
Photodynamic Therapy as an Oxidative Anti-Tumor Modality: Negative Effects of Nitric Oxide on Treatment Efficacy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
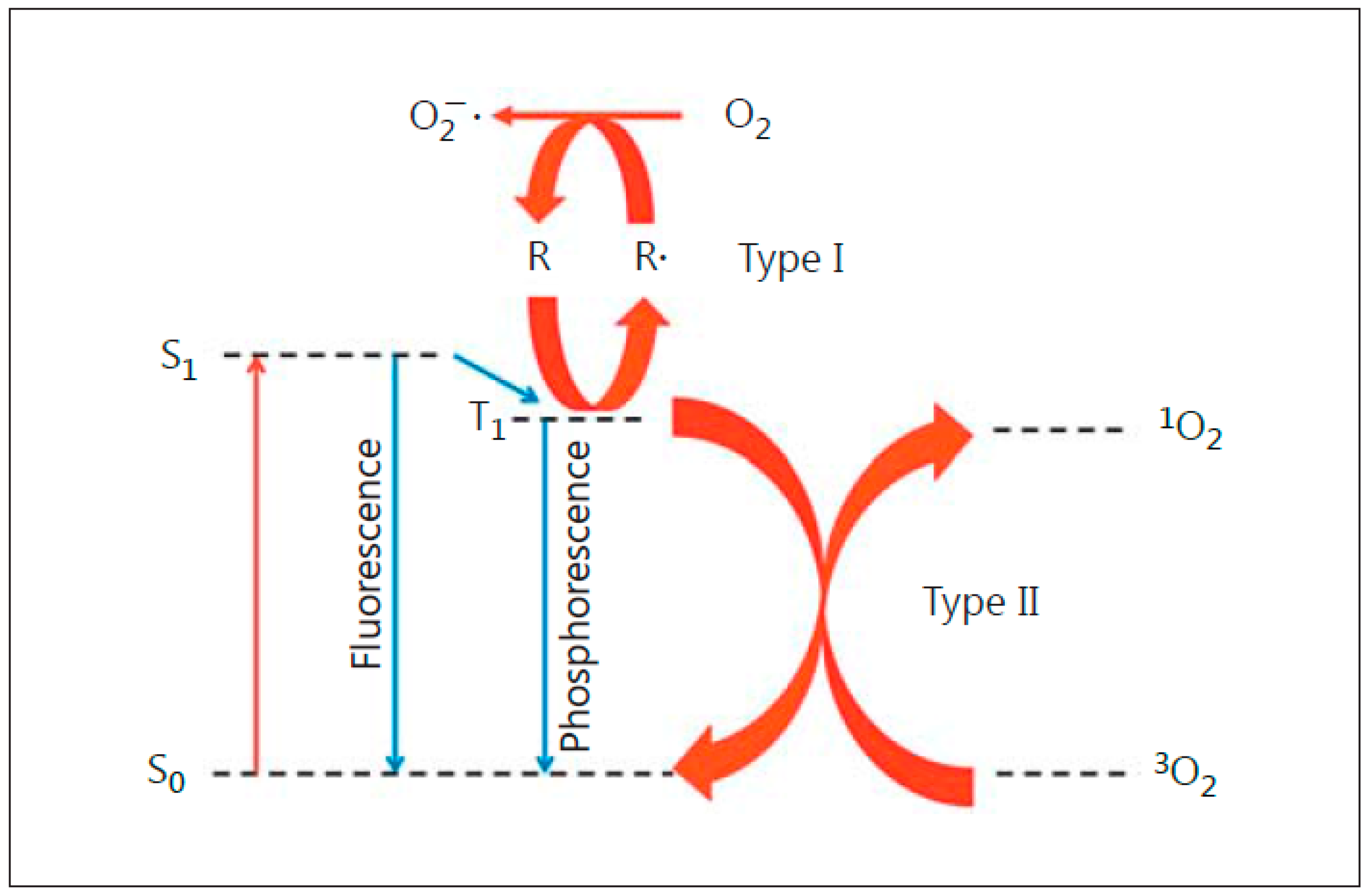
:1. Introduction: Photodynamic Therapy
2. Nitric Oxide and Its Role in Cancer
3. Role of Endogenous NO in Tumor Resistance to PDT
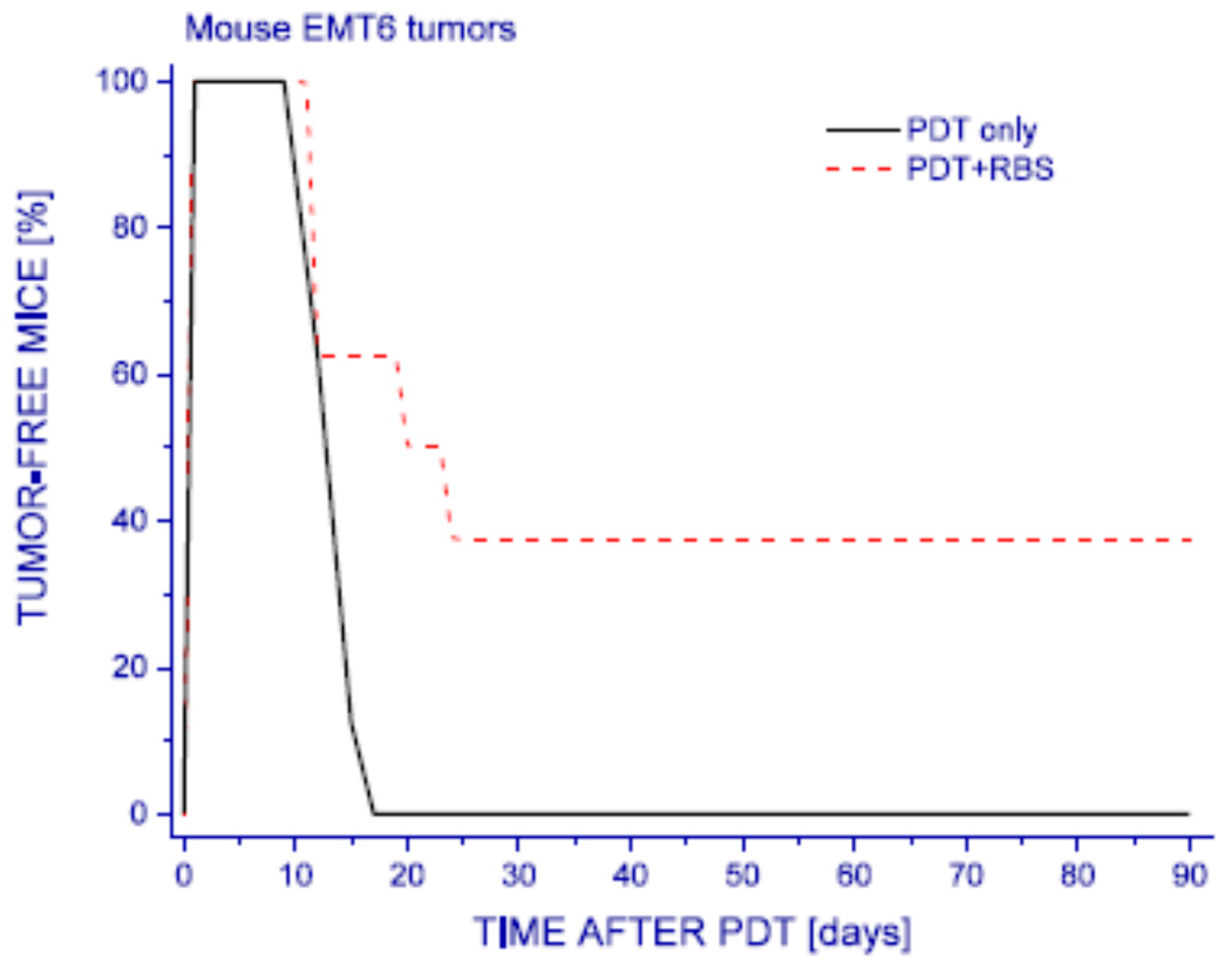
4. Possible Mechanisms of NO-Mediated Resistance: In Vivo Studies
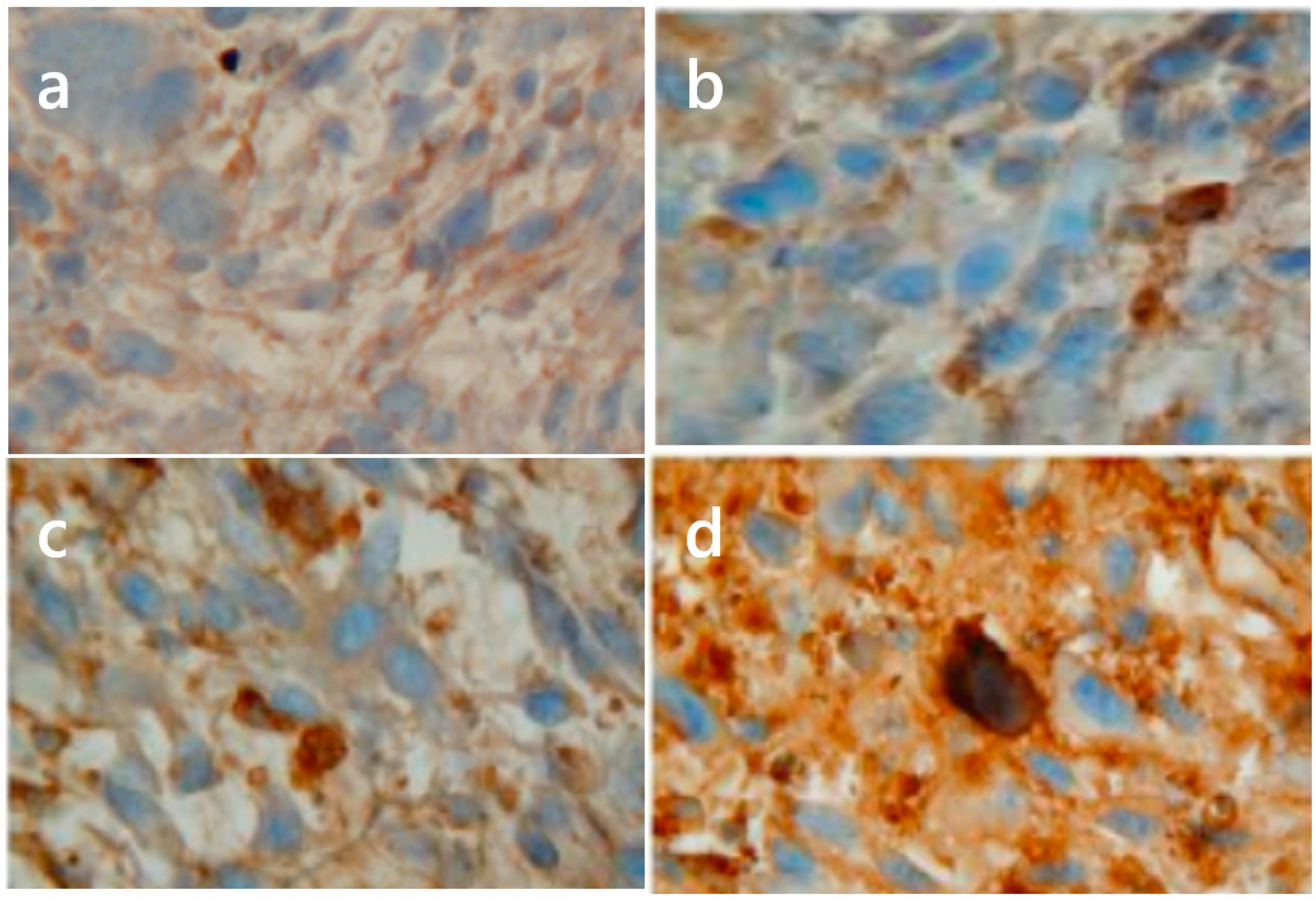
5. Role of iNOS/NO in Tumor Resistance to PDT: In Vitro and In Vivo Studies
6. Role of iNOS/NO in Hyper-Aggressiveness of PDT-Surviving Tumor Cells
7. Role of NO in PDT-Induced Bystander Effects
8. Mechanism of iNOS Upregulation by Photodynamic Stress
9. Pharmacologic Approaches for Mitigating NO’s Anti-PDT Effects
10. Summary and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Dougherty, T.J.; Grindey, G.B.; Fiel, R.; Weishaupt, K.R.; Boyle, D.G. Photoradiation therapy II: Cure of animal tumors with hematoporphyrin and light. J. Natl. Cancer Inst. 1975, 55, 115–121. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef] [Green Version]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Benov, L. Photodynamic therapy: Current status and future directions. Med. Pract. 2015, 24, 14–28. [Google Scholar] [CrossRef]
- Girotti, A.W. Photosensitized oxidation of membrane lipids: Reaction pathways, cytotoxic effects, and cytoprotective mechanisms. J. Photochem. Photobiol. B 2001, 63, 103–113. [Google Scholar] [CrossRef]
- Kennedy, J.C.; Pottier, R.H. Endogenous protoporphyrin IX, a clinically useful photosensitizer for photodynamic therapy. J. Photochem. Photobiol. B 1992, 14, 275–292. [Google Scholar] [CrossRef]
- Peng, Q.; Berg, K.; Moan, J.; Kongshaug, M.; Nesland, J.M. 5-Aminolevulinic acid-based photodynamic therapy: Principles and experimental research. Photochem. Photobiol. 1997, 65, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Kim, R. Recent advances in understanding the cell death pathways activated by anticancer therapy. Cancer 2005, 103, 1551–1560. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [Green Version]
- Hei, T.K.; Show, H.; Chai, Y.; Ponnaiya, B.; Ivanov, V.N. Radiation-induced non-targeted response: Mechanism and potential clinical implications. Curr. Mol. Pharmacol. 2011, 4, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Casas, A.; DiVenosa, G.; Hasan, T. Mechanisms of resistance to photodynamic therapy. Curr. Med. Chem. 2011, 18, 2486–2515. [Google Scholar] [CrossRef] [Green Version]
