
Pharmacokinetic Model Analysis of Supralingual, Oral and Intravenous Deliveries of Mycophenolic Acid

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals, Animals, and Instruments
2.2. PK Studies and Tongue Tissue Distribution
2.2.1. IV PK Study
2.2.2. Oral PK Study
2.2.3. Patch PK Study
2.2.4. Tongue Tissue Distribution
2.3. Sample Preparation and Analysis
2.3.1. Plasma Samples
2.3.2. Tongue Samples
2.3.3. LC-MS/MS Analysis
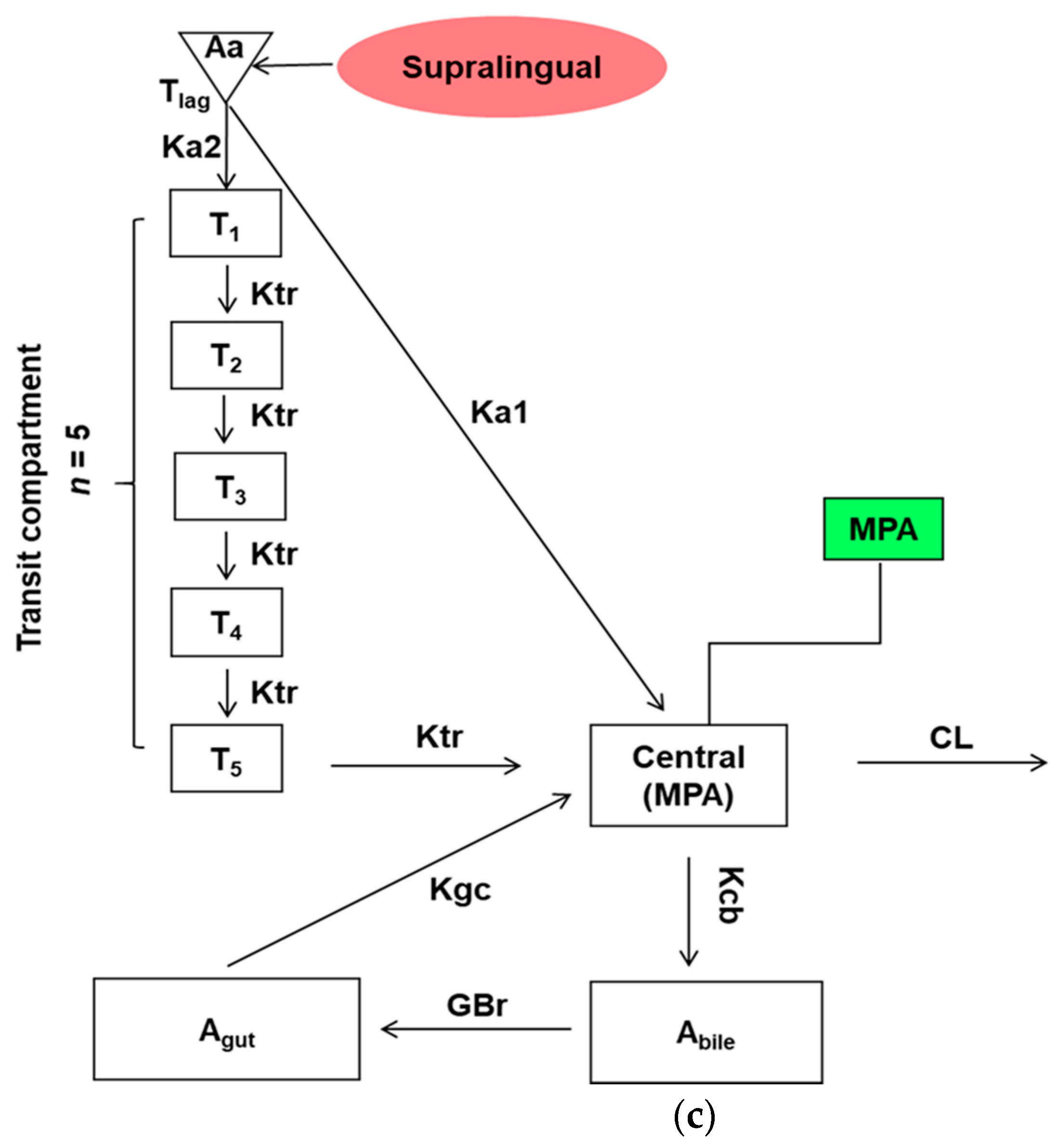
2.4. PK Model and Statistical Analyses
3. Results
3.1. Estimated PK Parameters
3.2. Individual EHR PK Modeling Analysis
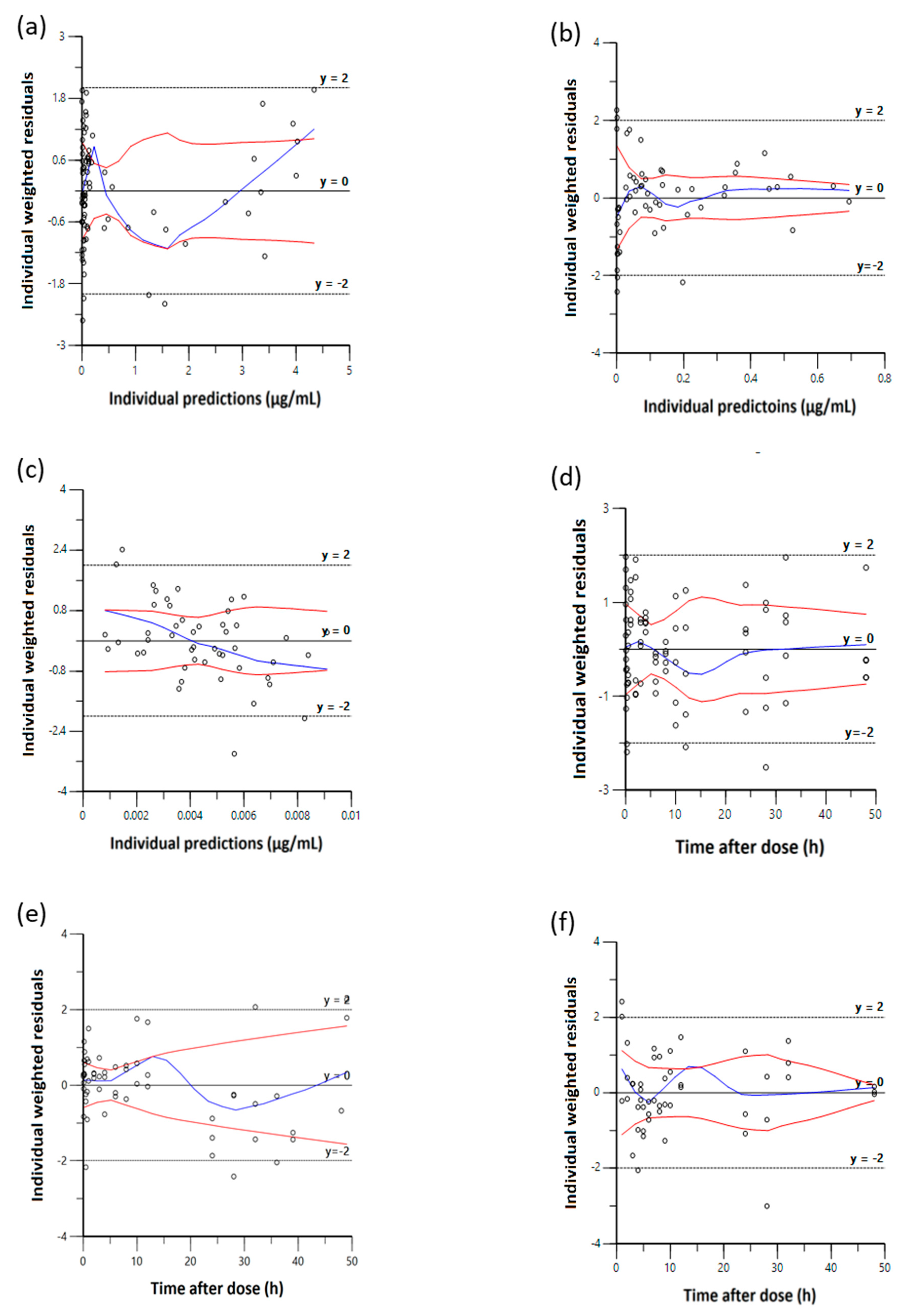
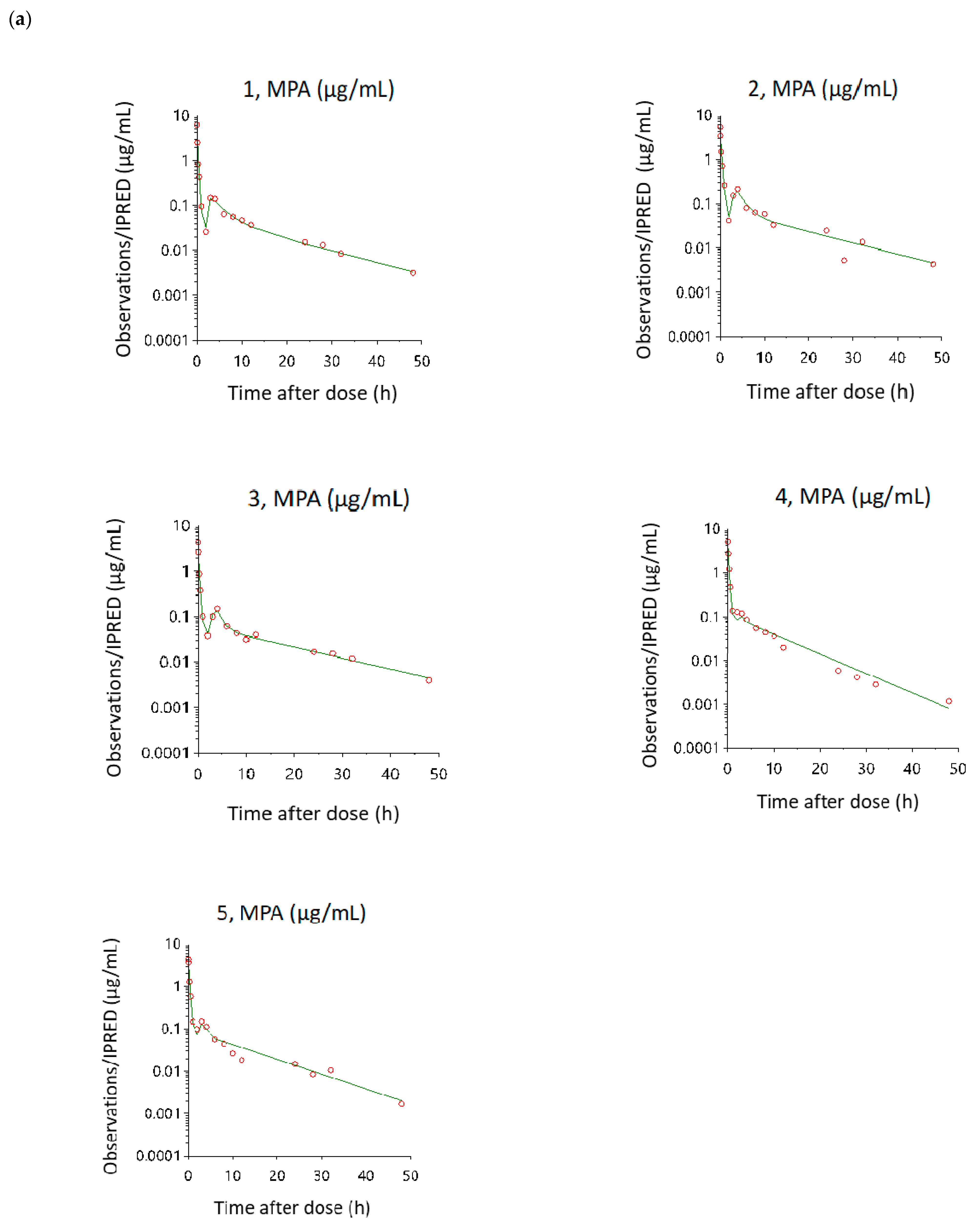
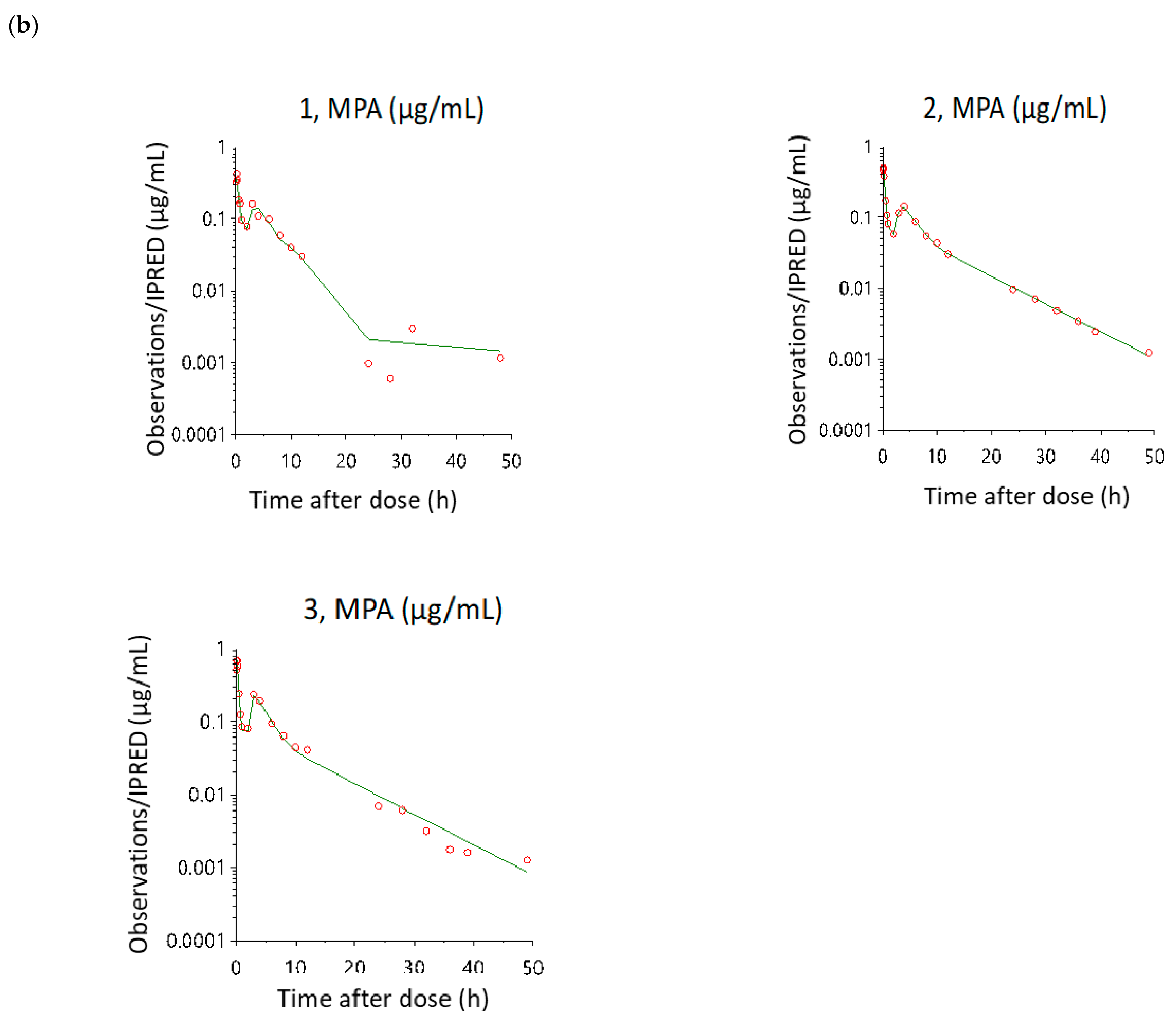
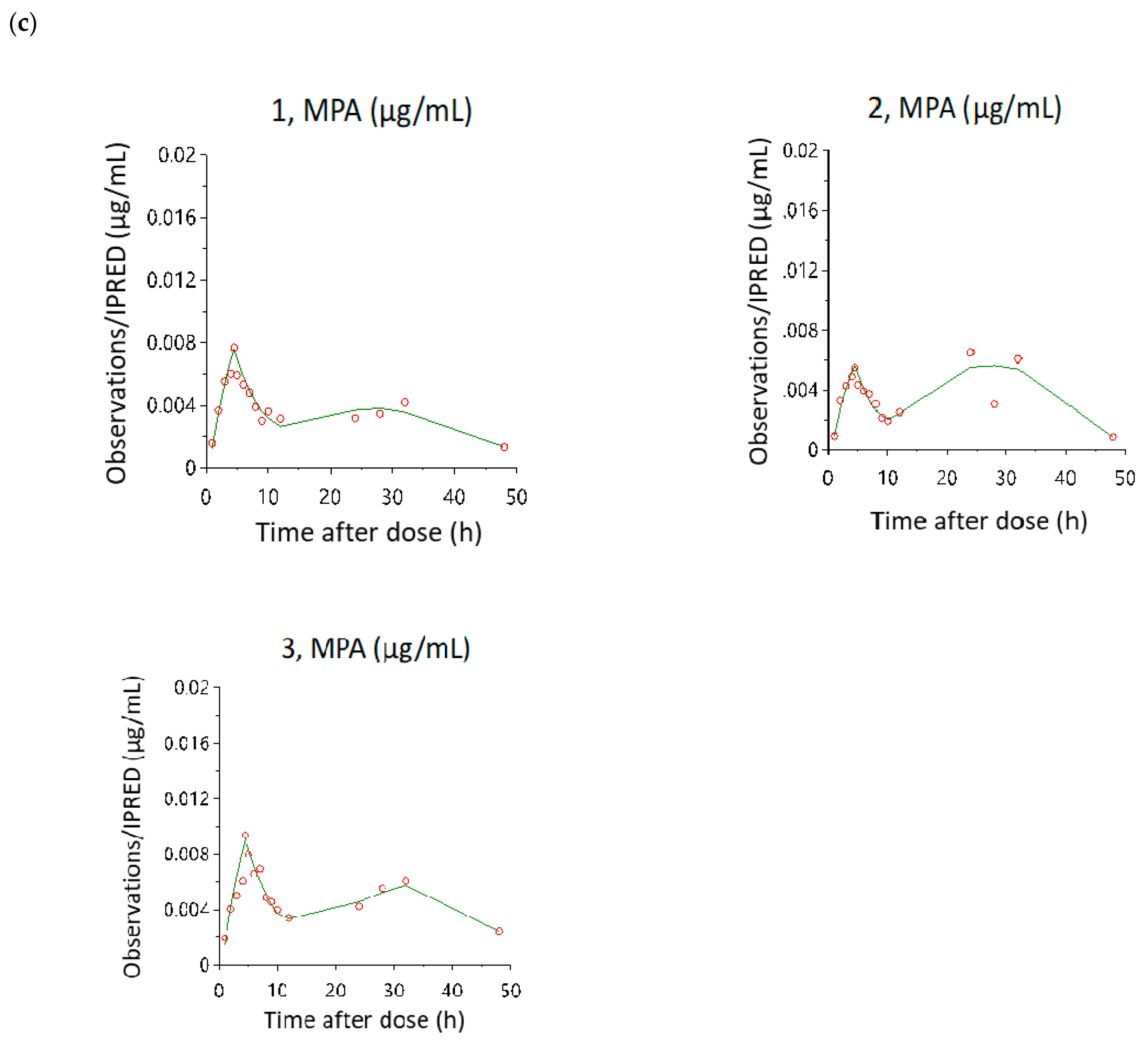
3.3. Model Evaluation
3.4. Tongue Distribution
4. Discussion
4.1. PK Parameters and EHR Phenomenon
4.2. EHR and PK Model Development
4.3. Tongue Tissue Distribution after Supralingal Application
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Inchingolo, F.; Santacroce, L.; Ballini, A.; Topi, S.; Dipalma, G.; Haxhirexha, K.; Bottalico, L.; Charitos, I.A. Oral cancer: A historical review. Int. J. Environ. Res. Public Health 2020, 17, 3168. [Google Scholar] [CrossRef] [PubMed]
- Samadi, F.M.; Suhail, S.; Sonam, M.; Ahmad, M.K.; Chandra, S.; Saleem, M. Telomerase in saliva: An assistant marker for oral squamous cell carcinoma. J. Oral Maxillofac. Pathol. 2019, 23, 187–191. [Google Scholar] [CrossRef]
- Treatment Options for Oral Cavity and Oropharyngeal Cancer by Stage. 2018. Available online: https://www.cancer.org/cancer/oral-cavity-and-oropharyngeal-cancer/treating/by-stage.html (accessed on 22 March 2021).
