. Br. J. Haematol. 2021, 193, 1026. [Google Scholar] [CrossRef] [PubMed]
- Rush, J.S.; Sweitzer, T.; Kent, C.; Decker, G.L.; Waechter, C.J. Biogenesis of the endoplasmic reticulum in activated B lymphocytes: Temporal relationships between the induction of protein N-glycosylation activity and the biosynthesis of membrane protein and phospholipid. Arch. Biochem. Biophys. 1991, 284, 63–70. [Google Scholar] [CrossRef]
- Fagone, P.; Sriburi, R.; Ward-Chapman, C.; Frank, M.; Wang, J.; Gunter, C.; Brewer, J.W.; Jackowski, S. Phospholipid biosynthesis program underlying membrane expansion during B-lymphocyte differentiation. J. Biol. Chem. 2007, 282, 7591–7605. [Google Scholar] [CrossRef] [Green Version]
- Fagone, P.; Gunter, C.; Sage, C.R.; Gunn, K.E.; Brewer, J.W.; Jackowski, S. CTP:phosphocholine cytidylyltransferase alpha is required for B-cell proliferation and class switch recombination. J. Biol. Chem. 2009, 284, 6847–6854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brewer, J.W.; Solodushko, V.; Aragon, I.; Barrington, R.A. Phosphatidylcholine as a metabolic cue for determining B cell fate and function. Cell. Immunol. 2016, 310, 78–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, S.; Nishino, I. Megaconial congenital muscular dystrophy due to loss-of-function mutations in choline kinase beta. Curr. Opin. Neurol. 2013, 26, 536–543. [Google Scholar] [CrossRef]
- Gruber, J.; See Too, W.C.; Wong, M.T.; Lavie, A.; McSorley, T.; Konrad, M. Balance of human choline kinase isoforms is critical for cell cycle regulation: Implications for the development of choline kinase-targeted cancer therapy. FEBS J. 2012, 279, 1915–1928. [Google Scholar] [CrossRef]
- Chu, W.C.; Chik, K.W.; Chan, Y.L.; Yeung, D.K.; Roebuck, D.J.; Howard, R.G.; Li, C.K.; Metreweli, C. White matter and cerebral metabolite changes in children undergoing treatment for acute lymphoblastic leukemia: Longitudinal study with MR imaging and 1H MR spectroscopy. Radiology 2003, 229, 659–669. [Google Scholar] [CrossRef]
- Huang, M.Q.; Nelson, D.S.; Pickup, S.; Qiao, H.; Delikatny, E.J.; Poptani, H.; Glickson, J.D. In vivo monitoring response to chemotherapy of human diffuse large B-cell lymphoma xenografts in SCID mice by 1H and 31P MRS. Acad. Radiol. 2007, 14, 1531–1539. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.C.; Huang, M.Q.; Nelson, D.S.; Pickup, S.; Wehrli, S.; Adegbola, O.; Poptani, H.; Delikatny, E.J.; Glickson, J.D. In vivo MRS markers of response to CHOP chemotherapy in the WSU-DLCL2 human diffuse large B-cell lymphoma xenograft. NMR Biomed. 2008, 21, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Delikatny, E.J.; Poptani, H.; Pickup, S.; Glickson, J.D. In vivo (1)H MRS of WSU-DLCL2 human non-Hodgkin’s lymphoma xenografts: Response to rituximab and rituximab plus CHOP. NMR Biomed. 2009, 22, 259–265. [Google Scholar] [CrossRef]
- Lee, S.C.; Poptani, H.; Pickup, S.; Jenkins, W.T.; Kim, S.; Koch, C.J.; Delikatny, E.J.; Glickson, J.D. Early detection of radiation therapy response in non-Hodgkin’s lymphoma xenografts by in vivo 1H magnetic resonance spectroscopy and imaging. NMR Biomed. 2010, 23, 624–632. [Google Scholar] [CrossRef]
