
Choline Kinase: An Unexpected Journey for a Precision Medicine Strategy in Human Diseases


Abstract
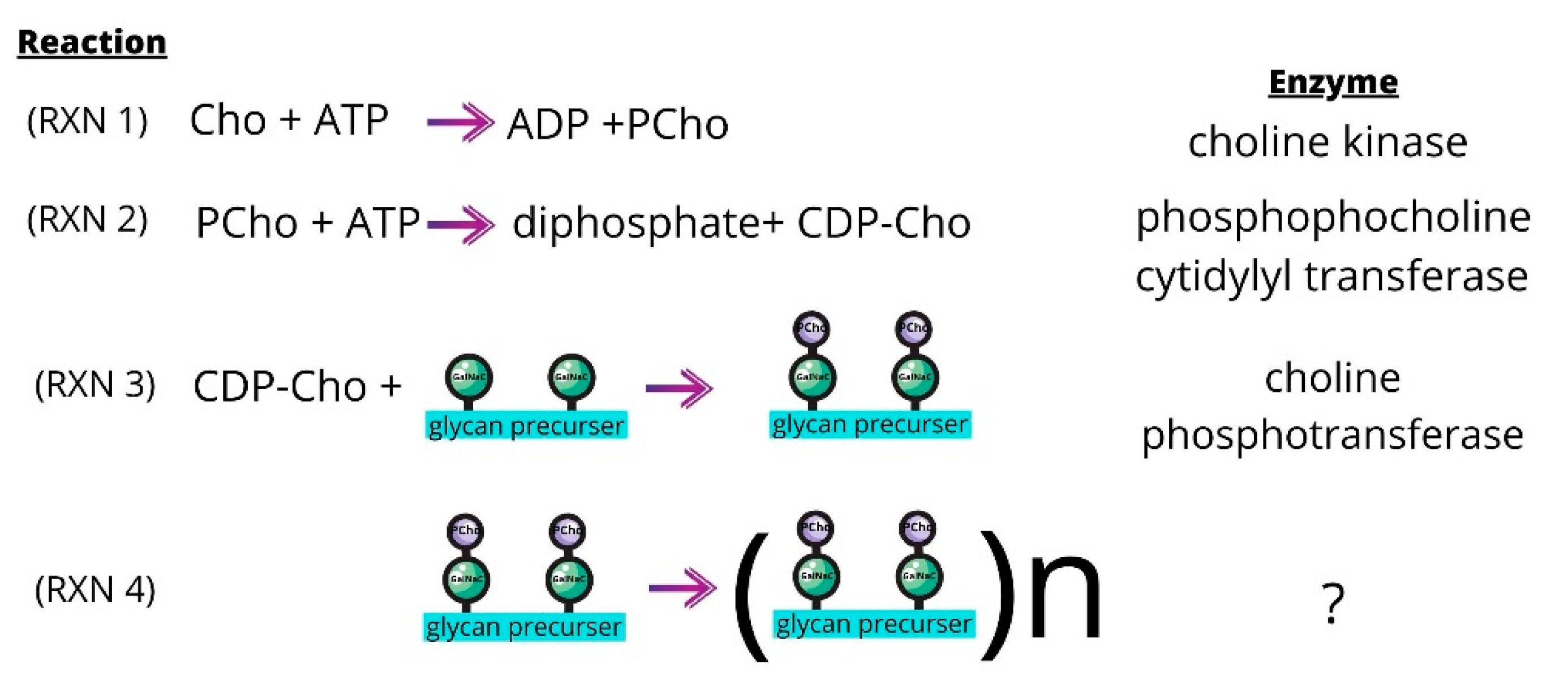
:1. Introduction
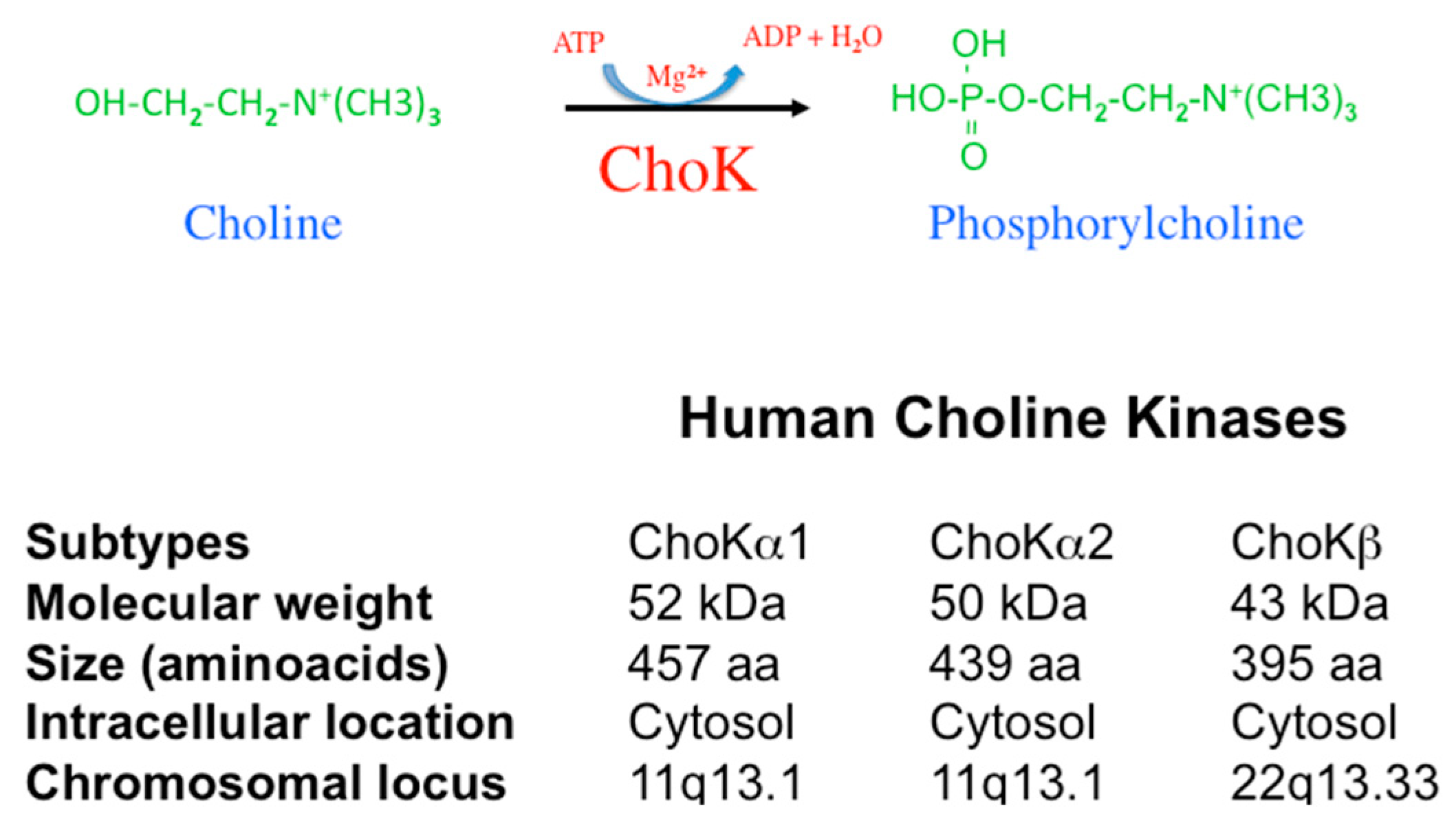
2. The ChoK Family in Humans
3. ChoKα in Cancer: A Promissing Therapeutic Target
4. Design of ChoKα Inhibitors as a Precision Medicine Strategy
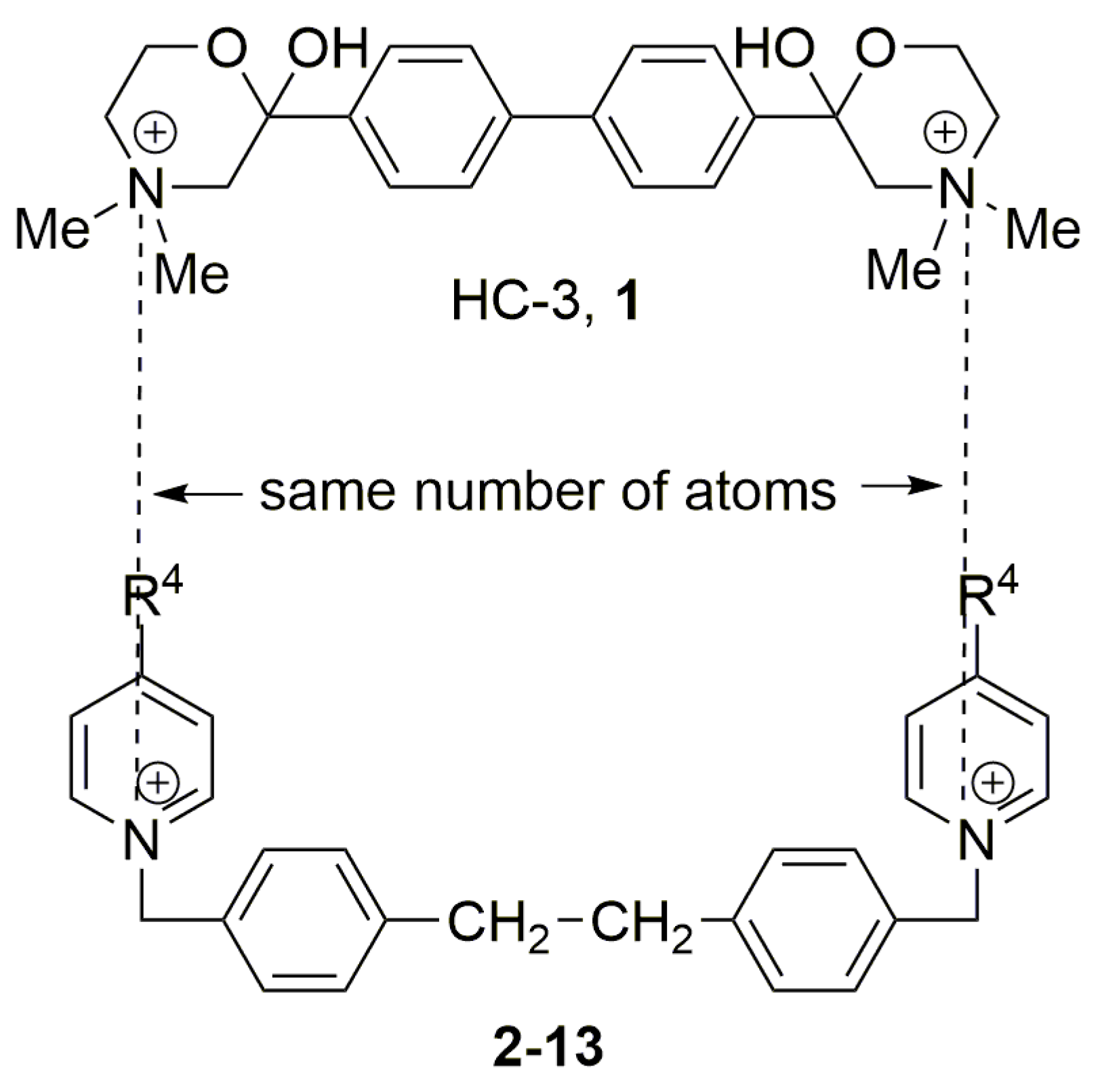
4.1. QSAR Studies between Ex Vivo ChoKα Inhibition and The Electronic Effects of Substituents at Position 4 of The Pyridinium Moieties
n = 5, r = 0.994, s = 0.052, F1,3 = 250.13 (significance at α < 0.001)
n = 10, r = 0.928, s = 0.181, F1,8 = 49.78 (significance at α < 0.001)
4.2. Ex Vivo ChoK Inhibition and Clog P: QSAR Studies
- (a)
- Cationic heads with a “zero electronic effect”, i.e., hydrogen at position 4, that allow the positive charge to be dispersed to a large extent. We used unsubstituted quinolinium and isoquinolinium rings to achieve this.
