
Untangling Dual-Targeting Therapeutic Mechanism of Epidermal Growth Factor Receptor (EGFR) Based on Reversed Allosteric Communication

Abstract
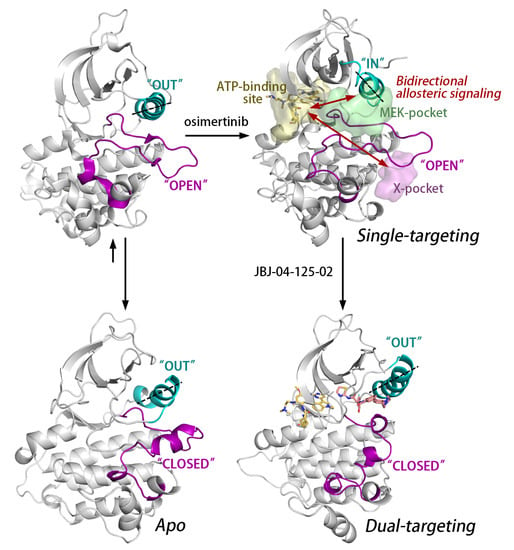
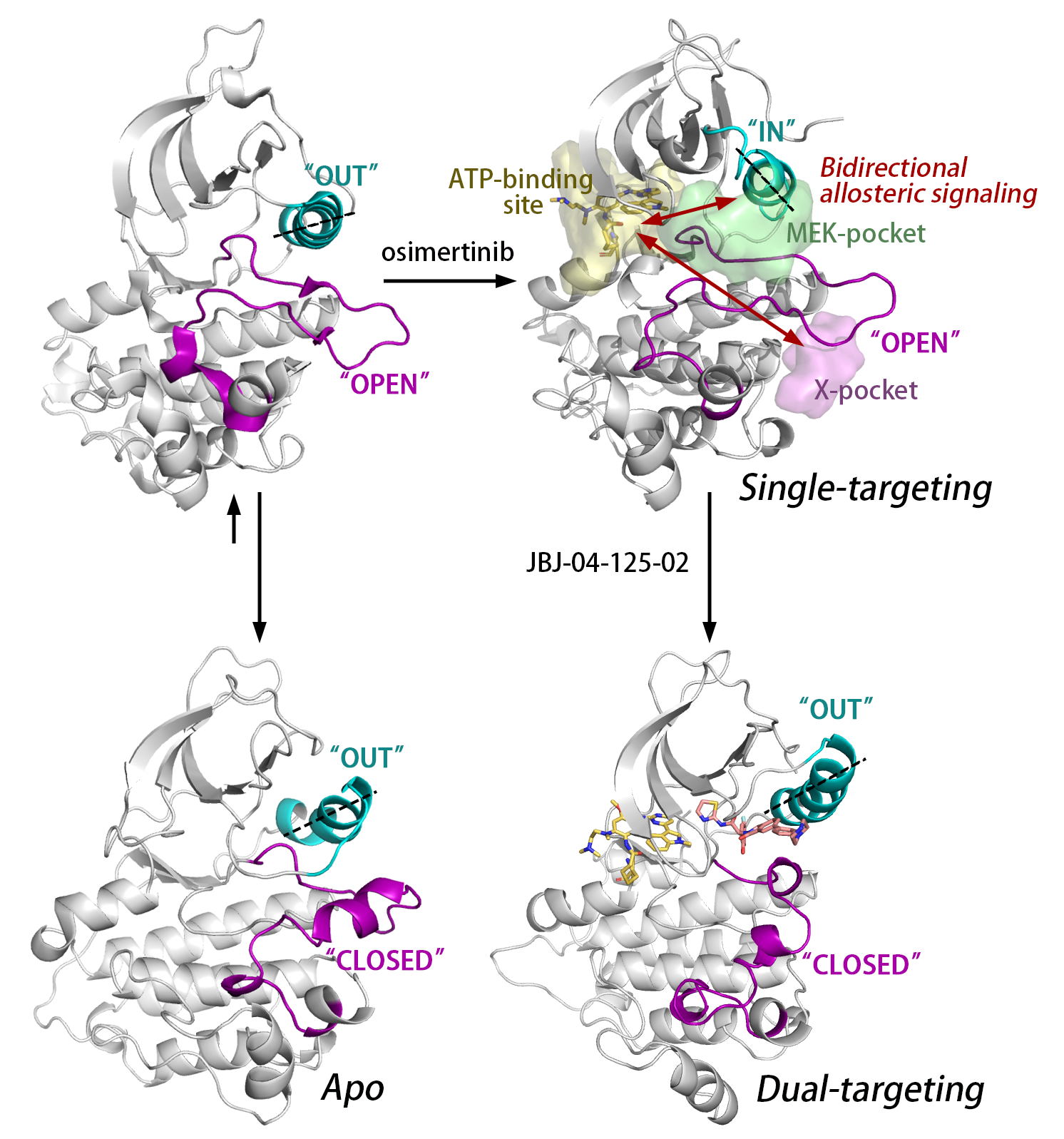
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Construction of Stimulated Systems
2.2. Covalent Docking
2.3. MD Simulations
2.4. Principal Component Analysis (PCA)
2.5. Community Network Analysis
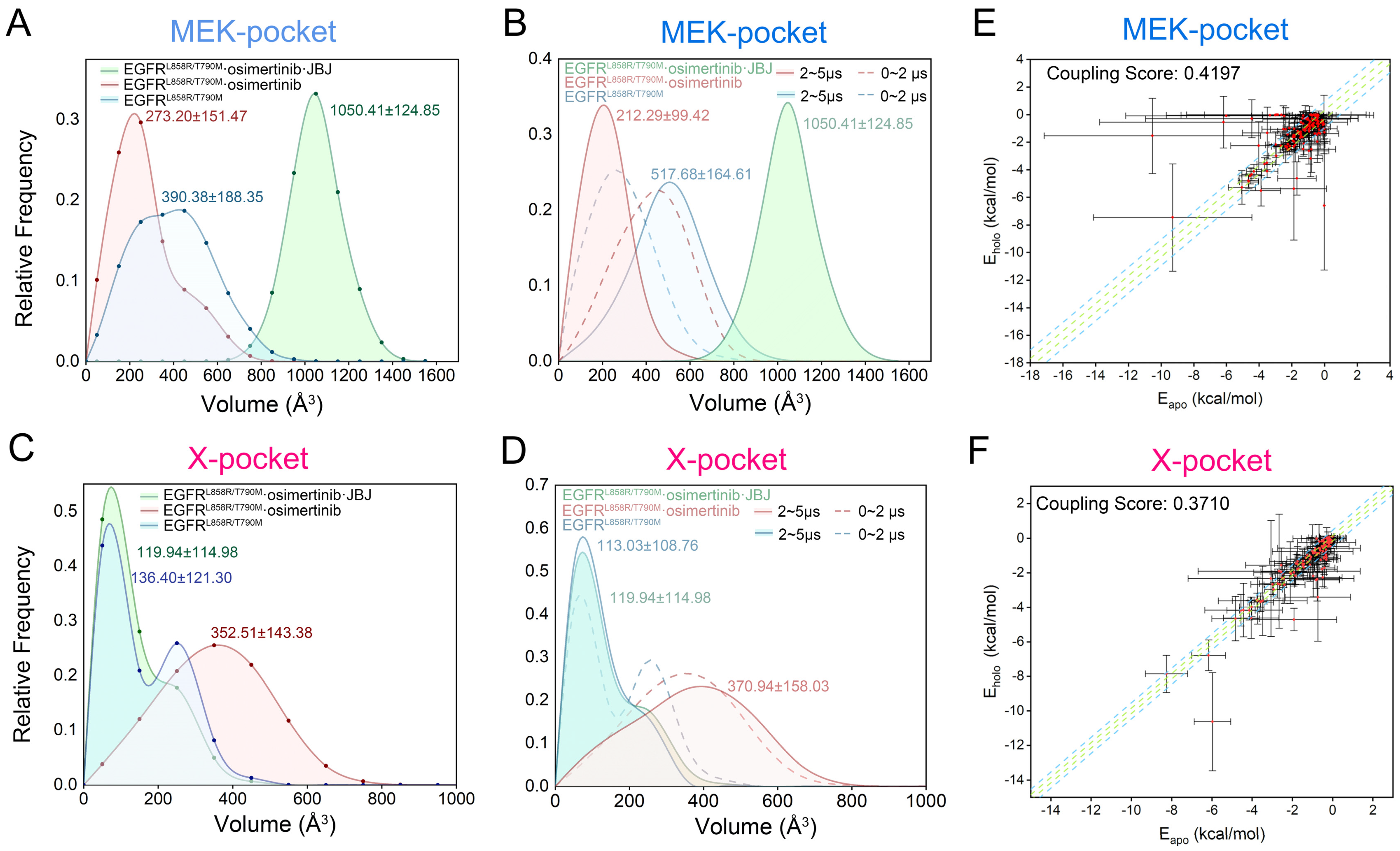
2.6. Energy Coupling Score Calculation
3. Results
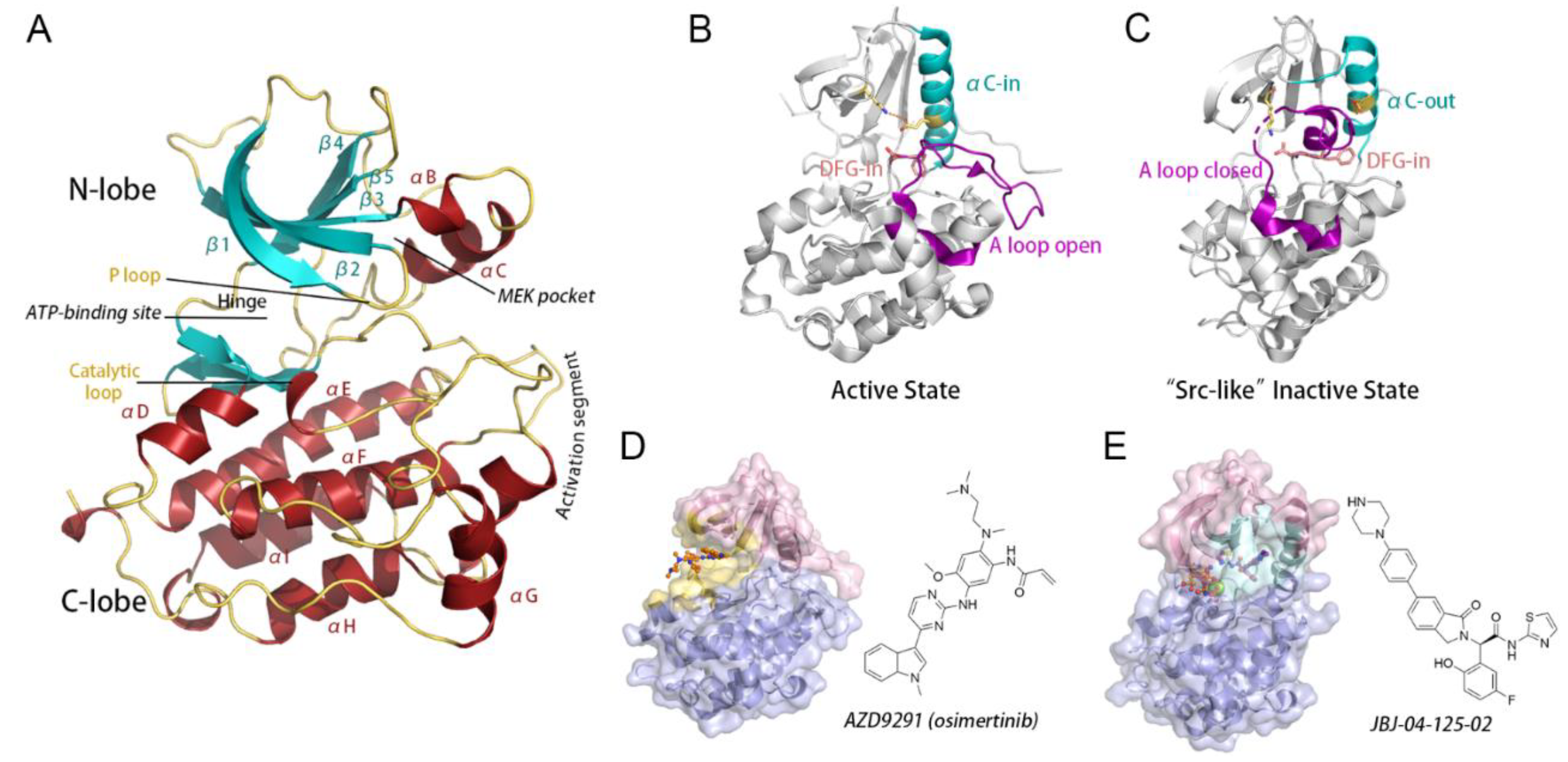
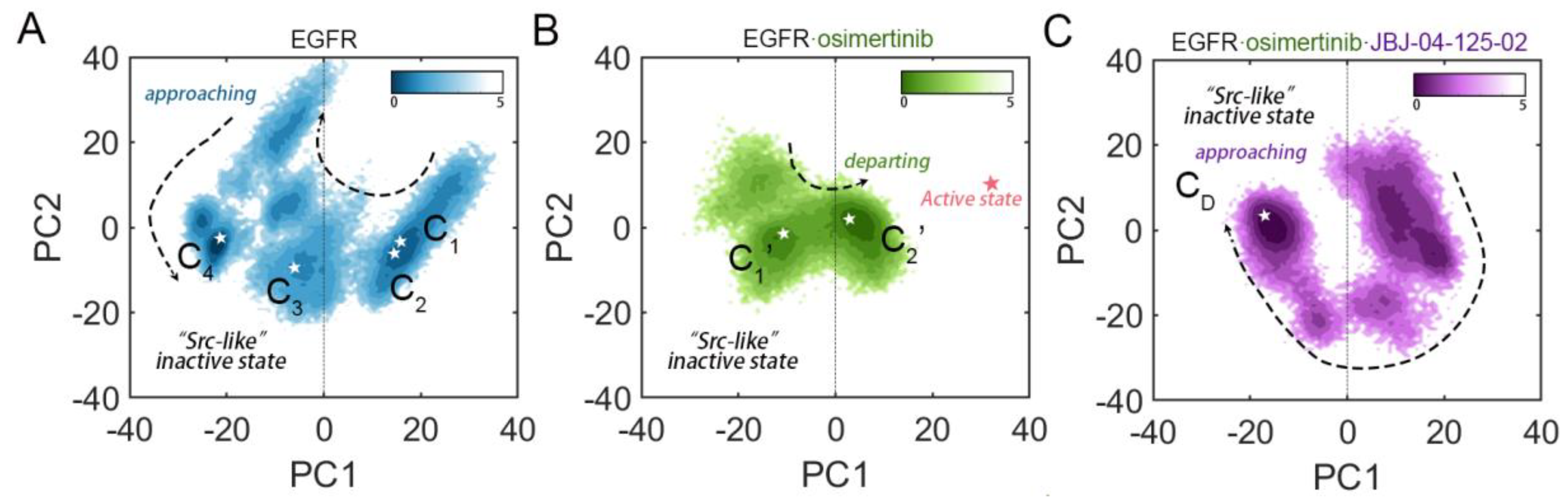
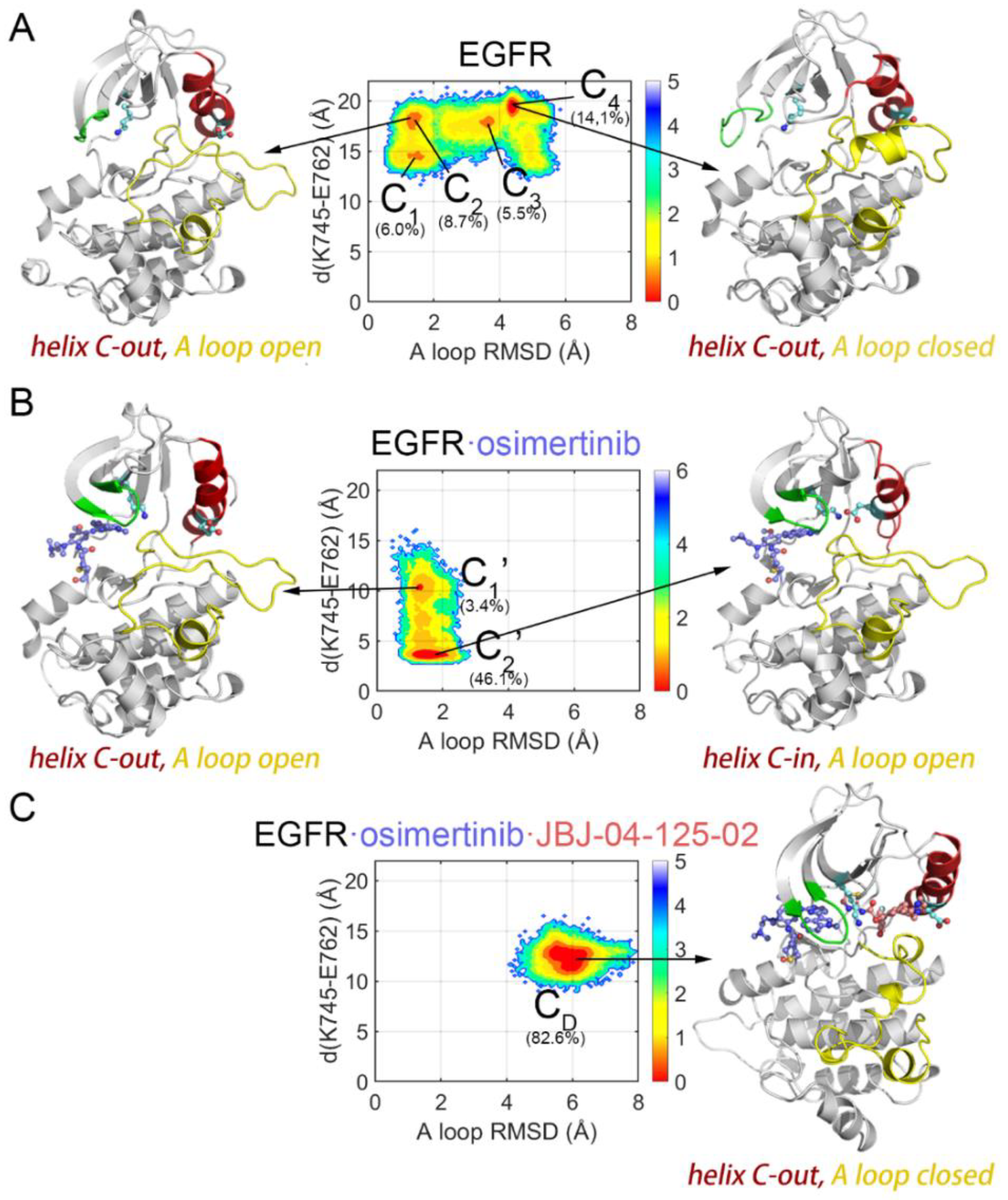
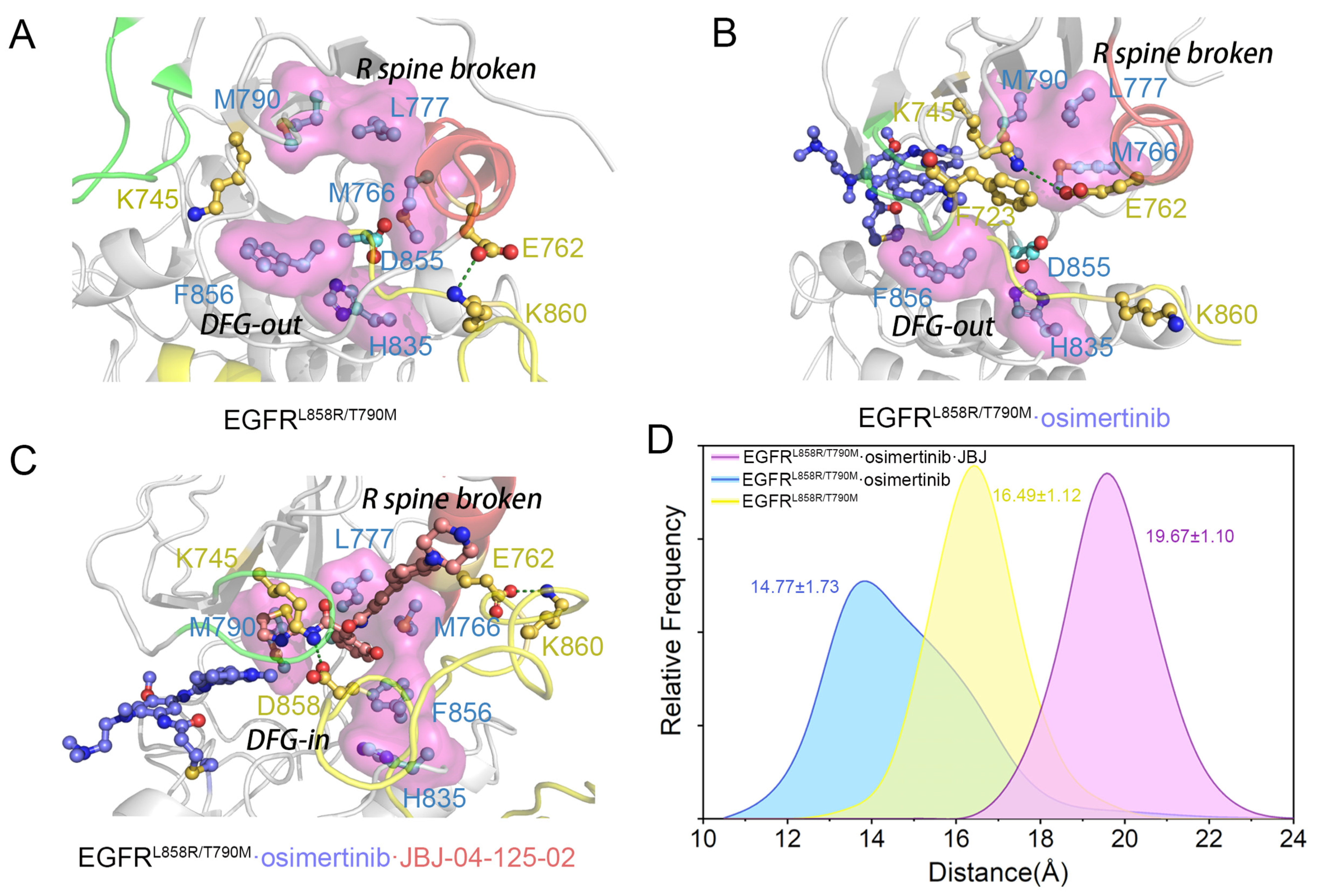
3.1. Orthosteric Osimertinib Binding Induced a Conformational Transition of EGFRL858R/T790M by Departing the “Src-like” Inactive State
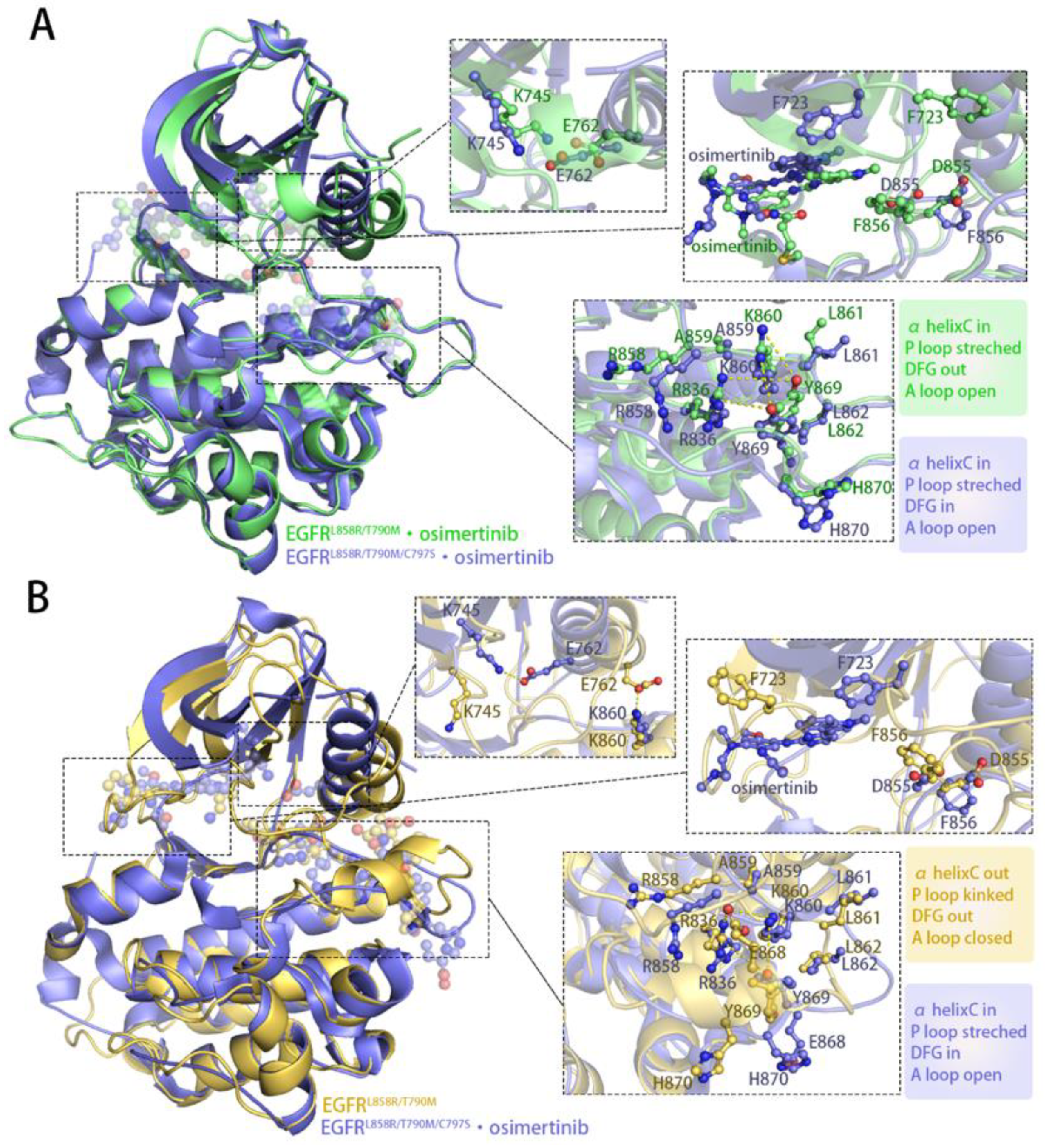
3.2. Osimertinib Binding Stabilized an Intermediate State Characterized by a Unique αC-in, A Loop Open, and DFG-out Conformation
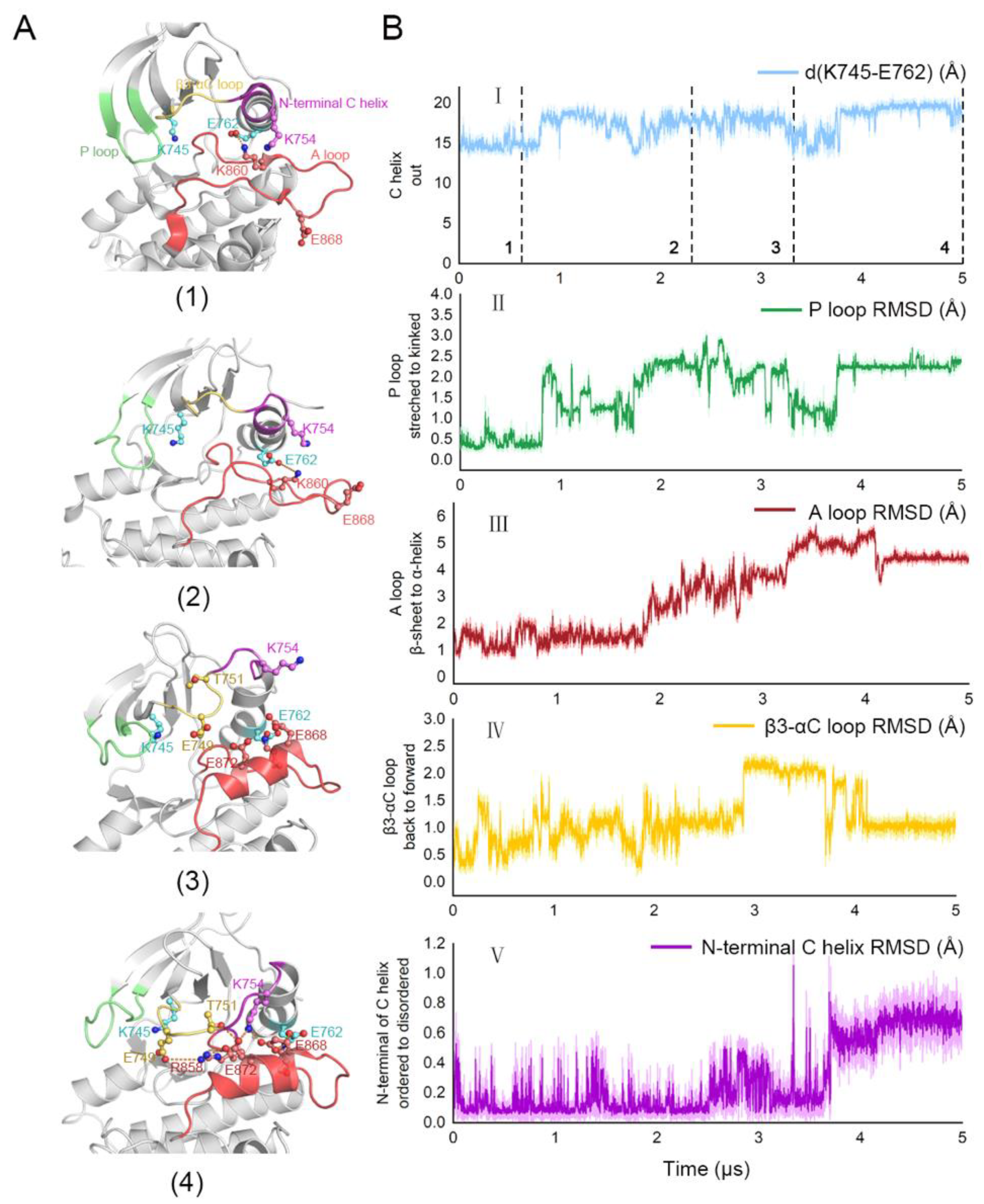
3.3. Stepwise Dynamics of EGFR Transition towards the “Src-like” Inactivation State
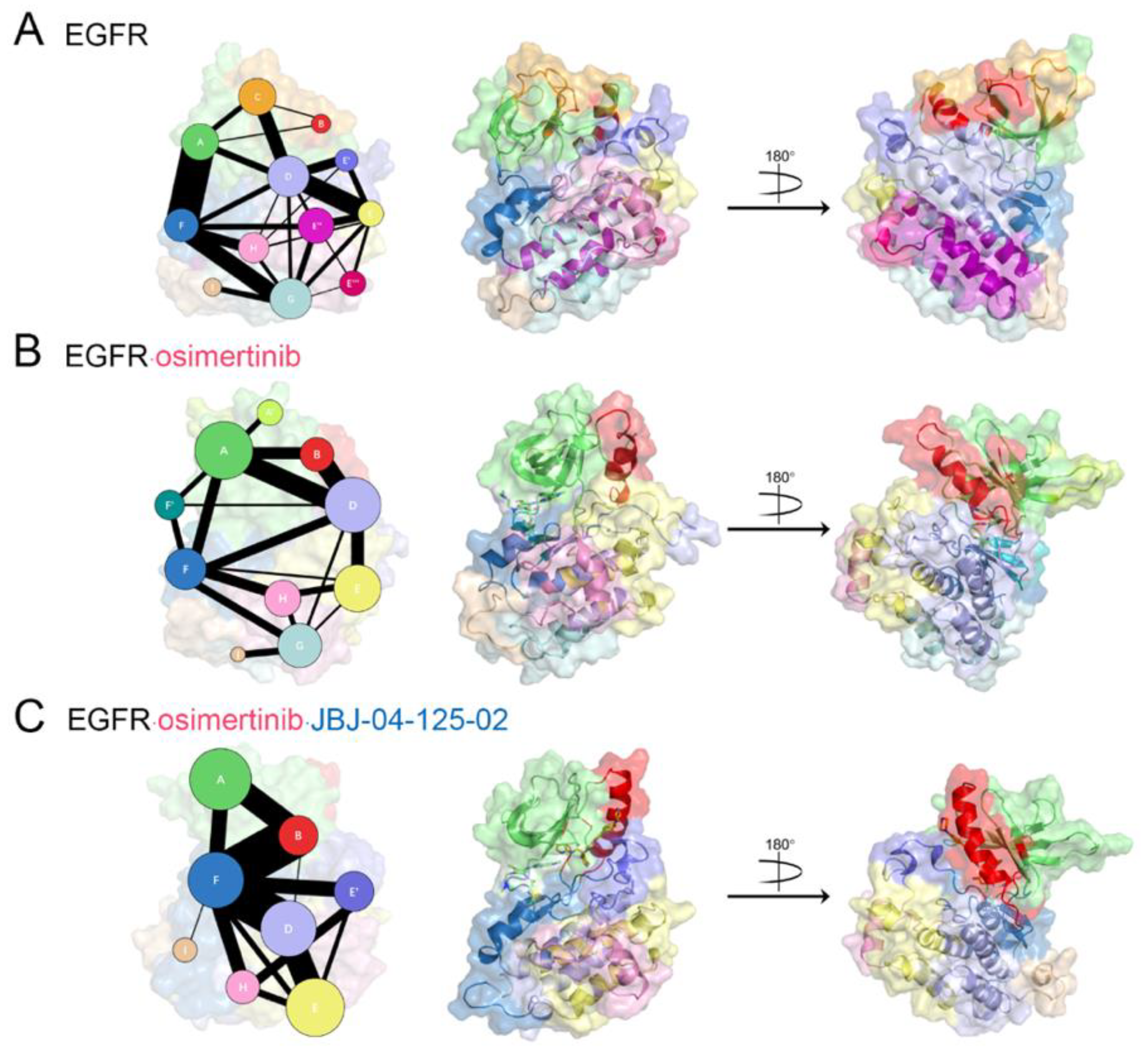
3.4. Reversed Allosteric Signaling Pathways within EGFRL858R/T790M Catalytic Domain
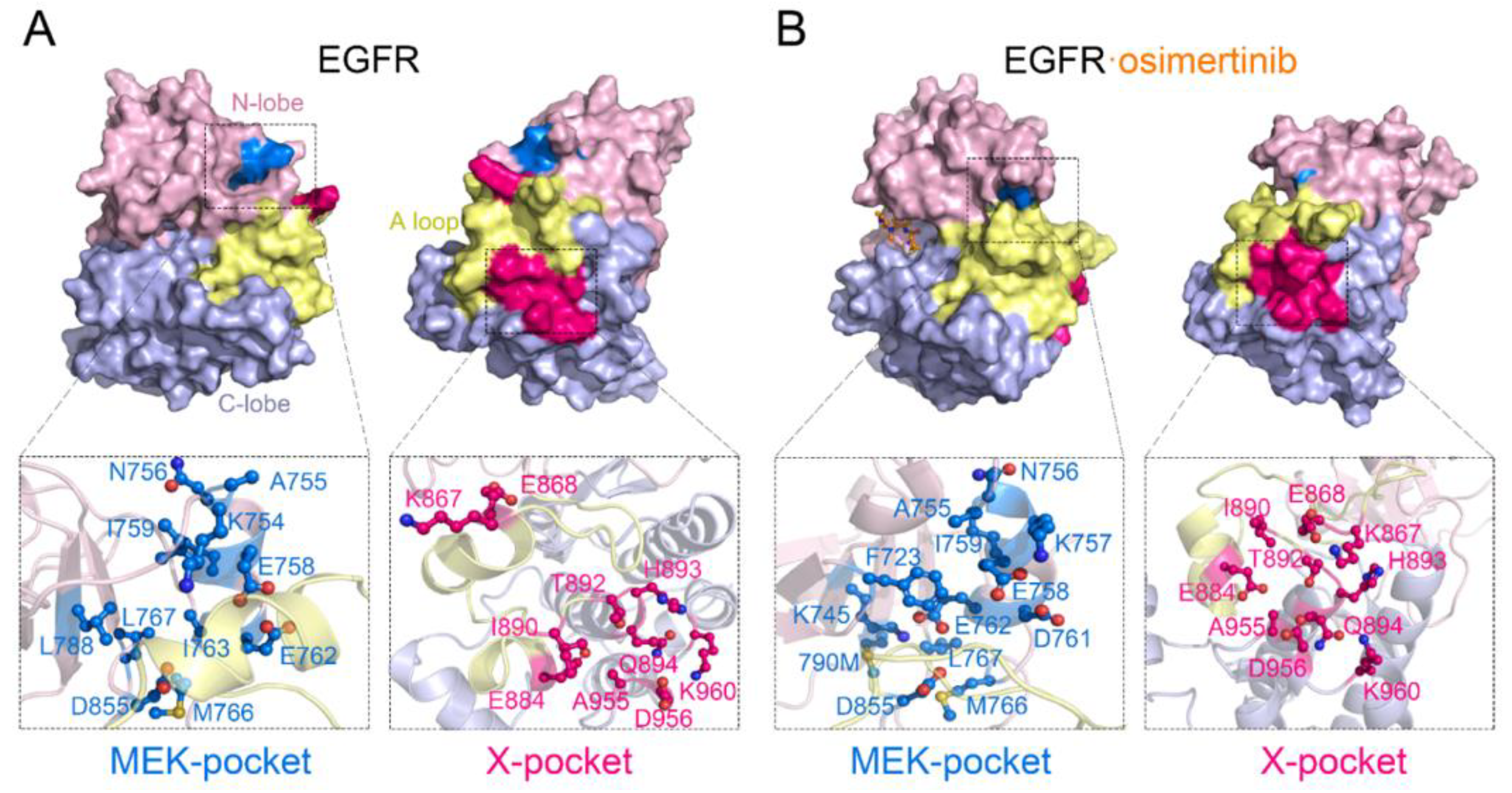
3.5. Identification of Potential Allosteric Pockets Based on Reversed Allosteric Communication
3.6. The Nonconservative Potential Allosteric Pocket Enables Specific Targeting of EGFR across the Human Kinome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lu, S.; Shen, Q.; Zhang, J. Allosteric Methods and Their Applications: Facilitating the Discovery of Allosteric Drugs and the Investigation of Allosteric Mechanisms. Acc. Chem. Res. 2019, 52, 492–500. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.J. Allostery in disease and in drug discovery. Cell 2013, 153, 293–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Changeux, J.P.; Christopoulos, A. Allosteric modulation as a unifying mechanism for receptor function and regulation. Cell 2017, 19, 4–21. [Google Scholar]
- Lu, S.; Li, S.; Zhang, J. Harnessing allostery: A novel approach to drug discovery. Med. Res. Rev. 2014, 34, 1242–1285. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Huang, W.; Wang, Q.; Shen, Q.; Li, S.; Nussinov, R.; Zhang, J. The Structural Basis of ATP as an Allosteric Modulator. PLoS Comput. Biol. 2014, 10, e1003831. [Google Scholar] [CrossRef]
