Application of a Physiologically Based Pharmacokinetic Model to Develop a Veterinary Amorphous Enrofloxacin Solid Dispersion

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animal
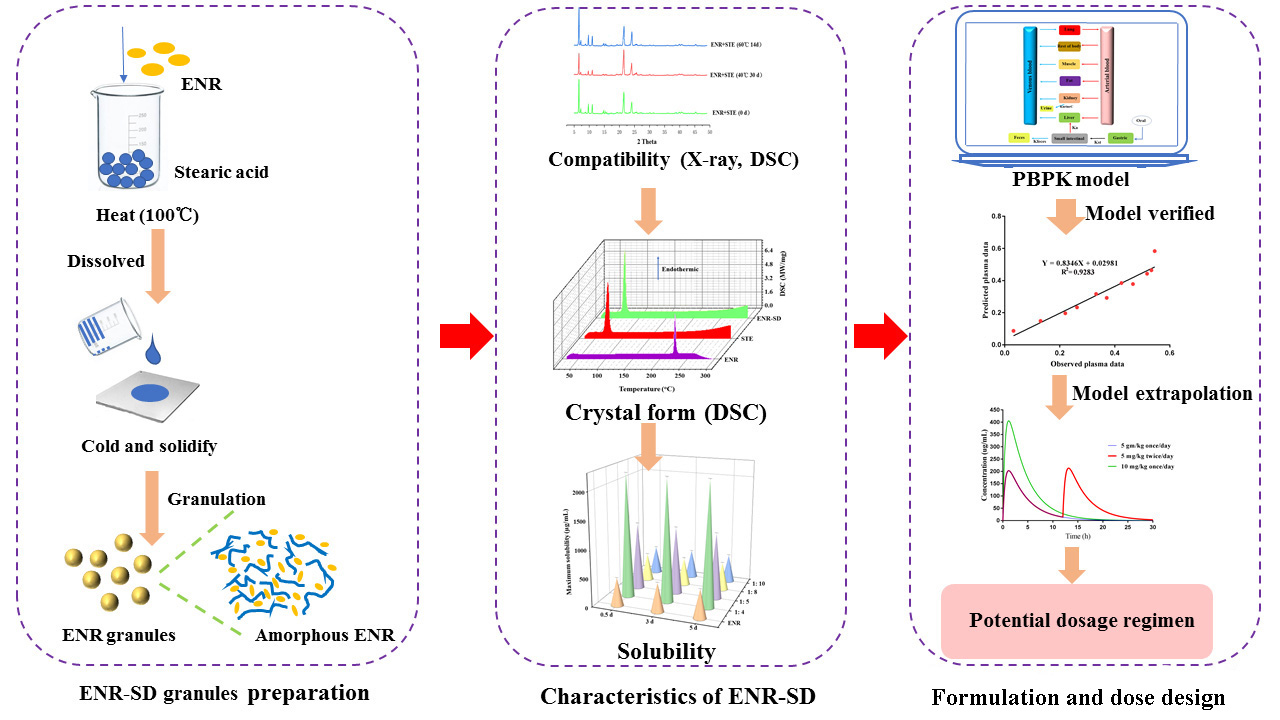
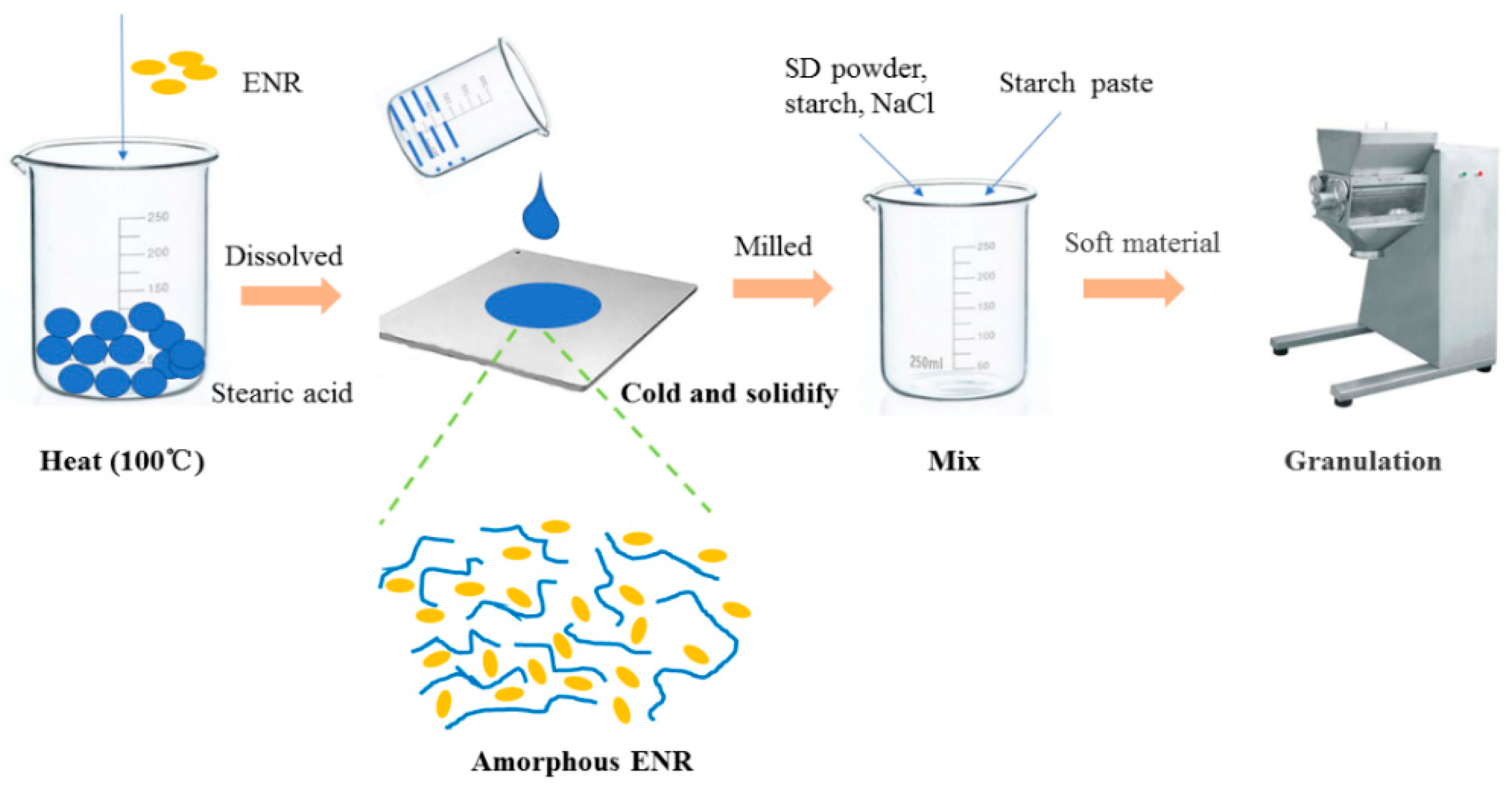
2.3. Preparation and Evaluation of ENR-SD Granules
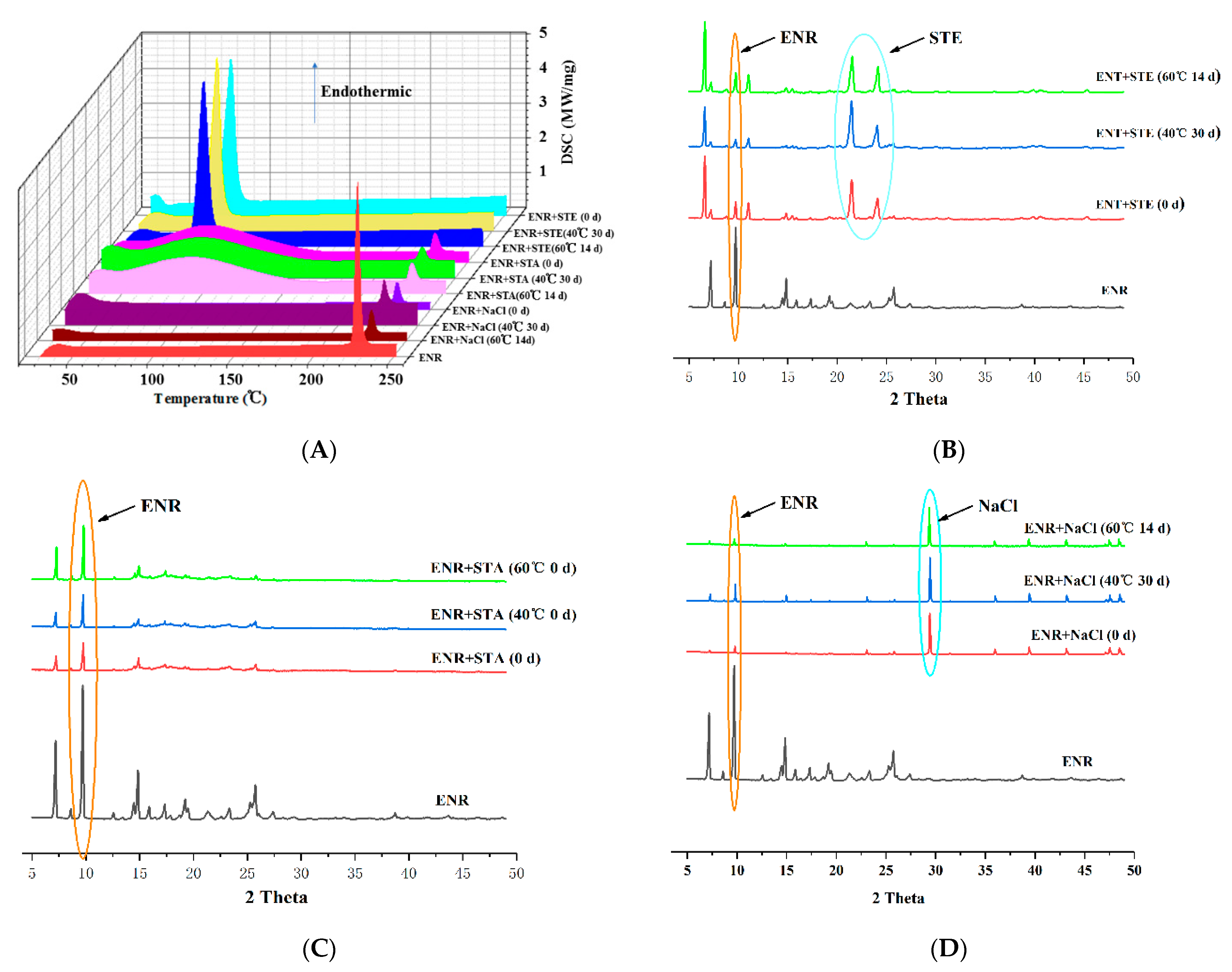
2.3.1. Compatibility Test
2.3.2. Preparation of ENR-Loaded Stearic Acid SD
2.3.3. Characteristics of ENR-Loaded Stearic Acid-SD
2.3.4. Preparation of ENR-SD Granules
2.3.5. In Vitro Release Test
2.4. Palatability Test
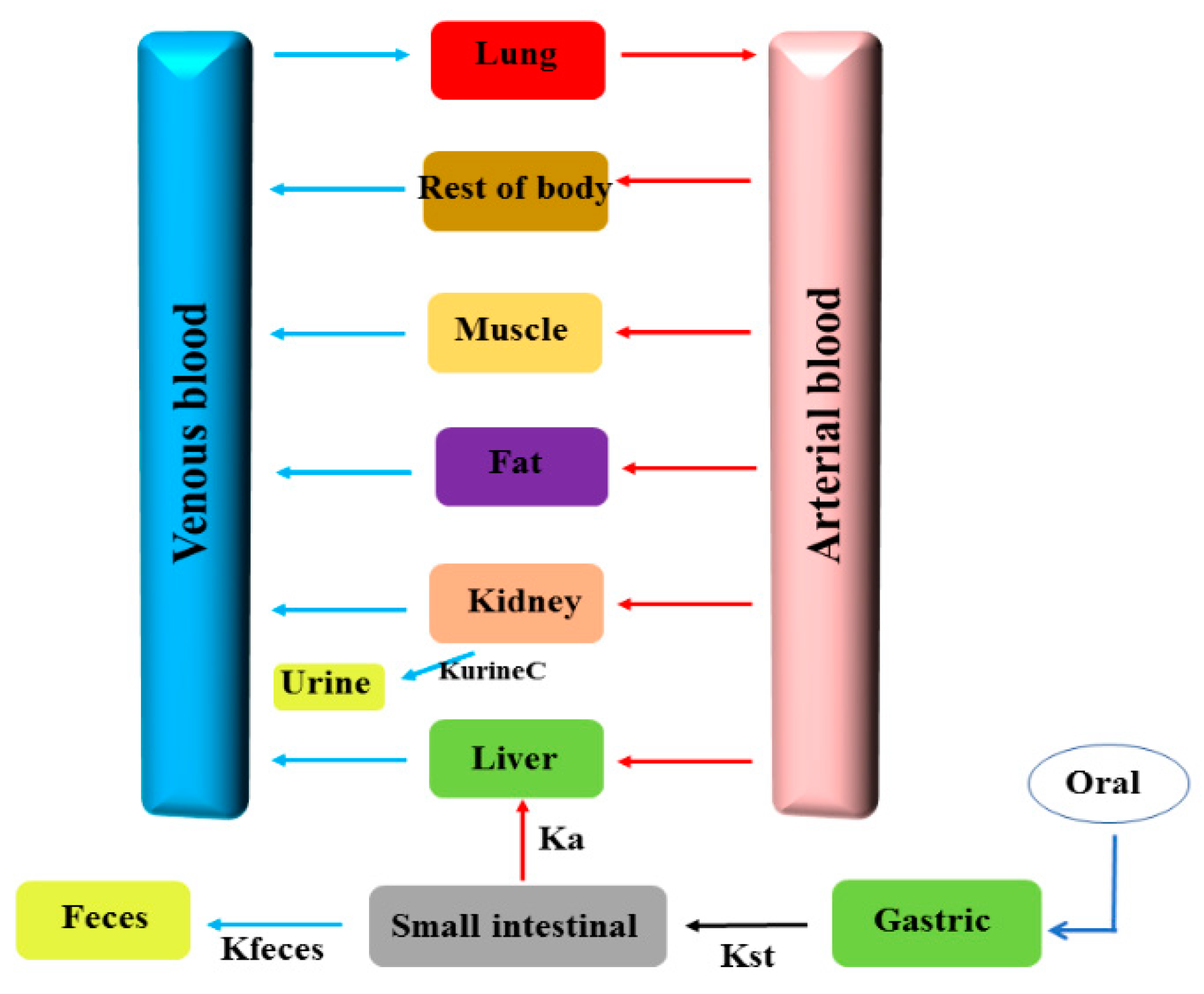
2.5. Development of the PBPK Model for ENR Granules in Pigs
2.6. Validation of the PBPK Model
2.7. Model Application
2.8. Parameter Sensitivity Analysis
3. Results and Discussion
3.1. Compatibility between ENR and Excipients
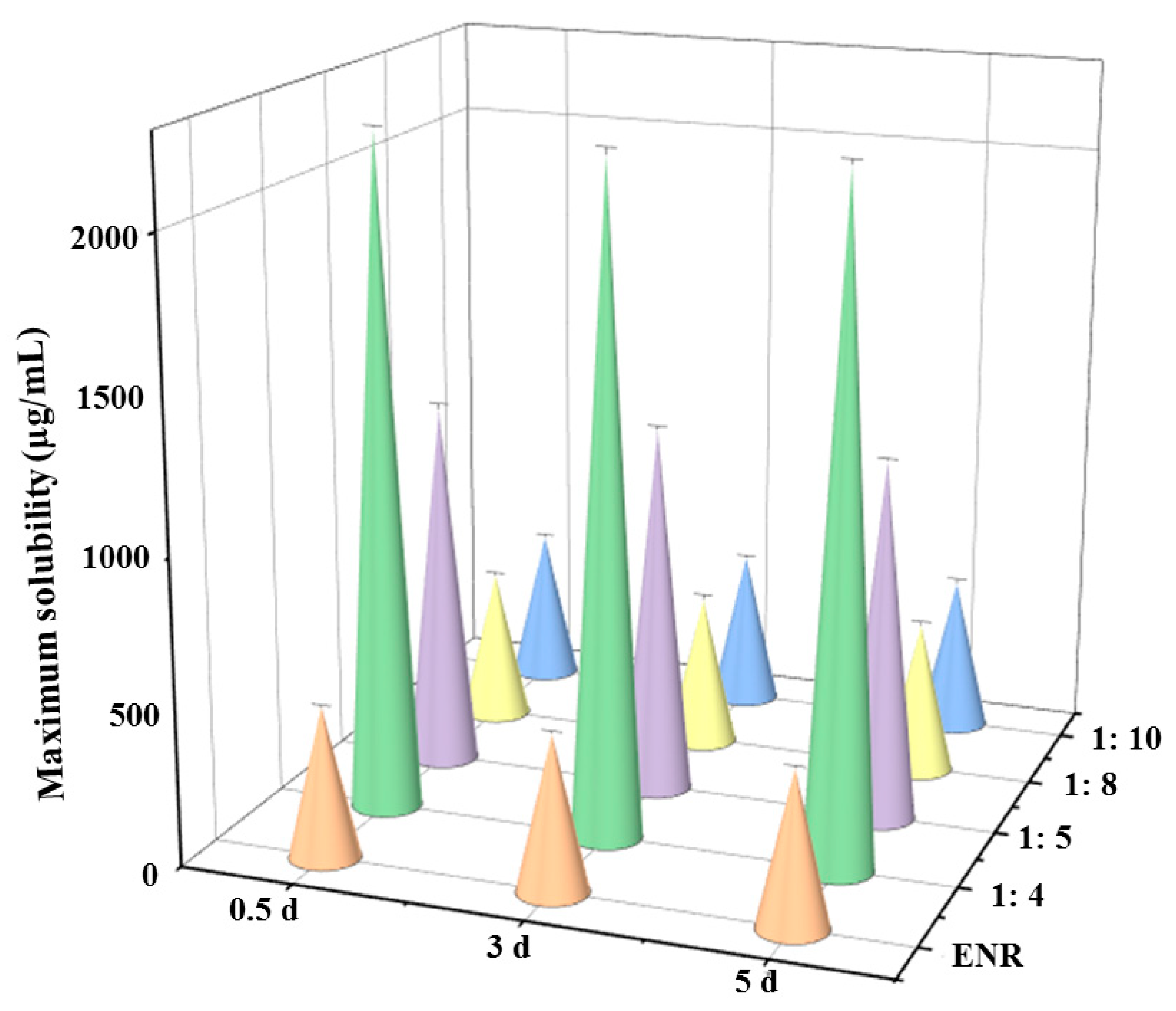
3.2. Solubility of ENR in SDs
3.3. Drug Crystals Form in the ENR-SD
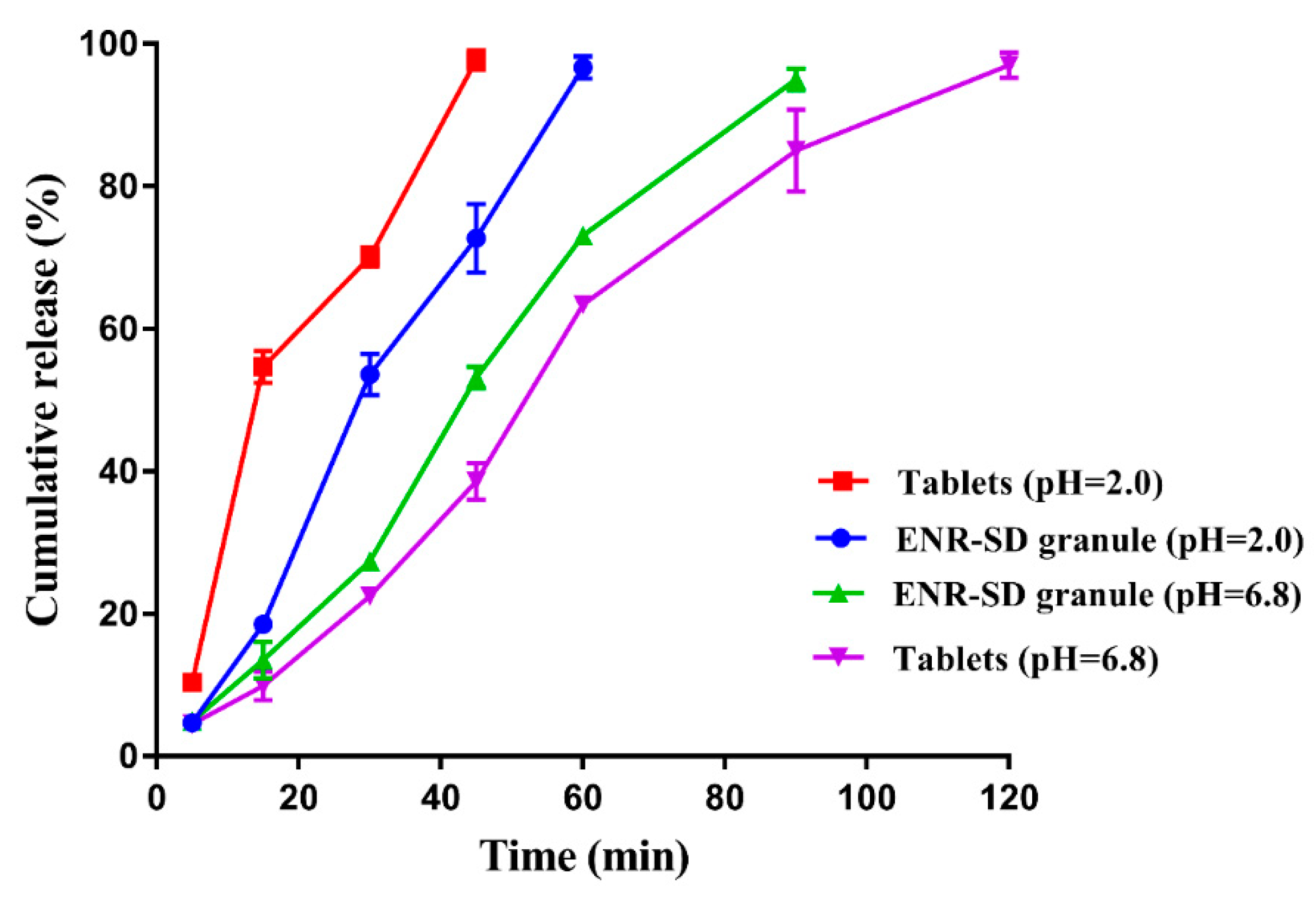
3.4. In Vitro Release Performance of ENR-SD Granules
3.5. Palatability of ENR-SD Granules
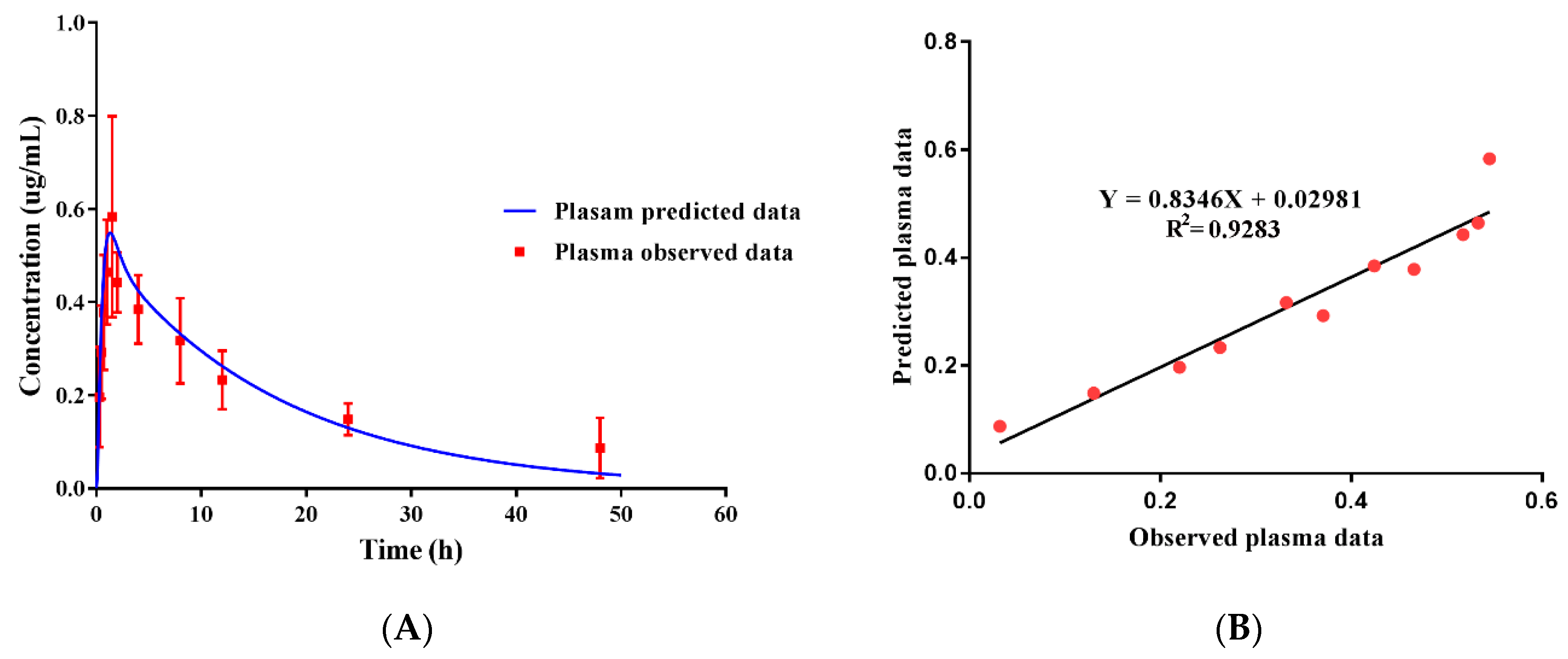
3.6. Pharmacokinetics and the Validation of the PBPK Model
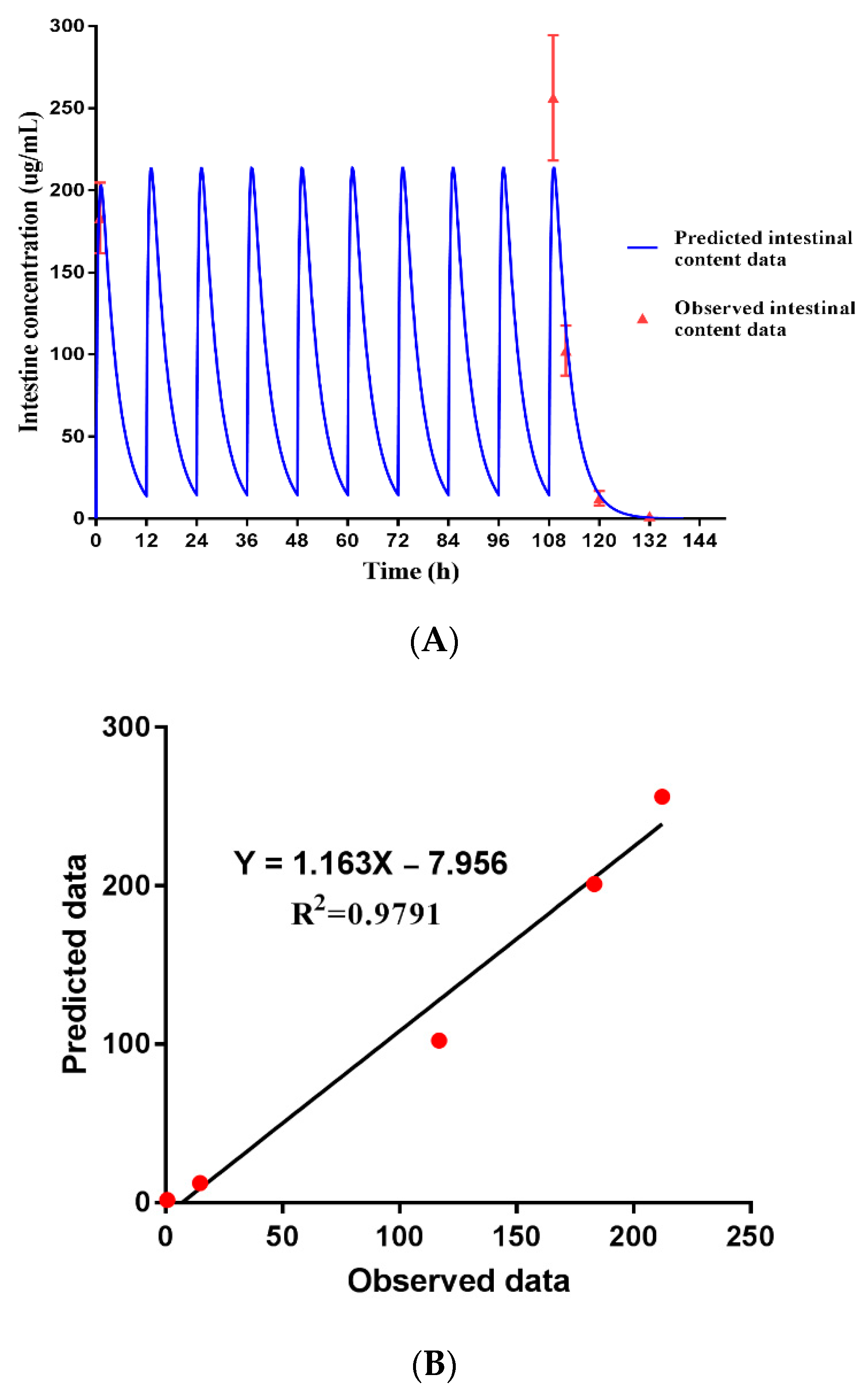
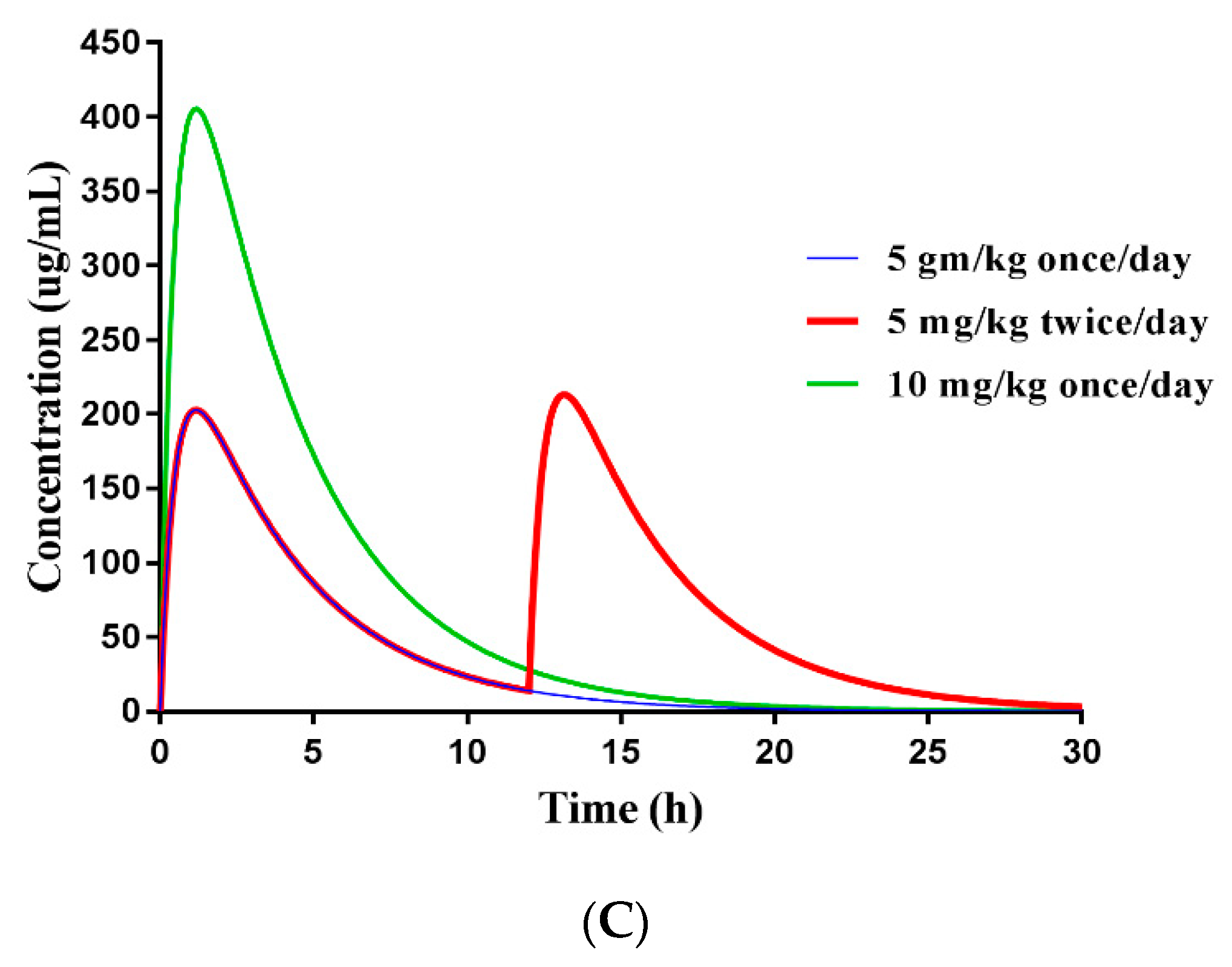
3.7. Dose Prediction of ENR-SD Granules
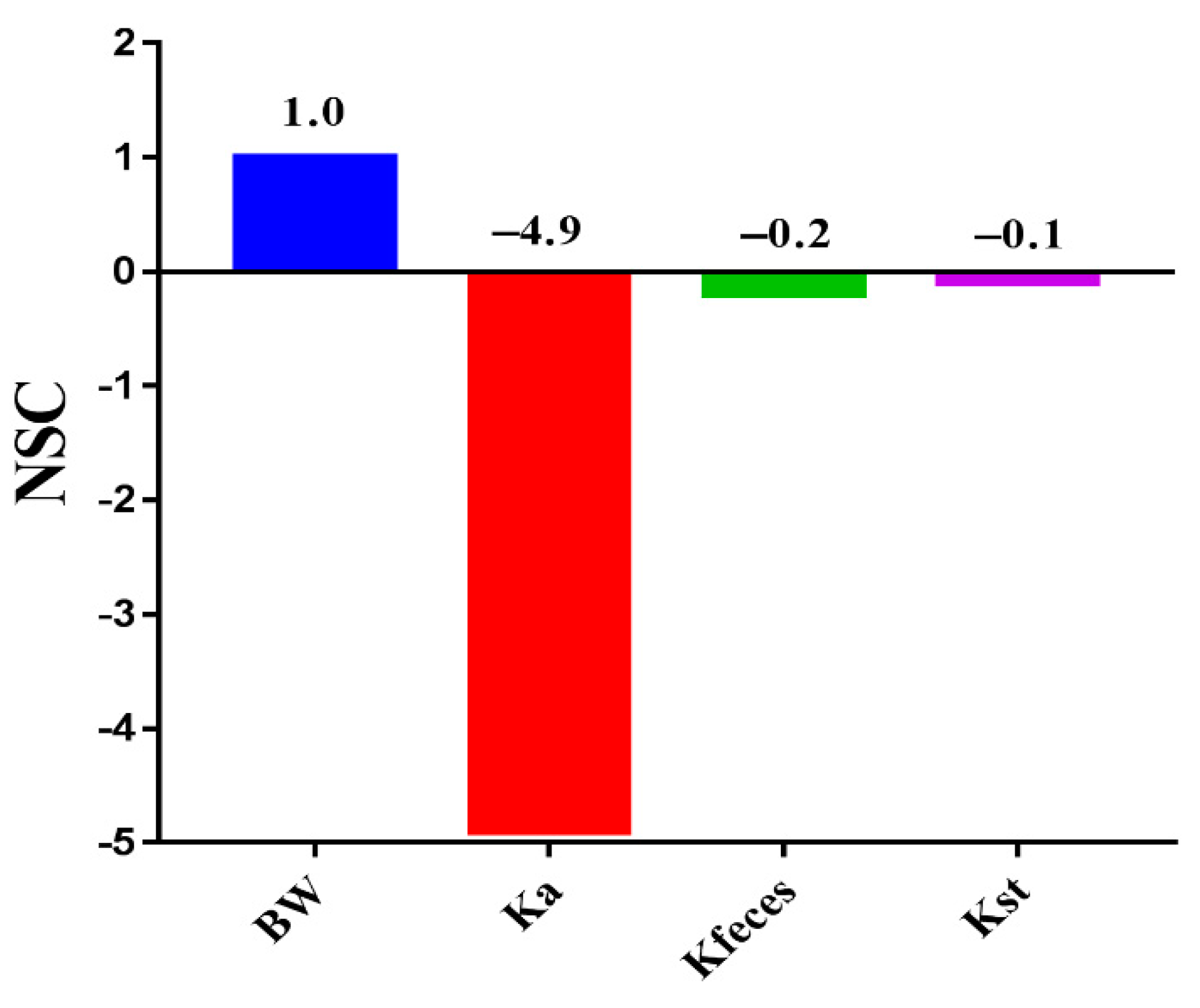
3.8. Sensitivity Parameters and Development Strategies of ENR-SD Granules
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Altekruse, S.F.; Stern, N.J.; Fields, P.I.; Swerdlow, D.L. Campylobacter jejuni—An emerging foodborne pathogen. Emerg. Infect. Dis. 1999, 5, 28–35. [Google Scholar] [CrossRef]
- Wernicki, A.; Nowaczek, A.; Urban-Chmiel, R. Bacteriophage therapy to combat bacterial infections in poultry. Virol. J. 2017, 14, 179. [Google Scholar] [CrossRef] [PubMed]
- Nhung, N.T.; Chansiripornchai, N.; Carrique-Mas, J.J. Antimicrobial resistance in bacterial poultry pathogens: A review. Front. Vet. Sci. 2017, 4, 126. [Google Scholar] [CrossRef] [Green Version]
- Burnham, P.M.; Hendrixson, D.R. Campylobacter jejuni: Collective components promoting a successful enteric lifestyle. Nat. Rev. Microbiol. 2018, 16, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.B.; Isaacson, R.E. Salmonella in swine: Microbiota interactions. Annu. Rev. Anim. Biosci. 2017, 5, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.; Rajora, V.R.; Arora, N. Prevalence of Salmonella in pigs and broilers in the Tarai region of Uttarakhand, India. Indian J. Med. Microbiol. 2014, 32, 99–101. [Google Scholar] [CrossRef] [PubMed]