- Palasuberniam, P.; Yang, X.; Kraus, D.; Jones, P.; Myers, K.A.; Chen, B. ABCG2 transporter inhibitor restores the sensitivity of triple negative breast cancer cells to aminolevulinic acid-mediated photodynamic therapy. Sci. Rep. 2015, 5, 13298. [Google Scholar] [CrossRef]
- Thomas, D.D.; Liu, X.; Kantrow, S.P.; Lancaster, J.R., Jr. The biological lifetime of nitric oxide: Implications for the perivascular dynamics of NO and O2. Proc. Natl. Acad. Sci. USA 2011, 98, 355–360. [Google Scholar] [CrossRef]
- Wink, D.A.; Mitchell, J.B. Chemical biology of nitric oxide: Insights into the regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Rad. Biol. Med. 1998, 25, 434–456. [Google Scholar] [CrossRef]
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free Rad. Biol. Med. 2008, 45, 18–31. [Google Scholar] [CrossRef] [Green Version]
- Gantner, B.N.; LaFond, K.M.; Bonini, M.G. Nitric oxide in cellular adaptation and disease. Redox Biol. 2020, 34, 101550. [Google Scholar] [CrossRef] [PubMed]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Burke, A.J.; Sullivan, F.J.; Giles, F.J.; Glynn, S.A. The yin and yang of nitric oxide in cancer progression. Carcinogenesis 2013, 34, 503–512. [Google Scholar] [CrossRef] [Green Version]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, T.A.; da Silva, R.S.; Miranda, K.M.; Switzer, C.H.; Wink, D.A.; Fukuto, J.M. Biological nitric oxide signaling: Chemistry and terminology. Br. J. Pharmacol. 2013, 169, 1417–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamm, A.; Przychodzen, P.; Kuban-Jankowska, A.; Jacewicz, D.; Dabrowska, A.M.; Nussberger, S.; Wozniak, M.; Gorska-Ponikowska, M. Nitric oxide and its derivatives in the cancer battlefield. Nitric Oxide 2019, 93, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Switzer, C.H.; Glynn, S.A.; Ridnour, L.A.; Cheng, R.Y.; Vitek, M.P.; Ambs, S.; Wink, D.A. Nitric oxide and protein phosphatase 2A provide novel therapeutic opportunities in ER-negative breast cancer. Trends Pharmacol. Sci. 2011, 32, 644–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, K.; Ambs, S.; Lupold, S.E.; Kapust, R.B.; Spillare, E.A.; Weinberg, W.C.; Felley-Bosco, E.; Wang, X.W.; Geller, D.A.; Tzeng, E.; et al. Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc. Natl. Acad. Sci. USA 1996, 93, 2442–2447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calmels, S.; Hainaut, P.; Ohshima, H. Nitric oxide induces conformational and functional modifications of wild-type p53 tumor suppressor protein. Cancer Res. 1997, 57, 3365–3369. [Google Scholar]
