
Growth Inhibitory Effects of Ester Derivatives of Menahydroquinone-4, the Reduced Form of Vitamin K2(20), on All-Trans Retinoic Acid-Resistant HL60 Cell Line

Abstract
:1. Introduction
2. Materials and Methods
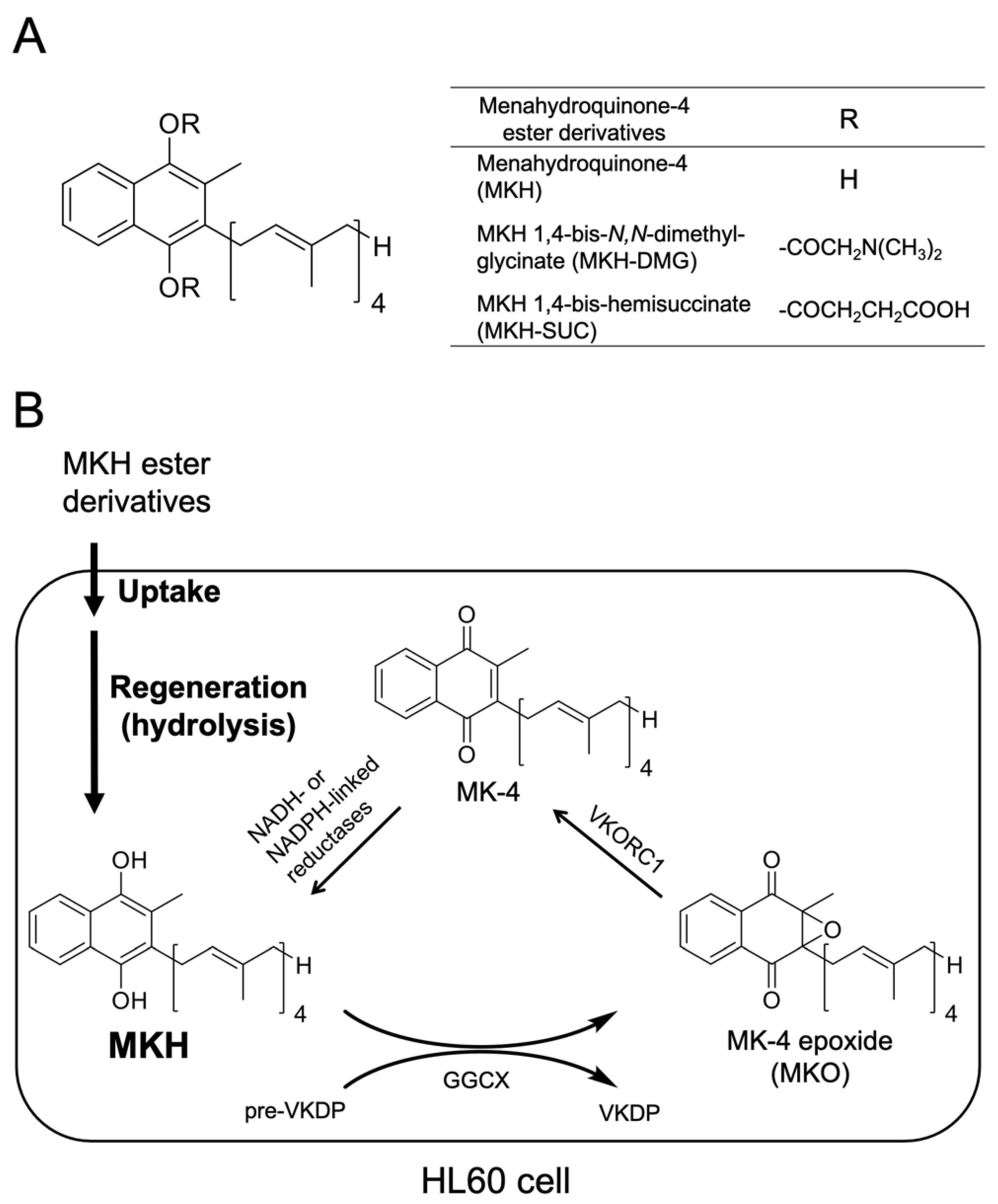
2.1. Chemicals
2.2. Cell Lines
2.3. Cell Viability Assay
2.4. Determination of Intracellular MK-4 and MKO Levels after Drug Treatment
2.5. LC-MS/MS
2.6. Cell Cycle Analysis
2.7. Apoptosis Detection
2.8. Mitochondrial Membrane Potential
2.9. Determination of Cytochrome C in Mitochondrial and Cytosolic Fractions
2.10. Western Blotting
2.11. Statistical Analysis
3. Results
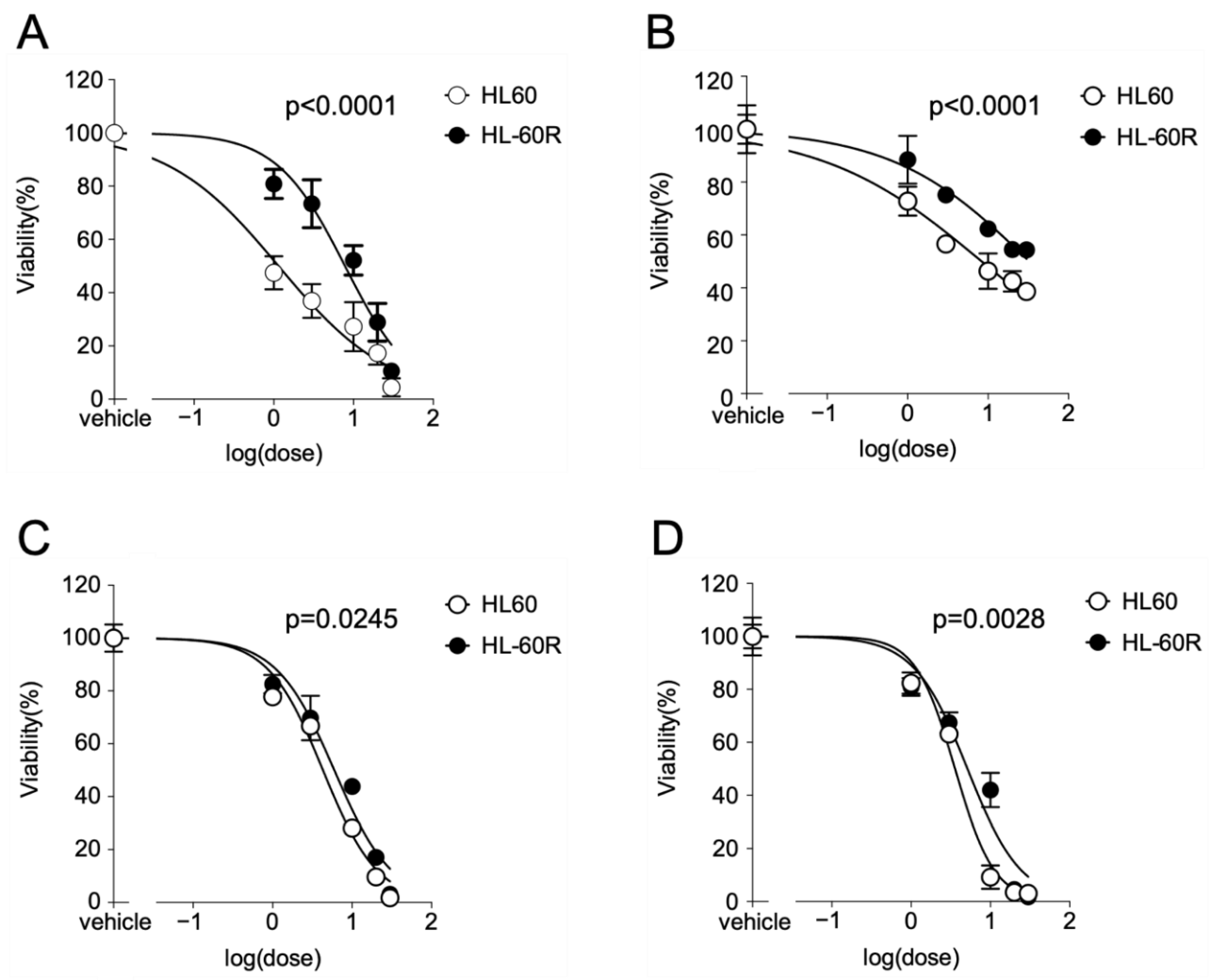
3.1. Growth-Inhibitory Effects of MKH Derivatives on HL60 and HL-60R Cells
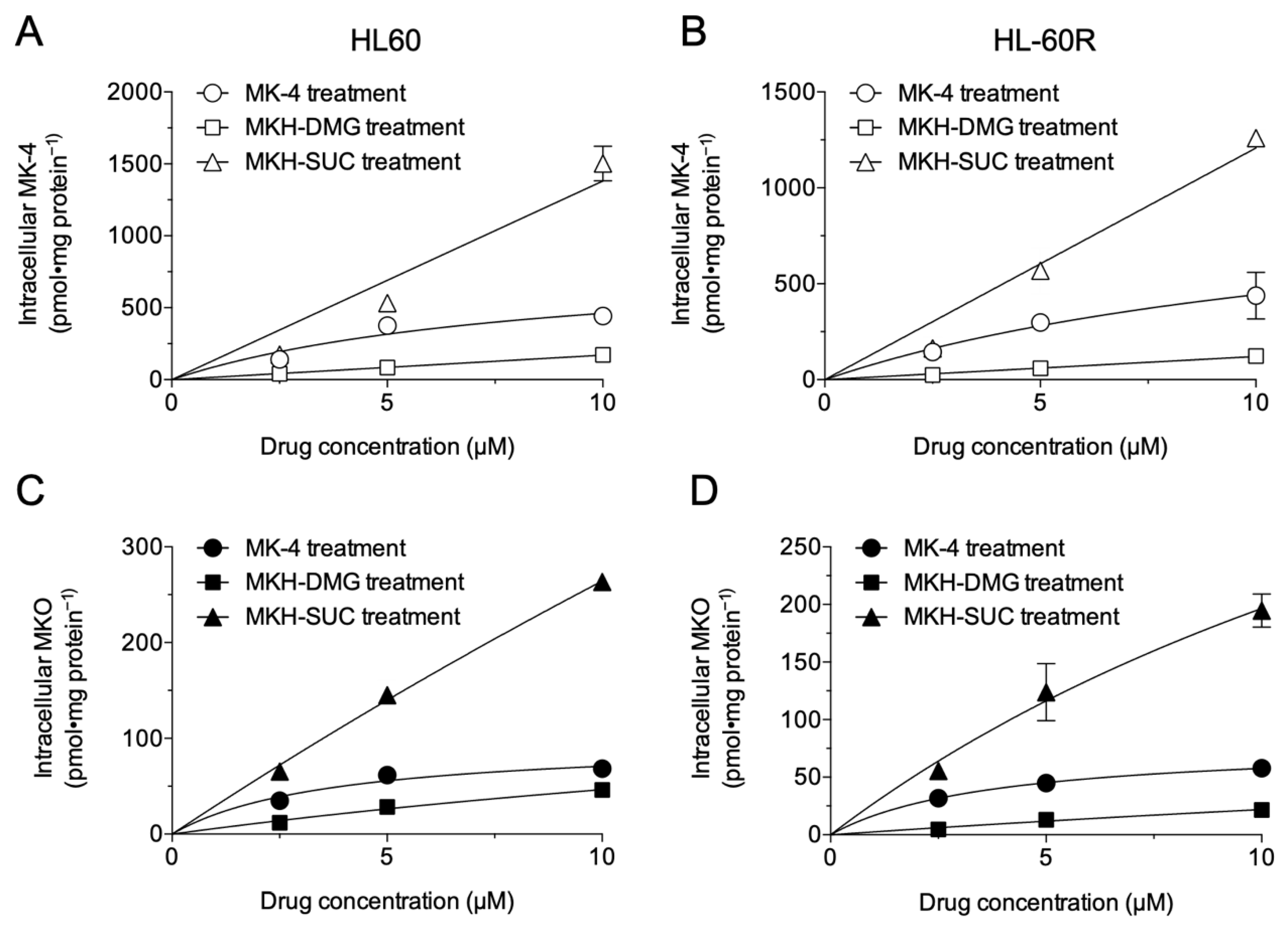
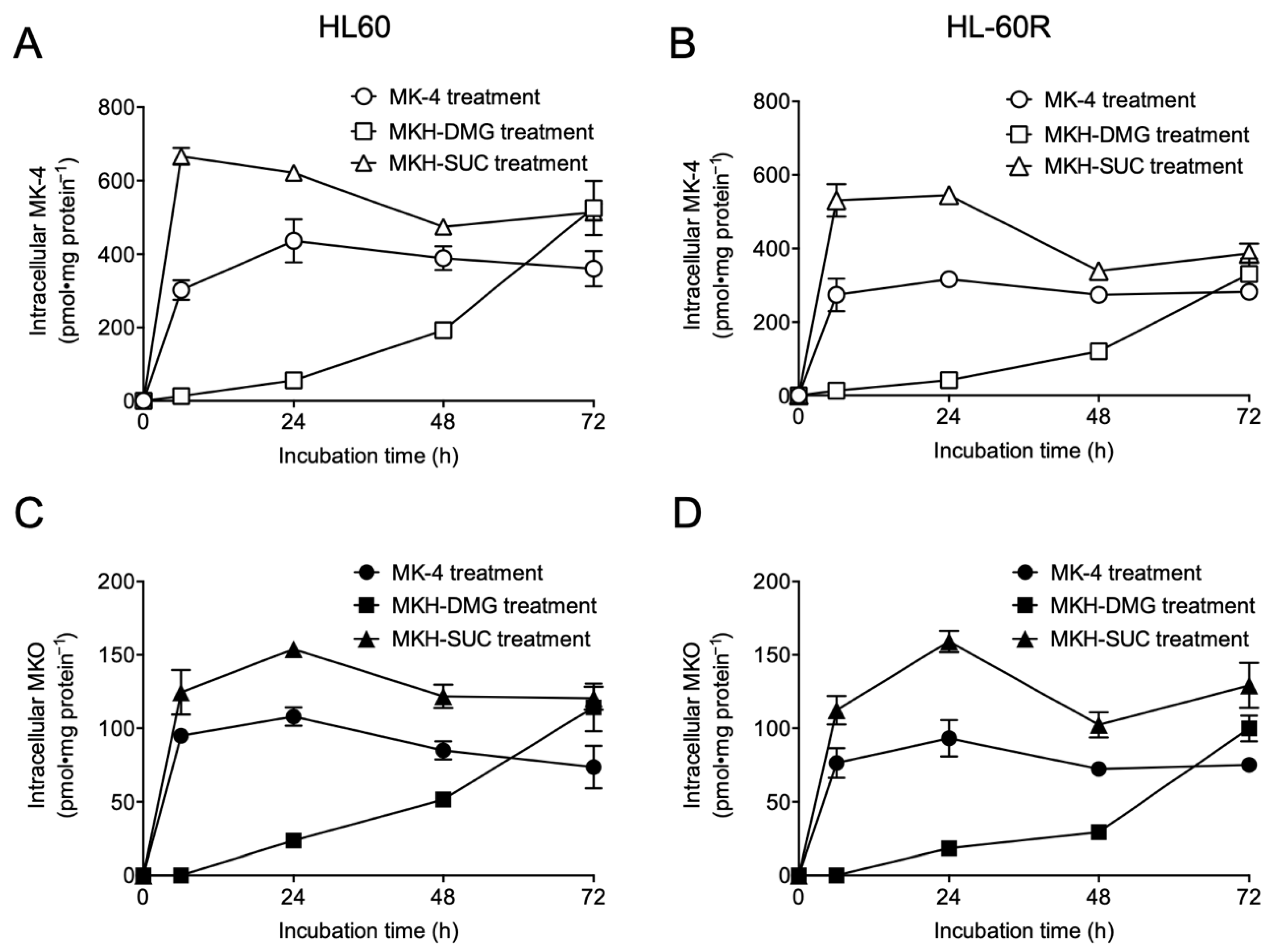
3.2. Intracellular Drug Levels in HL60 and HL-60R Cells Treated with MK-4 and MKH Derivatives
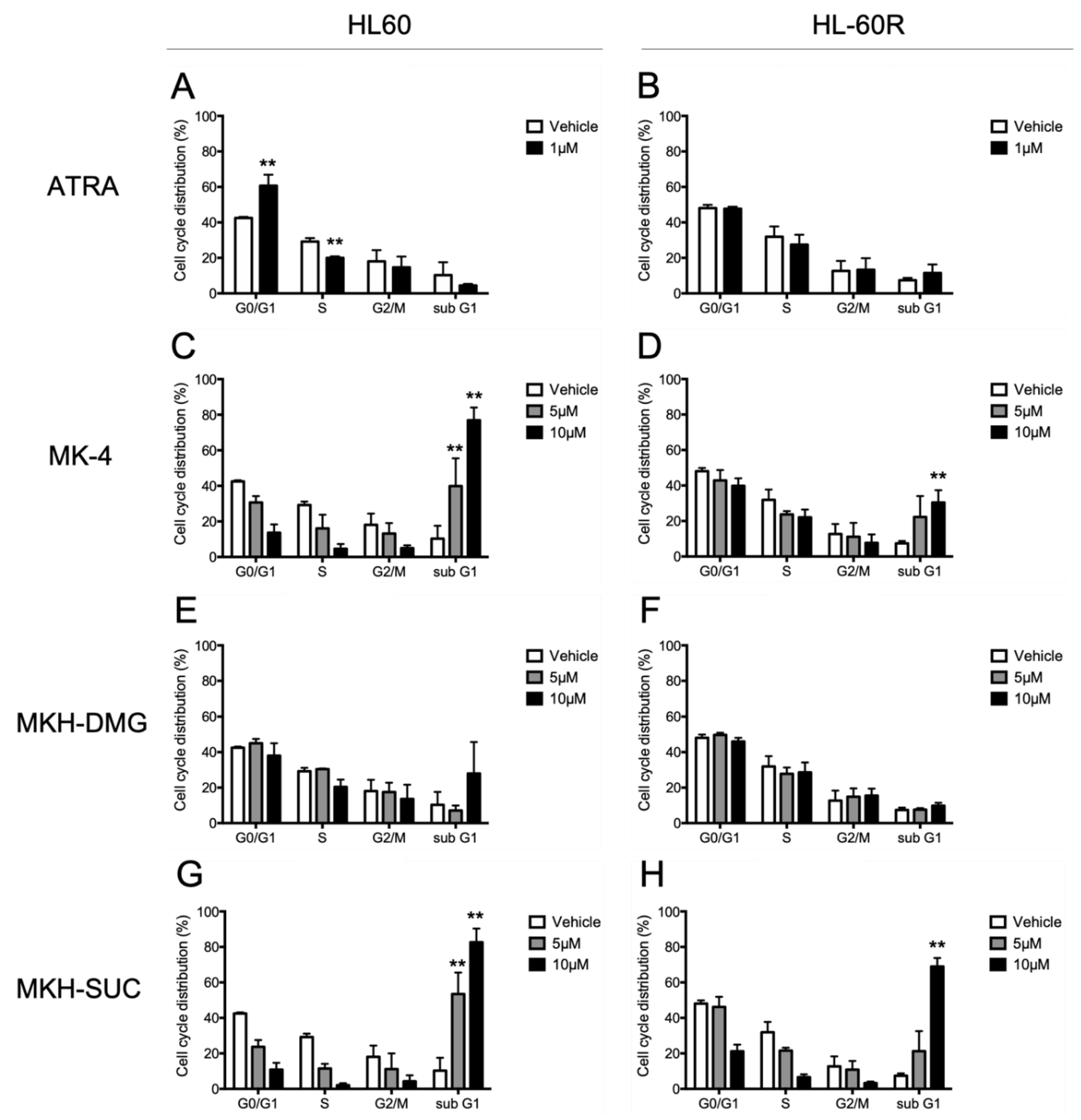
3.3. Effects of MKH Derivatives on the Cell Cycle of HL60 and HL-60R Cells
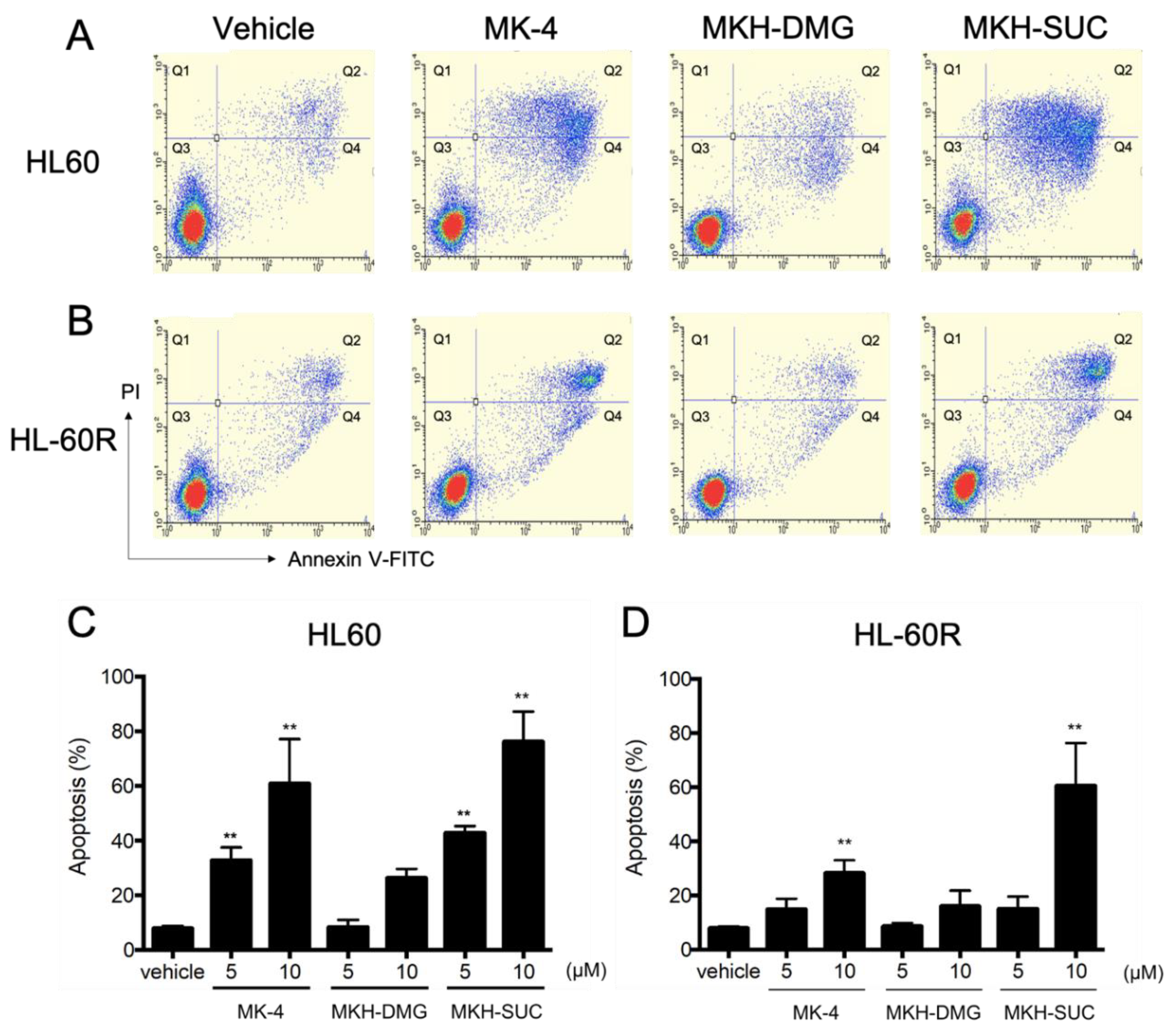
3.4. Effects of MKH Derivatives on the Cell Apoptosis of HL60 and HL-60R Cells
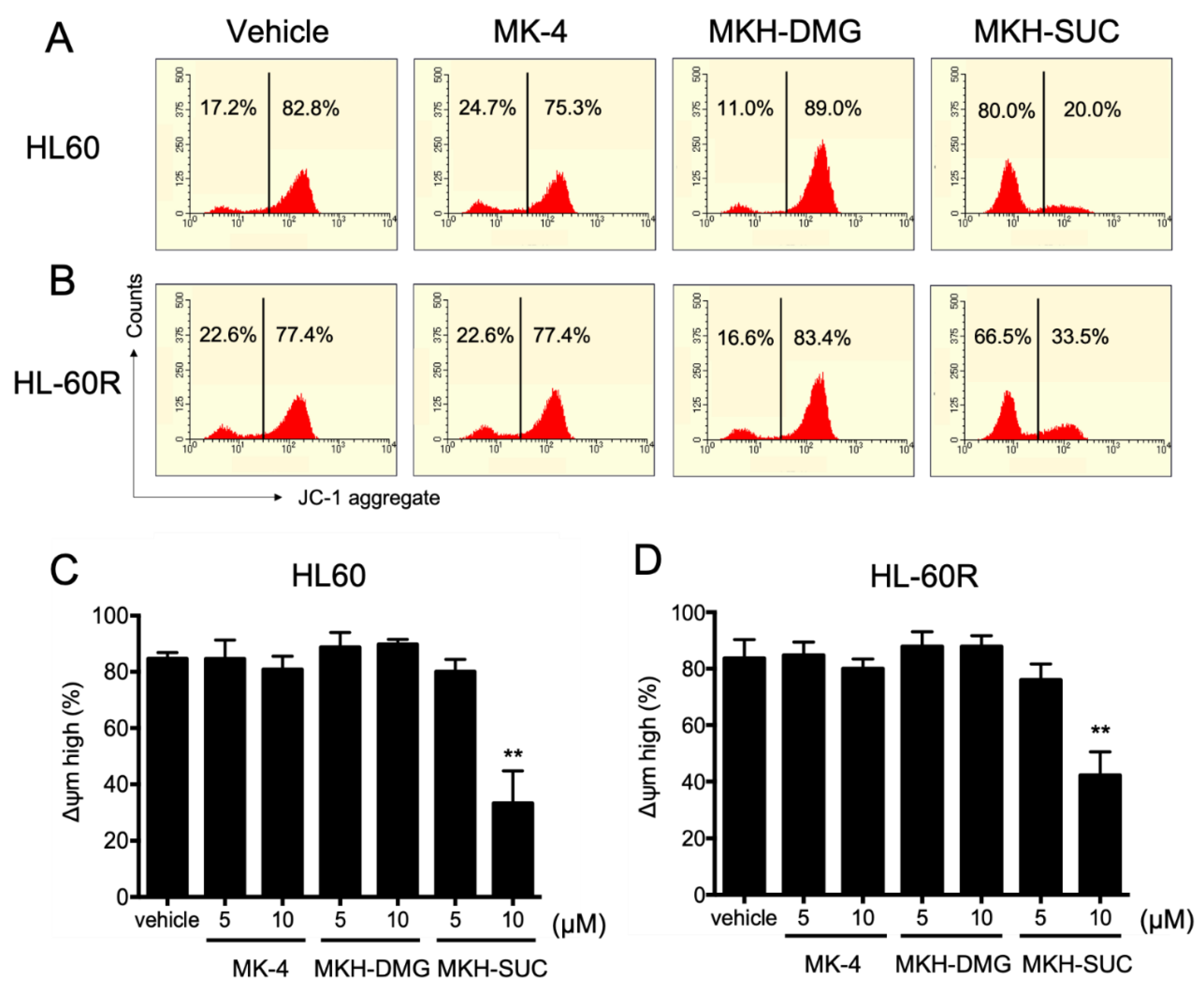
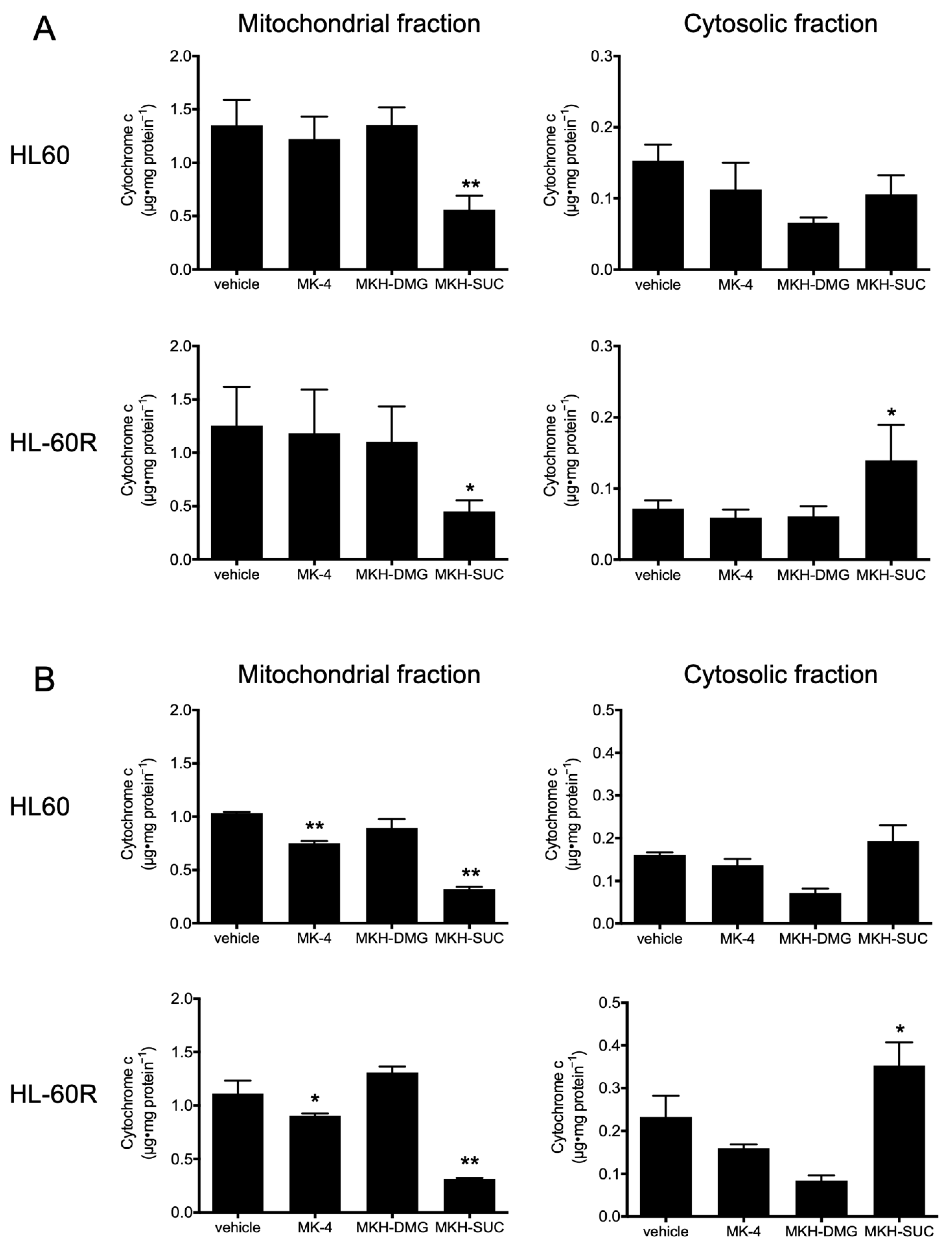
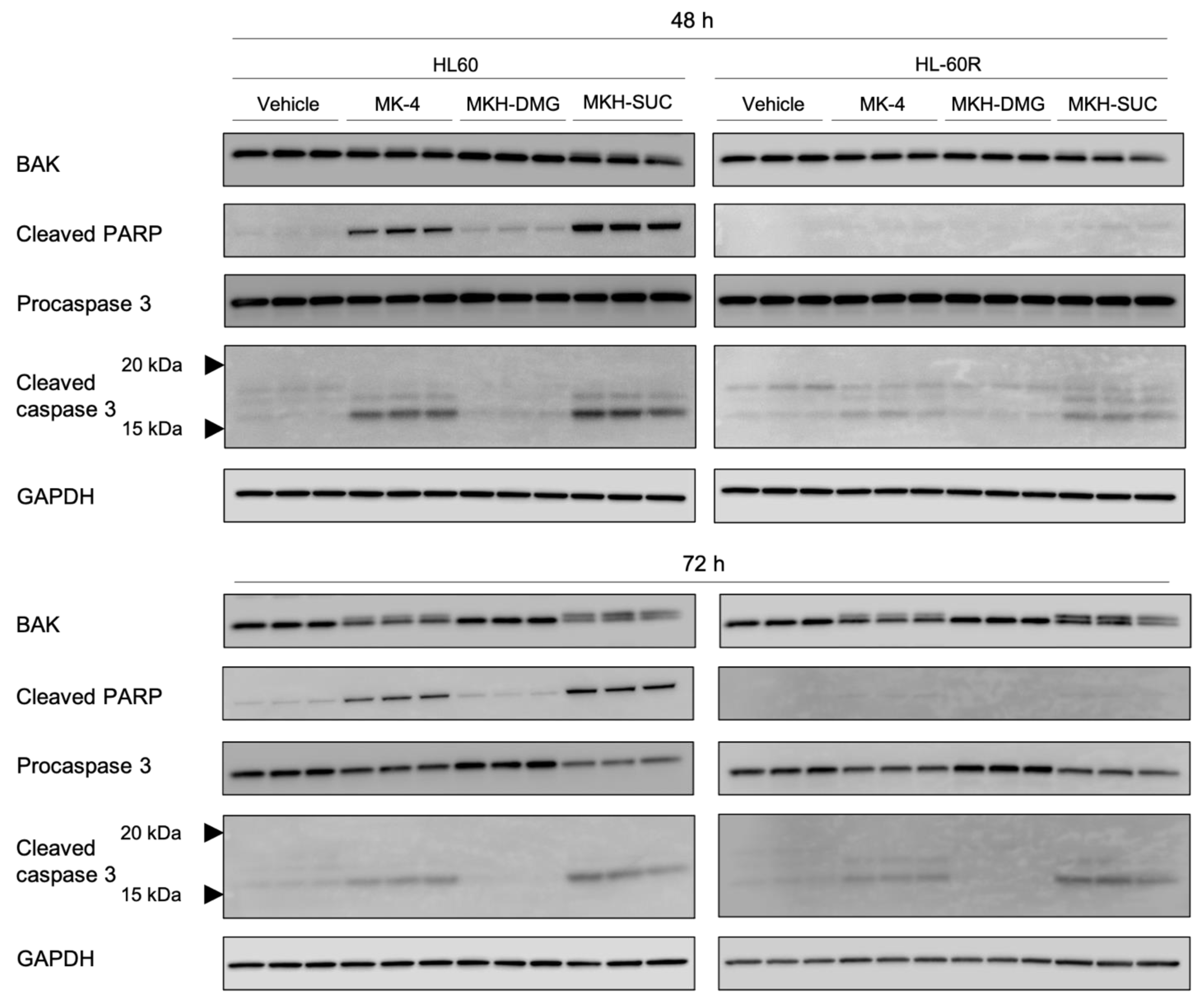