- EFSA. The European Union Summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2014. EFSA J. 2015, 13, 4329. [Google Scholar]
- Yang, F.; Kang, J.; Yang, F.; Zhao, Z.; Kong, T.; Zeng, Z. Preparation and evaluation of enrofloxacin microspheres and tissue distribution in rats. J. Vet. Sci. 2015, 16, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Yin, D.; Fu, H.; Deng, F.; Peng, G.; Shu, G.; Yuan, Z.; Shi, F.; Lin, J.; Zhao, L.; et al. Double-coated enrofloxacin microparticles with chitosan and alginate: Preparation, characterization and taste-masking effect study. Carbohydr. Polym. 2017, 170, 247–253. [Google Scholar] [CrossRef]
- Shin, E.; Lee, Y. Antimicrobial resistance of 114 porcine isolates of Campylobacter coli. Food Microbiol. 2007, 118, 223–227. [Google Scholar] [CrossRef]
- Cao, T.T.; Deng, G.H.; Fang, L.X.; Yang, R.S.; Sun, J.; Liu, Y.H.; Liao, X.P. Characterization of quinolone resistance in Salmonella enterica from farm animals in china. J. Food Prot. 2017, 80, 1742–1748. [Google Scholar] [CrossRef]
- Sang, K.; Hao, H.; Huang, L.; Wang, X.; Yuan, Z. Pharmacokinetic-pharmacodynamic modeling of enrofloxacin against Escherichia coli in broilers. Front. Vet. Sci. 2016, 2, 80. [Google Scholar] [CrossRef] [Green Version]
- Chun, M.K.; Choi, H.K. Preparation and characterization of enrofloxacin/carbopol complex in aqueous solution. Arch. Pharmacal. Res. 2004, 27, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Xie, X.; Liu, X.; Cui, Z.; Yang, X.; Yeung, K.; Pan, H.; Chu, P.K.; Wu, S. The controlled drug release by pH-sensitive molecularly imprinted nanospheres for enhanced antibacterial activity. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 77, 84–91. [Google Scholar] [CrossRef]
- Choonara, B.F.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; Toit, L.D.; Pillay, V.A. Review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. Biotechnol. Adv. 2014, 32, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Rani, S.; Rana, R.; Saraogi, G.K.; Kumar, V.; Gupta, U. Self-emulsifying oral lipid drug delivery systems: Advances and challenges. AAPS PharmSciTech 2019, 20, 129. [Google Scholar] [CrossRef] [PubMed]
- Blokhina, S.V.; Sharapova, A.V.; Ol’khovich, M.V.; Volkova, T.V.; Perlovich, G.L. Solubility, lipophilicity and membrane permeability of some fluoroquinolone antimicrobials. Eur. J. Pharm. Sci. 2016, 93, 29–37. [Google Scholar] [CrossRef]
