
Physiologically Based Pharmacokinetic Modelling of Cabozantinib to Simulate Enterohepatic Recirculation, Drug–Drug Interaction with Rifampin and Liver Impairment


Abstract
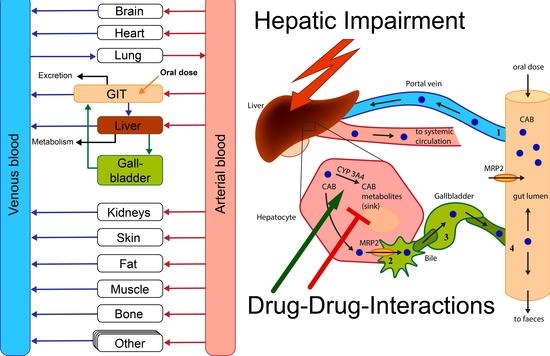
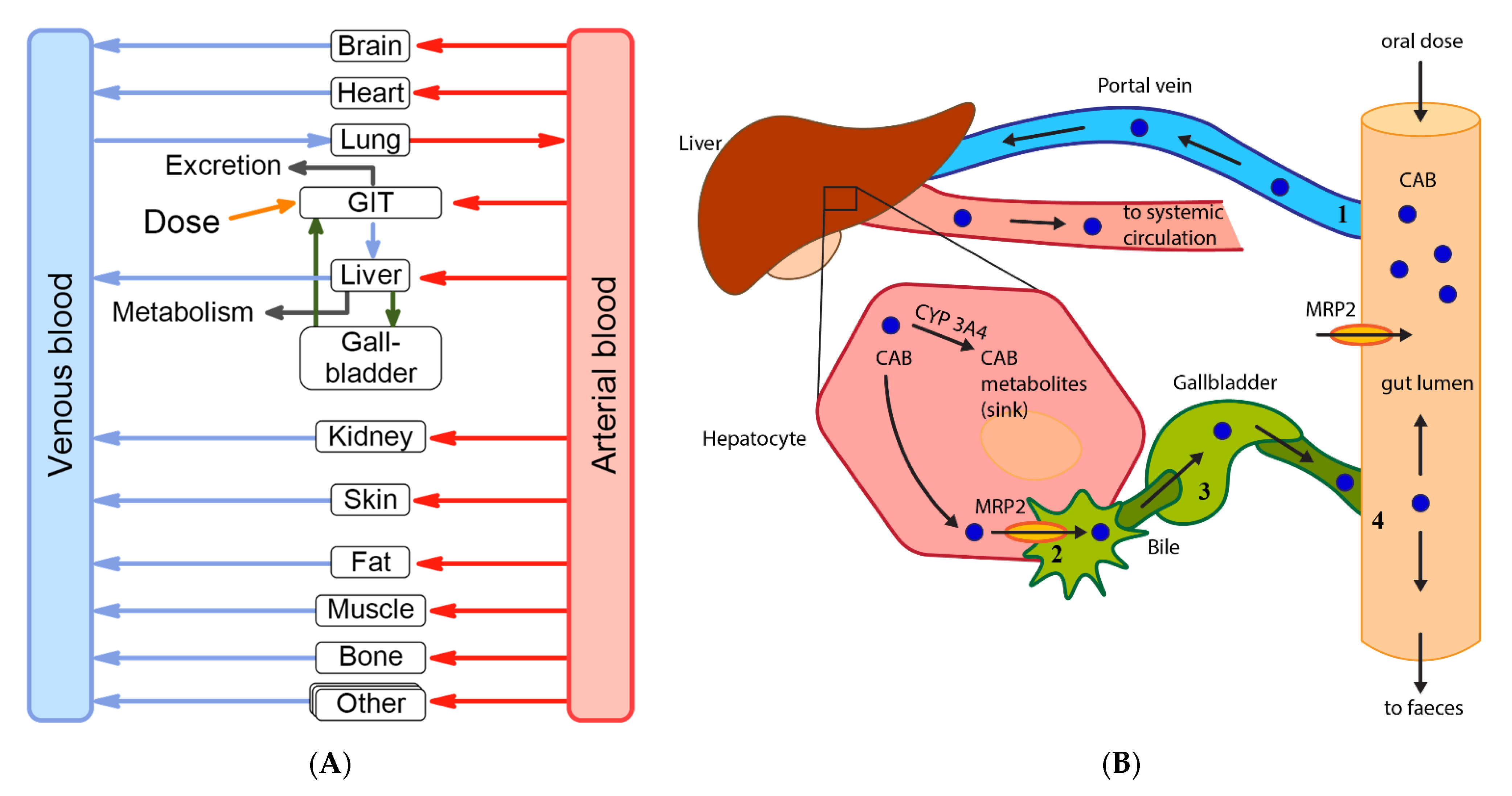
:
1. Introduction
2. Materials and Methods
2.1. Software
2.2. Clinical Data
2.3. Workflow and Model Development
2.4. Enterohepatic Circulation Modelling
2.5. Model Evaluation
2.6. DDI Interaction between CAB and RIF
2.7. Hepatic Impairment Simulations
3. Results
3.1. Rat Intravenous and Intragastric Simulations
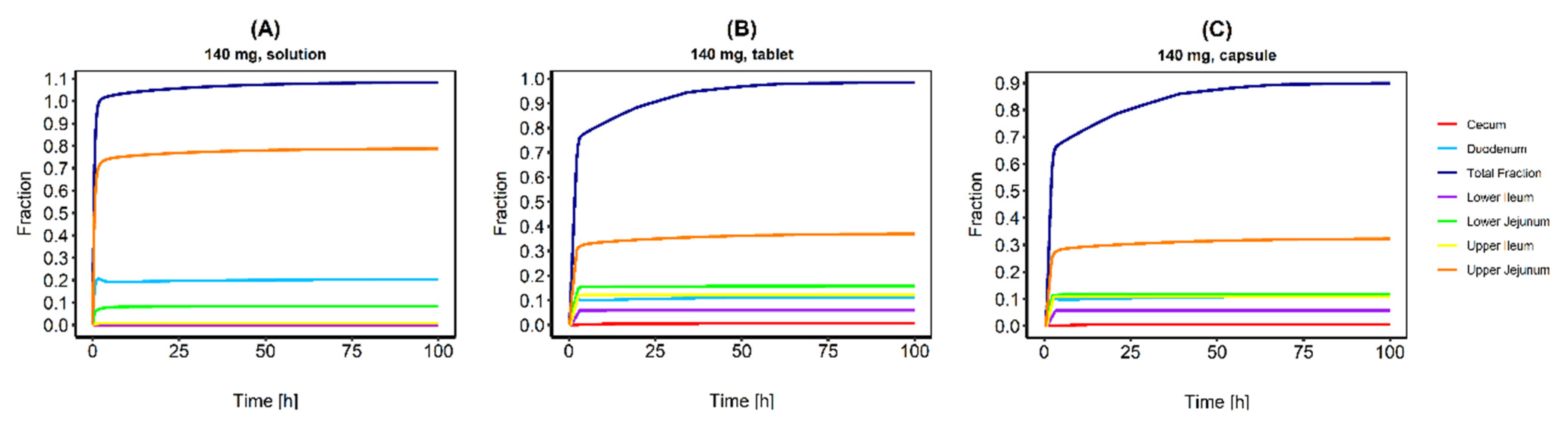
3.2. Human Peroral Simulations for Different CAB Formulations
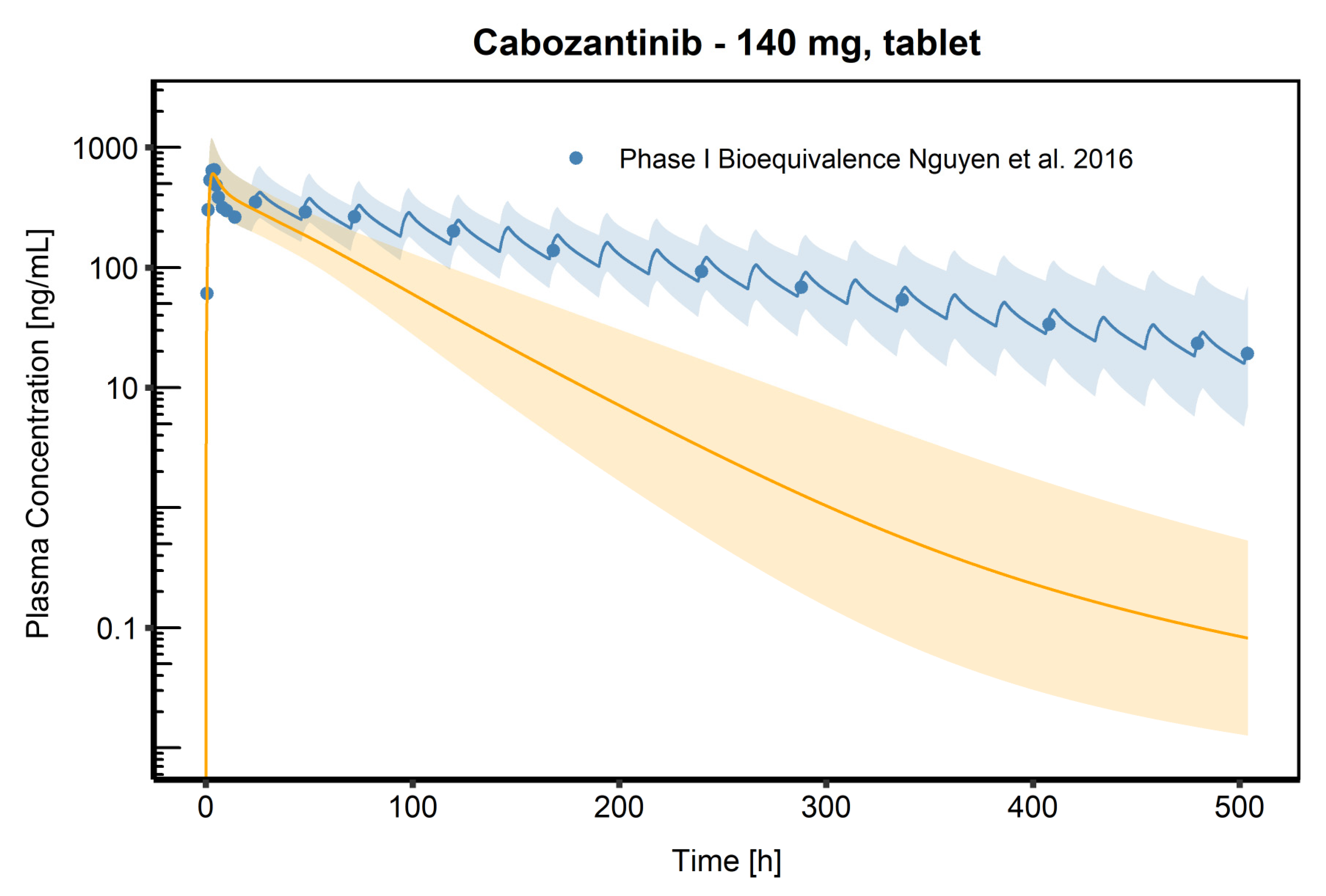
3.3. PBPK Model Evaluation