- (b)
- Aralkyl spacers with different number of methylene groups.
n = 7, r = 0.922, s = 0.151, F1,5 = 28.57 (significance at α < 0.005)
4.3. Combining The Electronic and The Lipophilic Effects in The Same Molecules
n = 19, r = 0.836, s = 0.241, F2,16 = 18.61 (significance at α < 0.001)
n = 19, r = 0.917, s = 0.181, F3,15 = 26.46 (significance at α < 0.001)
4.4. Influence on The Antiproliferative Activity Against The HT-29 Cancerous Cell Line
− 3.73 (± 0.71) σR
n = 40 (series A, B and C), r = 0.920, s = 0.223, F4,35 = 47.856, α < 0.001
n = 40, r = 0.916, s = 0.220, F1,38 = 198.854 (significance at α < 0.001)
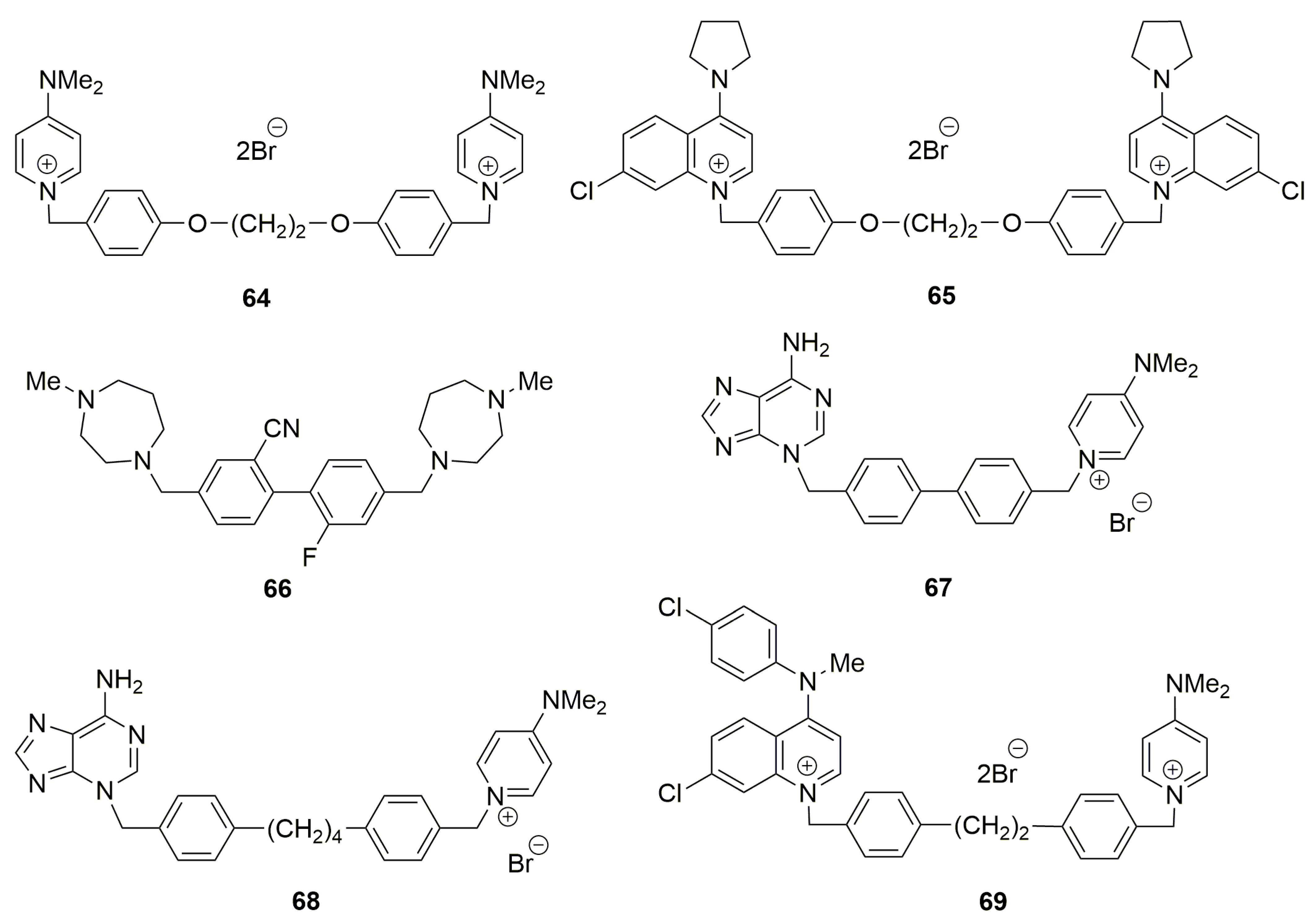
4.5. Other Symmetrical and Unsymmetrical ChoKα Inhibitors
5. ChoKIs Mechanism of Action
6. ChoKα in Drug Resistance
7. ChoKs, More Than Metabolism Enzymes?
8. ChoKs and ChoKIs of Protozoal Parasites
8.1. Plasmodium falciparum and ChoK
8.2. Leishmania infantum and Choline Kinase
8.3. Trypanosoma brucei and Choline Kinase
9. Bacterial Pathogens and Their ChoKs and ChoKIs
9.1. Streptococcus pneumoniae and Choline Kinase
9.2. Haemophilius influenzae ChoK
10. Rheumatoid Arthritis and Inflammation
11. Future Perspectives: ChoKIs Development for the Treatment of Cancer, Arthritis, Inflammation, Infections and Beyond
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ChoKα, ChoKβ | Choline Kinase α or β |
| PC | phosphatidylcholine |
| PCho | phosphorylcholine |
| ChoKI | ChoK inhibitor |
| ChoKαI | ChoKα-specific inhibitor |
| siRNA | small interfering RNA |
| 5-FU | 5-Fluorouracil |
| PI3K | phosphatidylinositol 3 kinase |
| MAPK | mitogen activated protein kinase |
| EGFR | epidermal growth factor receptor |
| NMR | nuclear magnetic resonance |
| MRS | magnetic resonance spectroscopy |
| PET | positron emission tomography |
| HR-MAS MRS | high-resolution magic angle spinning magnetic resonance spectroscopy |
| FLS | fibroblast-like synoviocytes |
| RA | rheumatoid arthritis |
References
- DeBerardinis, R.J.; Thompson, C.B. Cellular metabolism and disease: What do metabolic outliers teach us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacal, J.C. Choline kinase: A novel target to search for antitumoral drugs. IDrugs 2001, 4, 419–426. [Google Scholar]
- Lacal, J.C. Choline kinase as a precision medicine target for therapy in cancer, autoimmune diseases and malaria. Precis. Med. 2015, 1. [Google Scholar] [CrossRef]
- Aoyama, C.; Liao, H.; Ishidate, K. Structure and function of choline kinase isoforms in mammalian cells. Prog. Lipid Res. 2004, 43, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Fagone, P.; Jackowski, S. Phosphatidylcholine and the CDP–choline cycle. Biochim. Biophys. Acta 2013, 1831, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Qiu, H.; Wang, C.; Yuan, Y.; Tickner, J.; Xu, J.; Zou, J. Molecular structure and differential function of choline kinases CHKalpha and CHKbeta in musculoskeletal system and cancer. Cytokine Growth Factor Rev. 2017, 33, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Aoyama, C.; Young, S.G.; Vance, D.E. Early Embryonic Lethality Caused by Disruption of the Gene for Choline Kinase α, the First Enzyme in Phosphatidylcholine Biosynthesis. J. Biol. Chem. 2008, 283, 1456–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-C.; Liu, Y.-C.; Nakamura, Y. The Choline/Ethanolamine Kinase Family in Arabidopsis: Essential Role of CEK4 in Phospholipid Biosynthesis and Embryo Development. Plant Cell 2015, 27, 1497–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sher, R.B.; Aoyama, C.; Huebsch, K.A.; Ji, S.; Kerner, J.; Yang, Y.; Frankel, W.N.; Hoppel, C.L.; Wood, P.A.; Vance, D.E.; et al. A Rostrocaudal Muscular Dystrophy Caused by a Defect in Choline Kinase Beta, the First Enzyme in Phosphatidylcholine Biosynthesis. J. Biol. Chem. 2006, 281, 4938–4948. [Google Scholar] [CrossRef] [Green Version]
- Sayed-Zahid, A.A.; Sher, R.B.; Sukoff, R.; Anderson, L.C.; Patenaude, K.E.; Cox, G.A.; Sayed, A.A. Functional rescue in a mouse model of congenital muscular dystrophy with megaconial myopathy. Hum. Mol. Genet. 2019, 28, 2635–2647. [Google Scholar] [CrossRef]
- Kular, J.; Tickner, J.C.; Pavlos, N.J.; Viola, H.M.; Abel, T.; Lim, B.S.; Yang, X.; Chen, H.; Cook, R.; Hool, L.C.; et al. Choline Kinase Mutant Mice Exhibit Reduced Phosphocholine, Elevated Osteoclast Activity, and Low Bone Mass. J. Biol. Chem. 2015, 290, 1729–1742. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wu, G.; Van Der Veen, J.N.; Hermansson, M.; Vance, D.E. Phosphatidylcholine metabolism and choline kinase in human osteoblasts. Biochim. Biophys. Acta 2014, 1841, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, S.; Nishino, I. Megaconial congenital muscular dystrophy due to loss-of-function mutations in choline kinase β. Curr. Opin. Neurol. 2013, 26, 536–543. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Araguirang, G.E.; Ngo, A.H.; Lin, K.-T.; Angkawijaya, A.E.; Nakamura, Y. The Four Arabidopsis Choline/Ethanolamine Kinase Isozymes Play Distinct Roles in Metabolism and Development. Plant Physiol. 2020, 183, 152–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallego-Ortega, D.; De Molina, A.R.; Ramos, M.; Valdes-Mora, F.; Sarmentero-Estrada, J.; Lacal, J.C. Differential role of choline kinase alpha and beta isoforms in human carcinogenesis. Eur. J. Cancer Suppl. 2008, 6, 26. [Google Scholar] [CrossRef]
- Malito, E.; Sekulic, N.; Too, W.C.S.; Konrad, M.; Lavie, A. Elucidation of Human Choline Kinase Crystal Structures in Complex with the Products ADP or Phosphocholine. J. Mol. Biol. 2006, 364, 136–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, B.S.; Allali-Hassani, A.; Tempel, W.; MacKenzie, F.; Dimov, S.; Vedadi, M.; Park, H.W. Crystal Structures of Human Choline Kinase Isoforms in Complex with Hemicholinium-3: Single amino acid near the active site influences inhibitor sensitivity. J. Biol. Chem. 2010, 285, 16330–16340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyama, C.; Ohtani, A.; Ishidate, K. Expression and characterization of the active molecular forms of choline/ethanolamine kinase-alpha and -beta in mouse tissues, including carbon tetrachloride-induced liver. Biochem. J. 2002, 363, 777–784. [Google Scholar] [CrossRef]
- Gruber, J.; Too, W.C.S.; Wong, M.T.; Lavie, A.; McSorley, T.; Konrad, M. Balance of human choline kinase isoforms is critical for cell cycle regulation. FEBS J. 2012, 279, 1915–1928. [Google Scholar] [CrossRef]
- Ramírez de Molina, A.; Gutiérrez, R.; Ramos, M.A.; Silva, J.M.; Silva, J.; Bonilla, F.; Sánchez, J.J.; Lacal, J.C. Increased choline kinase activity in human breast carcinomas: Clinical evidence for a potential novel antitumoral strategy. Oncogene 2002, 21, 4317–4322. [Google Scholar] [CrossRef] [Green Version]
