
Amorphous Solid Dispersion Tablets Overcome Acalabrutinib pH Effect in Dogs

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
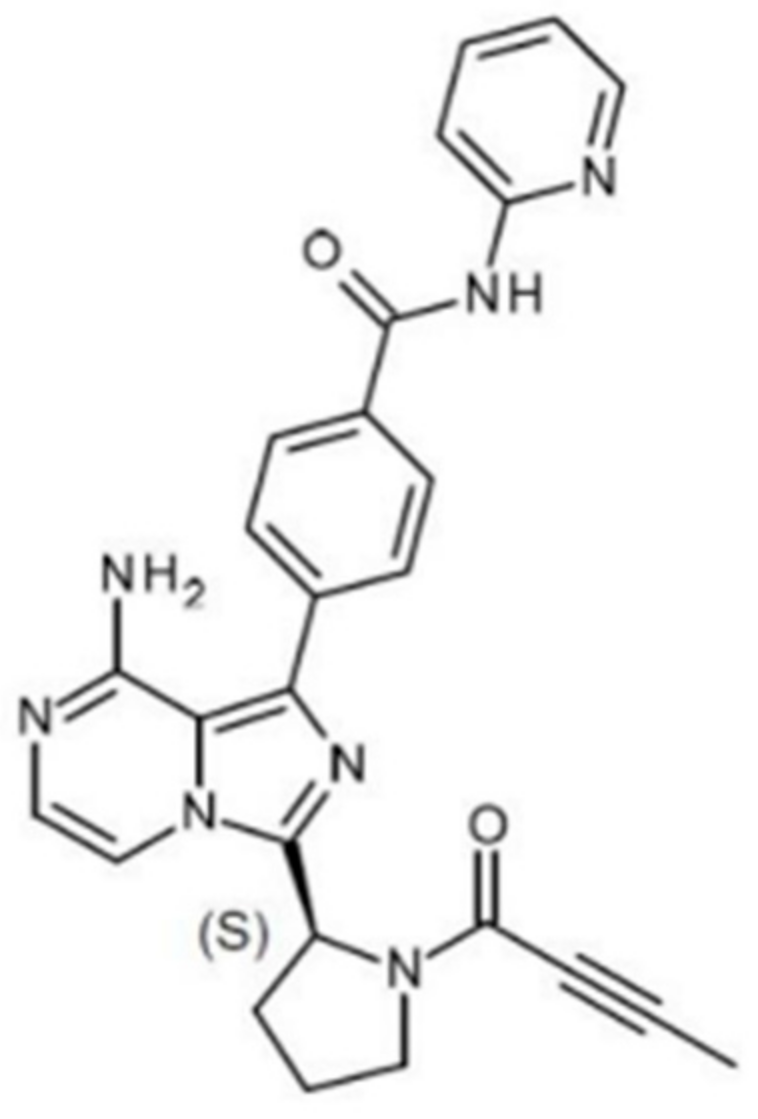
2.1. Material Sourcing