- Garzon, J.G.; Bassa, P.; Moragas, M.; Soler, M.; Riera, E. Incidental diagnosis of diffuse large B-cell lymphoma by 11C-choline PET/CT in a patient with biochemical recurrence of prostate cancer. Clin. Nucl. Med. 2014, 39, 742–743. [Google Scholar] [CrossRef]
- Goineau, A.; Colombie, M.; Rousseau, C.; Sadot-Lebouvier, S.; Supiot, S. Incidental Detection of a Hodgkin Lymphoma on 18F-Choline PET/CT and Comparison With 18F-FDG in a Patient With Prostate Cancer. Clin. Nucl. Med. 2015, 40, 670–671. [Google Scholar] [CrossRef]
- van Waarde, A.; Elsinga, P.H. Proliferation markers for the differential diagnosis of tumor and inflammation. Curr. Pharm. Des. 2008, 14, 3326–3339. [Google Scholar] [CrossRef]
- Musharraf, S.G.; Siddiqui, A.J.; Shamsi, T.; Choudhary, M.I.; Rahman, A.U. Serum metabonomics of acute leukemia using nuclear magnetic resonance spectroscopy. Sci. Rep. 2016, 6, 30693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Wang, L.; Fei, X.C.; Jiang, X.F.; Zheng, Z.; Zhao, Y.; Wang, C.F.; Li, B.; Chen, S.J.; Janin, A.; et al. MYC is a positive regulator of choline metabolism and impedes mitophagy-dependent necroptosis in diffuse large B-cell lymphoma. Blood Cancer J. 2017, 7, e582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Li, Q.; Xiang, J.; Zhang, H.; Sun, H.; Ruan, G.; Tang, Y. NMR-based plasma metabolomics of adult B-cell acute lymphoblastic leukemia. Mol. Omics 2021, 17, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Du, J.; Zhang, H.; Ruan, G.; Xiang, J.; Wang, L.; Sun, H.; Guan, A.; Shen, G.; Liu, Y.; et al. Serum Metabolomics of Burkitt Lymphoma Mouse Models. PLoS ONE 2017, 12, e0170896. [Google Scholar] [CrossRef] [PubMed]
- Michel, V.; Yuan, Z.; Ramsubir, S.; Bakovic, M. Choline transport for phospholipid synthesis. Exp. Biol. Med. 2006, 231, 490–504. [Google Scholar] [CrossRef]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [Green Version]
- Brune, V.; Tiacci, E.; Pfeil, I.; Doring, C.; Eckerle, S.; van Noesel, C.J.; Klapper, W.; Falini, B.; von Heydebreck, A.; Metzler, D.; et al. Origin and pathogenesis of nodular lymphocyte-predominant Hodgkin lymphoma as revealed by global gene expression analysis. J. Exp. Med. 2008, 205, 2251–2268. [Google Scholar] [CrossRef]
- Eckerle, S.; Brune, V.; Doring, C.; Tiacci, E.; Bohle, V.; Sundstrom, C.; Kodet, R.; Paulli, M.; Falini, B.; Klapper, W.; et al. Gene expression profiling of isolated tumour cells from anaplastic large cell lymphomas: Insights into its cellular origin, pathogenesis and relation to Hodgkin lymphoma. Leukemia 2009, 23, 2129–2138. [Google Scholar] [CrossRef] [Green Version]
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.; Alizadeh, A.A.; Widhopf, G.; Simon, R.; Davis, R.E.; Yu, X.; Yang, L.; Pickeral, O.K.; Rassenti, L.Z.; Powell, J.; et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J. Exp. Med. 2001, 194, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Basso, K.; Margolin, A.A.; Stolovitzky, G.; Klein, U.; Dalla-Favera, R.; Califano, A. Reverse engineering of regulatory networks in human B cells. Nat. Genet. 2005, 37, 382–390. [Google Scholar] [CrossRef]
- Gokhale, S.; Lu, W.; Zhu, S.; Liu, Y.; Hart, R.P.; Rabinowitz, J.D.; Xie, P. Elevated Choline Kinase alpha-Mediated Choline Metabolism Supports the Prolonged Survival of TRAF3-Deficient B Lymphocytes. J. Immunol. 2020, 204, 459–471. [Google Scholar] [CrossRef]