3.4. Simulations of DDI between CAB and RIF
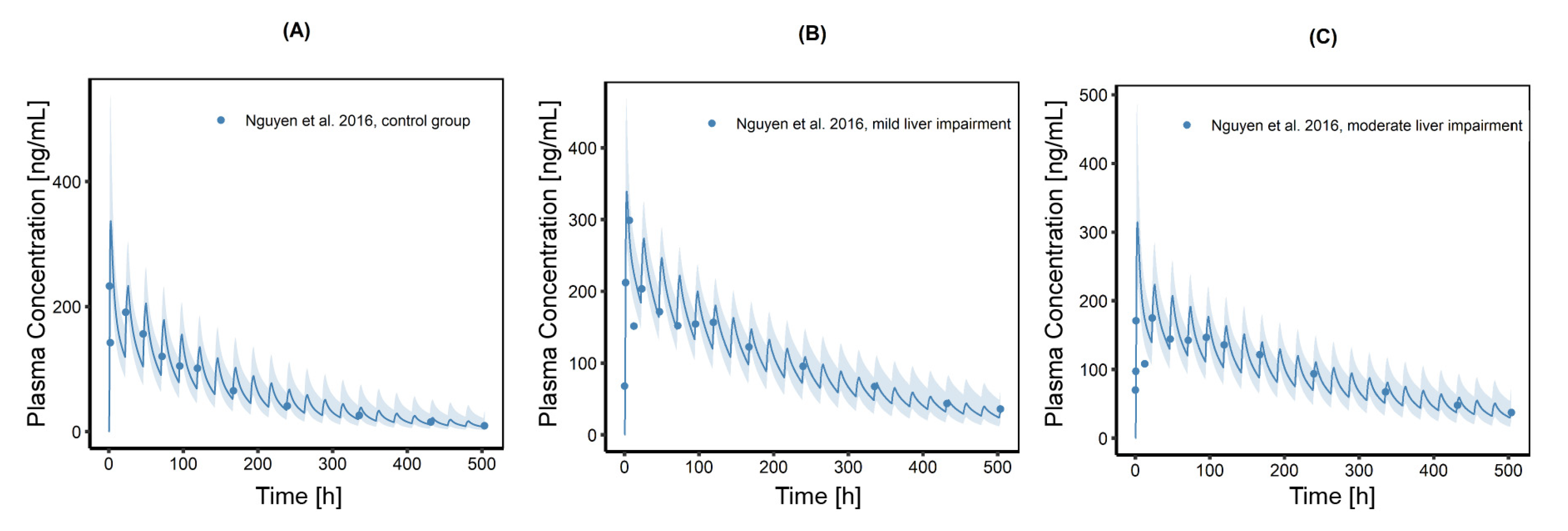
3.5. Investigation of Hepatic Impairment on CAB Plasma Exposure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kurzrock, R.; Sherman, S.I.; Ball, D.W.; Forastiere, A.A.; Cohen, R.B.; Mehra, R.; Pfister, D.G.; Cohen, E.E.; Janisch, L.; Nauling, F.; et al. Activity of XL184 (Cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J. Clin. Oncol. 2011, 29, 2660–2666. [Google Scholar] [CrossRef]
- Markowitz, J.N.; Fancher, K.M. Cabozantinib: A multitargeted oral tyrosine kinase inhibitor. Pharmacotherapy 2018, 38, 357–369. [Google Scholar] [CrossRef]