- Li, C.; Zhou, K.; Chen, D.; Xu, W.; Tao, Y.; Pan, Y.; Meng, K.; Shabbir, M.; Liu, Q.; Huang, L.; et al. Solid lipid nanoparticles with enteric coating for improving stability, palatability, and oral bioavailability of enrofloxacin. Int. J. Nanomed. 2019, 14, 1619–1631. [Google Scholar] [CrossRef] [Green Version]
- Karavas, E.; Georgarakis, E.; Sigalas, M.P.; Avgoustakis, K.; Bikiaris, D. Investigation of the release mechanism of a sparingly water-soluble drug from solid dispersions in hydrophilic carriers based on physical state of drug, particle size distribution and drug-polymer interactions. Eur. J. Pharm. Biopharm. 2007, 66, 334–347. [Google Scholar] [CrossRef]
- Bikiaris, D.N. Solid dispersions, part I: Recent evolutions and future opportunities in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1501–1519. [Google Scholar] [CrossRef]
- Tran, P.; Pyo, Y.C.; Kim, D.H.; Lee, S.E.; Kim, J.K.; Park, J.S. Overview of the manufacturing methods of solid dispersion technology for improving the solubility of poorly water-soluble drugs and application to anticancer drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Boyce, C.; Rhodes, T.; Liu, L.; Salituro, G.M.; Lee, K.J.; Bak, A.; Leung, D.H. A novel method for preparing stabilized amorphous solid dispersion drug formulations using acoustic fusion. Int. J. Pharm. 2020, 592, 120026. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, N.; Kuentz, M. Glass-forming ability of compounds in marketed amorphous drug products. Eur. J. Pharm. Biopharm. 2017, 112, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Zhu, W.; Kesisoglou, F. Physiologically based absorption modeling for amorphous solid dispersion formulations. Mol. Pharm. 2016, 13, 3206–3215. [Google Scholar] [CrossRef]
- Kambayashi, A.; Kiyota, T.; Fujiwara, M.; Dressman, J.B. PBPK modeling coupled with biorelevant dissolution to forecast the oral performance of amorphous solid dispersion formulations. Eur. J. Pharm. Sci. 2019, 135, 83–90. [Google Scholar] [CrossRef]
- Yang, F.; Wang, B.; Liu, Z.; Xia, X.; Wang, W.; Yin, D.; Sheng, L.; Li, Y. Prediction of a therapeutic dose for buagafuran, a potent anxiolytic agent by physiologically based pharmacokinetic/pharmacodynamic modeling starting from pharmacokinetics in rats and human. Front. Pharmacol. 2017, 8, 683. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, K.; Liu, F.; Li, Y.; Zhong, Z.; Hong, S.; Liu, X.; Liu, L. Predicting antitumor effect of deoxypodophyllotoxin in NCI-H460 tumor-bearing mice on the basis of in vitro pharmacodynamics and a physiologically based pharmacokinetic-pharmacodynamic model. Drug Metab. Dispos. 2018, 46, 897–907. [Google Scholar] [CrossRef] [Green Version]
