
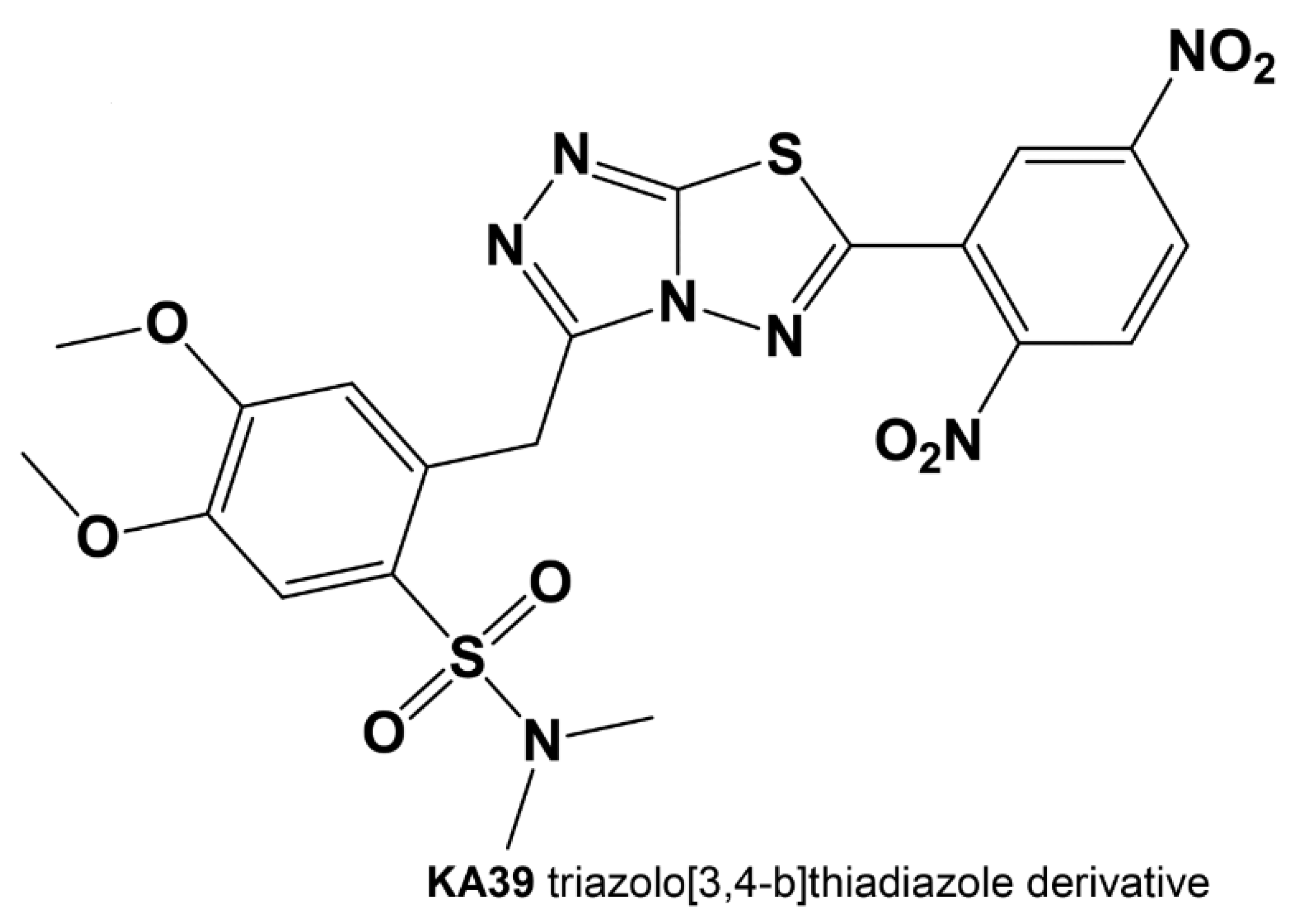
Effects of a Novel Thiadiazole Derivative with High Anticancer Activity on Cancer Cell Immunogenic Markers: Mismatch Repair System, PD-L1 Expression, and Tumor Mutation Burden

 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Cell Treatment
2.3. In Vitro Antiproliferative Activity
2.4. Flow Cytometric Analysis of Surface PD-L1 Expression
2.5. DNA Extraction
2.6. MSI Fragment Analysis
2.7. Tumor Mutation Burden Assay
2.8. Statistical Analysis
3. Results
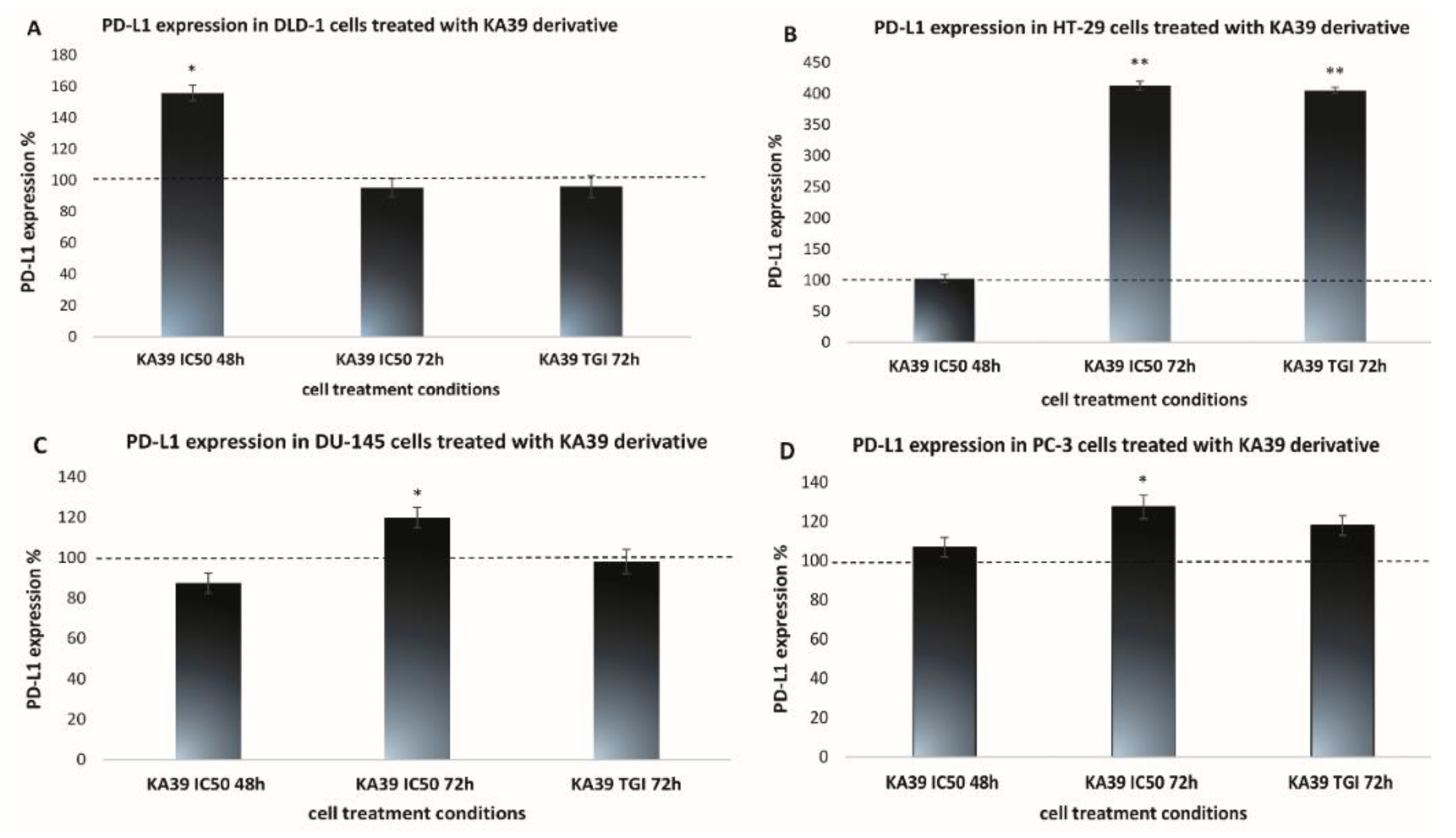
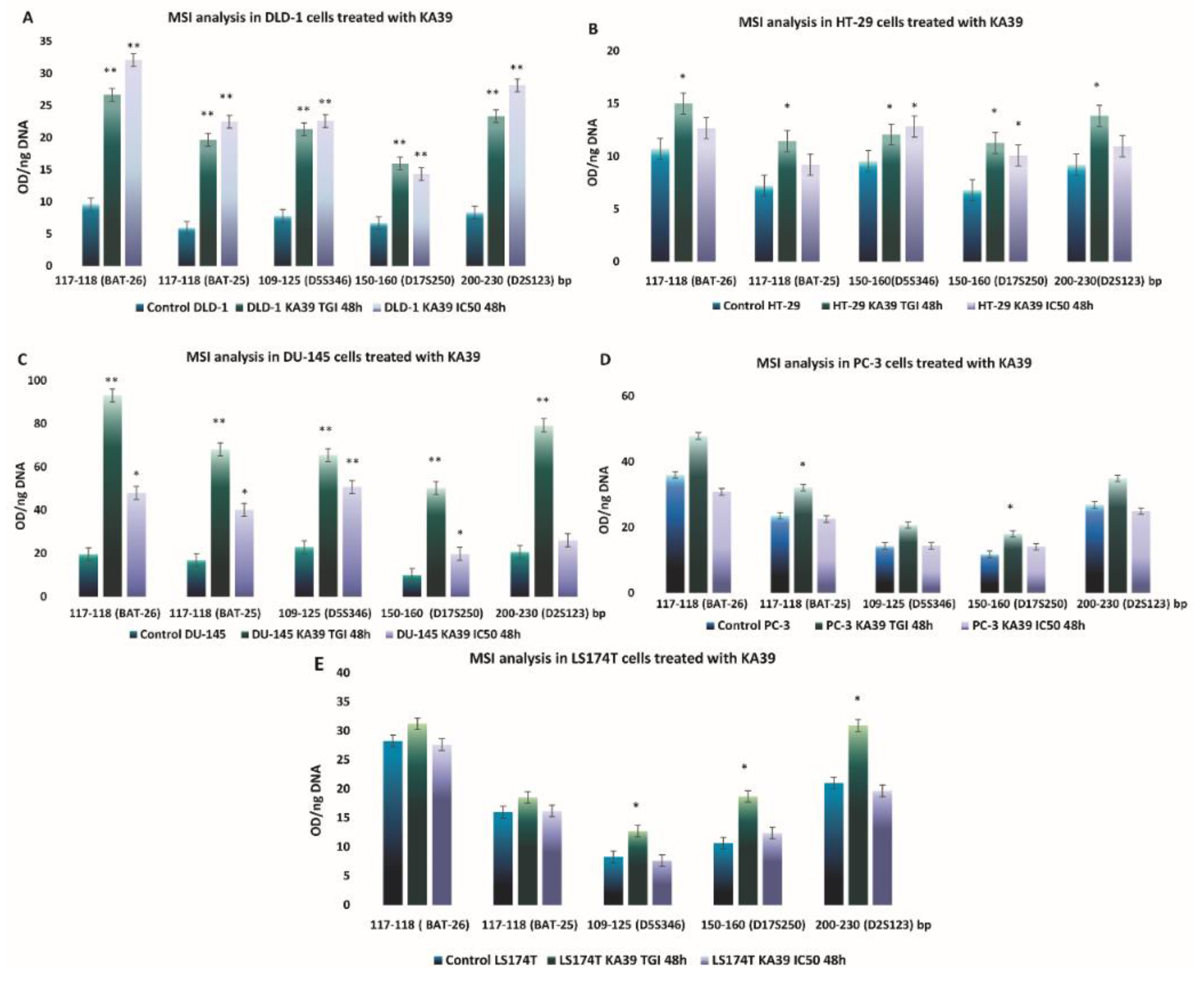
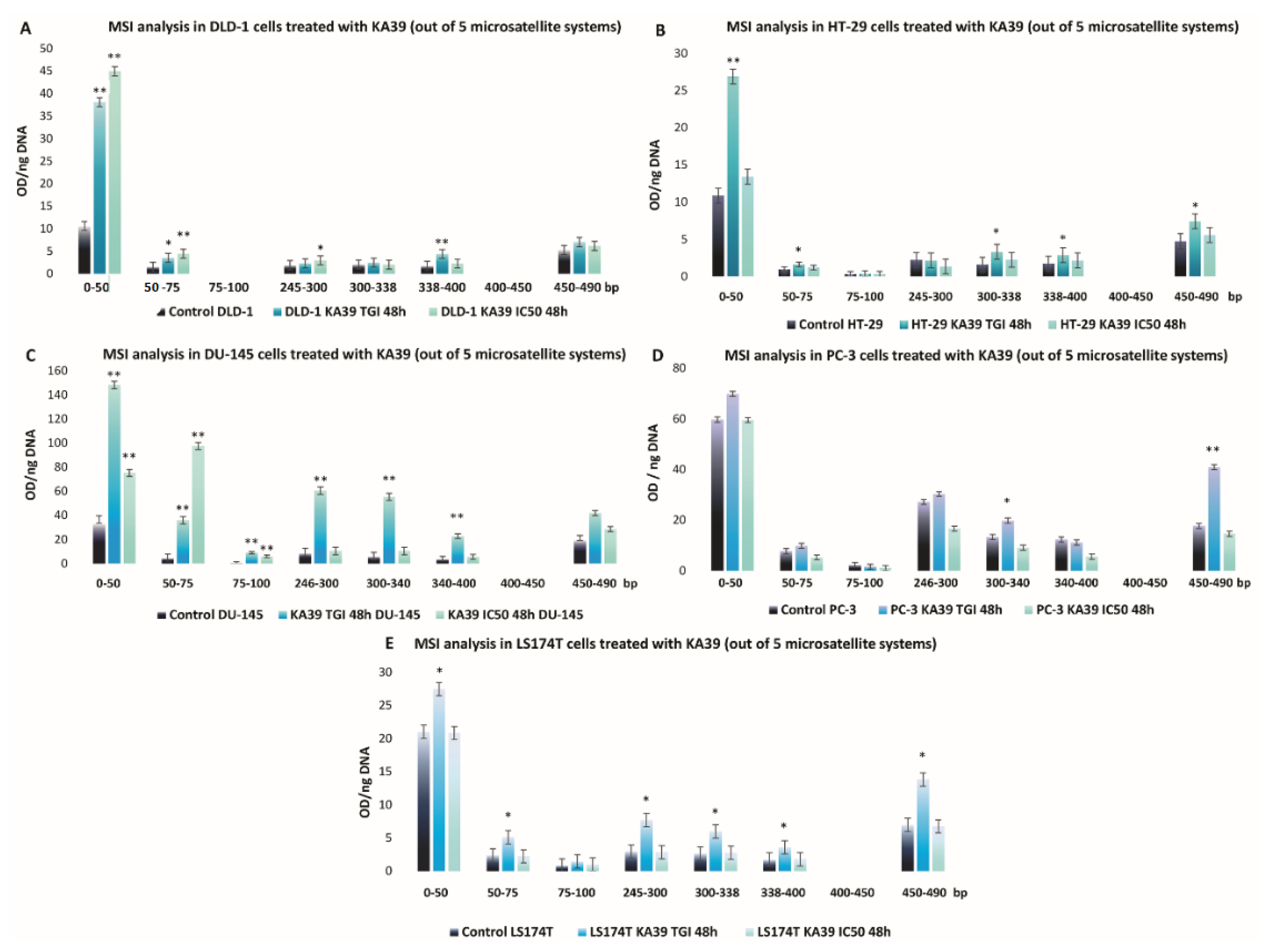
MSI, PD-L1 Expression, and TMB Analysis in Human Colorectal and Prostate Cancer Cells upon Treatment with KA39
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gomha, S.M.; Abdel-Aziz, H.M.; Khalil, K.D. Synthesis and SAR Study of the Novel Thiadiazole–Imidazole Derivatives as a New Anticancer Agents. Chem. Pharm. Bull. 2016, 64, 1356–1363. [Google Scholar] [CrossRef] [Green Version]
- Gomha, S.M.; Riyadh, S.M. Synthesis under Microwave Irradiation of [1,2,4]Triazolo[3,4-b][1,3,4]thiadiazoles and Other Diazoles Bearing Indole Moieties and Their Antimicrobial Evaluation. Molecules 2011, 16, 8244–8256. [Google Scholar] [CrossRef]
- Cirrincione, G.; Passannanti, A.; Diana, P.; Barraja, P.; Mingoia, F.; Lauria, A. Pyrrolo[2,3-d][1,2,3]triazoles as Potential Antineoplastic Agents. Heterocycles 1998, 48, 1229. [Google Scholar] [CrossRef]
- Charitos, G.; Trafalis, D.T.; Dalezis, P.; Potamitis, C.; Sarli, V.; Zoumpoulakis, P.; Camoutsis, C. Synthesis and anticancer activity of novel 3,6-disubstituted 1,2,4-triazolo-[3,4-b]-1,3,4-thiadiazole derivatives. Arab. J. Chem. 2019, 12, 4784–4794. [Google Scholar] [CrossRef] [Green Version]
- Sagredou, S.; Dalezis, P.; Nikoleousakos, N.; Nikolaou, M.; Voura, M.; Almpanakis, K.; I Panayiotidis, M.; Sarli, V.; Trafalis, D.T. 3,6-Disubstituted 1,2,4-Triazolo[3,4-b]Thiadiazoles with Anticancer Activity Targeting Topoisomerase II Alpha. OncoTargets Ther. 2020, 13, 7369–7386. [Google Scholar] [CrossRef]
- Trafalis, D.; Sagredou, S.; Dalezis, P.; Voura, M.; Fountoulaki, S.; Nikoleousakos, N.; Almpanakis, K.; Deligiorgi, M.; Sarli, V. Anticancer Activity of Triazolo-Thiadiazole Derivatives and Inhibition of AKT1 and AKT2 Activation. Pharmaceutics 2021, 13, 493. [Google Scholar] [CrossRef]