- Ambs, S.; Merriam, W.G.; Ogunfusika, M.O.; Bennett, W.P.; Ishibe, N.; Hussain, S.P.; Tzeng, E.E.; Geller, D.A.; Billiar, T.R.; Harris, C.C. p53 and vascular endothelial growth factor regulate tumor growth of NOS2-expressing human carcinoma cells. Nat. Med. 1998, 4, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Diers, A.R.; Keszler, A.; Hogg, N. Detection of S-nitrosothiols. Biochim. Bipphys. Acta 2014, 1840, 892–900. [Google Scholar] [CrossRef] [Green Version]
- Wynia-Smith, S.L.; Smith, B.C. Nitrosothiol formation and S-nitrosation signaling through nitric oxide synthases. Nitric Oxide 2017, 63, 52–60. [Google Scholar] [CrossRef]
- Rizza, S.; Filomeni, G. Exploiting S-nitrosylation for cancer therapy: Facts and perspectives. Biochem. J. 2020, 477, 3649–3672. [Google Scholar] [CrossRef]
- Park, H.-S.; Yu, J.-W.; Cho, J.-H.; Kim, M.-S.; Huh, S.-H.; Ryoo, K.; Choi, E.-K. Inhibition of apoptosis signal-regulating kinase 1 by nitric oxide through a thiol redox mechanism. J. Biol. Chem. 2004, 279, 7584–7590. [Google Scholar] [CrossRef] [Green Version]
- Azad, N.; Vallyathan, V.; Wang, L.; Tantishaiyakul, V.; Stehlik, C.; Leonard, S.S.; Rojanasakul, Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation: A novel antiapoptotic mechanism that suppresses apoptosis. J. Biol. Chem. 2006, 281, 34124–34134. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.; Sha, J.; Chen, X.; Xing, Y.; Yan, J.; Wang, Z. S-nitrosylation of mitogen activated protein kinase phosphatase-1 suppresses radiation-induced apoptosis. Cancer Lett. 2012, 314, 137–146. [Google Scholar] [CrossRef]
- Kwak, Y.D.; Ma, T.; Diao, S.; Zhang, X.; Chen, Y.; Hsu, J.; Lipton, S.A.; Masliah, E.; Xu, H.; Liao, F.F. NO signaling and S-nitrosylation regulate PTEN inhibition in neurodegeneration. Mol. Neurodegener. 2010, 5, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, B.W.; Sitnik-Busch, T.M.; Vaughn, L.A. Potentiation of photodynamic therapy antitumor activity in mice by nitric oxide synthase inhibition is fluence rate dependent. Photochem. Photobiol. 1999, 70, 64–71. [Google Scholar] [CrossRef]
- Korbelik, M.; Shibuya, H.; Cecic, I. Relevance of nitric oxide to the response of tumors to photodynamic therapy. SPIE Proc. 1998, 3247, 98–105. [Google Scholar]