- Alqahtani, S.; Kaddoumi, A. Development of physiologically based pharmacokinetic/pharmacodynamic model for indomethacin disposition in pregnancy. PLoS ONE 2015, 10, e0139762. [Google Scholar] [CrossRef] [Green Version]
- Schuck, E.L.; Dalhoff, A.; Stass, H.; Derendorf, H. Pharmacokinetic/pharmacodynamic (PK/PD) evaluation of a once-daily treatment using ciprofloxacin in an extended-release dose form. Infection 2005, 33, 22–28. [Google Scholar] [CrossRef]
- Ali, S.; Afrooz, H.; Hampel, R.; Mohamed, E.M.; Bhattacharya, R.; Cook, P.; Khan, M.A.; Rahman, Z. Blend of cellulose ester and enteric polymers for delayed and enteric coating of core tablets of hydrophilic and hydrophobic drugs. Int. J. Pharm. 2019, 567, 118462. [Google Scholar] [CrossRef]
- Ogawa, N.; Hiramatsu, T.; Suzuki, R.; Okamoto, R.; Shibagaki, K.; Fujita, K.; Takahashi, C.; Kawashima, Y.; Yamamoto, H. Improvement in the water solubility of drugs with a solid dispersion system by spray drying and hot-melt extrusion with using the amphiphilic polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol graft copolymer and d-mannitol. Eur. J. Pharm. Sci. 2018, 111, 205–214. [Google Scholar] [CrossRef]
- Hurley, D.; Davis, M.; Walker, G.M.; Lyons, J.G.; Higginbotham, C.L. The effect of cooling on the degree of crystallinity, solid-state properties, and dissolution rate of multi-component hot-melt extruded solid dispersions. Pharmaceutics 2020, 12, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, J.; Giri, B.R.; Song, E.S.; Bae, J.; Lee, J.; Kim, D.W. Spray-dried amorphous solid dispersions of atorvastatin calcium for improved supersaturation and oral bioavailability. Pharmaceutics 2019, 11, 461. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.S.; Illum, L.; Hinchcliffe, M. Gastrointestinal transit of dosage forms in the pig. J. Pharm. Pharmacol. 2001, 53, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Q.; Wang, A. In situ generation of sodium alginate/hydroxyapatite nanocomposite beads as drug-controlled release matrices. Acta Biomater. 2010, 6, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Yan, Y.; Chen, D.; Huang, L.; Li, C.; Meng, K.; Wang, S.; Algharib, S.A.; Yuan, Z.; Xie, S. Solid lipid nanoparticles for duodenum targeted oral delivery of tilmicosin. Pharmaceutics 2020, 12, 731. [Google Scholar] [CrossRef]