- Lu, S.; Qiu, Y.; Ni, D.; He, X.; Pu, J.; Zhang, J. Emergence of allosteric drug-resistance mutations: New challenges for allosteric drug discovery. Drug Discov. Today 2020, 25, 177–184. [Google Scholar] [CrossRef]
- Sutto, L.; Gervasio, F.L. Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 10616–10621. [Google Scholar] [CrossRef] [Green Version]
- Sultan, M.M.; Kiss, G.; Pande, V.S. Towards simple kinetic models of functional dynamics for a kinase subfamily. Nat. Chem. 2018, 10, 903–909. [Google Scholar] [CrossRef]
- Kannan, S.; Venkatachalam, G.; Lim, H.H.; Surana, U.; Verma, C. Conformational landscape of the epidermal growth factor receptor kinase reveals a mutant specific allosteric pocket. Chem. Sci. 2018, 9, 5212–5222. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Ni, D.; Li, X.; He, X.; Zhang, H.; Zhang, J.; Lu, S. Drugging K-RasG12C through covalent inhibitors: Mission possible? Pharmacol. Ther. 2019, 202, 1–17. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, F. Sticking it to KRAS: Covalent Inhibitors Enter the Clinic. Cancer Cell 2020, 37, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Jang, H.; Muratcioglu, S.; Gursoy, A.; Keskin, O.; Nussinov, R.; Zhang, J. Ras Conformational Ensembles, Allostery, and Signaling. Chem. Rev. 2016, 116, 6607–6665. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zhang, J. Small Molecule Allosteric Modulators of G-Protein-Coupled Receptors: Drug-Target Interactions. J. Med. Chem. 2019, 62, 24–45. [Google Scholar] [CrossRef]
- Suomivuori, C.-M.; Latorraca, N.R.; Wingler, L.M.; Eismann, S.; King, M.C.; Kleinhenz, A.L.W.; Skiba, M.A.; Staus, D.P.; Kruse, A.C.; Lefkowitz, R.J.; et al. Molecular mechanism of biased signaling in a prototypical G protein-coupled receptor. Science 2020, 367, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Ni, D.; Li, Y.; Qiu, Y.; Pu, J.; Lu, S.; Zhang, J. Combining Allosteric and Orthosteric Drugs to Overcome Drug Resistance. Trends Pharmacol. Sci. 2020, 41, 336–348. [Google Scholar] [CrossRef]