- Korbelik, M.; Parkins, C.S.; Shibuya, H.; Cecic, I.; Stratford, R.M.L.; Chaplin, D.J. Nitric oxide production by tumor tissue: Impact on the response to photodynamic therapy. Br. J. Cancer 2000, 82, 1835–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeves, K.L.; Reed, M.W.R.; Brown, N.J. The role of nitric oxide in the treatment of tumours with aminolaevulinic acid-induced photodynamic therapy. J. Photochem. Photobiol. B 2010, 101, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxinitrite: The good, the bad, and ugly. Am. J. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [Green Version]
- Shahani, M.; Sawa, A. Protein S-nitrosylation: Role for nitric oxide signaling in neuronal death. Biochim. Biophys. Acta 2012, 1820, 736–742. [Google Scholar] [CrossRef] [Green Version]
- Eiserich, J.P.; Hristova, M.; Cross, C.E.; Jones, A.D.; Freeman, B.A.; Halliwell, B.; van der Vliet, A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature 1988, 391, 393–407. [Google Scholar] [CrossRef]
- Krosl, G.; Korbelik, M.; Dougherty, G.J. Induction of immune cell infiltration into murine SCCVII tumor by Photofrin-based photodynamic therapy. Br. J. Cancer 1995, 71, 549–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbelik, M.; Cooper, P.D. Potentiation of photodynamic therapy by complement: The effect of γ-inulin. Br. J. Cancer 2007, 96, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Sluiter, W.; Korbelik, M.; Cecic, I.; de Wit, E. Neutrophil-mediated oxidative and non-oxidative stress in tumors treated by photodynamic therapy. In Proceedings of the 30th Annual Meeting of the American Society for Photobiology, Quebec City, QC, Canada, 13–17 July 2002. [Google Scholar]
- Cecic, I.; Korbelik, M. Mediators of peripheral blood neutrophilia induced by photodynamic therapy of solid tumors. Cancer Lett. 2002, 183, 43–51. [Google Scholar] [CrossRef]
- Bourassa, J.; DeGraff, W.; Kudo, S.; Wink, D.A.; Mitchell, J.B.; Ford, P.C. Photochemistry of Roussin’s Red Salt, Na2[Fe2S2(NO)4], and Roussin’s Black Salt, NH4[Fe4S3(NO)7]. In situ nitric oxide generation to sensitize γ-radiation induced cell death. J. Am. Chem. Soc. 1997, 119, 2853–2860. [Google Scholar] [CrossRef]