- Nowicki, T.; Hu-Lieskovan, S.; Ribas, A. Mechanisms of Resistance to PD-1 and PD-L1 Blockade. Cancer J. 2018, 24, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Jiao, D.; Xu, H.; Liu, Q.; Zhao, W.; Han, X.; Wu, K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol. Cancer 2018, 17, 129. [Google Scholar] [CrossRef]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.-Y.; Andre, F.; et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: A systematic review-based approach. Ann. Oncol. 2019, 30, 1232–1243. [Google Scholar] [CrossRef] [Green Version]
- Berg, K.C.G.; Eide, P.W.; Eilertsen, I.A.; Johannessen, B.; Bruun, J.; Danielsen, S.A.; Bjørnslett, M.; Meza-Zepeda, L.A.; Eknæs, M.; Lind, G.E.; et al. Multi-omics of 34 colorectal cancer cell lines—A resource for biomedical studies. Mol. Cancer 2017, 16, 116. [Google Scholar] [CrossRef]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknæs, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Fogh, J.; Trempe, G. New Human Tumor Cell Lines. In Human Tumor Cells In Vitro; Fogh, J., Ed.; Springer Science and Business Media LLC: Berlin, Germany, 1975; pp. 115–159. [Google Scholar]
- Laghi, L.; Bianchi, P.; Delconte, G.; Celesti, G.; Di Caro, G.; Pedroni, M.; Chiaravalli, A.M.; Jung, B.; Capella, C.; de Leon, M.P.; et al. MSH3 Protein Expression and Nodal Status in MLH1-Deficient Colorectal Cancers. Clin. Cancer Res. 2012, 18, 3142–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodo, S.; Colas, C.; Buhard, O.; Collura, A.; Tinat, J.; Lavoine, N.; Guilloux, A.; Chalastanis, A.; Lafitte, P.; Coulet, F.; et al. Diagnosis of Constitutional Mismatch Repair-Deficiency Syndrome Based on Microsatellite Instability and Lymphocyte Tolerance to Methylating Agents. Gastroenterology 2015, 149, 1017–1029.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, J.; Fraig, M.M.; Metcalf, J.; Turner, W.R.; Bissada, N.K.; Watson, D.K.; Schweinfest, C.W. Defects of DNA mismatch repair in human prostate cancer. Cancer Res. 2001, 61, 4112–4121. [Google Scholar] [PubMed]
- Fukuhara, S.; Chang, I.; Mitsui, Y.; Chiyomaru, T.; Yamamura, S.; Majid, S.; Saini, S.; Deng, G.; Gill, A.; Wong, D.K.; et al. Functional role of DNA mismatch repair gene PMS2 in prostate cancer cells. Oncotarget 2015, 6, 16341–16351. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, S.; Chang, I.; Mitsui, Y.; Chiyomaru, T.; Yamamura, S.; Majid, S.; Saini, S.; Hirata, H.; Deng, G.; Gill, A.; et al. DNA mismatch repair gene MLH1 induces apoptosis in prostate cancer cells. Oncotarget 2014, 5, 11297–11307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chetram, M.A.; Odero-Marah, V.; Hinton, C.V. Loss of PTEN Permits CXCR4-Mediated Tumorigenesis through ERK1/2 in Prostate Cancer Cells. Mol. Cancer Res. 2011, 9, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Vlietstra, R.J.; Van Alewijk, D.C.; Hermans, K.G.; Van Steenbrugge, G.J.; Trapman, J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998, 58, 2720–2723. [Google Scholar]