- Huang, J.; Yuan, Y.; Zhao, N.; Pu, D.; Tang, Q.; Zhang, S.; Luo, S.; Yang, X.; Wang, N.; Xiao, Y.; et al. Orthosteric-allosteric dual inhibitors of PfHT1 as selective anti-malarial agents. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, J.; Adrián, F.J.; Jahnke, W.; Cowan-Jacob, S.W.; Li, A.G.; Iacob, R.E.; Sim, T.; Powers, J.; Dierks, C.; Sun, F.; et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 2010, 463, 501–506. [Google Scholar] [CrossRef] [Green Version]
- To, C.; Jang, J.; Chen, T.; Park, E.; Mushajiang, M.; De Clercq, D.J.H.; Xu, M.; Wang, S.; Cameron, M.D.; Heppner, D.E.; et al. Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discov. 2019, 9, 926–943. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.-H.; O’Boyle, D.R., 2nd; Fridell, R.A.; Langley, D.R.; Wang, C.; Roberts, S.B.; Nower, P.; Johnson, B.M.; Moulin, F.; Nophsker, M.J.; et al. Resensitizing daclatasvir-resistant hepatitis C variants by allosteric modulation of NS5A. Nature 2015, 527, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.A.; Lay, C.; Li, W.; et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat. Med. 2018, 24, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Schulze, J.O.; Saladino, G.; Busschots, K.; Neimanis, S.; Süß, E.; Odadzic, D.; Zeuzem, S.; Hindie, V.; Herbrand, A.K.; Lisa, M.-N.; et al. Bidirectional Allosteric Communication between the ATP-Binding Site and the Regulatory PIF Pocket in PDK1 Protein Kinase. Cell Chem. Biol. 2016, 23, 1193–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroux, A.E.; Biondi, R.M. Renaissance of Allostery to Disrupt Protein Kinase Interactions. Trends Biochem. Sci. 2020, 45, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xie, J.; Lai, L. Correlation Between Allosteric and Orthosteric Sites. Adv. Exp. Med. Biol. 2019, 1163, 89–105. [Google Scholar] [PubMed]
- Ma, X.; Meng, H.; Lai, L. Motions of Allosteric and Orthosteric Ligand-Binding Sites in Proteins are Highly Correlated. J. Chem. Inf. Model. 2016, 56, 1725–1733. [Google Scholar] [CrossRef]
- Meng, H.; McClendon, C.L.; Dai, Z.; Li, K.; Zhang, X.; He, S.; Shang, E.; Liu, Y.; Lai, L. Discovery of Novel 15-Lipoxygenase Activators To Shift the Human Arachidonic Acid Metabolic Network toward Inflammation Resolution. J. Med. Chem. 2016, 59, 4202–4209. [Google Scholar] [CrossRef]