3.5. Effect of MKH Derivatives on Mitochondrial Apoptotic Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thomas, X. Acute Promyelocytic Leukemia: A History over 60 Years—From the Most Malignant to the most Curable Form of Acute Leukemia. Oncol. Ther. 2019, 7, 33–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.E.; Ye, Y.C.; Chen, S.R.; Chai, J.R.; Lu, J.X.; Zhoa, L.; Gu, L.J.; Wang, Z.Y. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masue, I. Molecular Mechanism of the Leukemogenesis and Differentiation-Induction in Acute Promyelocytic Leukemia. Jpn. J. Pediatr. Hematol. 2002, 16, 50–61. [Google Scholar]
- Kimitaka, T.; Hiroshi, T. Pharmacokinetics of all-trans retinoic acid in Japanese patients with acute promyelocytic leukemia. Vitamins 2011, 85, 631–644. [Google Scholar]
- Tomita, A.; Kiyoi, H.; Naoe, T. Mechanisms of action and resistance to all-trans retinoic acid (ATRA) and arsenic trioxide (As2O3) in acute promyelocytic leukemia. Int. J. Hematol. 2013, 97, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Honma, Y.; Takenaga, K.; Kasukabe, T.; Hozumi, M. Induction of differentiation of cultured human promyelocytic leukemia cells by retinoids. Biochem. Biophys. Res. Commun. 1980, 95, 507–512. [Google Scholar] [CrossRef]