- Burton, G.W.; Ingold, K.U. Vitamin E as an in vitro and in vivo antioxidant. Ann. N. Y. Acad. Sci. 1989, 570, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Rubbo, H.; Radi, R.; Trujillo, M.; Telleri, R.; Kalyanaraman, B.; Barnes, S.; Kirk, M.; Freeman, B.A. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation: Formation of novel nitrogen-containing oxidized lipid derivatives. J. Biol. Chem. 1994, 269, 26066–26075. [Google Scholar] [CrossRef]
- Rubbo, H.; Parthasarathy, S.; Barnes, S.; Kirk, M.; Kalyanaraman, B.; Freeman, B.A. Nitric oxide inhibition of lipoxygenase-dependent liposome and low-density lipoprotein oxidation: Termination of radical chain propagation reactions and formation of nitrogen-containing oxidized lipid derivatives. Arc. Biochem. Biophys. 1995, 324, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Kelley, E.E.; Wagner, B.A.; Buettner, G.R.; Burns, C.P. Nitric oxide inhibits iron-induced lipid peroxidation in HL-60 cells. Arch. Biochem. Biophys. 1999, 370, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korytowski, W.; Zareba, M.; Girotti, A.W. Inhibition of free radical-mediated cholesterol peroxidation by diazeniumdiolate-derived nitric oxide: Effect of release rate on mechanism of action in a membrane system. Chem. Res. Toxicol. 2000, 13, 1265–1274. [Google Scholar] [CrossRef]
- Korytowski, W.; Geiger, P.G.; Girotti, A.W. Lipid hydroperoxide analysis by high performance liquid chromatography with mercury cathode electrochemical detection. Methods Enzymol. 1999, 300, 23–33. [Google Scholar]
- Korytowski, W.; Wrona, M.; Girotti, A.W. Radiolabeled cholesterol as a reporter for assessing one-electron turnover of lipid hydroperoxides. Anal. Biochem. 1999, 270, 123–132. [Google Scholar] [CrossRef] [PubMed]
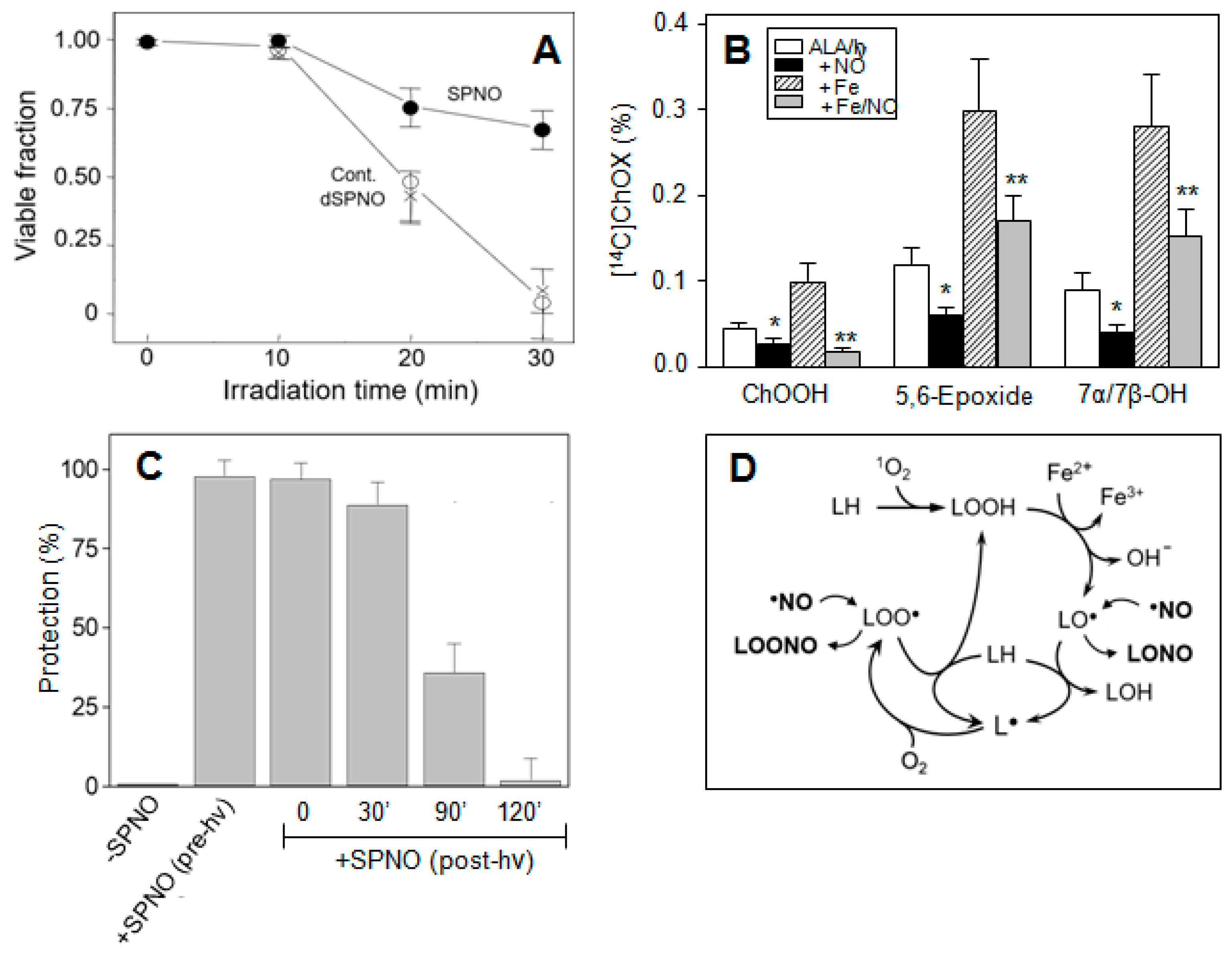
- Niziolek, M.; Korytowski, W.; Girotti, A.W. Nitric oxide inhibition of free radical-mediated lipid peroxidation in photodynamically treated membranes and cells. Free Radic. Biol. Med. 2003, 34, 997–1005. [Google Scholar] [CrossRef]
- Niziolek, M.; Korytowski, W.; Girotti, A.W. Chain-breaking antioxidant and cytoprotective action of nitric oxide on photodynamically stressed tumor cells. Photochem. Photobiol. 2003, 78, 262–270. [Google Scholar] [CrossRef]