- Ni, D.; Wei, J.; He, X.; Rehman, A.U.; Li, X.; Qiu, Y.; Pu, J.; Lu, S.; Zhang, J. Discovery of cryptic allosteric sites using reversed allosteric communication by a combined computational and experimental strategy. Chem. Sci. 2021. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Da Cunha Santos, G.; Shepherd, F.A.; Tsao, M.S. EGFR mutations and lung cancer. Annu. Rev. Pathol. 2011, 6, 49–69. [Google Scholar] [CrossRef] [Green Version]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Castellanos, E.; Feld, E.; Horn, L. Driven by Mutations: The Predictive Value of Mutation Subtype in EGFR-Mutated Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 612–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, S.; Song, X.; Han, B.; Cheng, Y.; Huang, C.; Yang, S.; Liu, X.; Liu, Y.; Lu, S.; et al. Gefitinib versus placebo as maintenance therapy in patients with locally advanced or metastatic non-small-cell lung cancer (INFORM; C-TONG 0804): A multicentre, double-blind randomised phase 3 trial. Lancet Oncol. 2012, 13, 466–475. [Google Scholar] [CrossRef]
- Sequist, L.V.; Yang, J.C.-H.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.-M.; Boyer, M.; et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.-K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.-H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [Green Version]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [Green Version]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Yun, C.H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niggenaber, J.; Heyden, L.; Grabe, T.; Müller, M.P.; Lategahn, J.; Rauh, D. Complex Crystal Structures of EGFR with Third-Generation Kinase Inhibitors and Simultaneously Bound Allosteric Ligands. ACS Med. Chem. Lett. 2020, 11, 2484–2490. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, D.J.H.; Heppner, D.E.; To, C.; Jang, J.; Park, E.; Yun, C.-H.; Mushajiang, M.; Shin, B.H.; Gero, T.W.; Scott, D.A.; et al. Discovery and Optimization of Dibenzodiazepinones as Allosteric Mutant-Selective EGFR Inhibitors. ACS Med. Chem. Lett. 2019, 10, 1549–1553. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, L.; Rastelli, G. αc Helix Displacement As a General Approach for Allosteric Modulation of Protein Kinases. Drug Discov. Today 2013, 18, 407–414. [Google Scholar] [CrossRef]