- Nguyen, S.; Hiorth, M. Advanced drug delivery systems for local treatment of the oral cavity. Ther. Deliv. 2015, 6, 595–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baykul, T.; Yilmaz, H.H.; Aydin, U.; Aydin, M.A.; Aksoy, M.; Yildirim, D. Early diagnosis of oral cancer. J. Int. Med. Res. 2010, 38, 737–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, J.A.; van der Voort Maarschalk, K. Understanding the oral mucosal absorption and resulting clinical pharmacokinetics of asenapine. AAPS PharmSciTech 2012, 13, 1110–1115. [Google Scholar] [CrossRef] [Green Version]
- Bruschi, M.L.; de Freitas, O. Oral bioadhesive drug delivery systems. Drug Dev. Ind. Pharm. 2005, 31, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.F.; Liu, F.; Brown, M.B. Advances in oral transmucosal drug delivery. J. Control Release 2011, 153, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nafee, N.A.; Ismail, F.A.; Boraie, N.A.; Mortada, L.M. Mucoadhesive buccal patches of miconazole nitrate: In vitro/in vivo performance and effect of ageing. Int. J. Pharm. 2003, 264, 1–14. [Google Scholar] [CrossRef]
- Radha, B. A detailed review on oral mucosal drug delivery system. Int. J. Pharm. Sci. Res. 2011, 659–681. [Google Scholar]
- Holpuch, A.S.; Phelps, M.P.; Desai, K.G.; Chen, W.; Koutras, G.M.; Han, B.B.; Warner, B.M.; Pei, P.; Seghi, G.A.; Tong, M.; et al. Evaluation of a mucoadhesive fenretinide patch for local intraoral delivery: A strategy to reintroduce fenretinide for oral cancer chemoprevention. Carcinogenesis 2012, 33, 1098–1105. [Google Scholar] [CrossRef]
- Desai, K.G.; Mallery, S.R.; Holpuch, A.S.; Schwendeman, S.P. Development and in vitro-in vivo evaluation of fenretinide-loaded oral mucoadhesive patches for site-specific chemoprevention of oral cancer. Pharm. Res. 2011, 28, 2599–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.; Tsai, H.; Wong, Y.; Hong, J.; Chang, S.; Lee, M. Preparation and characterization of gellan gum/glucosamine/clioquinol film as oral cancer treatment patch. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 82, 317–322. [Google Scholar] [CrossRef]
- Loprete, L.; Leuratti, C.; Frangione, V.; Radicioni, M. Pharmacokinetics of a novel sildenafil orodispersible film administered by the supralingual and the sublingual route to healthy men. Clin. Drug Investig. 2018, 38, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dun, B.; Sharma, A.; Xu, H.; Liu, H.; Bai, S.; Zeng, L.; She, J.X. Transcriptomic changes induced by mycophenolic acid in gastric cancer cells. Am. J. Transl. Res. 2013, 6, 28–42. [Google Scholar] [PubMed]
- Newbold, N. A review of enteric-coated mycophenolate sodium for renal transplant immunosuppression. Clin. Med. Insight 2009. [Google Scholar] [CrossRef] [Green Version]
- Hermann, L.L.; Coombs, K.M. Inhibition of reovirus by mycophenolic acid is associated with the M1 genome segment. J. Virol. 2004, 78, 6171–6179. [Google Scholar] [CrossRef] [Green Version]
- Buell, J.F.; Gross, T.G.; Woodle, E.S. Malignancy after transplantation. Transplantation 2005, 80, S254–S264. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, H.; Li, Y.; Wei, Q.; Li, H.; Yang, Y.; Lu, Y. Safety of mycophenolate mofetil versus azathioprine in renal transplantation: A systematic review. Transplant Proc. 2004, 36, 2068–2070. [Google Scholar] [CrossRef]
- Robert, Y.; Tsai, T.L. Mycophenolic Acid Analogues as Anti-Tumor Chemosensitizing Agents. WO2013044028A2, 21 September 2012. [Google Scholar]
- U.S. Food and Drug Administration. Risk Evaluation and Mitigation Strategy (REMS) under Review for CellCept and Myfotric. 2009. Available online: https://www.firstwordpharma.com/node/362407?tsid=17 (accessed on 22 March 2021).