- Ramírez de Molina, A.; Rodríguez-González, A.; Gutiérrez, R.; Martínez-Piñeiro, L.; Sánchez, J.J.; Bonilla, F.; Rosell, R.; Lacal, J.C. Overexpression of choline kinase is a frequent feature in human tumor-derived cell lines and in lung, prostate and colorectal human cancers. Biochem. Biophys. Res. Commun. 2002, 296, 580–583. [Google Scholar] [CrossRef]
- Ramírez de Molina, A.; Sarmentero-Estrada, J.; Belda-Iniesta, C.; Taron, M.; Ramírez de Molina, V.; Cejas, P.; Skrzypski, M.; Gallego-Ortega, D.; de Castro, J.; Casado, E.; et al. Expression of choline kinase alfa to predict outcome in patients with early-stage non-small-cell lung cancer: A retrospective study. Lancet Oncol. 2007, 8, 889–897. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, P.S.; Li, G.; Bao, W. CHKA mediates the poor prognosis of lung adenocarcinoma and acts as a prognostic indicator. Oncol Lett. 2016, 12, 1849–1853. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Wang, R.-Y.; Cai, J.; Feng, D.; Yang, G.-Z.; Xu, Q.-G.; Zhai, Y.-X.; Zhang, Y.; Zhou, W.-P.; Cai, Q.-P. Overexpression of CHKA contributes to tumor progression and metastasis and predicts poor prognosis in colorectal carcinoma. Oncotarget 2016, 7, 66660–66678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernando, E.; Sarmentero-Estrada, J.; Koppie, T.; Iniesta, C.B.; De Molina, A.R.; Cejas, P.; Ozu, C.; Le, C.; Sánchez, J.J.; González-Barón, M.; et al. A critical role for choline kinase-α in the aggressiveness of bladder carcinomas. Oncogene 2009, 28, 2425–2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challapalli, A.; Trousil, S.; Hazell, S.; Kozlowski, K.; Gudi, M.; O Aboagye, E.; Mangar, S. Exploiting altered patterns of choline kinase-alpha expression on human prostate tissue to prognosticate prostate cancer. J. Clin. Pathol. 2015, 68, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Burch, T.C.; Isaac, G.; Booher, C.L.; Rhim, J.S.; Rainville, P.; Langridge, J.; Baker, A.; Nyalwidhe, J.O. Comparative Metabolomic and Lipidomic Analysis of Phenotype Stratified Prostate Cells. PLoS ONE 2015, 10, e0134206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanni, S.; Aiello, A.; Salis, C.; Re, A.; Cencioni, C.; Bacci, L.; Pierconti, F.; Pinto, F.; Ripoli, C.; Ostano, P.; et al. Metabolic Reprogramming by Malat1 Depletion in Prostate Cancer (Stage 2). Cancers 2020, 13, 15. [Google Scholar] [CrossRef]
- Iorio, E.; Mezzanzanica, D.; Alberti, P.; Spadaro, F.; Ramoni, C.; D’Ascenzo, S.; Millimaggi, D.; Pavan, A.; Dolo, V.; Canevari, S.; et al. Alterations of Choline Phospholipid Metabolism in Ovarian Tumor Progression. Cancer Res. 2005, 65, 9369–9376. [Google Scholar] [CrossRef] [Green Version]
- Trousil, S.; Lee, P.; Pinato, D.J.; Ellis, J.K.; Dina, R.; Aboagye, E.O.; Keun, H.C.; Sharma, R. Alterations of Choline Phospholipid Metabolism in Endometrial Cancer Are Caused by Choline Kinase Alpha Overexpression and a Hyperactivated Deacylation Pathway. Cancer Res. 2014, 74, 6867–6877. [Google Scholar] [CrossRef] [Green Version]
- Mazarico, J.M.; Sánchez-Arévalo Lobo, V.J.; Favicchio, R.; Greenhalf, W.; Costello, E.; Carrillo-de Santa Pau, E.; Marqués, M.; Lacal, J.C.; Aboagye, E.; Real, F.X. Choline Kinase Alpha (CHKα) as a Therapeutic Target in Pancreatic Ductal Adenocarcinoma: Expression, Predictive Value, and Sensitivity to Inhibitors. Mol. Cancer Ther. 2016, 15, 323–333. [Google Scholar] [CrossRef] [Green Version]
- Mok, S.R.S.; Mohan, S.; Grewal, N.; Elfant, A.B.; Judge, T.A. A genetic database can be utilized to identify potential biomarkers for biphenotypic hepatocellular carcinoma-cholangiocarcinoma. J. Gastrointest. Oncol. 2016, 7, 570–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrillo-Reixach, J.; Torrens, L.; Simon-Coma, M.; Royo, L.; Domingo-Sàbat, M.; Abril-Fornaguera, J.; Akers, N.; Sala, M.; Ragull, S.; Arnal, M.; et al. Epigenetic footprint enables molecular risk stratification of hepatoblastoma with clinical implications. J. Hepatol. 2020, 73, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Kwee, S.A.; Hernandez, B.; Chan, O.; Wong, L. Choline Kinase Alpha and Hexokinase-2 Protein Expression in Hepatocellular Carcinoma: Association with Survival. PLoS ONE 2012, 7, e46591. [Google Scholar] [CrossRef]
- Lin, X.-M.; Hu, L.; Gu, J.; Wang, R.-Y.; Li, L.; Tang, J.; Zhang, B.-H.; Yan, X.-Z.; Zhu, Y.-J.; Hu, C.-L.; et al. Choline Kinase α Mediates Interactions Between the Epidermal Growth Factor Receptor and Mechanistic Target of Rapamycin Complex 2 in Hepatocellular Carcinoma Cells to Promote Drug Resistance and Xenograft Tumor Progression. Gastroenterology 2017, 152, 1187–1202. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Wang, S.; Zhang, T.; Zhang, E.Y.; Zhou, L.; Hu, C.; Yu, J.J.; Xu, G. Activation of choline kinase drives aberrant choline metabolism in esophageal squamous cell carcinomas. J. Pharm. Biomed. Anal. 2018, 155, 148–156. [Google Scholar] [CrossRef]
- Xiong, J.; Bian, J.; Wang, L.; Zhou, J.-Y.; Wang, Y.; Zhao, Y.; Wu, L.-L.; Hu, J.-J.; Li, B.; Chen, S.-J.; et al. Dysregulated choline metabolism in T-cell lymphoma: Role of choline kinase-α and therapeutic targeting. Blood Cancer J. 2015, 5, e287. [Google Scholar] [CrossRef] [Green Version]
- Gobeil Odai, K.; O’Dwyer, C.; Steenbergen, R.; Shaw, T.A.; Renner, T.M.; Ghorbani, P.; Rezaaifar, M.; Han, S.; Langlois, M.A.; Crawley, A.M.; et al. In Vitro Hepatitis C Virus Infection and Hepatic Choline Metabolism. Viruses. 2020, 12, 108. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhu, W.; Zhang, L.; Lei, H.; Wu, X.; Guo, L.; Chen, X.; Wang, Y.; Tang, H. The metabolic responses to hepatitis B virus infection shed new light on pathogenesis and targets for treatment. Sci. Rep. 2015, 5, 8421. [Google Scholar] [CrossRef] [Green Version]
- Ling, C.S.; Yin, K.B.; Cun, S.T.W.; Ling, F.L. Expression profiling of choline and ethanolamine kinases in MCF7, HCT116 and HepG2 cells, and the transcriptional regulation by epigenetic modification. Mol. Med. Rep. 2014, 11, 611–618. [Google Scholar] [CrossRef]
- Kumar, M.; Arlauckas, S.P.; Saksena, S.; Verma, G.; Ittyerah, R.; Pickup, S.; Popov, A.V.; Delikatny, E.J.; Poptani, H. Magnetic Resonance Spectroscopy for Detection of Choline Kinase Inhibition in the Treatment of Brain Tumors. Mol. Cancer Ther. 2015, 14, 899–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, F.; Zou, Y.; Sun, S.; Wang, Z.; Huang, L.; Ma, H. Knockdown of choline kinase α (CHKA) inhibits the proliferation, invasion and migration of human U87MG glioma cells. Chin. J. Cell. Mol. Immunol. 2020, 36, 724–728. [Google Scholar]
- Penet, M.-F.; Shah, T.; Bharti, S.; Krishnamachary, B.; Artemov, D.; Mironchik, Y.; Wildes, F.; Maitra, A.; Bhujwalla, Z.M. Metabolic Imaging of Pancreatic Ductal Adenocarcinoma Detects Altered Choline Metabolism. Clin. Cancer Res. 2015, 21, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Mariotto, E.; Bortolozzi, R.; Volpin, I.; Carta, D.; Serafin, V.; Accordi, B.; Basso, G.; Navarro, P.L.; López-Cara, L.C.; Viola, G. EB-3D a novel choline kinase inhibitor induces deregulation of the AMPK-mTOR pathway and apoptosis in leukemia T-cells. Biochem. Pharmacol. 2018, 155, 213–223. [Google Scholar] [CrossRef]