- Lotan, T.; Gurel, B.; Sutcliffe, S.; Esopi, D.; Liu, W.; Xu, J.; Hicks, J.L.; Park, B.H.; Humphreys, E.; Partin, A.W.; et al. PTEN Protein Loss by Immunostaining: Analytic Validation and Prognostic Indicator for a High Risk Surgical Cohort of Prostate Cancer Patients. Clin. Cancer Res. 2011, 17, 6563–6573. [Google Scholar] [CrossRef] [Green Version]
- Elkamhawy, A.; Park, J.-E.; Cho, N.-C.; Sim, T.; Pae, A.N.; Roh, E.J. Discovery of a broad spectrum antiproliferative agent with selectivity for DDR1 kinase: Cell line-based assay, kinase panel, molecular docking, and toxicity studies. J. Enzym. Inhib. Med. Chem. 2015, 31, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paull, K.D.; Shoemaker, R.H.; Hodes, L.; Monks, A.; Scudiero, D.A.; Rubinstein, L.; Plowman, J.; Boyd, M.R. Display and Analysis of Patterns of Differential Activity of Drugs Against Human Tumor Cell Lines: Development of Mean Graph and COMPARE Algorithm. J. Natl. Cancer Inst. 1989, 81, 1088–1092. [Google Scholar] [CrossRef]
- Dietmaier, W.; Wallinger, S.; Bocker, T.; Kullmann, F.; Fishel, R.; Rüschoff, J. Diagnostic microsatellite instability: Definition and correlation with mismatch repair protein expression. Cancer Res. 1997, 57, 4749–4756. [Google Scholar]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar] [PubMed]
- Constantinidou, A.; Alifieris, K.; Trafalis, D.T. Targeting Programmed Cell Death-1 (PD-1) and Ligand (PD-L1): A new era in cancer active immunotherapy. Pharmacol. Ther. 2019, 194, 84–106. [Google Scholar] [CrossRef]
- Gilad, Y.; Eliaz, Y.; Yu, Y.; Han, S.J.; O’Malley, B.W.; Lonard, D.M. Author Correction: Drug-induced PD-L1 expression and cell stress response in breast cancer cells can be balanced by drug combination. Sci. Rep. 2020, 10, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Su, D.-M.; Liang, M.; Fu, J. Chemopreventive agents induce programmed death-1-ligand 1 (PD-L1) surface expression in breast cancer cells and promote PD-L1-mediated T cell apoptosis. Mol. Immunol. 2008, 45, 1470–1476. [Google Scholar] [CrossRef]
- Qin, X.; Liu, C.; Zhou, Y.; Wang, G. Cisplatin induces programmed death-1-ligand 1(PD-L1) over-expression in hepatoma H22 cells via Erk/MAPK signaling pathway. Cell. Mol. Biol. 2010, 56, OL1366–OL1372. [Google Scholar]