- Upton, R.N. Organ weights and blood flows of sheep and pig for physiological pharmacokinetic modelling. J. Pharmacol. Toxicol. Methods 2008, 58, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Vahl, C.I.; Riviere, J.E. Human food safety implications of variation in food animal drug metabolism. Sci. Rep. 2016, 6, 27907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Li, M.; Wang, Y.S.; Tell, L.A.; Baynes, R.E.; Davis, J.L.; Vickroy, T.W.; Riviere, J.E. Physiological parameter values for physiologically based pharmacokinetic models in food-producing animals. Part. I: Cattle and swine. J. Vet. Pharmacol. Ther. 2020, 43, 385–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, D.; Lin, Z.; Fang, B.; Li, M.; Gehring, R.; Riviere, J.E.; Zeng, Z. Pharmacokinetics of mequindox and its marker residue 1,4-bisdesoxymequindox in swine following multiple oral gavage and intramuscular administration: An experimental study coupled with population physiologically based pharmacokinetic modeling. J. Agric. Food Chem. 2017, 65, 5768–5777. [Google Scholar] [CrossRef]
- Lautz, L.S.; Dorne, J.; Oldenkamp, R.; Hendriks, A.J.; Ragas, A. Generic physiologically based kinetic modelling for farm animals: Part I. Data collection of physiological parameters in swine, cattle and sheep. Toxicol. Lett. 2020, 319, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Craig, W.A. Does the dose matter? Clin. Infect. Dis. 2001, 33 (Suppl. S3), S233–S237. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Li, M.; Chou, W.C.; Lin, Z. A physiologically based pharmacokinetic model of doxycycline for predicting tissue residues and withdrawal intervals in grass carp (Ctenopharyngodon idella). Food Chem. Toxicol. 2020, 137, 111127. [Google Scholar] [CrossRef] [PubMed]
- Mesallati, H.; Umerska, A.; Tajber, L. Fluoroquinolone amorphous polymeric salts and dispersions for veterinary uses. Pharmaceutics 2019, 11, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corvis, Y.; Négrier, P.; Espeau, P. Physicochemical stability of solid dispersions of enantiomeric or racemic ibuprofen in stearic acid. J. Pharm. Sci. 2011, 100, 5235–5243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorat, A.A.; Suryanarayanan, R. Characterization of phosphate buffered saline (PBS) in frozen state and after freeze-drying. Pharm. Res. 2019, 36, 98. [Google Scholar] [CrossRef]
- Wang, Z.J.; Liang, C.L.; Li, G.M.; Yu, C.Y.; Yin, M. Stearic acid protects primary cultured cortical neurons against oxidative stress. Acta Pharmacol. Sin. 2007, 28, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.Y.; Zhang, Z.H.; Jiang, Y.R.; Ning, Q.; Liu, Q.Y.; Jia, X.B. Studies on sustained release solid dispersion of tripterine carried by HPMC-stearic acid. Zhongguo Zhong Yao Za Zhi. 2012, 37, 3052–3055. [Google Scholar]