- Cattaneo, D.; Cortinovis, M.; Baldelli, S.; Bitto, A.; Gotti, E.; Remuzzi, G.; Perico, N. Pharmacokinetics of mycophenolate sodium and comparison with the mofetil formulation in stable kidney transplant recipients. Clin. J. Am. Soc. Nephrol. 2007, 2, 1147–1155. [Google Scholar] [CrossRef] [Green Version]
- Davies, N.M.; Grinyó, J.; Heading, R.; Maes, B.; Meier-Kriesche, H.U.; Oellerich, M. Gastrointestinal side effects of mycophenolic acid in renal transplant patients: A reappraisal. Nephrol. Dial. Transplant 2007, 22, 2440–2448. [Google Scholar] [CrossRef] [Green Version]
- Le Guellec, C.; Bourgoin, H.; Büchler, M.; Le Meur, Y.; Lebranchu, Y.; Marquet, P.; Paintaud, G. Population pharmacokinetics and Bayesian estimation of mycophenolic acid concentrations in stable renal transplant patients. Clin. Pharmacokinet. 2004, 43, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Payen, S.; Zhang, D.; Maisin, A.; Popon, M.; Bensman, A.; Bouissou, F.; Loirat, C.; Gomeni, R.; Bressolle, F.; Jacqz-Aigrain, E. Population pharmacokinetics of mycophenolic acid in kidney transplant pediatric and adolescent patients. Ther. Drug Monit. 2005, 27, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Chow, D.S. Clinical pharmacokinetics of mycophenolic acid in hematopoietic stem cell transplantation recipients. Eur. J. Drug. Metab. Pharmacokinet. 2017, 42, 183–189. [Google Scholar] [CrossRef]
- Sherwin, C.M.; Sagcal-Gironella, A.C.; Fukuda, T.; Brunner, H.I.; Vinks, A.A. Development of population PK model with enterohepatic circulation for mycophenolic acid in atients with childhood-onset systemic lupus erythematosus. Br. J. Clin. Pharmacol. 2012, 73, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Z.; Ding, J.J.; Shen, J.; Liang, H.Q.; Zhong, L.J.; Wang, Y.; Zhong, M.K.; Lu, W.Y. Population pharmacokinetic modelling for enterohepatic circulation of mycophenolic acid in healthy Chinese and the influence of polymorphisms in UGT1A9. Br. J. Clin. Pharmacol. 2008, 65, 893–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colom, H.; Lloberas, N.; Andreu, F.; Caldés, A.; Torras, J.; Oppenheimer, F.; Sanchez-Plumed, J.; Gentil, M.A.; Kuypers, D.R.; Brunet, M.; et al. Pharmacokinetic modeling of enterohepatic circulation of mycophenolic acid in renal transplant recipients. Kidney Int. 2014, 85, 1434–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takekuma, Y.; Kakiuchi, H.; Yamazaki, K.; Miyauchi, S.; Kikukawa, T.; Kamo, N.; Ganapathy, V.; Sugawara, M. Difference between pharmacokinetics of mycophenolic acid (MPA) in rats and that in humans is caused by different affinities of MRP2 to a glucuronized form. J. Pharm. Pharm. Sci. 2007, 10, 71–85. [Google Scholar]
- Gao, J.W.; Peng, Z.H.; Li, X.Y.; Sun, B.; Guo, Y.K.; Liu, G.L. Simultaneous determination of mycophenolic acid and its metabolites by HPLC and pharmacokinetic studies in rat plasma and bile. Arch Pharm. Res. 2011, 34, 59–69. [Google Scholar] [CrossRef]
- Feturi, F.G.; Weinstock, M.; Zhao, W.; Zhang, W.; Schnider, J.T.; Erbas, V.E.; Oksuz, S.; Plock, J.A.; Rohan, L.; Spiess, A.M.; et al. Mycophenolic acid for topical immunosuppression in vascularized composite allotransplantation: Optimizing formulation and preliminary evaluation of bioavailability and pharmacokinetics. Front. Surg. 2018, 5, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dridi, I.; Ben-Cherif, W.; Aouam, K.; Ben-Attia, M.; Klouz, A.; einberg, A.; Boughattas, N.A. Circadian variation of mycophenolate mofetil pharmacokinetics in rats. Eur. J. Pharm. Sci. 2014, 58, 20–25. [Google Scholar] [CrossRef]
- Yoshimura, K.; Yano, I.; Kawanishi, M.; Nakagawa, S.; Yonezawa, A.; Matsubara, K. Pharmacokinetics and pharmacodynamics of mycophenolic acid in Nagase analbuminemic rats: Evaluation of protein binding effects using the modeling and simulation approach. Drug Metab. Pharmacokinet. 2015, 30, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.M. Immunosuppression with mycophenolic acid: One size does not fit all. J. Am. Soc. Nephrol. 2003, 14, 2414–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hummel, M.; Yonan, N.; Ross, H.; Miller, L.W.; Sechaud, R.; Balez, S.; Koelle, E.U.; Gerosa, G.; Group, E.S. Pharmacokinetics and variability of mycophenolic acid from enteric-coated mycophenolate sodium compared with mycophenolate mofetil in de novo heart transplant recipients. Clin. Transplant 2007, 21, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Tsai, R.Y.L.; Ma, J.; Bhupal, P.K.; Liu, X.; Liang, D.; Xie, H. Determination and validation of mycophenolic acid by a UPLC-MS/MS method: Applications to pharmacokinetics and tongue tissue distribution studies in rats. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2020, 1136, 121930. [Google Scholar] [CrossRef]
- Ekpenyong, O.; Cooper, C.; Ma, J.; Liang, D.; Olaleye, O.; Xie, H. A simple, sensitive and reliable LC-MS/MS method for the determination of 7-bromo-5-chloroquinolin-8-ol (CLBQ14), a potent and selective inhibitor of methionine aminopeptidases: Application to pharmacokinetic studies. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018, 1097, 35–43. [Google Scholar] [CrossRef]
- Malek, O. The Pharmacokinetics of Enterohepatic Circulaiton. 2015. Available online: https://conservancy.umn.edu/bitstream/handle/11299/182173/Okour_umn_0130E_16705.pdf?sequence=1&isAllowed=y (accessed on 22 March 2021).