- Ramirez de Molina, A.; Báñez-Coronel, M.; Gutiérrez, R.; Rodríguez-González, A.; Olmeda, D.; Megías, D. Choline kinase activation is a critical requirement for the proliferation of primary human mammary epithelial cells and breast tumor progression. Cancer Res. 2004, 64, 6732–6739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, T.; Wildes, F.; Penet, M.-F.; Jr, P.T.W.; Glunde, K.; Artemov, D.; Ackerstaff, E.; Gimi, B.; Kakkad, S.; Raman, V.; et al. Choline kinase overexpression increases invasiveness and drug resistance of human breast cancer cells. NMR Biomed. 2010, 23, 633–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokhale, S.; Lu, W.; Zhu, S.; Liu, Y.; Hart, R.P.; Rabinowitz, J.D.; Xie, P. Elevated Choline Kinase α–Mediated Choline Metabolism Supports the Prolonged Survival of TRAF3-Deficient B Lymphocytes. J. Immunol. 2020, 204, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Pera, B.; Krumsiek, J.; Assouline, S.E.; Marullo, R.; Patel, J.; Phillip, J.M.; Roman, L.; Mann, K.K.; Cerchietti, L. Metabolomic Profiling Reveals Cellular Reprogramming of B-Cell Lymphoma by a Lysine Deacetylase Inhibitor through the Choline Pathway. EBioMedicine 2018, 28, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadiya, M.; Mori, N.; Cao, M.D.; Mironchik, Y.; Kakkad, S.; Gribbestad, I.S.; Glunde, K.; Krishnamachary, B.; Bhujwalla, Z.M. Phospholipase D1 and choline kinase-α are interactive targets in breast cancer. Cancer Biol. Ther. 2014, 15, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Tian, L.; Jung, M.; Choi, S.K.; Sun, Y.; Kim, H.; Moon, W.K. Downregulation of Choline Kinase-Alpha Enhances Autophagy in Tamoxifen-Resistant Breast Cancer Cells. PLoS ONE 2015, 10, e0141110. [Google Scholar] [CrossRef] [Green Version]
- Glunde, K.; Raman, V.; Mori, N.; Bhujwalla, Z.M. RNA Interference-Mediated Choline Kinase Suppression in Breast Cancer Cells Induces Differentiation and Reduces Proliferation. Cancer Res. 2005, 65, 11034–11043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bañez-Coronel, M.; De Molina, A.R.; Rodríguez-González, A.; Sarmentero, J.; Ramos, M.A.; García-Cabezas, M.A.; García-Oroz, L.; Lacal, J.C. Choline kinase alpha depletion selectively kills tumoral cells. Curr. Cancer Drug Targets 2008, 8, 709–719. [Google Scholar] [CrossRef]
- Krishnamachary, B.; Glunde, K.; Wildes, F.; Mori, N.; Takagi, T.; Raman, V.; Bhujwalla, Z.M. Noninvasive detection of lentiviral-mediated choline kinase targeting in a human breast cancer xenograft. Cancer Res. 2009, 69, 3464–3471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Cueva, A.; Ramírez de Molina, A.; Álvarez-Ayerza, N.; Ramos, M.A.; Cebrián, A.; Del Pulgar, T.G.; Lacal, J.C. Combined 5-FU and ChoKα inhibitors as a new alternative therapy of colorectal cancer: Evidence in human tumor-derived cell lines and mouse xenographs. PLoS ONE 2013, 8, e64961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, N.; Glunde, K.; Takagi, T.; Raman, V.; Bhujwalla, Z.M. Choline Kinase Down-regulation Increases the Effect of 5-Fluorouracil in Breast Cancer Cells. Cancer Res. 2007, 67, 11284–11290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariotto, E.; Viola, G.; Ronca, R.; Persano, L.; Aveic, S.; Bhujwalla, Z.M.; Mori, N.; Accordi, B.; Serafin, V.; López-Cara, L.C.; et al. Choline Kinase Alpha Inhibition by EB-3D Triggers Cellular Senescence, Reduces Tumor Growth and Metastatic Dissemination in Breast Cancer. Cancers 2018, 10, 391. [Google Scholar] [CrossRef] [Green Version]
- Lacal, J.C.; Andera, L. Choline kinase inhibitors synergize with TRAIL in the treatment of colorectal tumors and overcomes TRAIL resistance. Cancer Transl. Med. 2016, 2, 163. [Google Scholar] [CrossRef]
- Rizzo, A.; Satta, A.; Garrone, G.; Cavalleri, A.; Napoli, A.; Raspagliesi, F.; Figini, M.; De Cecco, L.; Iorio, E.; Tomassetti, A.; et al. Choline kinase alpha impairment overcomes TRAIL resistance in ovarian cancer cells. J. Exp. Clin. Cancer Res. 2021, 40, 1–13. [Google Scholar] [CrossRef]
- Ramírez de Molina, A.; de la Cueva, A.; Machado-Pinilla, R.; Rodríguez-Fanjul, V.; Gómez del Pulgar, T.; Cebrián, A.; Perona, R.; Lacal, J.C. Acid ceramidase as a chemotherapeutic target to overcome resistance to the antitumoral effect of choline kinase α inhibition. Curr. Cancer Drug Targets 2012, 12, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-González, A.; Ramírez de Molina, A.; Báñez-Coronel, M.; Megías, D.; Núñez, M.C.; Lacal, J.C. Inhibition of choline kinase renders a highly selective cytotoxic effect in tumor cells through a mitochondrial independent mechanism. Int. J. Oncol. 2005, 26, 999–1008. [Google Scholar]
- Rodriguez-Gonzalez, A.; De Molina, A.R.; Fernández, F.; Lacal, J.C. Choline kinase inhibition induces the increase in ceramides resulting in a highly specific and selective cytotoxic antitumoral strategy as a potential mechanism of action. Oncogene 2004, 23, 8247–8259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-López, E.; Menchén, L.E.; Seco, E.; Gómez del Pulgar, T.; Moyer, M.P.; Lacal, J.C.; Cebrian, A. Endoplasmic reticulum stress participates in the cytotoxic effect of choline kinase α inhibitors in tumor cells. Cell Death Dis. 2013, 4, e933. [Google Scholar]
- Arlauckas, S.P.; Kumar, M.; Popov, A.V.; Poptani, H.; Delikatny, E.J. Near infrared fluorescent imaging of choline kinase alpha expression and inhibition in breast tumors. Oncotarget 2017, 8, 16518–16530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, J.; Núñez, M.C.; Rodríguez, V.; Gallo, M.A.; Espinosa, A. QSAR of 1,1′-(1,2-ethylenebisbenzyl)bis(4-substitutedpyridinium) dibromides as choline kinase inhibitors: A different approach for antiproliferative drug design. Bioorg. Med. Chem. Lett. 2000, 10, 767–770. [Google Scholar] [CrossRef]
- Cannon, J.G. Structure-activity aspects of hemicholinium-3 (HC-3) and its analogs and congeners. Med. Res. Rev. 1994, 14, 505–531. [Google Scholar] [CrossRef]
- Sánchez-Martín, R.; Campos, J.; Conejo-García, A.; Cruz-López, O.; Báñez-Coronel, M.; Rodríguez-González, A.; Gallo, M.A.; Lacal, J.C.; Espinosa, A. Symmetrical Bis-Quinolinium Compounds: New Human Choline Kinase Inhibitors with Antiproliferative Activity against the HT-29 Cell Line. J. Med. Chem. 2005, 48, 3354–3363. [Google Scholar] [CrossRef] [PubMed]
- Viswanadhan, V.N.; Ghose, A.K.; Revankar, G.R.; Robins, R.K. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J. Chem. Inf. Model. 1989, 29, 163–172. [Google Scholar] [CrossRef]
- Pallas Frame Module, a Prediction Tool of Physicochemical Parameters. Available online: www.compudrug.com (accessed on 15 April 2021).