- Neul, C.; Schaeffeler, E.; Sparreboom, A.; Laufer, S.; Schwab, M.; Nies, A.T. Impact of Membrane Drug Transporters on Resistance to Small-Molecule Tyrosine Kinase Inhibitors. Trends Pharm. Sci. 2016, 37, 904–932. [Google Scholar] [CrossRef] [PubMed]
- Grüllich, C. Cabozantinib: A MET, RET, and VEGFR2 tyrosine kinase inhibitor. In Small Molecules in Oncology; Springer: Berlin/Heidelberg, Germany, 2014; pp. 207–214. [Google Scholar]
- Fallahi, P.; Ferrari, S.M.; Di Bari, F.; Materazzi, G.; Benvenga, S.; Miccoli, P.; Antonelli, A. Cabozantinib in thyroid cancer. Recent Pat. Anticancer Drug Discov. 2015, 10, 259–269. [Google Scholar] [CrossRef]
- Singh, H.; Brave, M.; Beaver, J.A.; Cheng, J.; Tang, S.; Zahalka, E.; Palmby, T.R.; Venugopal, R.; Song, P.; Liu, Q. US Food and Drug Administration approval: Cabozantinib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res. 2017, 23, 330–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannir, N.M.; Schwab, G.; Grünwald, V. Cabozantinib: An active novel multikinase inhibitor in renal cell carcinoma. Curr. Oncol. Rep. 2017, 19, 14. [Google Scholar] [CrossRef] [Green Version]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Cabozantinib: A Review in Advanced Hepatocellular Carcinoma. Target. Oncol. 2019, 14, 107–113. [Google Scholar] [CrossRef]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [Green Version]
- U.S. National Library of Medicine. ClinicalTrials.gov. Search Term: “Cabozantinib”. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=Cabozantinib&cntry=&state=&city=&dist= (accessed on 9 September 2020).