- Doi, T.; Ishikawa, T.; Okayama, T.; Oka, K.; Mizushima, K.; Yasuda, T.; Sakamoto, N.; Katada, K.; Uchiyama, K.; Handa, O.; et al. The JAK/STAT pathway is involved in the upregulation of PD-L1 expression in pancreatic cancer cell lines. Oncol. Rep. 2017, 37, 1545–1554. [Google Scholar] [CrossRef] [Green Version]
- Crane, C.A.; Panner, A.; Murray, J.C.; Wilson, S.P.; Xu, H.; Chen, L.; Simko, J.P.; Waldman, F.M.; Pieper, R.O.; Parsa, A.T. PI(3) kinase is associated with a mechanism of immunoresistance in breast and prostate cancer. Oncogene 2008, 28, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]
- Sato, H.; Jeggo, P.A.; Shibata, A. Regulation of programmed death-ligand 1 expression in response to DNA damage in cancer cells: Implications for precision medicine. Cancer Sci. 2019, 110, 3415–3423. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, J.; Hu, J.; Zhang, H.; Xu, F.; He, W.; Wang, X.; Li, M.; Lu, W.; Zeng, G.; et al. cGAS/STING axis mediates a topoisomerase II inhibitor–induced tumor immunogenicity. J. Clin. Investig. 2019, 129, 4850–4862. [Google Scholar] [CrossRef] [PubMed]
- Grabosch, S.; Bulatović, M.; Zeng, F.; Ma, T.; Zhang, L.; Ross, M.; Brozick, J.; Fang, Y.; Tseng, G.; Kim, E.; et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene 2019, 38, 2380–2393. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, T.; Mimura, K.; Okayama, H.; Nakayama, Y.; Saito, K.; Yamada, L.; Endo, E.; Sakamoto, W.; Fujita, S.; Endo, H.; et al. A subset of patients with MSS/MSI-low-colorectal cancer showed increased CD8(+) TILs together with up-regulated IFN-γ. Oncol. Lett. 2019, 18, 5977–5985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Li, H.-R.; Thompson, H.J.; Zhu, Z.; Jiang, W. Hypersensitivity of Tumor Cell Lines with Microsatellite Instability to DNA Double Strand Break Producing Chemotherapeutic Agent Bleomycin. Cancer Res. 2004, 64, 4760–4767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S.; et al. Programmed Cell Death 1 (PD-1) and Its Ligand (PD-L1) in Common Cancers and Their Correlation with Molecular Cancer Type. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2965–2970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Mills, A.M.; Dill, E.A.; Moskaluk, C.A.; Dziegielewski, J.; Bullock, T.N.; Dillon, P.M. The Relationship between Mismatch Repair Deficiency and PD-L1 Expression in Breast Carcinoma. Am. J. Surg. Pathol. 2018, 42, 183–191. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef]