- Breitman, T.R.; Selonick, S.E.; Collins, S.J. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. USA 1980, 77, 2936–2940. [Google Scholar] [CrossRef] [Green Version]
- Dalton, W.T.; Ahearn, J.M.; McCredie, K.B.; Freireich, E.J.; Stass, S.A.; Trujillo, J.M. HL-60 cell line was derived from a patient with FAB-M2 and not FAB-M3. Blood 1988, 71, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Yaguchi, M.; Miyazawa, K.; Katagiri, T.; Nishimaki, J.; Kizaki, M.; Tohyama, K. Vitamin K2 and its derivatives induce apoptosis in leukemia cells and enhance the effect of all-trans retinoic acid. Leukemia 1997, 11, 779–787. [Google Scholar] [CrossRef] [Green Version]
- Yaguchi, M.; Miyazawa, K.; Otawa, M.; Katagiri, T.; Nishimaki, J.; Uchida, Y.; Iwase, O.; Gotoh, A.; Kawanishi, Y.; Toyama, K. Vitamin K2 selectively induces apoptosis of blastic cells in myelodysplastic syndrome: Flow cytometric detection of apoptotic cells using APO2.7 monoclonal antibody. Leukemia 1998, 12, 1392–1397. [Google Scholar] [CrossRef] [Green Version]
- Sakai, I.; Hashimoto, S.; Yoda, M.; Hida, T.; Ohsawa, S.; Nakajo, S.; Nakaya, K. Novel Role of Vitamin K2: A Potent Inducer of Differentiation of Various Human Myeloid Leukemia Cell Lines. Biochem. Biophys. Res. Commun. 1994, 205, 1305–1310. [Google Scholar] [CrossRef]