- Sherwin, C.M.; Fukuda, T.; Brunner, H.I.; Goebel, J.; Vinks, A.A. The evolution of population pharmacokinetic models to describe the enterohepatic recycling of mycophenolic acid in solid organ transplantation and autoimmune disease. Clin. Pharmacokinet. 2011, 50, 1–24. [Google Scholar] [CrossRef]
- Administration U.S.F. Myfortic. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/050791s002lbl.pdf (accessed on 22 March 2021).
- Choudhary, A.; Sriphrapradang, C.; Refetoff, S.; Antal, Z. Familial dysalbuminemic hyperthyroxinemia in a 4-year-old girl with hyperactivity, palpitations and advanced dental age: How gold standard assays may be misleading. J. Pediatr. Endocrinol. Metab. 2015, 28, 241–245. [Google Scholar] [CrossRef]
- Yoshimura, K.; Yano, I.; Yamamoto, T.; Kawanishi, M.; Isomoto, Y.; Yonezawa, A.; Kondo, T.; Takaori-Kondo, A.; Matsubara, K. Population pharmacokinetics and pharmacodynamics of mycophenolic acid using the prospective data in patients undergoing hematopoietic stem cell transplantation. Bone Marrow Transplant 2018, 53, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Staatz, C.E.; Tett, S.E. Clinical pharmacokinetics and pharmacodynamics of mycophenolate in solid organ transplant recipients. Clin. Pharmacokinet. 2007, 46, 13–58. [Google Scholar] [CrossRef]
- Calmet, F.H.; Yarur, A.J.; Pukazhendhi, G.; Ahmad, J.; Bhamidimarri, K.R. Endoscopic and histological features of mycophenolate mofetil colitis in patients after solid organ transplantation. Ann. Gastroenterol. 2015, 28, 366–373. [Google Scholar]
- Atcheson, B.A.; Taylor, P.J.; Mudge, D.W.; Johnson, D.W.; Hawley, C.M.; Campbell, S.B.; Isbel, N.M.; Pillans, P.I.; Tett, S.E. Mycophenolic acid pharmacokinetics and related outcomes early after renal transplant. Br. J. Clin. Pharmacol. 2005, 59, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Sobiak, J.; Resztak, M.; Głyda, M.; Szczepaniak, P.; Chrzanowska, M. Pharmacokinetics of mycophenolate sodium co-administered with tacrolimus in the first year after renal transplantation. Eur. J. Drug Metab. Pharmacokinet. 2016, 41, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.C.; Zhou, P.J.; Wang, X.H.; Françoise, B.; Xu, D.; Zhang, W.X.; Chen, B. Population pharmacokinetics and Bayesian estimation of mycophenolic acid concentrations in Chinese adult renal transplant recipients. Acta Pharmacol. Sin. 2017, 38, 1566–1579. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Fukuda, T.; Cox, S.; de Vries, M.T.; Hooper, D.K.; Goebel, J.; Vinks, A.A. Population pharmacokinetic-pharmacodynamic modelling of mycophenolic acid in paediatric renal transplant recipients in the early post-transplant period. Br. J. Clin. Pharmacol. 2014, 78, 1102–1112. [Google Scholar] [CrossRef]
- de Winter, B.C.; Neumann, I.; van Hest, R.M.; van Gelder, T.; Mathot, R.A. Limited sampling strategies for therapeutic drug monitoring of mycophenolate mofetil therapy in patients with autoimmune disease. Ther. Drug Monit. 2009, 31, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Yau, W.P.; Vathsala, A.; Lou, H.X.; Zhou, S.; Chan, E. Mechanism-based enterohepatic circulation model of mycophenolic acid and its glucuronide metabolite: Assessment of impact of cyclosporine dose in Asian renal transplant patients. J. Clin. Pharmacol. 2009, 49, 684–699. [Google Scholar] [CrossRef] [PubMed]