- Gradia, S.; Subramanian, D.; Wilson, T.; Acharya, S.; Makhov, A.; Griffith, J.; Fishel, R. hMSH2–hMSH6 Forms a Hydrolysis-Independent Sliding Clamp on Mismatched DNA. Mol. Cell 1999, 3, 255–261. [Google Scholar] [CrossRef]
- Martin, S.A.; Lord, C.J.; Ashworth, A. Therapeutic Targeting of the DNA Mismatch Repair Pathway. Clin. Cancer Res. 2010, 16, 5107–5113. [Google Scholar] [CrossRef] [Green Version]
- Iyer, R.R.; Pluciennik, A.; Burdett, V.; Modrich, P.L. DNA Mismatch Repair: Functions and Mechanisms. Chem. Rev. 2006, 106, 302–323. [Google Scholar] [CrossRef]
- Boland, C.R.; Koi, M.; Chang, D.K.; Carethers, J.M. The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch Syndrome: From bench to bedside. Fam. Cancer 2007, 7, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.C.; Lee, C.; Dahiya, R. DNA mismatch repair enzyme activity and gene expression in prostate cancer. Biochem. Biophys. Res. Commun. 2001, 285, 409–413. [Google Scholar] [CrossRef]
- Di Pietro, M.; Bellver, J.S.; Menigatti, M.; Bannwart, F.; Schnider, A.; Russell, A.; Truninger, K.; Jiricny, J.; Marra, G. Defective DNA Mismatch Repair Determines a Characteristic Transcriptional Profile in Proximal Colon Cancers. Gastroenterology 2005, 129, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Aebi, S.; Fink, D.; Gordon, R.; Kim, H.K.; Zheng, H.; Fink, J.L.; Howell, S.B. Resistance to cytotoxic drugs in DNA mismatch repair-deficient cells. Clin. Cancer Res. 1997, 3, 1763–1767. [Google Scholar] [PubMed]
- Irving, J.A.; Hall, A.G. Mismatch repair defects as a cause of resistance to cytotoxic drugs. Expert Rev. Anticancer Ther. 2001, 1, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Alas, M.M.D.L.; Aebi, S.; Fink, D.; Howell, S.B.; Los, G. Loss of DNA Mismatch Repair: Effects on the Rate of Mutation to Drug Resistance. J. Natl. Cancer Inst. 1997, 89, 1537–1541. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, V.A.; Walt, H.; Carpini, R.D.; Haller, U.; Fink, D. Resistance to topoisomerase poisons due to loss of DNA mismatch repair. Int. J. Cancer 2001, 93, 571–576. [Google Scholar] [CrossRef]
- Liu, Q.; Turner, K.M.; Yung, W.K.A.; Chen, K.; Zhang, W. Role of AKT signaling in DNA repair and clinical response to cancer therapy. Neuro-Oncology 2014, 16, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Zhang, Y.; Cai, J.; Wang, J.; Ding, H.; Zhou, J.; Fang, F.; Wang, Z. A novel function of protein kinase B as an inducer of the mismatch repair gene hPMS2 degradation. Cell. Signal. 2013, 25, 1498–1504. [Google Scholar] [CrossRef] [PubMed]