- Karasawa, S.; Azuma, M.; Kasama, T.; Sakamoto, S.; Kabe, Y.; Imai, T.; Yamaguchi, Y.; Miyazawa, K.; Handa, H. Vitamin K2 Covalently Binds to Bak and Induces Bak-Mediated Apoptosis. Mol. Pharmacol. 2012, 83, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazawa, K.; Yaguchi, M.; Funato, K.; Gotoh, A.; Kawanishi, Y.; Nishizawa, Y.; Yuo, A.; Ohyashiki, K. Apoptosis/differentiation-inducing effects of vitamin K2 on HL-60 cells: Dichotomous nature of vitamin K2 in leukemia cells. Leukemia 2001, 15, 1111–1117. [Google Scholar] [CrossRef] [Green Version]
- Miyazawa, S.; Moriya, S.; Kokuba, H.; Hino, H.; Takano, N.; Miyazawa, K. Vitamin K2 induces non-apoptotic cell death along with autophagosome formation in breast cancer cell lines. Breast Cancer 2020, 27, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Miyazawa, K.; Naito, M.; Toyotake, J.; Tauchi, T.; Itoh, M.; Yuo, A.; Hayashi, Y.; Georgescu, M.-M.; Kondo, Y.; et al. Vitamin K2 induces autophagy and apoptosis simultaneously in leukemia cells. Autophagy 2008, 4, 629–640. [Google Scholar] [CrossRef]
- Funato, K.; Miyazawa, K.; Yaguchi, M.; Gotoh, A.; Ohyashiki, K. Combination of 22-oxa-1,25-dihydroxyvitamin D3, a vitamin D3 derivative, with vitamin K2 (VK2) synergistically enhances cell differentiation but suppresses VK2-inducing apoptosis in HL-60 cells. Leukemia 2002, 16, 