- Gajiwala, K.S.; Feng, J.; Ferre, R.; Ryan, K.; Brodsky, O.; Weinrich, S.; Kath, J.C.; Stewart, A. Insights into the aberrant activity of mutant EGFR kinase domain and drug recognition. Structure 2013, 21, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.J.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins 1993, 17, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.W. Algorithm 97, Shortest Path Algorithms. Commun. ACM 1962, 5, 345. [Google Scholar] [CrossRef]
- Newman, M.E.J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, Y.; Arkhipov, A.; Kim, E.T.; Pan, A.C.; Shawa, D.E. Transitions to catalytically inactive conformations in EGFR kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 7270–7275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.-E.; Ayaz, P.; Zhu, S.-J.; Zhao, P.; Liang, L.; Zhang, C.H.; Wu, Y.-C.; Li, J.-L.; Choi, H.G.; Huang, X.; et al. Structural Basis of AZD9291 Selectivity for EGFR T790M. J. Med. Chem. 2020, 63, 8502–8511. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Saleh, T.; Rossi, P.; Kalodimos, C.G. Conformational states dynamically populated by a kinase determine its function. Science 2020, 370, 6513. [Google Scholar] [CrossRef]
- Kashima, K.; Kawauchi, H.; Tanimura, H.; Tachibana, Y.; Chiba, T.; Torizawa, T.; Sakamoto, H. CH7233163 overcomes osimertinib resistant EGFR-Del19/T790M/C797S mutation. Mol. Cancer Ther. 2020, 19, 2288–2297. [Google Scholar] [CrossRef]
- Yosaatmadja, Y.; Silva, S.; Dickson, J.M.; Patterson, A.V.; Smaill, J.B.; Flanagan, J.U.; McKeage, M.J.; Squire, C.J. Binding mode of the breakthrough inhibitor AZD9291 to epidermal growth factor receptor revealed. J. Struct. Biol. 2015, 192, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Chaikuad, A.; Gray, N.S.; Knapp, S. The ins and outs of selective kinase inhibitor development. Nat. Chem. Biol. 2015, 11, 818–821. [Google Scholar] [CrossRef]
- Okuzumi, T.; Fiedler, D.; Zhang, C.; Gray, D.C.; Aizenstein, B.; Hoffman, R.; Shokat, K.M. Inhibitor hijacking of Akt activation. Nat. Chem. Biol. 2009, 5, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Kornev, A.P.; Taylor, S.S. Dynamics-Driven Allostery in Protein Kinases. Trends Biochem. Sci. 2015, 40, 628–647. [Google Scholar] [CrossRef] [Green Version]