- Wen, T.; Niu, B.; Wu, Q.; Zhou, Y.; Pan, X.; Quan, G.; Wu, C. Fenofibrate solid dispersion processed by hot-melt extrusion: Elevated bioavailability and its cell transport mechanism. Curr. Drug Deliv. 2019, 16, 538–547. [Google Scholar] [CrossRef]
- Ponnammal, P.; Kanaujia, P.; Yani, Y.; Ng, W.K.; Tan, R. Orally disintegrating tablets containing melt extruded amorphous solid dispersion of tacrolimus for dissolution enhancement. Pharmaceutics 2018, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Taylor, L.S. Tailoring supersaturation from amorphous solid dispersions. J. Control. Release 2018, 279, 114–125. [Google Scholar] [CrossRef]
- Weerapol, Y.; Tubtimsri, S.; Jansakul, C.; Sriamornsak, P. Improved dissolution of Kaempferia parviflora extract for oral administration by preparing solid dispersion via solvent evaporation. Asian J. Pharm. Sci. 2017, 12, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Łyszczarz, E.; Hofmanová, J.; Szafraniec-Szczęsny, J.; Jachowicz, R. Orodispersible films containing ball milled aripiprazole-poloxamer®407 solid dispersions. Int. J. Pharm. 2020, 575, 118955. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Liu, Q.; Yang, B.; Xiong, J.; Li, K.; Ahmed, S.; Hong, L.; Chen, P.; He, Q.; Cao, J. Clinical efficacy and residue depletion of 10% enrofloxacin enteric-coated granules in pigs. Front. Pharmacol. 2017, 8, 294. [Google Scholar] [CrossRef]
- Hao, H.; Pan, H.; Ahmad, I.; Cheng, G.; Wang, Y.; Dai, M.; Tao, Y.; Chen, D.; Peng, D.; Liu, Z.; et al. Susceptibility breakpoint for enrofloxacin against swine Salmonella spp. J. Clin. Microbiol. 2013, 51, 3070–3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hao, H.; Huang, L.; Liu, Z.; Chen, D.; Yuan, Z. Pharmacokinetic and pharmacodynamic integration and modeling of enrofloxacin in swine for Escherichia coli. Front. Microbiol. 2016, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Wattanaphansak, S.; Pereira, C.; Kaenson, W.; Assavacheep, P.; Tantilertcharoen, R.; Resende, T.P.; Barrera-Zarate, J.A.; de Oliveira-Lee, J.; Klein, U.; Gebhart, C.J.; et al. Isolation and in vitro antimicrobial susceptibility of porcine Lawsonia intracellularis from Brazil and Thailand. BMC Microbiol. 2019, 19, 27. [Google Scholar] [CrossRef]
- Lee, S.J.; Awji, E.G.; Park, N.H.; Park, S.C. Using in vitro dynamic models to evaluate fluoroquinolone activity against emergence of resistant Salmonella enterica serovar typhimurium. Antimicrob. Agents Chemother. 2017, 61, e01756-16. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Liu, C.; Wu, Y.; Zhang, X.; Fan, J.; Cao, Y. A multiscale physiologically-based pharmacokinetic model for doxorubicin to explore its mechanisms of cytotoxicity and cardiotoxicity in human physiological contexts. Pharm. Res. 2018, 35, 174. [Google Scholar] [CrossRef]