- Fumet, J.-D.; Truntzer, C.; Yarchoan, M.; Ghiringhelli, F. Tumour mutational burden as a biomarker for immunotherapy: Current data and emerging concepts. Eur. J. Cancer 2020, 131, 40–50. [Google Scholar] [CrossRef]
- Krieger, T.; Pearson, I.; Bell, J.; Doherty, J.; Robbins, P. Targeted literature review on use of tumor mutational burden status and programmed cell death ligand 1 expression to predict outcomes of checkpoint inhibitor treatment. Diagn. Pathol. 2020, 15, 6. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Cell Lines | GI50 (μΜ) | TGI (μΜ) | IC50 (μΜ) |
|---|---|---|---|
| DLD-1 | 3 ± 0.52 | 5 ± 0.76 | 9 ± 0.76 |
| HT-29 | 11.5 ± 0.8 | 15.9 ± 0.55 | 19.5 ± 0.9 |
| LS174T | 12 ± 1.52 | 16.5 ± 1.25 | 21.5 ± 1.5 |
| PC-3 | 5 ± 0.15 | 8.4 ± 0.1 | 12 ± 0.1 |
| DU-145 | 5.8 ± 0.2 | 8 ± 0.4 | 10.3 ± 1.8 |
| Cancer Type | Human Cell Line Designation | MSI Status | MMR Deficiency | PTEN | References |
|---|---|---|---|---|---|
| Colorectal adenocarcinoma, Dukes’ type C | DLD-1 | MSI-H | MSH6 deficiency | Wild type | [10,11,12] |
| Colorectal adenocarcinoma | HT-29 | MSS | - | Wild type | [10,11,13] |
| Colorectal adenocarcinoma, Dukes’ type B | LS174T | MSI-H | MLH1 deficiency | Wild type | [10,11,14,15] |
| Prostate carcinoma | DU-145 | MSI-H | PMS2 and MLH1 deficiency | Wild type | [14,16,17,18,19] |
| Prostate adenocarcinoma, grade IV | PC-3 | MSS | - | PTEN deficiency (homologous deletion) | [16,20,21] |
| Repeat Type | Chromosomal Location | Repeat Motif | Primer Sequence (5′→3′) | Size (bp) |
|---|---|---|---|---|
| Mononucleotide | ||||
| BAT25 | 4q12 | TTTT.T.TTTT.(T)7.A(T)25 | TCGCCTCCAAGAATGTAAGT | ~90 |
| TCTGCATTTTAACTATGGCTC | ||||
| BAT26 | 2p | (T)5…..(A)26 | TGACTACTTTTGACTTCAGCC | ~80–100 |
| AACCATTCAACATTTTTAACCC | ||||
| Dinucleotide (non-complex) | ||||
| D5S346 (APC) | 5q21/22 | (CA)26 | ACTCACTCTAGTGATAAATCG | 96–122 |
| AGCAGATAAGACAGTATTACTAGTT | ||||
| Dinucleotide (complex) | ||||
| D17S250 (Mfd15CA) | 17q11.2-q12 | (TA)7………………(CA)24 | GGAAGAATCAAATAGACAAT | ~150 |
| GCTGGCCATATATATATTTAAACC | ||||
| D2S123 (AFM093xh3) | 2p16 | (CA)13TA(CA)15(T/GA)7 | AAACAGGATGCCTGCCTTTA | 197–227 |
| GGACTTTCCACCTATGGGAC |
| ABL2 | CD79A | EPHB1 | GRM8 | LIFR | MYH9 | PMS1 | SOX2 | WAS | GNAS | ATRX | TSC2 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ACVR2A | CD79B | EPHB4 | GUCY1A2 | LPHN3 | NCOA1 | POT1 | SSX1 | WHSC1 | HFN1A | BAP1 | WT1 |
| ADAMTS20 | CDC73 | EPHB6 | HCAR1 | LPP | NCOA2 | POU5F1 | STK36 | WRN | HRAS | CDK12 | |
| AFF1 | CDH1 | ERCC1 | HIF1A | LRP1B | NCOA4 | PPARG | SUFU | XPA | IDH1 | CDKN2A | |
| AFF3 | CDH11 | ERCC3 | HLF | LTF | NFKB1 | PPP2R1A | SYK | XPC | IDH2 | CDKN2B | |