- Charton, M. Electrical Effect Substituent Constants for Correlation Analysis. Prog. Phys. Org. Chem. 2007, 13, 119–251. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A. Substituent Constant for Correlation Analysis in Chemistry and Biology; Wiley: New York, NY, USA, 1979; pp. 51–52. [Google Scholar]
- Campos, J.; Núñez, M.C.; Sánchez, R.M.; Gómez-Vidal, J.A.; Rodríguez-González, A.; Báñez, M.; Gallo, M.A.; Lacal, J.C.; Espinosa, A. Quantitative structure–activity relationships for a series of symmetrical bisquaternary anticancer compounds. Bioorg. Med. Chem. 2002, 10, 2215–2231. [Google Scholar] [CrossRef]
- Campos, J.; Núñez, C.; Díaz, J.J.; Sanchez, R.M.; Gallo, M.A.; Espinosa, A. Anticancer bisquaternary heterocyclic compounds: A ras-ional design. Il Farm. 2003, 58, 221–229. [Google Scholar] [CrossRef]
- Gupta, S.P. QSAR studies on enzyme inhibitors. Chem. Rev. 1987, 87, 1183–1253. [Google Scholar] [CrossRef]
- Kearney, P.C.; Mizoue, L.S.; Kumpf, R.A.; Forman, J.; McCurdy, A.; Dougherty, D.A. Molecular recognition in aqueous media. New binding studies provide further insights into the cation-.pi. interaction and related phenomena. J. Am. Chem. Soc. 1993, 115, 9907–9919. [Google Scholar] [CrossRef]
- Kubinyi, H. QSAR: Hansch Analysis and Related Approaches. In Methods and Principles in Medicinal Chemistry; Mannhold, R., Krogsgaard-Larsen, P., Timmerman, H., Eds.; VCH: Weinheim, Germany, 1993; Volume 1, pp. 40–41. [Google Scholar]
- Blaney, J.M.; Hansch, C. Quantitative Drug Design. In Comprehensive Medicinal Chemistry. The Rational Design, Mechanistic Study & Therapeutic Application of Chemical Compounds; Ramsden, C.A., Hansch, C., Sammes, P.G., Taylor, J.B., Eds.; Pergamon Press: Oxford, UK, 1990; Volume 4, pp. 459–496. [Google Scholar]
- Hansch, C.; Blaney, J.M. Chemische Struktur und Biologische Aktivität von Wirkstoffen. Methoden der Quantitativen Struktur-Wirkung-Analyse; Seydel, J.K., Schaper, K.-J., Eds.; Verlag Chemie: Weinheim, Germany, 1979; pp. 185–208. [Google Scholar]
- Lacal, J.C.; Campos, J. Preclinical Characterization of RSM-932A, a Novel Anticancer Drug Targeting the Human Choline Kinase Alpha, an Enzyme Involved in Increased Lipid Metabolism of Cancer Cells. Mol. Cancer Ther. 2015, 14, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Study of Intravenous TCD-717 in Patients with Advanced Solid Tumours. ClinicalTrials.gov Identifier: NCT01215864. Available online: http://clinicaltrials.gov/ct2/show/NCT01215864 (accessed on 15 April 2021).
- Schiaffino-Ortega, S.; Baglioni, E.; Mariotto, E.; Bortolozzi, R.; Serrán-Aguilera, L.; Ríos-Marco, P.; Carrasco-Jimenez, M.P.; Gallo, M.A.; Hurtado-Guerrero, R.; Marco, C.; et al. Design, synthesis, crystallization and biological evaluation of new symmetrical biscationic compounds as selective inhibitors of human Choline Kinase α1 (ChoKα1). Sci. Rep. 2016, 6, 23793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zech, S.G.; Kohlmann, A.; Zhou, T.; Li, F.; Squillace, R.M.; Parillon, L.E.; Greenfield, M.T.; Miller, D.P.; Qi, J.; Thomas, R.M.; et al. Novel Small Molecule Inhibitors of Choline Kinase Identified by Fragment-Based Drug Discovery. J. Med. Chem. 2016, 59, 671–686. [Google Scholar] [CrossRef]
- Rubio-Ruiz, B.; Serrán-Aguilera, L.; Hurtado-Guerrero, R.; Conejo-García, A. Recent advances in the design of choline kinase α inhibitors and the molecular basis of their inhibition. Med. Res. Rev. 2021, 41, 902–927. [Google Scholar] [CrossRef] [PubMed]
- Rubbini, G.; Buades-Martín, A.B.; Kimatrai-Salvador, M.; Entrena, A.; Gallo-Mezo, M.Á.; Ríos-Marco, P.; Marco, C.; Mattiuzzo, E.; Bortolozzi, R.; Mariotto, E.; et al. Lead optimization-hit expansion of new asymmetrical pyridinium/quinolinium compounds as choline kinase α1 inhibitors. Future Med. Chem. 2018, 10, 1769–1786. [Google Scholar] [CrossRef]
- Falcon, S.C.; Hudson, C.S.; Huang, Y.; Mortimore, M.; Golec, J.M.; Charlton, P.A.; Weber, P.; Sundaram, H. A non-catalytic role of choline kinase alpha is important in promoting cancer cell survival. Oncogenesis 2013, 2, e38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, N.; Wildes, F.; Kakkad, S.; Jacob, D.; Solaiyappan, M.; Glunde, K.; Bhujwalla, Z.M. Choline kinase-α protein and phosphatidylcholine but not phosphocholine are required for breast cancer cell survival. NMR Biomed. 2015, 28, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Asim, M.; Massie, C.E.; Orafidiya, F.; Pértega-Gomes, N.; Warren, A.Y.; Esmaeili, M.; Selth, L.A.; Zecchini, H.I.; Luko, K.; Qureshi, A.; et al. Choline Kinase Alpha as an Androgen Receptor Chaperone and Prostate Cancer Therapeutic Target. J. Natl. Cancer Inst. 2016, 108, djv371. [Google Scholar] [CrossRef] [Green Version]
- Asim, M.; Massie, C.E.; Neal, D.E. Kinase joins the chaperone club: Androgen-regulated kinome reveals choline kinase alpha as a potential drug target in prostate cancer. Mol. Cell Oncol. 2016, 3, e1140262. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, T.; Moneriz, C.; Diez, A.; Bautista, J.M.; Del Pulgar, T.G.; Cebrián, A.; Lacal, J.C. Antiplasmodial Activity and Mechanism of Action of RSM-932A, a Promising Synergistic Inhibitor of Plasmodium falciparum Choline Kinase. Antimicrob. Agents Chemother. 2013, 57, 5878–5888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, C.S.; Knegtel, R.M.; Brown, K.; Charlton, P.A.; Pollard, J.R. Kinetic and mechanistic characterisation of Choline Kinase-α. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Kall, S.L.; Delikatny, E.J.; Lavie, A. Identification of a Unique Inhibitor-Binding Site on Choline Kinase α. Biochemistry 2018, 57, 1316–1325. [Google Scholar] [CrossRef]
- Koch, K.; Hartmann, R.; Schröter, F.; Suwala, A.K.; Maciaczyk, D.; Krüger, A.C.; Willbold, D.; Kahlert, U.D.; Maciaczyk, J. Reciprocal regulation of the cholinic phenotype and epithelial-mesenchymal transition in glioblastoma cells. Oncotarget 2016, 7, 73414–73431. [Google Scholar] [CrossRef] [Green Version]
- Granata, A.; Nicoletti, R.; Perego, P.; Iorio, E.; Krishnamachary, B.; Benigni, F.; Ricci, A.; Podo, F.; Bhujwalla, Z.M.; Canevari, S.; et al. Global metabolic profile identifies choline kinase alpha as a key regulator of glutathione-dependent antioxidant cell defense in ovarian carcinoma. Oncotarget 2015, 6, 11216–11230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.C.; Kanchara, K.; Nakamura, Y. Arabidopsis CHOLINE/ETHANOLAMINE KINASE 1 (CEK1) is a primary choline ekinase localized at the endoplasmic reticulum (ER) and involved in ER stress tolerance. New Phytol. 2019, 223, 1904–1917. [Google Scholar] [CrossRef]
- Trousil, S.; Kaliszczak, M.; Schug, Z.; Nguyen, Q.-D.; Tomasi, G.; Favicchio, R.; Brickute, D.; Fortt, R.; Twyman, F.J.; Carroll, L.; et al. The novel choline kinase inhibitor ICL-CCIC-0019 reprograms cellular metabolism and inhibits cancer cell growth. Oncotarget 2016, 7, 37103–37120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez de Molina, A.; Gallego-Ortega, D.; Sarmentero-Estrada, J.; Lagares, D.; Gómez del Pulgar, T.; Bandrés, E.; García-Foncillas, J.; Lacal, J.C. Choline kinase as a link connecting phospholipid metabolism and cell cycle regulation: Implications in cancer therapy. Int. J. Biochem. Cell Biol. 2008, 40, 1753–1763. [Google Scholar]
- Moestue, S.A.; Borgan, E.; Huuse, E.M.; Lindholm, E.M.; Sitter, B.; Børresen-Dale, A.L.; Engebraaten, O.; Maelandsmo, G.M.; Gribbestad, I.S. Disctinct choline metabolic profiles are associated with differences in gene expression for basal-like and luminal-like breast cancer xenograft models. BMC Cancer 2010, 10, 433. [Google Scholar] [CrossRef] [Green Version]
- Grinde, M.T.; Skrbo, N.; Moestue, S.A.; A Rødland, E.; Borgan, E.; Kristian, A.; Sitter, B.; Bathen, T.F.; Børresen-Dale, A.-L.; Mælandsmo, G.M.; et al. Interplay of choline metabolites and genes in patient-derived breast cancer xenografts. Breast Cancer Res. 2014, 16, R5. [Google Scholar] [CrossRef] [Green Version]
- Miyake, T.; Parsons, S.J. Functional interactions between choline kinase-α, epidermal growth factor receptor and c-Src in breast cancer cell proliferation. Oncogene 2012, 31, 1431–1441. [Google Scholar] [CrossRef] [Green Version]