1519–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setoguchi, S.; Watase, D.; Matsunaga, K.; Matsubara, M.; Kubo, Y.; Kusuda, M.; Nagata-Akaho, N.; Enjoji, M.; Nakashima, M.; Takeshita, M.; et al. Enhanced Antitumor Effects of Novel Intracellular Delivery of an Active Form of Menaquinone-4, Menahydroquinone-4, into Hepatocellular Carcinoma. Cancer Prev. Res. 2015, 8, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setoguchi, S.; Watase, D.; Matsunaga, K.; Yamakawa, H.; Goto, S.; Terada, K.; Ohe, K.; Enjoji, M.; Karube, Y.; Takata, J. Antitumor Effects and Delivery Profiles of Menahydroquinone-4 Prodrugs with Ionic or Nonionic Promoiety to Hepatocellular Carcinoma Cells. Molecules 2018, 23, 1738. [Google Scholar] [CrossRef] [Green Version]
- Takata, J.; Karube, Y.; Hanada, M.; Matsunaga, K.; Matsushima, Y.; Sendo, T.; Aoyama, T. Vitamin K Prodrugs: 1. Synthesis of Amino Acid Esters of Menahydroquinone-4 and Enzymatic Reconversion to an Active Form. Pharm. Res. 1995, 12, 18–23. [Google Scholar] [CrossRef]
- Takata, J.; Karube, Y.; Hanada, M.; Matsunaga, K.; Matsushima, Y.; Sendo, T.; Oishi, R. Vitamin K Prodrugs: 2. Water-Soluble Prodrugs of Menahydroquinone-4 for Systemic Site-Specific Delivery. Pharm. Res. 1995, 12, 1973–1979. [Google Scholar] [CrossRef]