| AKAP9 | CDH2 | ERCC4 | HOOK3 | LTK | NFKB2 | PRDM1 | SYNE1 | XPO1 | JAK2 | CEBPA | |
| APC | CDH20 | ERCC5 | HSP90AA1 | MAF | NIN | PRKAR1A | TAF1 | XRCC2 | KOR | CHEK1 | |
| ARID2 | CDH5 | ERG | HSP90AB1 | MAFB | NKX2-1 | PRKDC | TAF1L | ZNF384 | KIT | CHEK2 | |
| ARNT | CDK8 | ETS1 | ICK | MAGEA1 | NLRP1 | PSIP1 | TAL1 | ZNF521 | KRAS | CREBBP | |
| ATF1 | CDKN2C | ETV1 | IGF1R | MAGl1 | NOTCH4 | PTGS2 | TBX22 | ABL1 | MAP2K1 | DNMT3A | |
| AURKA | CIC | ETV4 | IGF2 | MALT1 | NSD1 | PTPRD | TCF12 | AKT1 | MAP2K2 | FANCA | |
| AURKB | CKS1B | EXT1 | IGF2R | MAML2 | NUMA1 | PTPRT | TCF3 | AKT2 | MAP2K4 | FANCD2 | |
| AURKC | CMPK1 | EXT2 | IKBKB | MAP3K7 | NUP214 | RALGDS | TCF7L1 | AKT3 | MAPK1 | FBXW7 | |
| BAI3 COL | COL1A1 | FAM123B | IKBKE | MAPK8 | NUP98 | RARA | TCF7L2 | ALK | MET | MLH1 | |
| BCL10 | CRBN | FANCC | IKZF1 | MARK1 | PAK3 | RECQL4 | TCL1A | AR | MPL | MSH2 | |
| BCL11A | CREB1 | FANCF | IL2 | MARK4 | PARP1 | REL | TET1 | AXL | MTOR | MSH6 | |
| BCL11B | CRKL | FANCG | IL21R | MBD1 | PAX3 | RHOH | TFE3 | BRAF | MYC | NBN | |
| BCL2 | CRTC1 | FANCJ | IL6ST | MCL1 | PAX5 | RNASEL | TGFBR2 | CBL | MYCN | NF1 | |
| BCL2L1 | CSMD3 | FAS | IL7R | MDM2 | PAX7 | RNF2 | TGM7 | CCND1 | NFE2L2 | NF2 | |
| BCL2L2 | CTNNA1 | FH | ING4 | MDM4 | PAX8 | RNF213 | THBS1 | CDK4 | NRAS | NOTCH1 | |
| BCL3 | CTNNB1 | FLCN | IRF4 | MEN1 | PBRM1 | RPS6KA2 | TIMP3 | CDK6 | NTRK1 | NOTCH2 | |
| BCL6 | CYLD | FLl1 | IRS2 | MITF | PBX1 | RRM1 | TLR4 | CSF1R | NTRK3 | NPM1 | |
| BCL9 | CYP2C19 | FLT1 | ITGA10 | MLL | PDE4DIP | RUNX1T1 | TLX1 | DDR2 | PDGFRA | PALB2 | |
| BCR | CYP2D6 | FLT4 | ITGA9 | MLL2 | PDGFB | SAMD9 | TNFAIP3 | EGFR | PDGFRB | PIK3R1 | |
| BIRC2 | DAXX | FN1 | ITGB2 | MLL3 | PER1 | SBDS | TNFRSF14 | ERBB2 | PIK3CA | PMS2 | |
| BIRC3 | DCC | FOXL2 | ITGB3 | MLLT10 | PGAP3 | SDHA | TNK2 | ERBB3 | PIK3CB | PTCH1 | |
| BIRC5 | DDB2 | FOXO1 | JAK1 | MMP2 | PHOX2B | SDHB | TOP1 | ERBB4 | PTPN11 | PTEN | |
| BLM | DDIT3 | FOXO3 | JAK3 | MN1 | PIK3C2B | SDHC | TPR | ERCC2 | RAF1 | RADSO | |
| BLNK | DEK | FOXP1 | JUN | MRE11A | PIK3CD | SOHD | TRIM24 | ESR1 | RET | RB1 | |
| BMPR1A | DICER1 | FOXP4 | KAT6A | MTR | PIK3CG | SEPT9 | TRIM33 | EZH2 | ROS1 | RUNX1 | |
| BRD3 | DPYD | FZR1 | KAT6B | MTRR | PIK3R2 | SGK1 | TRIP11 | FGFR1 | SF3B1 | SETD2 | |
| BTK | DST | G6PD | KDM5C | MUC1 | PIM1 | SH2D1A | TRRAP | FGFR2 | SMO | SMARCA4 | |
| BUB1B | EML4 | GATA1 | KDM6A | MUTYH | PKHD1 | SMAD2 | TSHR | FGFR3 | SRC | SMARCB1 | |
| CARD11 | EP300 | GATA2 | KEAP1 | MYB | PLAG1 | SMAD4 | UBR5 | FGFR4 | ARID1A | STK11 | |