- Lacy, S.A.; Miles, D.R.; Nguyen, L.T. Clinical Pharmacokinetics and Pharmacodynamics of Cabozantinib. Clin. Pharm. 2017, 56, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Lacy, S.; Hsu, B.; Miles, D.; Aftab, D.; Wang, R.; Nguyen, L. Metabolism and Disposition of Cabozantinib in Healthy Male Volunteers and Pharmacologic Characterization of Its Major Metabolites. Drug Metab. Dispos. 2015, 43, 1190–1207. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.J.; Wu, H.J.; Sun, L.H.; Xu, X.; Mo, L.Y.; Zhang, L.; Zhang, J.Y.; Wu, C.Y. A sensitive LC-MS/MS method for simultaneous determination of cabozantinib and its metabolite cabozantinib N-oxide in rat plasma and its application in a pharmacokinetic study. Biomed. Chromatogr. 2018, 32, e4227. [Google Scholar] [CrossRef] [PubMed]
- Lehr, T.; Staab, A.; Tillmann, C.; Trommeshauser, D.; Schaefer, H.-G.; Kloft, C. A quantitative enterohepatic circulation model. Clin. Pharm. 2009, 48, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Metsugi, Y.; Miyaji, Y.; Ogawara, K.-I.; Higaki, K.; Kimura, T. Appearance of double peaks in plasma concentration–time profile after oral administration depends on gastric emptying profile and weight function. Pharm. Res. 2008, 25, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Suttle, A.B.; Pollack, G.M.; Brouwer, K.L. Use of a pharmacokinetic model incorporating discontinuous gastrointestinal absorption to examine the occurrence of double peaks in oral concentration–time profiles. Pharm. Res. 1992, 9, 350–356. [Google Scholar] [CrossRef]
- Nguyen, L.; Holland, J.; Miles, D.; Engel, C.; Benrimoh, N.; O’Reilly, T.; Lacy, S. Pharmacokinetic (PK) drug interaction studies of cabozantinib: Effect of CYP3A inducer rifampin and inhibitor ketoconazole on cabozantinib plasma PK and effect of cabozantinib on CYP2C8 probe substrate rosiglitazone plasma PK. J. Clin. Pharm. 2015, 55, 1012–1023. [Google Scholar] [CrossRef]
- European Medicines Agency. Cabometyx Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/cabometyx-epar-product-information_en.pdf (accessed on 9 March 2021).
- U.S. Food and Drug Administration. Physiologically Based Pharmacokinetic Analyses–Format and Content–Guidance for Industry. 2018. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/physiologically-based-pharmacokinetic-analyses-format-and-content-guidance-industry (accessed on 2 March 2021).
- Peters, S.A. Physiologically-Based Pharmacokinetic (PBPK) Modeling and Simulations: Principles, Methods, and Applications in the Pharmaceutical Industry, 1st ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Jones, H.; Rowland-Yeo, K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, W.; Willmann, S. Physiology-based pharmacokinetic modeling: Ready to be used. Drug Discov. Today Technol. 2004, 1, 449–456. [Google Scholar] [CrossRef]
- Jones, H.M.; Parrott, N.; Jorga, K.; Lavé, T. A novel strategy for physiologically based predictions of human pharmacokinetics. Clin. Pharm. 2006, 45, 511–542. [Google Scholar] [CrossRef]
- Open Systems Pharmacology. PK-Sim®. Version 7.4.0. Available online: https://github.com/Open-Systems-Pharmacology/Suite/releases/tag/7.4.0 (accessed on 26 September 2019).
- Eissing, T.; Kuepfer, L.; Becker, C.; Block, M.; Coboeken, K.; Gaub, T.; Goerlitz, L.; Jaeger, J.; Loosen, R.; Ludewig, B.; et al. A computational systems biology software platform for multiscale modeling and simulation: Integrating whole-body physiology, disease biology, and molecular reaction networks. Front. Physiol. 2011, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Willmann, S.; Lippert, J.; Sevestre, M.; Solodenko, J.; Fois, F.; Schmitt, W. PK-Sim®: A physiologically based pharmacokinetic ‘whole-body’ model. Biosilico 2003, 1, 121–124. [Google Scholar] [CrossRef]
- Kuepfer, L.; Niederalt, C.; Wendl, T.; Schlender, J.F.; Willmann, S.; Lippert, J.; Block, M.; Eissing, T.; Teutonico, D. Applied Concepts in PBPK Modeling: How to Build a PBPK/PD Model. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 516–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Open Systems Pharmacology. PK-Sim® Software Manual. Available online: https://docs.open-systems-pharmacology.org/ (accessed on 26 September 2019).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.eea.europa.eu/data-and-maps/indicators/oxygen-consuming-substances-in-rivers/r-development-core-team-2006 (accessed on 30 April 2021).