- Kall, S.L.; Whitlatch, K.; Smithgall, T.E.; Lavie, A. Molecular basis for the interaction between human choline kinase alpha and the SH3 domain of the c-Src tyrosine kinase. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.J.; Chen, Y.H.; Tsai, K.W.; Yeah, H.Y.; Yeh, C.Y.; Tu, Y.T.; Yang, C.Y. Involvement of MicroRNA-1-FAM83A Axis Dysfunction in the Growth and Motility of Lung Cancer Cells. Int. J. Mol. Sci. 2020, 21, 8833. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Few, L.L.; Konrad, M.; Too, W.C.S. Phosphorylation of Human Choline Kinase Beta by Protein Kinase A: Its Impact on Activity and Inhibition. PLoS ONE 2016, 11, e0154702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, S.; Safaei, J.; Pelech, S. Evolutionary Ancestry of Eukaryotic Protein Kinases and Choline Kinases. J. Biol. Chem. 2016, 291, 5199–5205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, B.T.; Gallego-Ortega, D.; De Molina, A.R.; Ullrich, A.; Lacal, J.C.; Downward, J. Regulation of Akt(ser473) phosphorylation by Choline kinase in breast carcinoma cells. Mol. Cancer 2009, 8, 131. [Google Scholar] [CrossRef] [Green Version]
- Yalcin, A.; Clem, B.; Makoni, S.; Clem, A.; Nelson, K.; Thornburg, J.; Siow, D.; Lane, A.N.; E Brock, S.; Goswami, U.; et al. Selective inhibition of choline kinase simultaneously attenuates MAPK and PI3K/AKT signaling. Oncogene 2009, 29, 139–149. [Google Scholar] [CrossRef]
- Clem, B.F.; Clem, A.L.; Yalcin, A.; Goswami, U.; Arumugam, S.; Telang, S.; O Trent, J.; Chesney, J. A novel small molecule antagonist of choline kinase-α that simultaneously suppresses MAPK and PI3K/AKT signaling. Oncogene 2011, 30, 3370–3380. [Google Scholar] [CrossRef] [Green Version]
- Ramírez de Molina, A.; Penalva, V.; Lucas, L.; Lacal, J.C. Regulation of Choline kinase activity by Ras proteins involves Ral-GDS and PI3K. Oncogene 2002, 21, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Glunde, K.; Shah, T.; Winnard, P.T., Jr.; Raman, V.; Takagi, T.; Vesuna, F.; Artemov, D.; Bhujwalla, Z.M. Hypoxia regulates choline kinase expression through hypoxia-inducible factor-1 alpha signaling in a human prostate cancer model. Cancer Res. 2008, 68, 172–180. [Google Scholar]
- Bansal, A.; Harris, R.A.; DeGrado, T.R. Choline phosphorylation and regulation of transcription of choline kinase α in hypoxia. J. Lipid Res. 2012, 53, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanath, P.; Radoul, M.; Izquierdo-Garcia, J.L.; Luchman, H.A.; Cairncross, J.G.; Pieper, R.O.; Phillips, J.J.; Ronen, S.M. Mutant IDH1 gliomas downregulate phosphocholine and phosphoethanolamine synthesis in a 2-hydroxyglutarate-dependent manner. Cancer Metab. 2018, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Marcucci, H.; Paoletti, L.; Jackowski, S.; Banchio, C. Phosphatidylcholine Biosynthesis during Neuronal Differentiation and Its Role in Cell Fate Determination. J. Biol. Chem. 2010, 285, 25382–25393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domizi, P.; Aoyama, C.; Banchio, C. Choline kinase alpha expression during RA-induced neuronal differentiation: Role of C/EBPβ. Biochim. Biophys. Acta 2014, 1841, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Domizi, P.; Malizia, F.; Chazarreta-Cifre, L.; Diacovich, L.; Banchio, C. KDM2B regulates choline kinase expression and neuronal differentiation of neuroblastoma cells. PLoS ONE 2019, 14, e0210207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadiya, P.; Fatima, S.; Baghel, T.; Mir, S.S.; Nazir, A. A Systematic RNAi Screen of Neuroprotective Genes Identifies Novel Modulators of Alpha-Synuclein-Associated Effects in Transgenic Caenorhabditis elegans. Mol. Neurobiol. 2015, 53, 6288–6300. [Google Scholar] [CrossRef]
- Wong, M.T.; Chen, S.S. Hepatitis C virus subverts human choline kinase-alpha to bridge phosphatidylinositol-4-kinase IIIalpha (PI4KIIIalpha) and NS5A and upregulates PI4KIIIalpha activation, thereby promoting the translocation of the ternary complex to the endoplasmic reticulum for viral replication. J. Virol. 2017, 91, e00355-17. [Google Scholar] [PubMed] [Green Version]
- Wong, M.-T.; Chen, S.S. Human Choline Kinase-α Promotes Hepatitis C Virus RNA Replication through Modulation of Membranous Viral Replication Complex Formation. J. Virol. 2016, 90, 9075–9095. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Sher, R.B.; Cox, G.A.; Vance, D.E. Differential expression of choline kinase isoforms in skeletal muscle explains the phenotypic variability in the rostrocaudal muscular dystrophy mouse. Biochim. Biophys. Acta 2010, 1801, 446–454. [Google Scholar] [CrossRef]
- Wu, G.; Sher, R.B.; Cox, G.A.; Vance, D.E. Understanding the muscular dystrophy caused by deletion of choline kinase beta in mice. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2009, 1791, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, S.; Hatakeyama, H.; Karahashi, M.; Koumura, T.; Nonaka, I.; Hayashi, Y.K.; Noguchi, S.; Sher, R.B.; Nakagawa, Y.; Manfredi, G.; et al. Muscle choline kinase beta defect causes mitochondrial dysfunction and increased mitophagy. Hum. Mol. Genet. 2011, 20, 3841–3851. [Google Scholar] [CrossRef] [PubMed]
- Panaite, P.-A.; Gantelet, E.; Kraftsik, R.; Gourdon, G.; Kuntzer, T.; Barakat-Walter, I. Myotonic Dystrophy Transgenic Mice Exhibit Pathologic Abnormalities in Diaphragm Neuromuscular Junctions and Phrenic Nerves. J. Neuropathol. Exp. Neurol. 2008, 67, 763–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuhashi, S.; Ohkuma, A.; Talim, B.; Karahashi, M.; Koumura, T.; Aoyama, C.; Kurihara, M.; Quinlivan, R.; Sewry, C.; Mitsuhashi, H.; et al. A congenital muscular dystrophy with mitochondrial structural abnormalities caused by defective de novo phosphatidylcholine biosynthesis. Am. J. Hum. Genet. 2011, 88, 845–851. [Google Scholar] [CrossRef] [Green Version]
- Fujii, Y.; Sugiura, C.; Fukuda, C.; Maegaki, Y.; Ohno, K. Sequential neuroradiological and neurophysiological studies in a Japanese girl with merosin-deficient congenital muscular dystrophy. Brain Dev. 2011, 33, 140–144. [Google Scholar] [CrossRef]
- Gutiérrez Ríos, P.; Kalra, A.A.; Wilson, J.D.; Tanji, K.; Akman, H.O.; Area Gómez, E.; Schon, E.A.; DiMauro, S. Congenital megaconial myopathy due to a novel defect in the choline kinase Beta gene. Arch. Neurol. 2012, 69, 657–661. [Google Scholar]
- Quinlivan, R.; Mitsuahashi, S.; Sewry, C.; Cirak, S.; Aoyama, C.; Mooore, D.; Abbs, S.; Robb, S.; Newton, T.; Moss, C.; et al. Muscular dystrophy with large mitochondria associated with mutations in the CHKB gene in three British patients: Extending the clinical and patho-logical phenotype. Neuromuscul. Disord. 2013, 23, 549–556. [Google Scholar] [CrossRef]
- Castro-Gago, M.; Dacruz-Alvarez, D.; Pintos-Martínez, E.; Beiras-Iglesias, A.; Delmiro, A.; Arenas, J.; Martín, M.A.; Martínez-Azorín, F. Exome sequencing identifies a CHKB mutation in Spanish patient with megaconial congenital muscular dystrophy and mtDNA depletion. Eur. J. Paediatr. Neurol. 2014, 18, 796–800. [Google Scholar] [CrossRef]
- Cabrera-Serrano, M.; Junckerstorff, R.C.; Atkinson, V.; Sivadorai, P.; Allcock, R.J.; Lamont, P.; Laing, N.G. Novel CHKB mutation expands the megaconial muscular dystrophy phenotype. Muscle Nerve 2015, 51, 140–143. [Google Scholar] [CrossRef]
- Oliveira, J.; Negrão, L.; Fineza, I.; Taipa, R.; Melo-Pires, M.; Fortuna, A.M.; Gonçalves, A.R.; Froufe, H.; Egas, C.; Santos, R.; et al. New splicing mutation in the choline kinase beta (CHKB) gene causing a muscular dystrophy detected by whole-exome sequencing. J. Hum. Genet. 2015, 60, 305–312. [Google Scholar] [CrossRef]
- Castro-Gago, M.; Dacruz-Alvarez, D.; Pintos-Martínez, E.; Beiras-Iglesias, A.; Arenas, J.; Martín, M.A.; Martínez-Azorín, F. Congenital neurogenic muscular atrophy in megaconial myopathy due to a mutation in CHKB gene. Brain Dev. 2016, 38, 167–172. [Google Scholar] [CrossRef]
- Haliloglu, G.; Talim, B.; Sel, C.G.; Topaloglu, H. Clinical characteristics of megaconial congenital muscular dystrophy due to choline kinase beta gene defects in a series of 15 patients. J. Inherit. Metab. Dis. 2015, 38, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.V.; Yao, F.; Howenstine, A.; Takenaka, R.; Hyatt, J.A.; Sears, K.E.; Shewchuk, B.M. Emergent Coordination of the CHKB and CPT1B Genes in Eutherian Mammals: Implications for the Origin of Brown Adipose Tissue. J. Mol. Biol. 2020, 432, 6127–6145. [Google Scholar] [CrossRef]
- Gréchez-Cassiau, A.; Feillet, C.; Guérin, S.; Delaunay, F. The hepatic circadian clock regulates the choline kinase a gene through the BMAL1-REV-ERBα axis. Chronobiol. Int. 2015, 32, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Raikundalia, S.; Sa’Domm, S.A.F.M.; Few, L.L.; Too, W.C.S. MicroRNA 367 3p induces apoptosis and suppresses migration of MCF 7 cells by downregulating the expression of human choline kinase α. Oncol Lett. 2021, 21, 183. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Yao, Y.; Jiang, Z.; Zhou, H.; Xie, K.; Luo, J.; Shen, Y. FAM83A and FAM83A-AS1 both play oncogenic roles in lung adenocarcinoma. Oncol Lett. 2021, 21, 297. [Google Scholar] [CrossRef] [PubMed]
- WHO Human African Trypanosomiasis (Sleeping Sickness). Available online: http://www.who.int/topics/trypanosomiasis_african/en/ (accessed on 23 May 2021).