- Bhowmick, R.; Girotti, A.W. Cytoprotective induction of nitric oxide synthase in a cellular model of 5-aminolevulinic acid-based photodynamic therapy. Free Radic. Biol. Med. 2010, 48, 1296–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhowmick, R.; Girotti, A.W. Pro-survival and pro-growth effects of stress-induced nitric oxide in a prostate cancer photodynamic therapy model. Cancer Lett. 2014, 343, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Fahey, J.M.; Girotti, A.W. Accelerated migration and invasion of prostate cancer cells after a photodynamic therapy-like challenge: Role of nitric oxide. Nitric Oxide 2015, 49, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahey, J.M.; Emmer, J.V.; Korytowski, W.; Hogg, N.; Girotti, A.W. Antagonistic effects of endogenous nitric oxide in a glioblastoma photodynamic therapy model. Photochem. Photobiol. 2016, 92, 842–853. [Google Scholar] [CrossRef] [Green Version]
- Fahey, J.M.; Girotti, A.W. Nitric oxide-mediated resistance to photodynamic therapy in a human breast tumor xenograft model: Improved outcome with NOS2 inhibitors. Nitric Oxide 2017, 62, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Stamenkovic, I. Matrix metalloproteinases in tumor invasion and metastasis. Semin. Cancer Biol. 2000, 10, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, M.; Koivunen, E. Gelatinase-mediated migration and invasion of cancer cells. Biochim. Biophys. Acta 2005, 1755, 37–69. [Google Scholar] [CrossRef] [Green Version]
- Azzam, E.I.; de Toledo, S.M.; Little, J.B. Stress signaling from irradiated to non-irradiated cells. Curr. Cancer Drug Targets 2006, 4, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R. Emerging role of radiation-induced bystander effects: Cell communications and carcinogenesis. Genome Integr. 2010, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- Hei, T.K.; Zho, H.; Ivanov, V.N.; Hong, M.; Lieberman, H.B.; Brenner, d.J.; Amundson, S.A.; Geard, C.R. Mechanism of radiation-induced bystander effects: A unifying model. J. Pham. Pharmacol. 2008, 60, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Shao, C.; Folkard, M.; Prise, K.M. Role of TGF-beta1 and nitric oxide in the bystander response of irradiated glioma cells. Oncogene 2008, 27, 434–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelle, E.; Mammone, T.; Maes, D.; Frenkel, K. Keratinocytes act as a source of reactive oxygen species by transferring hydrogen peroxide to melanocytes. J. Investig. Dermatol. 2005, 124, 793–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, H.; Hayashi, S.; Hatashita, M.; Ohnishi, K.; Shioura, H.; Ohtsubo, T.; Kitai, R.; Ohnishi, T.; Kano, E. Induction of radioresistance by a nitric oxide-mediated bystander effect. Radiat. Res. 2001, 155, 387–396. [Google Scholar] [CrossRef]