- Brightman, F.A.; Leahy, D.E.; Searle, G.E.; Thomas, S. Application of a generic physiologically based pharmacokinetic model to the estimation of xenobiotic levels in rat plasma. Drug Metab. Dispos. 2006, 34, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jedlitschky, G.; Hoffmann, U.; Kroemer, H.K. Structure and function of the MRP2 (ABCC2) protein and its role in drug disposition. Expert Opin. Drug Metab. Toxicol. 2006, 2, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Baier, V.; Cordes, H.; Thiel, C.; Castell, J.V.; Neumann, U.P.; Blank, L.M.; Kuepfer, L. A Physiology-Based Model of Human Bile Acid Metabolism for Predicting Bile Acid Tissue Levels After Drug Administration in Healthy Subjects and BRIC Type 2 Patients. Front. Physiol. 2019, 10, 1192. [Google Scholar] [CrossRef] [Green Version]
- European Medicines Agency. Guideline on the Reporting of Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulation. 2018. Available online: https://www.ema.europa.eu/en/reporting-physiologically-based-pharmacokinetic-pbpk-modelling-simulation (accessed on 12 March 2020).
- Kuemmel, C.; Yang, Y.; Zhang, X.; Florian, J.; Zhu, H.; Tegenge, M.; Huang, S.M.; Wang, Y.; Morrison, T.; Zineh, I. Consideration of a credibility assessment framework in model-informed drug development: Potential application to physiologically-based pharmacokinetic modeling and simulation. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Hanke, N.; Frechen, S.; Moj, D.; Britz, H.; Eissing, T.; Wendl, T.; Lehr, T. PBPK models for CYP3A4 and P-gp DDI prediction: A modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil, and digoxin. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 647–659. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.; Holland, J.; Ramies, D.; Mamelok, R.; Benrimoh, N.; Ciric, S.; Marbury, T.; Preston, R.A.; Heuman, D.M.; Gavis, E.; et al. Effect of Renal and Hepatic Impairment on the Pharmacokinetics of Cabozantinib. J. Clin. Pharm. 2016, 56, 1130–1140. [Google Scholar] [CrossRef]
- Edginton, A.N.; Willmann, S. Physiology-based simulations of a pathological condition. Clin. Pharm. 2008, 47, 743–752. [Google Scholar] [CrossRef]
- Johnson, T.N.; Boussery, K.; Rowland-Yeo, K.; Tucker, G.T.; Rostami-Hodjegan, A. A semi-mechanistic model to predict the effects of liver cirrhosis on drug clearance. Clin. Pharm. 2010, 49, 189–206. [Google Scholar] [CrossRef]
- George, J.; Murray, M.; Byth, K.; Farrell, G.C. Differential alterations of cytochrome P450 proteins in livers from patients with severe chronic liver disease. Hepatology 1995, 21, 120–128. [Google Scholar]
- Martignoni, M.; Groothuis, G.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef] [PubMed]
- Colclough, N.; Ruston, L.; Wood, J.M.; MacFaul, P.A. Species differences in drug plasma protein binding. MedChemComm 2014, 5, 963–967. [Google Scholar] [CrossRef]
- Lin, Q.M.; Li, Y.H.; Lu, X.R.; Wang, R.; Pang, N.H.; Xu, R.A.; Cai, J.P.; Hu, G.X. Characterization of Genetic Variation in CYP3A4 on the Metabolism of Cabozantinib in Vitro. Chem. Res. Toxicol. 2019, 32, 1583–1590. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. CABOMETYX (cabozantinib) Tablets. Chemistry Review(s). 2015. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/208692Orig1s000ChemR.pdf (accessed on 15 January 2020).
- Ipsen Biopharmaceuticals Canada Inc. CABOMETYX Product Monograph. 2019. Available online: https://ipsen.com/websites/IPSENCOM-PROD/wp-content/uploads/sites/18/2019/11/21094828/Cabometyx-PM-EN-07Nov2019.pdf (accessed on 15 January 2020).
- Deo, A.K.; Prasad, B.; Balogh, L.; Lai, Y.; Unadkat, J.D. Interindividual variability in hepatic expression of the multidrug resistance-associated protein 2 (MRP2/ABCC2): Quantification by liquid chromatography/tandem mass spectrometry. Drug Metab. Dispos. 2012, 40, 852–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically based pharmacokinetic modeling 1: Predicting the tissue distribution of moderate-to-strong bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.; Rowland, M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef]
- Kroiss, M.; Megerle, F.; Kurlbaum, M.; Zimmermann, S.; Wendler, J.; Jimenez, C.; Lapa, C.; Quinkler, M.; Scherf-Clavel, O.; Habra, M.A. Objective response and prolonged disease control of advanced adrenocortical carcinoma with cabozantinib. J. Clin. Endocrinol. Metab. 2020, 105, 1461–1468. [Google Scholar] [CrossRef]
- Abbiati, R.A.; Manca, D. Enterohepatic Circulation Effect in Physiologically Based Pharmacokinetic Models: The Sorafenib Case. Ind. Eng. Chem. Res. 2017, 56, 3156–3166. [Google Scholar] [CrossRef]
- European Medicines Agency. Assessment Report Cometriq. 2013. Available online: https://www.ema.europa.eu/en/documents/assessment-report/cometriq-epar-public-assessment-report_en.pdf (accessed on 9 October 2019).