| CASC5 | EP400 | GATA3 | KLF6 | MYCL1 | PLCG1 | SMUG1 | UGT1A1 | FLT3 | ASXL1 | TET2 | |
| CCND2 | EPHA3 | GDNF | LAMP1 | MYD88 | PLEKHG5 | SOCS1 | USP9X | GNA11 | ATM | TP53 | |
| CCNE1 | EPHA7 | GPR124 | LCK | MYH11 | PML | SOX11 | VHL | GNAQ | ATR | TSC1 |
| Cancer Cell Lines | PD-L1 Expression | ||||
|---|---|---|---|---|---|
| Control 48 h | KA39 IC50 48 h | KA39 IC50 72 h | Control 72 h | KA39 TGI 72 h | |
| DLD-1 | 12.92 ± 0.64 | 20.14 ± 1.0 | 12.3 ± 0.61 | 23.19 ± 1.39 | 22.27 ± 1.11 |
| HT-29 | 19.56 ± 1.36 | 20.13 ± 1.61 | 80.82 ± 4.04 | 19.4 ± 1.35 | 78.56 ± 5.49 |
| DU-145 | 27.99 ± 1.67 | 24.48 ± 2.2 | 33.55 ± 2.34 | 23.46 ± 1.17 | 23.02 ± 1.61 |
| PC-3 | 25.91 ± 1.81 | 27.66 ± 2.7 | 32.98 ± 3.2 | 23.93 ± 1.91 | 28.21 ± 2.53 |
| Cancer Cell Lines | Tumor Mutation Burden (TMB) | |||
|---|---|---|---|---|
| Control 48 h | KA39 IC50 (μΜ) 48 h | |||
| Non-Synonymous Mutations | Synonymous Mutations | Non-Synonymous Mutations | Synonymous Mutations | |
| DLD-1 | 204.17 Muts/Mb | 31.18 Muts/Mb | 198.28 Muts/Mb | 80.93 Muts/Mb |
| HT-29 | 10.05 Muts/Mb | 1.67 Muts/Mb | 10.01 Muts/Mb | 1.67 Muts/Mb |
| LS174T | 67.48 Muts/Mb | 3.48 Muts/Mb | 68.06 Muts/Mb | 3.62 Muts/Mb |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sagredou, S.; Dalezis, P.; Papadopoulou, E.; Voura, M.; Deligiorgi, M.V.; Nikolaou, M.; Panayiotidis, M.I.; Nasioulas, G.; Sarli, V.; Trafalis, D.T. Effects of a Novel Thiadiazole Derivative with High Anticancer Activity on Cancer Cell Immunogenic Markers: Mismatch Repair System, PD-L1 Expression, and Tumor Mutation Burden. Pharmaceutics 2021, 13, 885. https://doi.org/10.3390/pharmaceutics13060885
Sagredou S, Dalezis P, Papadopoulou E, Voura M, Deligiorgi MV, Nikolaou M, Panayiotidis MI, Nasioulas G, Sarli V, Trafalis DT. Effects of a Novel Thiadiazole Derivative with High Anticancer Activity on Cancer Cell Immunogenic Markers: Mismatch Repair System, PD-L1 Expression, and Tumor Mutation Burden. Pharmaceutics. 2021; 13(6):885. https://doi.org/10.3390/pharmaceutics13060885
Chicago/Turabian StyleSagredou, Sofia, Panagiotis Dalezis, Eirini Papadopoulou, Maria Voura, Maria V. Deligiorgi, Michail Nikolaou, Mihalis I. Panayiotidis, George Nasioulas, Vasiliki Sarli, and Dimitrios T. Trafalis. 2021. "Effects of a Novel Thiadiazole Derivative with High Anticancer Activity on Cancer Cell Immunogenic Markers: Mismatch Repair System, PD-L1 Expression, and Tumor Mutation Burden" Pharmaceutics 13, no. 6: 885. https://doi.org/10.3390/pharmaceutics13060885