- Shao, C.; Stewart, V.; Folkard, M.; Michael, B.D.; Prise, K.M. Nitric oxide-mediated signaling in the bystander response of individually targeted glioma cells. Cancer Res. 2003, 63, 8437–8442. [Google Scholar]
- Yakovlev, V.A. Role of nitric oxide in the radiation-induced bystander effect. Redox Biol. 2015, 6, 396–400. [Google Scholar] [CrossRef] [Green Version]
- Dahle, J.; Bagdonas, S.; Kaalhus, O.; Olsen, G.; Steen, H.B.; Moan, J. The by stander effect in photodynamic inactivation of cells. Biochim. Biophys. Acta 2000, 1475, 273–280. [Google Scholar] [CrossRef]
- Chakraborty, A.; Held, K.D.; Prise, K.M.; Liber, H.L.; Redmond, R.W. Bystander effects induced by diffusing mediators after photodynamic stress. Radiat. Res. 2009, 172, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Rubio, N.; Rajadurai, A.; Held, K.D.; Prise, K.M.; Liber, H.L.; Redmond, R.W. Real-time imaging of novel spatial and temporal responses to photodynamic stress. Free Radic. Biol. Med. 2009, 47, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Bazak, J.; Fahey, J.M.; Wawak, K.; Korytowski, W.; Girotti, A.W. Enhanced aggressiveness of bystander cells in an anti-tumor photodynamic therapy model: Role of nitric oxide produced by targeted cells. Free Radic. Biol. Med. 2017, 102, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Bazak, J.; Korytowski, W.; Girotti, A.W. Bystander effects of nitric oxide in cellular models of anti-tumor photodynamic therapy. Cancers 2019, 11, 1674. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, R.; Girotti, A.W. Cytoprotective signaling associated with nitric oxide upregulation in tumor cells subjected to photodynamic therapy-like oxidative stress. Free Radic. Biol. Med. 2013, 57, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahey, J.M.; Korytowski, W.; Girotti, A.W. Upstream signaling events leading ot elevated production of pro-survival nitric oxide in photodynamically-challenged glioblastoma cells. Free Radic. Biol. Med. 2019, 137, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Zhow, Z.; Huang, B.; Wu, X.; Zhang, H.; Qi, J.; Bradner, J.; Nair, S.; Chen, L.F. Brd4 maintains constitutively active NF-κB in cancer cells by binding to acetylated Rel A. Oncogene 2014, 33, 2395–2404. [Google Scholar]
- Kleszcz, R.; Paluszczak, J.; Baer-Dubowska, D. Targeting aberrant cancer metabolism: The role of sirtuins. Pharmacol. Rep. 2015, 67, 1068–1080. [Google Scholar] [CrossRef] [PubMed]