- European Medicines Agency. CHMP Assessment Report Cabometyx. 2016. Available online: https://www.ema.europa.eu/en/documents/assessment-report/cabometyx-epar-public-assessment-report_en.pdf (accessed on 9 October 2019).
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2012, 64, 138–153. [Google Scholar] [CrossRef]
- D’Angelo, A.; Sobhani, N.; Bagby, S.; Casadei-Gardini, A.; Roviello, G. Cabozantinib as a second-line treatment option in hepatocellular carcinoma. Expert Rev. Clin. Pharm. 2020, 13, 623–629. [Google Scholar] [CrossRef]
- Fuhr, L.M.; Marok, F.Z.; Hanke, N.; Selzer, D.; Lehr, T. Pharmacokinetics of the CYP3A4 and CYP2B6 Inducer Carbamazepine and Its Drug–Drug Interaction Potential: A Physiologically Based Pharmacokinetic Modeling Approach. Pharmaceutics 2021, 13, 270. [Google Scholar] [CrossRef] [PubMed]
- Castellano, D.; Maroto, J.P.; Benzaghou, F.; Taguieva, N.; Nguyen, L.; Clary, D.O.; Jonasch, E. Exposure-response modeling of cabozantinib in patients with renal cell carcinoma: Implications for patient care. Cancer Treat. Rev. 2020, 89. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.S.; Magnusson, B.M.; Burczynski, F.J.; Weiss, M. Enterohepatic circulation. Clin. Pharm. 2002, 41, 751–790. [Google Scholar] [CrossRef]
- van Erp, N.P.; Gelderblom, H.; Guchelaar, H.-J. Clinical pharmacokinetics of tyrosine kinase inhibitors. Cancer Treat. Rev. 2009, 35, 692–706. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.; Jumbe, N.L.; Lacy, S.; Nguyen, L. Population pharmacokinetic model of cabozantinib in patients with medullary thyroid carcinoma and its application to an exposure-response analysis. Clin. Pharmacokinet. 2016, 55, 93–105. [Google Scholar] [CrossRef]
- Spinella, R.; Sawhney, R.; Jalan, R. Albumin in chronic liver disease: Structure, functions and therapeutic implications. Hepatol. Int. 2016, 10, 124–132. [Google Scholar] [CrossRef]
- Carvalho, J.R.; Machado, M.V. New insights about albumin and liver disease. Ann. Hepatol. 2018, 17, 547–560. [Google Scholar] [CrossRef]
- Cheeti, S.; Budha, N.R.; Rajan, S.; Dresser, M.J.; Jin, J.Y. A physiologically based pharmacokinetic (PBPK) approach to evaluate pharmacokinetics in patients with cancer. Biopharm. Drug Dispos. 2013, 34, 141–154. [Google Scholar] [CrossRef]
- Oswald, M.; Kullak-Ublick, G.A.; Paumgartner, G.; Beuers, U. Expression of hepatic transporters OATP-C and MRP2 in primary sclerosing cholangitis. Liver 2001, 21, 247–253. [Google Scholar] [CrossRef]
- Pauli-Magnus, C.; Meier, P.J. Hepatobiliary transporters and drug-induced cholestasis. Hepatology 2006, 44, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Borst, P.; Zelcer, N.; Van De Wetering, K. MRP2 and 3 in health and disease. Cancer Lett. 2006, 234, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Schrenk, D.; Baus, P.R.; Ermel, N.; Klein, C.; Vorderstemann, B.; Kauffmann, H.-M. Up-regulation of transporters of the MRP family by drugs and toxins. Toxicol. Lett. 2001, 120, 51–57. [Google Scholar] [CrossRef]
- Kullak-Ublick, G.A.; Stieger, B.; Meier, P.J. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology 2004, 126, 322–342. [Google Scholar] [CrossRef] [PubMed]
- Mottino, A.D.; Catania, V.A. Hepatic drug transporters and nuclear receptors: Regulation by therapeutic agents. World J. Gastroenterol. WJG 2008, 14, 7068. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Unit | Value Used in PBPK Model | Literature Value [Reference] | Description |