- Dixit, A.; Verkhivker, G.M. The energy landscape analysis of cancer mutations in protein kinases. PLoS ONE 2011, 6, e26071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Zhang, X.; Peng, C.; Wang, J.; Xu, Z.; Chen, K.; Shi, J.; Zhu, W. D3Pockets: A Method and Web Server for Systematic Analysis of Protein Pocket Dynamics. J. Chem. Inf. Model. 2019, 59, 3353–3358. [Google Scholar] [CrossRef]
- Palomo, V.; Soteras, I.; Perez, D.I.; Perez, C.; Gil, C.; Campillo, N.E.; Martinez, A. Exploring the binding sites of glycogen synthase kinase 3. Identification and characterization of allosteric modulation cavities. J. Med. Chem. 2011, 54, 8461–8470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomo, V.; Perez, D.I.; Roca, C.; Anderson, C.; Rodríguez-Muela, N.; Perez, C.; Morales-Garcia, J.A.; Reyes, J.A.; Campillo, N.E.; Perez-Castillo, A.M.; et al. Subtly Modulating Glycogen Synthase Kinase 3 β: Allosteric Inhibitor Development and Their Potential for the Treatment of Chronic Diseases. J. Med. Chem. 2017, 60, 4983–5001. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, J.; Chen, Y.; Mei, Z.; Xiao, Y. Screening of inhibitors of glycogen synthase kinase-3β from traditional Chinese medicines using enzyme-immobilized magnetic beads combined with high-performance liquid chromatography. J. Chromatogr. A 2015, 1425, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, P.; Bidon-Chanal, A.; Luque, F.J.; Barril, X. MDpocket: Open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics 2011, 27, 3276–3285. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Wang, Q.; Tang, B.; Lai, L. Identifying Allosteric Binding Sites in Proteins with a Two-State Go Model for Novel Allosteric Effector Discovery. J. Chem. Theory Comput. 2012, 8, 2962–2971. [Google Scholar] [CrossRef] [PubMed]
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.C.; Hawkins, N.J.; Sanglard, D.; Gurr, S.J. Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science 2018, 360, 739–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, P.; Moorthy, V.; Paulin, S.; Hill, S.R.; Sprenger, M.; Garner, S.; Simão, M.; Guerra, R.; Magrini, N.; Swaminathan, S. The drugs don’t work: WHO’s role in advancing new antibiotics. Lancet 2018, 392, 264–266. [Google Scholar] [CrossRef]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.A.; VanSchouwen, B.; Akimoto, M.; Melacini, G. Allosteric inhibition explained through conformational ensembles sampling distinct “mixed” states. Comput. Struct. Biotechnol. J. 2020, 18, 3803–3818. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Zhu, Y.; Long, J.; Ye, F.; Hu, G. Both intra and inter-domain interactions define the intrinsic dynamics and allosteric mechanism in DNMT1s. Comput. Struct. Biotechnol. J. 2020, 18, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ji, D.; Lei, C.; Chen, Y.; Qiu, Y.; Li, X.; Li, M.; Ni, D.; Pu, J.; Zhang, J.; et al. Mechanistic insights into the effect of phosphorylation on Ras conformational dynamics and its interactions with cell signaling proteins. Comput. Struct. Biotechnol. J. 2021, 19, 1184–1199. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Zhang, M.; Nussinov, R. The quaternary assembly of KRas4B with Raf-1 at the membrane. Comput. Struct. Biotechnol. J. 2020, 18, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, Y.Y.; Li, S.P.; Yang, W.D.; Sun, L.F.; Jang, M.Q.; Wu, X.L.; Wang, Q.C.; Chen, L.; Wu, Y.K. Ca2+-based allosteric switches and shape shifting in RGLG1 VWA domain. Comput. Struct. Biotechnol. J. 2020, 18, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Maryam, A.; Vedithi, S.C.; Khalid, R.R.; Alsulami, A.F.; Torres, P.H.M.; Siddiqi, A.R.; Blundell, T.L. The Molecular Organization of Human cGMP Specific Phosphodiesterase 6 (PDE6): Structural Implications of Somatic Mutations in Cancer and Retinitis Pigmentosa. Comput. Struct. Biotechnol. J. 2019, 17, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, T.; Li, S.; Tong, L.; Li, J.; Su, Z.; Feng, F.; Sun, D.; Tong, Y.; Wang, X.; et al. Discovery of Potent and Noncovalent Reversible EGFR Kinase Inhibitors of EGFR(L858R/T790M/C797S). ACS Med. Chem. Lett. 2019, 10, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Wittlinger, F.; Heppner, D.E.; To, C.; Günther, M.; Shin, B.H.; Rana, J.K.; Schmoker, A.M.; Beyett, T.S.; Jänne, P.A.; Eck, M.J.; et al. Molecular design of a “two-in-one” orthosteric-allosteric chimeric mutant. ChemRxiv 2020, 1–15. [Google Scholar] [CrossRef]
- Wagner, J.R.; Lee, C.T.; Durrant, J.D.; Malmstrom, R.D.; Feher, V.A.; Amaro, R.E. Emerging Computational Methods for the Rational Discovery of Allosteric Drugs. Chem. Rev. 2016, 116, 6370–6390. [Google Scholar] [CrossRef]