- Mori, J.; Suzuki, S.; Hara, M.; Kaneko, A.; Yamashita, K.; Kumagai, M.; Sakuma, T.; Kakizawa, T.; Yamazaki, M.; Takeda, T.; et al. Characterization of Two Novel Retinoic Acid-resistant Cell Lines Derived from HL-60 Cells Following Long-term Culture with all-trans-Retinoic Acid. Jpn. J. Cancer Res. 1999, 90, 660–668. [Google Scholar] [CrossRef]
- Goto, S.; Setoguchi, S.; Yamakawa, H.; Watase, D.; Terada, K.; Matsunaga, K.; Karube, Y.; Takata, J. Prodrugs for Skin Delivery of Menahydroquinone-4, an Active Form of Vitamin K-2(20), Could Overcome the Photoinstability and Phototoxicity of Vitamin K-2(20). Int. J. Mol. Sci. 2019, 20, 2548. [Google Scholar] [CrossRef] [Green Version]
- Ishii, M.; Shimomura, M.; Hasegawa, J.; Asano, Y.; Yamato, Y. Multiple dose pharmacokinetic study of soft gelatin capsule of menatetrenone (Ea-0167) in elderly and young volunteers. Jpn. Pharmacol. Ther. 1995, 23, 2637–2642. [Google Scholar]
- Sano, Y.; Tadano, K.; Kaneko, K.; Kikuchi, K.; Yuzuriha, T. Distribution of menaquinone-4, a therapeutic agent for osteoporosis, in bone and other tissues od rats. J. Nutr. Sci. Vitaminol. 1995, 41, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Sano, Y.; Tadano, K.; Kaneko, K.; Kikuchi, K.; Yuzuriha, T. Metabolic Fate of Menaquinone-4 in Dogs. (I). Absorption, Distribution, Metabolism and Excretion after a Single Oral Administration. Drug Metab. Pharmacokinet. 