- Xie, P. TRAF molecules in cell signaling and in human diseases. J. Mol. Signal. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Lalani, A.I.; Luo, C.; Han, Y.; Xie, P. TRAF3: A novel tumor suppressor gene in macrophages. Macrophage (Houst) 2015, 2, e1009. [Google Scholar]
- Lalani, A.I.; Zhu, S.; Gokhale, S.; Jin, J.; Xie, P. TRAF molecules in inflammation and inflammatory diseases. Curr. Pharmacol. Rep. 2018, 4, 64–90. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Jin, J.; Gokhale, S.; Lu, A.; Shan, H.; Feng, J.; Xie, P. Genetic Alterations of TRAF Proteins in Human Cancers. Front. Immunol. 2018, 9, 2111. [Google Scholar] [CrossRef]
- Bishop, G.A.; Xie, P. Multiple roles of TRAF3 signaling in lymphocyte function. Immunol. Res. 2007, 39, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Kraus, Z.J.; Stunz, L.L.; Bishop, G.A. Roles of TRAF molecules in B lymphocyte function. Cytokine Growth Factor Rev. 2008, 19, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.R.; Edwards, S.K.; Xie, P. Targeting TRAF3 Downstream Signaling Pathways in B cell Neoplasms. J. Cancer Sci. Ther. 2015, 7, 67–74. [Google Scholar]
- Xie, P.; Stunz, L.L.; Larison, K.D.; Yang, B.; Bishop, G.A. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity 2007, 27, 253–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardam, S.; Sierro, F.; Basten, A.; Mackay, F.; Brink, R. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity 2008, 28, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Moore, C.R.; Liu, Y.; Shao, C.S.; Covey, L.R.; Morse, H.C., 3rd; Xie, P. Specific deletion of TRAF3 in B lymphocytes leads to B lymphoma development in mice. Leukemia 2012, 26, 1122–1127. [Google Scholar] [CrossRef] [Green Version]
- De Silva, N.S.; Silva, K.; Anderson, M.M.; Bhagat, G.; Klein, U. Impairment of Mature B Cell Maintenance upon Combined Deletion of the Alternative NF-kappaB Transcription Factors RELB and NF-kappaB2 in B Cells. J. Immunol. 2016, 196, 2591–2601. [Google Scholar] [CrossRef] [Green Version]
- Heise, N.; De Silva, N.S.; Silva, K.; Carette, A.; Simonetti, G.; Pasparakis, M.; Klein, U. Germinal center B cell maintenance and differentiation are controlled by distinct NF-kappaB transcription factor subunits. J. Exp. Med. 2014, 211, 2103–2118. [Google Scholar] [CrossRef] [PubMed]
- Almaden, J.V.; Liu, Y.C.; Yang, E.; Otero, D.C.; Birnbaum, H.; Davis-Turak, J.; Asagiri, M.; David, M.; Goldrath, A.W.; Hoffmann, A. B-cell survival and development controlled by the coordination of NF-kappaB family members RelB and cRel. Blood 2016, 127, 1276–1286. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, C.; Ishidate, K.; Sugimoto, H.; Vance, D.E. Induction of choline kinase alpha by carbon tetrachloride (CCl4) occurs via increased binding of c-jun to an AP-1 element. Biochim. Biophys. Acta 2007, 1771, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Domizi, P.; Aoyama, C.; Banchio, C. Choline kinase alpha expression during RA-induced neuronal differentiation: Role of C/EBPbeta. Biochim. Biophys. Acta 2014, 1841, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Harris, R.A.; DeGrado, T.R. Choline phosphorylation and regulation of transcription of choline kinase alpha in hypoxia. J. Lipid. Res. 2012, 53, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.S.; Woods, S.M.; Ronen, S.M. Metabolic consequences of treatment with AKT inhibitor perifosine in breast cancer cells. NMR Biomed. 