- Dondorp, A.M.; Nosten, F.; Yi, P.; Das, D.; Phyo, A.P.; Tarning, J.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; Lee, S.J.; et al. Artemisinin Resistance inPlasmodium falciparumMalaria. N. Engl. J. Med. 2009, 361, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Pessi, G.; Kociubinski, G.; Mamoun, C.B. A pathway for phosphatidylcholine biosynthesis in Plasmodium falciparum involving phosphoethanolamine methylation. Proc. Natl. Acad. Sci. USA 2004, 101, 6206–6211. [Google Scholar] [CrossRef] [Green Version]
- Ben Mamoun, C.; Prigge, S.T.; Vial, H. Targeting the lipid metabolic pathways for the treatment of malaria. Drug Dev. Res. 2009, 71, 44–55. [Google Scholar] [CrossRef] [Green Version]
- Choubey, V.; Guha, M.; Maity, P.; Kumar, S.; Raghunandan, R.; Maulik, P.R.; Mitra, K.; Halder, U.C.; Bandyopadhyay, U. Molecular characterization and localization of Plasmodium falciparum choline kinase. Biochim. Biophys. Acta Gen. Subj. 2006, 1760, 1027–1038. [Google Scholar] [CrossRef]
- Choubey, V.; Maity, P.; Guha, M.; Kumar, S.; Srivastava, K.; Puri, S.K.; Bandyopadhyay, U. Inhibition of Plasmodium falciparum Choline Kinase by Hexadecyltrimethylammonium Bromide: A Possible Antimalarial Mechanism. Antimicrob. Agents Chemother. 2006, 51, 696–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrán-Aguilera, L.; Denton, H.; Rubio-Ruiz, B.; López-Gutiérrez, B.; Entrena, A.; Izquierdo, L.; Smith, T.K.; Conejo-García, A.; Hurtado-Guerrero, R. Plasmodium falciparum Choline Kinase Inhibition Leads to a Major Decrease in Phosphatidylethanolamine Causing Parasite Death. Sci. Rep. 2016, 6, 33189. [Google Scholar] [CrossRef] [Green Version]
- Torretta, A.; Lopez-Cara, L.C.; Parisini, E. Crystal Structure of the Apo and the ADP-Bound Form of Choline Kinase from Plasmodium falciparum. Crystals 2020, 10, 613. [Google Scholar] [CrossRef]
- Walker, J.A.; Friesen, J.D.; Peters, S.J.; Jones, M.A.; Friesen, J.A. Development of a new and reliable assay for choline kinase using (31)P NMR. Heliyon 2019, 5, e02585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CDC. Parasites—Leishmaniasis; Center for Disease Control: Atlanta, GA, USA, 2020. [Google Scholar]
- Crowther, G.J.; Napuli, A.J.; Gilligan, J.H.; Gagaring, K.; Borboa, R.; Francek, C.; Chen, Z.; Dagostino, E.F.; Stockmyer, J.B.; Wang, Y.; et al. Identification of inhibitors for putative malaria drug targets among novel antimalarial compounds. Mol. Biochem. Parasitol. 2011, 175, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibellini, F.; Hunter, W.N.; Smith, T.K. Biochemical characterization of the initial steps of the Kennedy pathway in Trypanosoma brucei: The ethanolamine and choline kinases. Biochem. J. 2008, 415, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Major, L.L.; Denton, H.; Smith, T.K. Coupled Enzyme Activity and Thermal Shift Screening of the Maybridge Rule of 3 Fragment Library against Trypanosoma brucei Choline Kinase; A Genetically Validated Drug Target. In Drug Discovery; El-Shemy, H.A., Ed.; IntechOpen: London, UK, 2013. [Google Scholar]
- Delespaux, V.; De Koning, H.P. Drugs and drug resistance in African trypanosomiasis. Drug Resist. Updates 2007, 10, 30–50. [Google Scholar] [CrossRef]
- Tomasz, A. Choline in the Cell Wall of a Bacterium: Novel Type of Polymer-Linked Choline in Pneumococcus. Science 1967, 157, 694–697. [Google Scholar] [CrossRef]
- Kharat, A.S.; Tomasz, A. Drastic reduction in the virulence of Streptococcus pneumoniae expressing type 2 capsular polysaccharide but lacking choline residues in the cell wall. Mol. Microbiol. 2006, 60, 93–107. [Google Scholar] [CrossRef]
- Young, N.M.; Foote, S.J.; Wakarchuk, W.W. Review of phosphocholine substituents on bacterial pathogen glycans: Synthesis, structures and interactions with host proteins. Mol. Immunol. 2013, 56, 563–573. [Google Scholar] [CrossRef]
- Whiting, G.C.; Gillespie, S.H. Incorporation of choline into Streptococcus pneumoniae cell wall antigens: Evidence for choline kinase activity. FEMS Microbiol. Lett. 1996, 138, 141–145. [Google Scholar] [CrossRef]
- Gründling, A.; Schneewind, O. Synthesis of glycerol phosphate lipoteichoic acid in Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2007, 104, 8478–8483. [Google Scholar] [CrossRef] [Green Version]
- Denapaite, D.; Bruckner, R.; Hakenbeck, R.; Vollmer, W. Biosynthesis of teichoic acids in Streptococcus pneumoniae and closely related species: Lessons from genomes. Microb. Drug Resist. 2012, 18, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Heß, N.; Waldow, F.; Kohler, T.P.; Rohde, M.; Kreikemeyer, B.; Gómez-Mejia, A.; Hain, T.; Schwudke, D.; Vollmer, W.; Hammerschmidt, S.; et al. Lipoteichoic acid deficiency permits normal growth but impairs virulence of Streptococcus pneumoniae. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, I. Role of lipoteichoic acid in infection and inflammation. Lancet Infect. Dis. 2002, 2, 171–179. [Google Scholar] [CrossRef]
- Zhang, J.R.; Idanpaan-Heikkila, I.; Fischer, W.; Tuomanen, E.I. Pneumococcal licD2 gene is involved in phosphorylcholine metabolism. Mol. Microbiol. 1999, 31, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Maestro, B.; Sanz, J.M. Choline Binding Proteins from Streptococcus pneumoniae: A Dual Role as Enzybiotics and Targets for the Design of New Antimicrobials. Antibiotics 2016, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Rosenow, C.; Ryan, P.; Weiser, J.N.; Johnson, S.; Fontan, P.; Ortqvist, A.; Masure, H.R. Contribution of novel choline-binding proteins to adherence, colonization and immunogenicity of Streptococcus pneumoniae. Mol. Microbiol. 1997, 25, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Gosink, K.K.; Mann, E.R.; Guglielmo, C.; Tuomanen, E.I.; Masure, H.R. Role of Novel Choline Binding Proteins in Virulence of Streptococcus pneumoniae. Infect. Immun. 2000, 68, 5690–5695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, T.; Lacal, J.C.; Ibrahim, S.A. A dual choline/phosphocholine colorimetric method for measuring the relative strength of inhibitors of choline kinases of Gram-positive pathogens. Food Sci. Appl. Biotechnol. 2018, 1, 131–139. [Google Scholar] [CrossRef]
- Zimmerman, T.; Ibrahim, S. Choline Kinase, A Novel Drug Target for the Inhibition of Streptococcus pneumoniae. Antibiotics 2017, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, T.; Ibrahim, S.A. Parallel Colorimetric Quantification of Choline and Phosphocholine as a Method for Studying Choline Kinase Activity in Complex Mixtures. Antibiotics 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiser, J.N.; Shchepetov, M.; Chong, S.T. Decoration of lipopolysaccharide with phosphorylcholine: A phase-variable characteristic of Haemophilus influenzae. Infect. Immun. 1997, 65, 943–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Goldfine, H.; Lysenko, E.; Weiser, J.N. The transfer of choline from the host to the bacterial cell surface requires glpQ in Haemophilus influenzae. Mol. Microbiol. 2008, 41, 1029–1036. [Google Scholar] [CrossRef] [PubMed]
- Lysenko, E.S.; Gould, J.; Bals, R.; Wilson, J.; Weiser, J.N. Bacterial Phosphorylcholine Decreases Susceptibility to the Antimicrobial Peptide LL-37/hCAP18 Expressed in the Upper Respiratory Tract. Infect. Immun. 2000, 68, 1664–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, S.; Snow, J.; Li, J.; Zola, T.A.; Weiser, J.N. Phosphorylcholine Allows for Evasion of Bactericidal Antibody by Haemophilus influenzae. PLoS Pathog. 2012, 8, e1002521. [Google Scholar] [CrossRef]