- Korbelik, M. Role of cell stress signaling networks in cancer cell death and antitumor immune response following proteotoxic injury inflicted by photodynamic therapy. Lasers Surg. Med. 2018, 50, 491–498. [Google Scholar] [CrossRef]
- Hansel, T.T.; Kharitonov, S.A.; Donnelly, L.E.; Erin, E.M.; Currie, M.G.; Moore, W.M.; Manning, P.T.; Recker, D.P.; Barnes, P.J. A selective inhibitor of inducible nitric oxide synthase inhibits exhaled breath nitric oxide in healthy volunteers and asthmatics. FASEB J. 2003, 17, 1298–1317. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Richards, D.; Knowles, R.G.; Schwartz, S.; Woodcock, A.; Langley, S.; O’Connor, B.J. Selective inducible nitric oxide synthase inhibition has no effect on allergen challenge in asthma. Am. J. Respir. Crit. Care Med. 2007, 176, 988–993. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysing acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.M.; Stancill, J.S.; Smith, B.C.; Girotti, A.W. Nitric oxide antagonism to glioblastoma photodynamic therapy and mitigation thereof by BET bromodomain inhibitor JQ1. J. Biol. Chem. 2018, 293, 5345–5359. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, Y.; Mei, H.; Yin, Z.; Geng, Y.; Zhang, T.; Wu, G.; Lin, Z. The BET bromodomain inhibitor JQ1 radiosensitizes non-small cell lung cancer cells by upregulating p21. Cancer Lett. 2017, 391, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.L.; Garcia, P.L.; Yoon, K.J. Developing effective combination therapy for pancreatic cancer: An overview. Pharmacol. Res. 2020, 155, 104740. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girotti, A.W.; Fahey, J.M.; Korbelik, M. Photodynamic Therapy as an Oxidative Anti-Tumor Modality: Negative Effects of Nitric Oxide on Treatment Efficacy. Pharmaceutics 2021, 13, 593. https://doi.org/10.3390/pharmaceutics13050593
Girotti AW, Fahey JM, Korbelik M. Photodynamic Therapy as an Oxidative Anti-Tumor Modality: Negative Effects of Nitric Oxide on Treatment Efficacy. Pharmaceutics. 2021; 13(5):593. https://doi.org/10.3390/pharmaceutics13050593
Chicago/Turabian StyleGirotti, Albert W., Jonathan M. Fahey, and Mladen Korbelik. 2021. "Photodynamic Therapy as an Oxidative Anti-Tumor Modality: Negative Effects of Nitric Oxide on Treatment Efficacy" Pharmaceutics 13, no. 5: 593. https://doi.org/10.3390/pharmaceutics13050593