1997, 12, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Wang, Y. Apoptosis of human leukemic HL-60 cells induced to differentiate by treatment with RA or DMSO. Cell Res. 1995, 5, 181–186. [Google Scholar] [CrossRef] [Green Version]
- Dimberg, A.; Bahram, F.; Karlberg, I.; Larsson, L.-G.; Nilsson, K.; Öberg, F. Retinoic acid–induced cell cycle arrest of human myeloid cell lines is associated with sequential down-regulation of c-Myc and cyclin E and posttranscriptional up-regulation of p27Kip1. Blood 2002, 99, 2199–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.-T.; Zhou, T.-T.; Liu, B.; Bao, J.-K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) (95% CI a) | Resistance Index b | |
|---|---|---|---|
| HL60 Cells | HL-60R Cells | ||
| ATRA | 1.15 (0.416–2.52) | 7.83 (4.72–13.0) | 6.79 |
| MK-4 | 8.33 c (5.25–13.2) | 32.5 c (20.2–52.3) | 3.94 |
| MKH-DMG | 4.30 (2.89–6.40) | 5.97 (3.74–9.53) | 1.39 |
| MKH-SUC | 3.58 (2.66–4.83) | 5.14 (2.81–9.40) | 1.44 |
| Compound | AUC0–72h a (nmol·h·mg Protein−1) in HL60 Cells | AUC0–72h a (nmol·h·mg Protein−1) in HL-60R Cells | ||
|---|---|---|---|---|
| MK-4 | MKO | MK-4 | MKO | |
| MK-4 | 26.4 ± 0.80 | 6.33 ± 0.32 | 19.9 ± 0.40 | 5.52 ± 0.03 |
| MKH-DMG | 12.3 ± 1.29 | 3.11 ± 0.36 | 7.90 ± 0.66 | 2.29 ± 0.15 |