- van Hest, R.M.; Mathot, R.A.; Pescovitz, M.D.; Gordon, R.; Mamelok, R.D.; van Gelder, T. Explaining variability in mycophenolic acid exposure to optimize mycophenolate mofetil dosing: A population pharmacokinetic meta-analysis of mycophenolic acid in renal transplant recipients. J. Am. Soc. Nephrol. 2006, 17, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Dong, M.; Fukuda, T.; Vinks, A.A. Optimization of mycophenolic acid therapy using clinical pharmacometrics. Drug Metab. Pharmacokinet. 2014, 29, 4–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Dunlavey, M.; Guzy, S.; Teuscher, N. A distributed delay approach for modeling delayed outcomes in pharmacokinetics and pharmacodynamics studies. J. Pharmacokinet. Pharmacodyn. 2018, 4, 285–308. [Google Scholar] [CrossRef]
- Joomi, L. Population pharmacokinetic analysis of the multiple peaks phenomenon in sumatriptan. Transl. Clin. Pharmacol. 2015, 32, 66–74. [Google Scholar]
- Reddy, K.S.; Yadav, A.; Arora, M.; Nazar, G.P. Integrating tobacco control into health and development agendas. Tob. Control 2012, 21, 281–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, R.B. Oral cavity as a site for bioadhesive drug delivery. Adv. Drug Deliv. Rev. 1994, 13, 43–74. [Google Scholar] [CrossRef]
- Kurosaki, Y.; Takatori, T.; Nishimura, H.; Nakayama, T.; Kimura, T. Regional variation in oral mucosal drug absorption: Permeability and degree of keratinization in hamster oral cavity. Pharm. Res. 1991, 8, 1297–1301. [Google Scholar] [CrossRef] [PubMed]
- Squier, C.A. The permeability of oral mucosa. Crit. Rev. Oral Biol. Med. 1991, 2, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, Y.; Kuotsu, K.; Bandyopadhyay, A.K. Buccal bioadhesive drug delivery--a promising option for orally less efficient drugs. J. Control. Release 2006, 114, 1540. [Google Scholar] [CrossRef]
- Narang, N. Sublingual mucosa as a route for systemic drug delivery. Int. J. Pharm. Pharm. Sci. 2011, 3, 18–22. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Unit | IV (n = 5) Mean | Oral (n = 3) Mean | Supralingual (n = 3) Mean |
|---|---|---|---|---|
| Dose | mg/kg | 0.5 | 0.5 | 0.5 |
| Tau | hr | 1.46 ± 0.535 | 4.54 ± 4.69 | 31.5 ± 11.6 ***,**** |
| Half-life | hr | 10.5 ± 1.20 | 7.40 ± 2.07 | 11.5 ± 2.98 |
| CL/Fabs | mL/(kg × h) | 117 ± 92.2 | 132 ± 73.2 | 250 ± 333 |
| CL2/Fabs | mL/(kg × h) | 224 ± 65.5 | 274 ± 162 | NA |
| Kcb | 1/h | 1.37 ± 1.02 | 10.3 ± 0.832 **,*** | 2.28 ± 3.62 |
| Kgc | 1/h | 1.97 ± 2.02 | 7.78 ± 8.52 | 32.4 ± 52.3 |
| V/Fabs | mL/kg | 110 ± 10.8 | 29.3 ± 9.82 | 3090 ± 49.1 ***,**** |
| V2/Fabs | mL/kg | 1740 ± 508 | 3000 ± 1690 | NA |
| Ka1 | 1/h | NA | 0.997 ± 0.313 | 21.6 ± 14.6 |
| Ka2 | 1/h | NA | 1.53 ± 0.366 | 37.3 ± 22.7 |
| Ktr | 1/h | NA | 1.25 ± 0.118 | 0.193 ± 0.040 *** |
| AUC0–48 | ng × h/mL | 2170 ± 355 | 1570 ± 218 ** | 165 ± 21.0 ***,**** |
| Fabs | % | NA | 72.4 | 7.60 |
| EHR | % | 54.1 ± 10.3 | 69.2 ± 9.56 | 96.6 ± 49.5 |
| MPA Concentration in Tongue Tissue after Patch Removal * | MPA Concentration | Percentage to Dose |
|---|---|---|
| 0 h (n = 4) | 42.8 ± 10.0 (µg/g) | 3.8 ± 0.6% |
| 20 h (n = 3) | 0.8 ± 0.6 (µg/g) | 0.11 ± 0.09% |
| Patch MPA residue ** (n = 7) | 34.4 ± 5.2 µg | 20.6 ± 2.8% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, X.; Wu, L.; Tsai, R.Y.L.; Ma, J.; Liu, X.; Chow, D.S.-L.; Liang, D.; Xie, H. Pharmacokinetic Model Analysis of Supralingual, Oral and Intravenous Deliveries of Mycophenolic Acid. Pharmaceutics 2021, 13, 574. https://doi.org/10.3390/pharmaceutics13040574
Gao X, Wu L, Tsai RYL, Ma J, Liu X, Chow DS-L, Liang D, Xie H. Pharmacokinetic Model Analysis of Supralingual, Oral and Intravenous Deliveries of Mycophenolic Acid. Pharmaceutics. 2021; 13(4):574. https://doi.org/10.3390/pharmaceutics13040574
Chicago/Turabian StyleGao, Xiuqing, Lei Wu, Robert Y. L. Tsai, Jing Ma, Xiaohua Liu, Diana S.-L. Chow, Dong Liang, and Huan Xie. 2021. "Pharmacokinetic Model Analysis of Supralingual, Oral and Intravenous Deliveries of Mycophenolic Acid" Pharmaceutics 13, no. 4: 574. https://doi.org/10.3390/pharmaceutics13040574