- Qiu, Y.; Li, X.; He, X.; Pu, J.; Zhang, J.; Lu, S. Computational methods-guided design of modulators targeting protein-protein interactions (PPIs). Eur. J. Med. Chem. 2020, 207, 112764. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Li, Q.; Gao, W.; Lu, S.; Shen, Q.; Liu, X.; Wu, Y.; Wang, B.; Lin, H.; Chen, G.; et al. AlloDriver: A method for the identification and analysis of cancer driver targets. Nucleic Acids Res. 2019, 47, W315–W321. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Song, K.; Liu, X.; Lu, S.; Shen, Q.; Wang, R.; Gao, J.; Hong, Y.; Li, Q.; Ni, D.; et al. AlloFinder: A strategy for allosteric modulator discovery and allosterome analyses. Nucleic Acids Res. 2018, 46, W451–W458. [Google Scholar] [CrossRef]
- Tan, Z.W.; Tee, W.-V.; Guarnera, E.; Booth, L.; Berezovsky, I.N. AlloMAPS: Allosteric mutation analysis and polymorphism of signaling database. Nucleic Acids Res. 2019, 47, D265–D270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wodak, S.J.; Paci, E.; Dokholyan, N.V.; Berezovsky, I.N.; Horovitz, A.; Li, J.; Hilser, V.J.; Bahar, I.; Karanicolas, J.; Stock, G.; et al. Allostery in Its Many Disguises: From Theory to Applications. Structure 2019, 27, 566–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- East, K.W.; Skeens, E.; Cui, J.Y.; Belato, H.B.; Mitchell, B.; Hsu, R.; Batista, V.S.; Palermo, G.; Lisi, G.P. NMR and computational methods for molecular resolution of allosteric pathways in enzyme complexes. Biophys. Rev. 2020, 12, 155–174. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, Y.; Yin, X.; Li, X.; Wang, Y.; Fu, Q.; Huang, R.; Lu, S. Untangling Dual-Targeting Therapeutic Mechanism of Epidermal Growth Factor Receptor (EGFR) Based on Reversed Allosteric Communication. Pharmaceutics 2021, 13, 747. https://doi.org/10.3390/pharmaceutics13050747
Qiu Y, Yin X, Li X, Wang Y, Fu Q, Huang R, Lu S. Untangling Dual-Targeting Therapeutic Mechanism of Epidermal Growth Factor Receptor (EGFR) Based on Reversed Allosteric Communication. Pharmaceutics. 2021; 13(5):747. https://doi.org/10.3390/pharmaceutics13050747
Chicago/Turabian StyleQiu, Yuran, Xiaolan Yin, Xinyi Li, Yuanhao Wang, Qiang Fu, Renhua Huang, and Shaoyong Lu. 2021. "Untangling Dual-Targeting Therapeutic Mechanism of Epidermal Growth Factor Receptor (EGFR) Based on Reversed Allosteric Communication" Pharmaceutics 13, no. 5: 747. https://doi.org/10.3390/pharmaceutics13050747