- Kuepfer, L.; Niederalt, C.; Wendl, T.; Schlender, J.F.; Willmann, S.; Lippert, J.; Block, M.; Eissing, T.; Teutonico, D. Applied concepts in pbpk modeling, how to build a PBPK/PD model. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 516–531. [Google Scholar] [CrossRef] [Green Version]
- Lenhard, J.R.; Smith, N.M.; Bulman, Z.P.; Tao, X.; Thamlikitkul, V.; Shin, B.S.; Nation, R.L.; Li, J.; Bulitta, J.B.; Tsuji, B.T. High-dose ampicillin-sulbactam combinations combat polymyxin-resistant Acinetobacter baumannii in a hollow-fiber infection model. Antimicrob. Agents Chemother. 2017, 61, e01268-16. [Google Scholar] [CrossRef] [Green Version]
- Pires de Mello, C.P.; Tao, X.; Kim, T.H.; Vicchiarelli, M.; Bulitta, J.B.; Kaushik, A.; Brown, A.N. Clinical Regimens of Favipiravir Inhibit Zika Virus Replication in the Hollow-Fiber Infection Model. Antimicrob. Agents Chemother. 2018, 62, e00967-18. [Google Scholar] [CrossRef] [Green Version]
- García-Fernández, E.; Koch, G.; Wagner, R.M.; Fekete, A.; Stengel, S.T.; Schneider, J.; Mielich-Süss, B.; Geibel, S.; Markert, S.M.; Stigloher, C.; et al. Membrane microdomain disassembly inhibits mrsa antibiotic resistance. Cell 2017, 171, 1354–1367.e20. [Google Scholar] [CrossRef]
- Huang, F.C.; Huang, S.C. Differential effects of statins on inflammatory interleukin-8 and antimicrobial peptide human β-defensin 2 responses in salmonella-infected intestinal epithelial cells. Int. J. Mol. Sci. 2018, 19, 1650. [Google Scholar] [CrossRef] [Green Version]
- Griffith, D.C.; Sabet, M.; Tarazi, Z.; Lomovskaya, O.; Dudley, M.N. Pharmacokinetics/Pharmacodynamics of vaborbactam, a novel beta-lactamase inhibitor, in combination with meropenem. Antimicrob Agents Chemother. 2018, 63, e01659-18. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.W.; Yu, H.H.; Zhao, J.; Han, M.L.; Zhu, Y.; Akter, J.; Wickremasinghe, H.; Walpola, H.; Wirth, V.; Rao, G.G.; et al. Polymyxin B in Combination with Enrofloxacin Exerts Synergistic Killing against Extensively Drug-Resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2018, 62, e00028-18. [Google Scholar] [CrossRef] [Green Version]
- Bhutto, Z.A.; He, F.; Zloh, M.; Yang, J.; Huang, J.; Guo, T.; Wang, L. Use of quercetin in animal feed: Effects on the P-gp expression and pharmacokinetics of orally administrated enrofloxacin in chicken. Sci. Rep. 2018, 8, 4400. [Google Scholar] [CrossRef] [Green Version]