|---|---|---|---|---|
| MW | [g/mol] | 501.50 | 501.50 [44] | Molecular weight |
| pKa [base] | 6.32 | 6.32 [45] | Acid dissociation constant | |
| fup | 0.24 | 0.24 [13] | Fraction unbound in plasma | |
| logP | 4.40 a | 5.15 [45] | Lipophilicity | |
| Solubility (pH 6.5) | [10−3 mg/mL] | 7.72 a | 0.00 [44] | Solubility |
| KM CYP3A4 | [µmol/L] | 0.97 a | 21.32 [43] | Michaelis-Menten constant CYP3A4 |
| kcat CYP3A4 | [1/min] | 0.67 | 0.66 [43] | Katalytic rate constant CYP3A4 |
| KM MRP2 | [µmol/L] | 10 a | -- | Michaelis-Menten constant MRP2 |
| kcat MRP2 | [1/min] | 2111.11 a | -- | Transport rate constant MRP2 |
| Reference concentration MRP2 | [µmol protein/l in the tissue of highest expression] | 0.09 | 0.06 [46] | Liver reference concentration |
| Partition coefficients | Rodgers and Rowland | [47,48] | Calculation method cell to plasma coefficients | |
| Cellular permeabilities | PKSim® Standard | [29] | Calculation method permeation across cell membranes | |
| Transcellular intestinal permeability | [10−4 cm/min] | 1.70 a | -- | Intestinal permeability via transcellular route |
| Tablet Weibull time | [min] | 36.00 a | Dissolution time (50% dissolved) fasted state | |
| Tablet Weibull shape | 1.29 a | Shape parameter of Weibull function | ||
| Capsule Weibull time | [min] | 45.00 | Dissolution time (50% dissolved) fasted state | |
| Capsule Weibull shape | 5.00 | Shape parameter of Weibull function | ||
| Emptying half-time | [min] | 41.44 | 69.98 [33] | Half-time for gallbladder emptying (exponential release) |
| EHC continuous fraction | 0.1 | Fraction of biliary secreted compound continuously entering duodenum | ||
| Gallbladder ejection fraction | 0.45 | Fraction discretely ejected into the duodenum | ||
| Refilling time | [min] | 241.00 | 419 [33] | Time to complete gallbladder refilling |
| PK Parameter | CAB with RIF | CAB without RIF | Ratio |
|---|---|---|---|
| AUClast | 13,624.61 | 59,738.08 | 0.23 |
| Cmax | 480.46 | 551.17 | 0.87 |
| Parameter | Control Group | Mild Hepatic Impairment | Moderate Hepatic Impairment |
|---|---|---|---|
| Hematocrit value b | 0.47 a | 0.39 b | 0.37 b |
| Blood flow b | |||
| portal | 1.00 | 0.40 | 0.36 |
| renal | 1.00 | 0.88 | 0.65 |
| hepatic arterial | 1.00 | 1.30 | 2.30 |
| other organs | 1.00 | 1.75 | 2.25 |
| Liver volume fractionb | 1.00 | 0.81 | 0.65 |
| CYP3A4 activity | 1.00 | 1.00 | 0.40 |
| Plasma protein scale factor | 1.00 a | 0.85 | 1.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerner, B.; Scherf-Clavel, O. Physiologically Based Pharmacokinetic Modelling of Cabozantinib to Simulate Enterohepatic Recirculation, Drug–Drug Interaction with Rifampin and Liver Impairment. Pharmaceutics 2021, 13, 778. https://doi.org/10.3390/pharmaceutics13060778
Gerner B, Scherf-Clavel O. Physiologically Based Pharmacokinetic Modelling of Cabozantinib to Simulate Enterohepatic Recirculation, Drug–Drug Interaction with Rifampin and Liver Impairment. Pharmaceutics. 2021; 13(6):778. https://doi.org/10.3390/pharmaceutics13060778
Chicago/Turabian StyleGerner, Bettina, and Oliver Scherf-Clavel. 2021. "Physiologically Based Pharmacokinetic Modelling of Cabozantinib to Simulate Enterohepatic Recirculation, Drug–Drug Interaction with Rifampin and Liver Impairment" Pharmaceutics 13, no. 6: 778. https://doi.org/10.3390/pharmaceutics13060778