2012, 25, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Beloueche-Babari, M.; Casals Galobart, T.; Delgado-Goni, T.; Wantuch, S.; Parkes, H.G.; Tandy, D.; Harker, J.A.; Leach, M.O. Monocarboxylate transporter 1 blockade with AZD3965 inhibits lipid biosynthesis and increases tumour immune cell infiltration. Br. J. Cancer 2020, 122, 895–903. [Google Scholar] [CrossRef] [Green Version]
- Westin, J.R. Status of PI3K/Akt/mTOR pathway inhibitors in lymphoma. Clin. Lymphoma Myeloma Leuk. 2014, 14, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blachly, J.S.; Baiocchi, R.A. Targeting PI3-kinase (PI3K), AKT and mTOR axis in lymphoma. Br. J. Haematol. 2014, 167, 19–32. [Google Scholar] [CrossRef]
- Wullenkord, R.; Friedrichs, B.; Erdmann, T.; Lenz, G. Therapeutic potential of PI3K signaling in distinct entities of B-cell lymphoma. Expert Rev. Hematol. 2019, 12, 1053–1062. [Google Scholar] [CrossRef]
- Neri, A.; Knowles, D.M.; Greco, A.; McCormick, F.; Dalla-Favera, R. Analysis of RAS oncogene mutations in human lymphoid malignancies. Proc. Natl. Acad. Sci. USA 1988, 85, 9268–9272. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Yoo, N.J.; Soung, Y.H.; Kim, H.S.; Park, W.S.; Kim, S.Y.; Lee, J.H.; Park, J.Y.; Cho, Y.G.; Kim, C.J.; et al. BRAF mutations in non-Hodgkin’s lymphoma. Br. J. Cancer 2003, 89, 1958–1960. [Google Scholar] [CrossRef] [Green Version]
- Pera, B.; Krumsiek, J.; Assouline, S.E.; Marullo, R.; Patel, J.; Phillip, J.M.; Roman, L.; Mann, K.K.; Cerchietti, L. Metabolomic Profiling Reveals Cellular Reprogramming of B-Cell Lymphoma by a Lysine Deacetylase Inhibitor through the Choline Pathway. EBioMedicine 2018, 28, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelzaher, E.; Mostafa, M.F. Lysophosphatidylcholine acyltransferase 1 (LPCAT1) upregulation in breast carcinoma contributes to tumor progression and predicts early tumor recurrence. Tumour Biol. 2015, 36, 5473–5483. [Google Scholar] [CrossRef] [PubMed]
- Grupp, K.; Sanader, S.; Sirma, H.; Simon, R.; Koop, C.; Prien, K.; Hube-Magg, C.; Salomon, G.; Graefen, M.; Heinzer, H.; et al. High lysophosphatidylcholine acyltransferase 1 expression independently predicts high risk for biochemical recurrence in prostate cancers. Mol. Oncol. 2013, 7, 1001–1011. [Google Scholar] [CrossRef]
- Uehara, T.; Kikuchi, H.; Miyazaki, S.; Iino, I.; Setoguchi, T.; Hiramatsu, Y.; Ohta, M.; Kamiya, K.; Morita, Y.; Tanaka, H.; et al. Overexpression of Lysophosphatidylcholine Acyltransferase 1 and Concomitant Lipid Alterations in Gastric Cancer. Ann. Surg. Oncol. 2016, 23 (Suppl. 2), 206–213. [Google Scholar] [CrossRef] [Green Version]
- Endsley, M.P.; Thill, R.; Choudhry, I.; Williams, C.L.; Kajdacsy-Balla, A.; Campbell, W.B.; Nithipatikom, K. Expression and function of fatty acid amide hydrolase in prostate cancer. Int. J. Cancer 2008, 123, 1318–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.; Zheng, Y.; Jia, Y.; Li, P.; Wang, Y. Decreased LIPF expression is correlated with DGKA and predicts poor outcome of gastric cancer. Oncol. Rep. 2016, 36, 1852–1860. [Google Scholar] [CrossRef] [Green Version]