- Giacone, D.V.; Carvalho, V.F.M.; Costa, S.K.P.; Lopes, L.B. Evidence that P-glycoprotein inhibitor (elacridar)-loaded nanocarriers improve epidermal targeting of an anticancer drug via absorptive cutaneous transporters inhibition. J. Pharm. Sci. 2018, 107, 698–705. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Before Experiment (kg/Group) | During Experiment (kg/Group) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1d | 2d | 3d | ± SD | 1d | 2d | 3d | 4d | 5d | ± SD | |
| Control | 1.25 | 1.32 | 1.37 | 1.31 ± 0.05 | 1.30 | 1.28 | 1.35 | 1.38 | 1.42 | 1.38 ± 0.03 |
| ENR granules | 1.30 | 1.35 | 1.33 | 1.33 ± 0.02 | 1.25 | 1.30 | 1.32 | 1.37 | 1.40 | 1.36 ± 0.03 |
| Parameters | Unit | Value |
|---|---|---|
| Vd | mL/Kg | 30,568 ± 15,686 |
| MRT | h | 15.37 ± 4.55 |
| AUC0-last | µg h/mL | 7.96 ± 1.22 |
| T1/2β | h | 12.58 ± 5.89 |
| Tmax | h | 1.42 ± 0.34 |
| Cmax | µg/mL | 0.64 ± 0.21 |
| Pathogens | MIC90 (µg/mL) | Dose Regimen | AUC24h (µg h/mL) | AUCI |
|---|---|---|---|---|
| Salmonella (n = 291) a,b | 8.0 | 2.5 mg/kg, twice/day | 957 | 120 |
| Salmonella (n = 291) a,b | 8.0 | 5.0 mg/kg, once/day | 1065 | 133 |
| E. coli (n = 918) c | 0.25 | 5.0 mg/kg, once/day | 1065 | 4260 |
| L. intracellularis (n = 1) d | 8.0 | 5.0 mg/kg, once/day | 1065 | 133 |
| Camp. Jejuni (n = 114) e | 16.0 | 7.5 mg/kg, once/day | 1542 | 96 |
| Camp. Jejuni (n = 114) e | 16.0 | 5.0 mg/kg, twice/day | 1884 | 118 |
| Camp. Jejuni (n = 114) e | 16.0 | 10.0 mg/kg, once/day | 2130 | 133 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, K.; Huo, M.; Ma, W.; Mi, K.; Xu, X.; Algharib, S.A.; Xie, S.; Huang, L. Application of a Physiologically Based Pharmacokinetic Model to Develop a Veterinary Amorphous Enrofloxacin Solid Dispersion. Pharmaceutics 2021, 13, 602. https://doi.org/10.3390/pharmaceutics13050602
Zhou K, Huo M, Ma W, Mi K, Xu X, Algharib SA, Xie S, Huang L. Application of a Physiologically Based Pharmacokinetic Model to Develop a Veterinary Amorphous Enrofloxacin Solid Dispersion. Pharmaceutics. 2021; 13(5):602. https://doi.org/10.3390/pharmaceutics13050602
Chicago/Turabian StyleZhou, Kaixiang, Meixia Huo, Wenjin Ma, Kun Mi, Xiangyue Xu, Samah Attia Algharib, Shuyu Xie, and Lingli Huang. 2021. "Application of a Physiologically Based Pharmacokinetic Model to Develop a Veterinary Amorphous Enrofloxacin Solid Dispersion" Pharmaceutics 13, no. 5: 602. https://doi.org/10.3390/pharmaceutics13050602