- High, N.J.; Fan, F.; Schwartzman, J.D. Haemophilus influenzae. In Molecular Medical Microbiology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 3, pp. 1709–1728. [Google Scholar]
- Guma, M.; Sanchez-Lopez, E.; Lodi, A.; Garcia-Carbonell, R.; Tiziani, S.; Karin, M.; Lacal, J.C.; Firestein, G.S. Choline kinase inhibition in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1399–1407. [Google Scholar] [CrossRef]
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nat. Cell Biol. 2003, 423, 356–361. [Google Scholar] [CrossRef]
- Meier, F.M.; McInnes, I.B. Small-molecule therapeutics in rheumatoid arthritis: Scientific rationale, efficacy and safety. Best Pr. Res. Clin. Rheumatol. 2014, 28, 605–624. [Google Scholar] [CrossRef]
- Al-Saffar, N.M.S.; Jackson, L.E.; Reynaud, F.I.; Workman, P.; Ramírez de Molina, A.; Lacal, J.C.; Workman, P.; Leach, M.O. The novel phosphatidylinositide 3-kinase inhibitor PI103 downregulates choline kinase resulting in phosphocholine depletion detected by Magnetic Resonance Spectroscopy. Cancer Res. 2010, 70, 5507–5517. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-López, E.; Zhong, Z.; Stubelius, A.; Sweeney, S.R.; Antonucci, L.; Liu-Bryan, R.; Lodi, A.; Terkeltaub, R.; Lacal, J.C.; Murphy, A.N.; et al. Choline uptake and metabolism modulate macrophage IL-1β production. Cell Metab. 2019, 29, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, T.; Chasten, V.; Lacal, J.C.; Ibrahim, S.A. Identification and validation of novel and more effective choline kinase inhibitors against Streptococcus pneumoniae. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, T.; Lacal, J.C.; Ibrahim, S.A. Choline Kinase Emerges as a Promising Drug Target in Gram-Positive Bacteria. Front. Microbiol. 2019, 6, 2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, T.; Gyawali, R.; Ibrahim, S. Autolyse the cell in order to save it? Inducing, then blocking, autolysis as a strategy for delaying cell death in the probiotic Lactobacillus reuteri. Biotechnol. Lett. 2017, 39, 1547–1551. [Google Scholar] [CrossRef]
- Obanla, T.; Adjei-Fremah, S.; Gyawali, R.; Zimmerman, T.; Worku, M.; Ibrahim, S.A. Effects of Long Term Exposure to Aspirin on Growth, Functionality and Protein Profile of Lactobacillus rhamnosus (LGG) (ATCC 53103). J. Food Res. 2016, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Pacheco-Torres, J.; Penet, M.-F.; Mironchik, Y.; Krishnamachary, B.; Bhujwalla, Z.M. The PD-L1 metabolic interactome intersects with choline metabolism and inflammation. Cancer Metab. 2021, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Siu, K.-L.; Yuen, K.-S.; Castano-Rodriguez, C.; Ye, Z.-W.; Yeung, M.-L.; Fung, S.-Y.; Yuan, S.; Chan, C.-P.; Yuen, K.-Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Comp. | R4 | (IC50)ex vivo (μM) a | (IC50)HT-29 (μM) a | σR b | clog P c | πR4 d |
| 2, MN58B | –NMe2 | 17.0 | 2.00 | −0.88 | −2.83 | 0.18 e |
| 3 | –NH2 | 23.0 | 4.00 | −0.80 | −4.67 | −1.23 e |
| 4 | –CH2OH | 100 | >100 | −0.07 | −4.25 | −1.03 e |
| 5 | –CH3 | 100 | 20.0 | −0.16 | −1.82 | 0.56 e |
| 6 | –COOH | 136.7 | >1000 | 0.11 | −3.50 | −0.32 e |
| 7 | –C≡N | >1000 | 200 | 0.08 | −3.15 | −0.57 e |
| 8 | –N(Allyl)2 | 17.0 | 0.55 | −0.80 f | −0.44 | 1.34 e |
| 9 | -pyrrolidino | 20.0 | 1.00 | −0.85 f | −1.94 | 0.59 e |
| 10 | -piperidino | 9.60 | 0.40 | −0.89 f | −0.93 | 0.85 e |
| 11 | -perhydroazepino | 15.0 | 0.40 | −0.86 f | 0.09 | 1.60 e |
| 12 | –NMePh | 6.4 | 0.34 | −0.78 f | 0.37 | 1.67 e |
 | ||||||
|---|---|---|---|---|---|---|
| Comp. | X | Y | n | (IC50)ex vivo (μM) a | (IC50)HT-29 (μM) a | Clog P b |
| 13 | =N+– | =CH– | 0 | 50 | 10 | − |
| 14 | =N+– | =CH– | 1 | 100 | 6.02 | −0.86 |
| 15 | =N+– | =CH– | 2 | 34 | 4 | −0.43 |
| 16 | =N+– | =CH– | 3 | 9 | 2.5 | 0.08 |
| 17 | =CH– | =N+– | 0 | >100 | ND c | −0.85 |
| 18 | =CH– | =N+– | 1 | 60 | 20 | −1.00 |
| 19 | =CH– | =N+– | 2 | 60 | 20 | −0.57 |
| 20 | =CH– | =N+– | 3 | 20 | 2 | −0.06 |
 | ||||||
|---|---|---|---|---|---|---|
| Comp. | R4 | (IC50)ex vivo (μM) a | (IC50)HT-29 (μM) a | σR b | clog P c | πR4 d |
| 21 | –NH2 | 10.0 | 2.00 | −0.80 | −2.13 | −1.23 |
| 22 | –NHCOBut | 10.5 | 4.74 | −0.35 e | 1.40 | 2.18 |
 | |||||
|---|---|---|---|---|---|
| Comp. | Linker | R3 | R4 | (IC50)ex vivo (μM) a,b | (IC50)HT-29 (μM) a,c |
| 23 | Biphenyl-3,3′-diyl | H | Amino | 1.20 | 1.90 |
| 24 | Me | Amino | 11.9 | 4.40 | |
| 25 | H | Dimethylamino | 4.40 | 1.60 | |
| 26 | H | Perhydroazepino | 0.50 | 0.50 | |
| 27 | H | Anilino | 1.30 | 1.60 | |
| 28 | H | N-Methylanilino | 0.40 | 0.80 | |
| 29 | H | 4-Chloro-N-methylanilino | 2.10 | 1.50 | |
| 30 | Biphenyl-4,4′-diyl | H | Amino | 81.1 | 2.20 |
| 31 | Me | Amino | >200 | 3.30 | |
| 32 | H | Dimethylamino | 39.7 | 1.70 | |
| 33 | H | Perhydroazepino | 2.20 | 0.50 | |
| 34 | H | Anilino | 17.8 | 0.70 | |
| 35 | H | N-Methylanilino | 3.00 | 0.60 | |
| 36 | H | 4-Chloro-N-methylanilino | 2.00 | 1.20 | |
| 37 | Bibenzyl-4,4′-diyl | H | Amino | 80.0 | 2.00 |
| 38 | H | Dimethylamino | 10.2 | 0.50 | |
| 39 | H | Perhydroazepino | 0.60 | 0.30 | |
| 40 | H | Anilino | 2.30 | 0.30 | |
| 41 | H | N-Methylanilino | 1.40 | 0.70 | |
| 42 | H | 4-Chloro-N-methylanilino | 4.80 | 0.70 | |
 | ||||
|---|---|---|---|---|
| Comp. | Linker | R4 | (IC50)ex vivo (μM) a,b | (IC50)HT-29 (μM) a,c |
| 43 | Biphenyl-3,3′-diyl | Amino | 20.6 | 1.90 |
| 44 | Dimethylamino | 9.60 | 0.70 | |
| 45 | Pyrrolidino | 1.20 | 0.40 | |
| 46 | N-Methylanilino | 3.10 | 1.00 | |
| 47 | 4-Chloro-N-methylanilino | 5.70 | 1.90 | |
| 48 | Biphenyl-4,4′-diyl | Amino | 63.3 | 3.20 |
| 49 | Dimethylamino | 20.6 | 0.80 | |
| 50 | Pyrrolidino | 19.8 | 2.40 | |
| 51 | N-Methylanilino | 11.4 | 0.50 | |
| 52, RSM-932A | 4-Chloro-N-methylanilino | 11.4 | 1.20 | |
| 53 | Bibenzyl-4,4′-diyl | Amino | 80.0 | 2.00 |
| 54 | Dimethylamino | 9.00 | 0.27 | |
| 55 | Pyrrolidino | 1.00 | 0.20 | |
| 56 | N-Methylanilino | 3.50 | 0.50 | |
| 57 | 4-Chloro-N-methylanilino | 5.70 | 0.80 | |
 | ||||
|---|---|---|---|---|
| Comp. | Linker | R4 | (IC50)ex vivo (μM) a,b | (IC50)HT-29 (μM) a,c |
| 58 | Biphenyl-3,3′-diyl | N-Methylanilino | 56.8 | 3.80 |
| 59 | 4-Chloro-N-methylanilino | 147 | 33.0 | |
| 60 | Biphenyl-4,4′-diyl | N-Methylanilino | 96.1 | 25.2 |
| 61 | 4-Chloro-N-methylanilino | 46.1 | 19.4 | |
| 62 | Bibenzyl-4,4′-diyl | N-Methylanilino | 133 | 7.00 |
| 63 | 4-Chloro-N-methylanilino | 57.5 | 19.5 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lacal, J.C.; Zimmerman, T.; Campos, J.M. Choline Kinase: An Unexpected Journey for a Precision Medicine Strategy in Human Diseases. Pharmaceutics 2021, 13, 788. https://doi.org/10.3390/pharmaceutics13060788
Lacal JC, Zimmerman T, Campos JM. Choline Kinase: An Unexpected Journey for a Precision Medicine Strategy in Human Diseases. Pharmaceutics. 2021; 13(6):788. https://doi.org/10.3390/pharmaceutics13060788
Chicago/Turabian StyleLacal, Juan Carlos, Tahl Zimmerman, and Joaquín M. Campos. 2021. "Choline Kinase: An Unexpected Journey for a Precision Medicine Strategy in Human Diseases" Pharmaceutics 13, no. 6: 788. https://doi.org/10.3390/pharmaceutics13060788