| MKH-SUC | 38.6 ± 0.50 | 9.10 ± 0.22 | 30.6 ± 1.24 | 8.70 ± 0.46 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamakawa, H.; Setoguchi, S.; Goto, S.; Watase, D.; Terada, K.; Nagata-Akaho, N.; Toki, E.; Koga, M.; Matsunaga, K.; Karube, Y.; et al. Growth Inhibitory Effects of Ester Derivatives of Menahydroquinone-4, the Reduced Form of Vitamin K2(20), on All-Trans Retinoic Acid-Resistant HL60 Cell Line. Pharmaceutics 2021, 13, 758. https://doi.org/10.3390/pharmaceutics13050758
Yamakawa H, Setoguchi S, Goto S, Watase D, Terada K, Nagata-Akaho N, Toki E, Koga M, Matsunaga K, Karube Y, et al. Growth Inhibitory Effects of Ester Derivatives of Menahydroquinone-4, the Reduced Form of Vitamin K2(20), on All-Trans Retinoic Acid-Resistant HL60 Cell Line. Pharmaceutics. 2021; 13(5):758. https://doi.org/10.3390/pharmaceutics13050758
Chicago/Turabian StyleYamakawa, Hirofumi, Shuichi Setoguchi, Shotaro Goto, Daisuke Watase, Kazuki Terada, Nami Nagata-Akaho, Erina Toki, Mitsuhisa Koga, Kazuhisa Matsunaga, Yoshiharu Karube, and et al. 2021. "Growth Inhibitory Effects of Ester Derivatives of Menahydroquinone-4, the Reduced Form of Vitamin K2(20), on All-Trans Retinoic Acid-Resistant HL60 Cell Line" Pharmaceutics 13, no. 5: 758. https://doi.org/10.3390/pharmaceutics13050758