- Caprini, E.; Cristofoletti, C.; Arcelli, D.; Fadda, P.; Citterich, M.H.; Sampogna, F.; Magrelli, A.; Censi, F.; Torreri, P.; Frontani, M.; et al. Identification of key regions and genes important in the pathogenesis of sezary syndrome by combining genomic and expression microarrays. Cancer Res. 2009, 69, 8438–8446. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Bian, J.; Wang, L.; Zhou, J.Y.; Wang, Y.; Zhao, Y.; Wu, L.L.; Hu, J.J.; Li, B.; Chen, S.J.; et al. Dysregulated choline metabolism in T-cell lymphoma: Role of choline kinase-alpha and therapeutic targeting. Blood Cancer J. 2015, 5, 287. [Google Scholar] [CrossRef] [Green Version]
- Mariotto, E.; Bortolozzi, R.; Volpin, I.; Carta, D.; Serafin, V.; Accordi, B.; Basso, G.; Navarro, P.L.; Lopez-Cara, L.C.; Viola, G. EB-3D a novel choline kinase inhibitor induces deregulation of the AMPK-mTOR pathway and apoptosis in leukemia T-cells. Biochem. Pharmacol. 2018, 155, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Piccaluga, P.P.; Agostinelli, C.; Califano, A.; Rossi, M.; Basso, K.; Zupo, S.; Went, P.; Klein, U.; Zinzani, P.L.; Baccarani, M.; et al. Gene expression analysis of peripheral T cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets. J. Clin. Investig. 2007, 117, 823–834. [Google Scholar] [CrossRef]
- Choi, Y.L.; Tsukasaki, K.; O’Neill, M.C.; Yamada, Y.; Onimaru, Y.; Matsumoto, K.; Ohashi, J.; Yamashita, Y.; Tsutsumi, S.; Kaneda, R.; et al. A genomic analysis of adult T-cell leukemia. Oncogene 2007, 26, 1245–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Gonzalez, A.; Ramirez de Molina, A.; Fernandez, F.; Lacal, J.C. Choline kinase inhibition induces the increase in ceramides resulting in a highly specific and selective cytotoxic antitumoral strategy as a potential mechanism of action. Oncogene 2004, 23, 8247–8259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Wang, S.; Chen, J.; Yu, Z. Histone Deacetylases (HDACs) Guided Novel Therapies for T-cell lymphomas. Int. J. Med. Sci. 2019, 16, 424–442. [Google Scholar] [CrossRef] [Green Version]
- Fernandis, A.Z.; Wenk, M.R. Membrane lipids as signaling molecules. Curr. Opin. Lipidol. 2007, 18, 121–128. [Google Scholar] [CrossRef]
- Sunshine, H.; Iruela-Arispe, M.L. Membrane lipids and cell signaling. Curr. Opin. Lipidol. 2017, 28, 408–413. [Google Scholar] [CrossRef]
- Mori, N.; Wildes, F.; Kakkad, S.; Jacob, D.; Solaiyappan, M.; Glunde, K.; Bhujwalla, Z.M. Choline kinase-alpha protein and phosphatidylcholine but not phosphocholine are required for breast cancer cell survival. NMR Biomed. 2015, 28, 1697–1706. [Google Scholar] [CrossRef]
- Krishna, S.; Zhong, X.P. Regulation of Lipid Signaling by Diacylglycerol Kinases during T Cell Development and Function. Front. Immunol. 2013, 4, 178. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gokhale, S.; Xie, P. ChoK-Full of Potential: Choline Kinase in B Cell and T Cell Malignancies. Pharmaceutics 2021, 13, 911. https://doi.org/10.3390/pharmaceutics13060911
Gokhale S, Xie P. ChoK-Full of Potential: Choline Kinase in B Cell and T Cell Malignancies. Pharmaceutics. 2021; 13(6):911. https://doi.org/10.3390/pharmaceutics13060911
Chicago/Turabian StyleGokhale, Samantha, and Ping Xie. 2021. "ChoK-Full of Potential: Choline Kinase in B Cell and T Cell Malignancies" Pharmaceutics 13, no. 6: 911. https://doi.org/10.3390/pharmaceutics13060911