2.2. ASD Manufacturing and Characterization
2.3. ASD Tablet Manufacturing and Characterization
2.4. Stability
2.5. In Vitro Dissolution Testing
2.6. Pharmacokinetic (PK) Study
3. Results
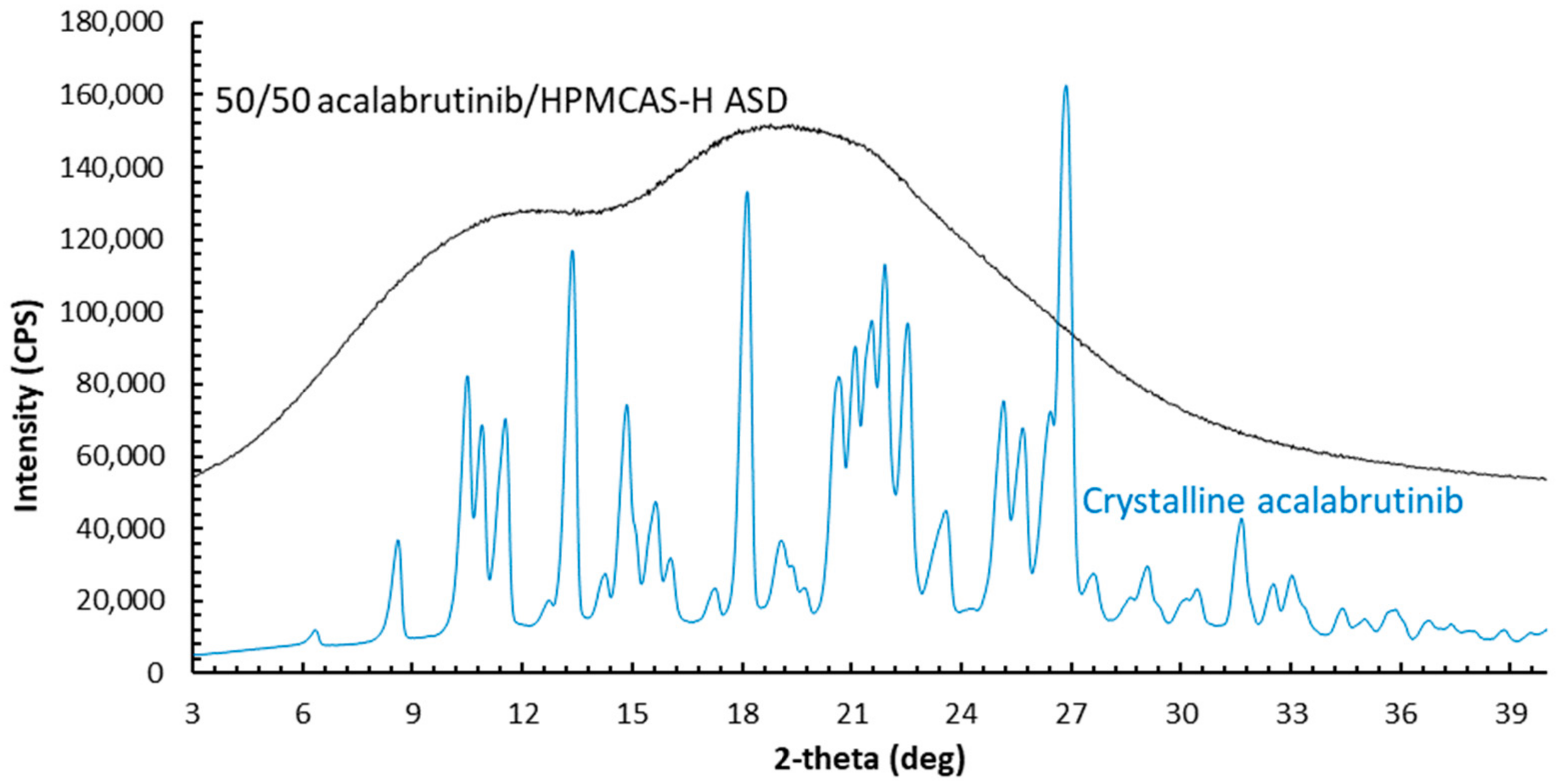
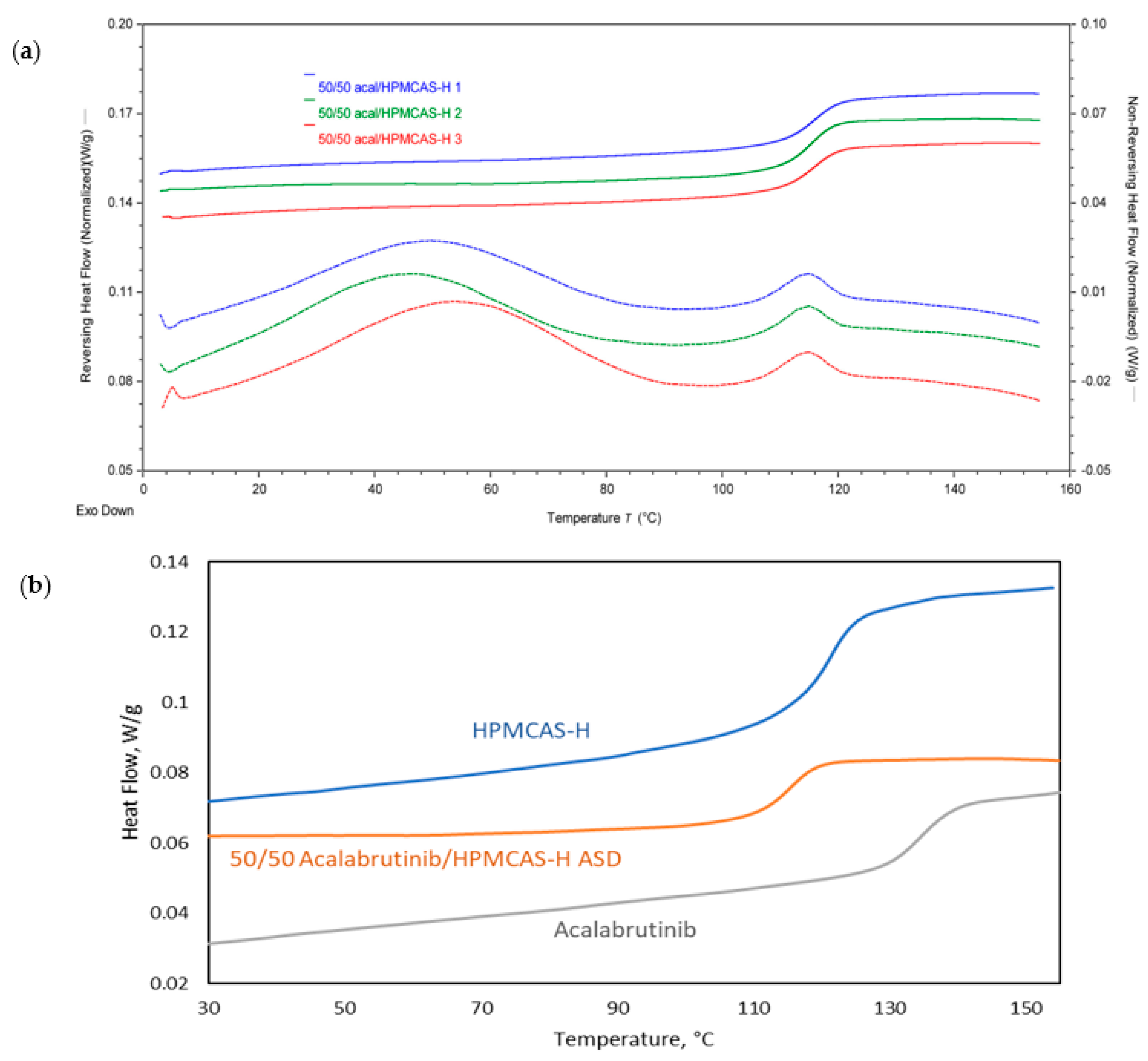
3.1. ASD and Tablet Manufacturing and Characterization
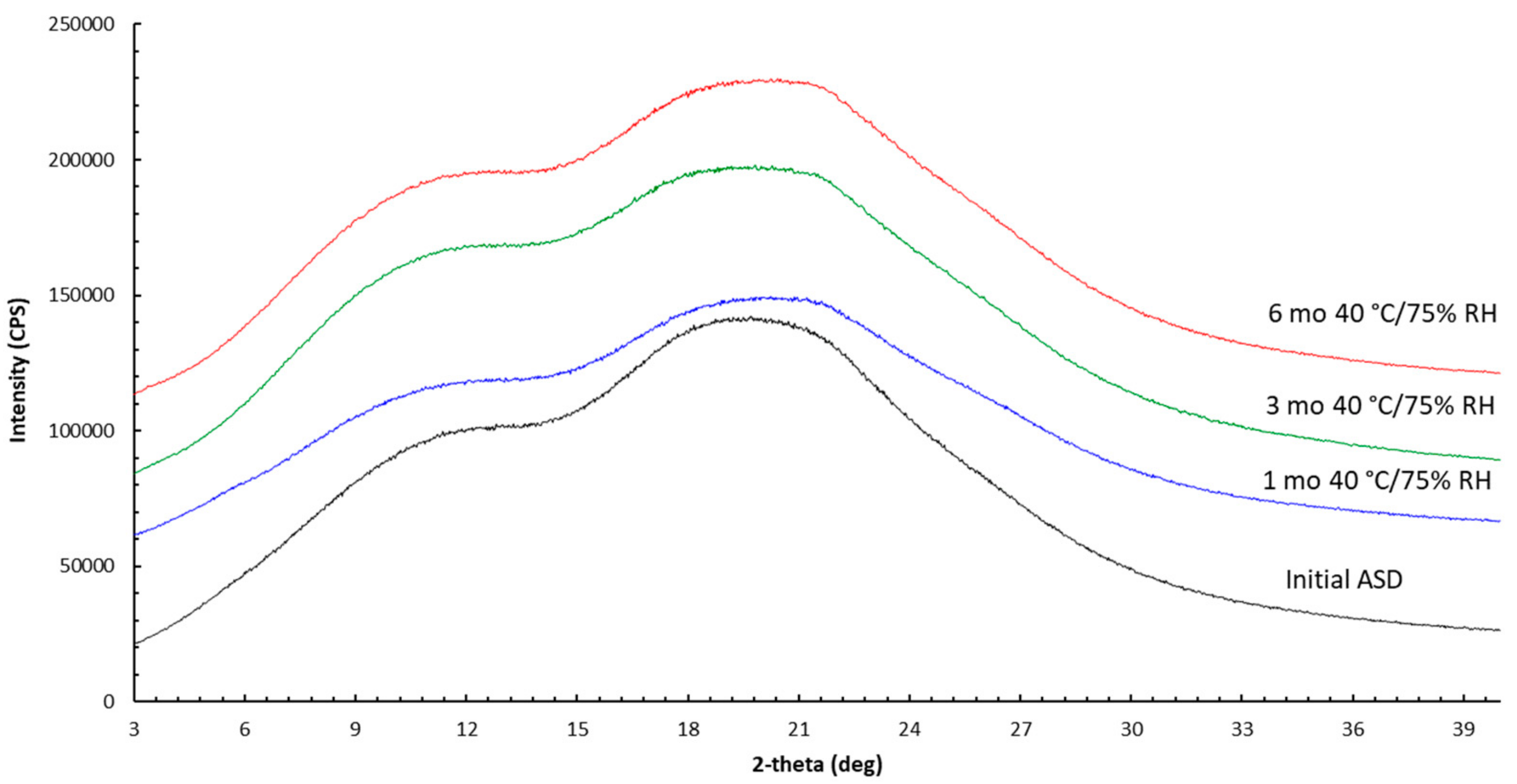
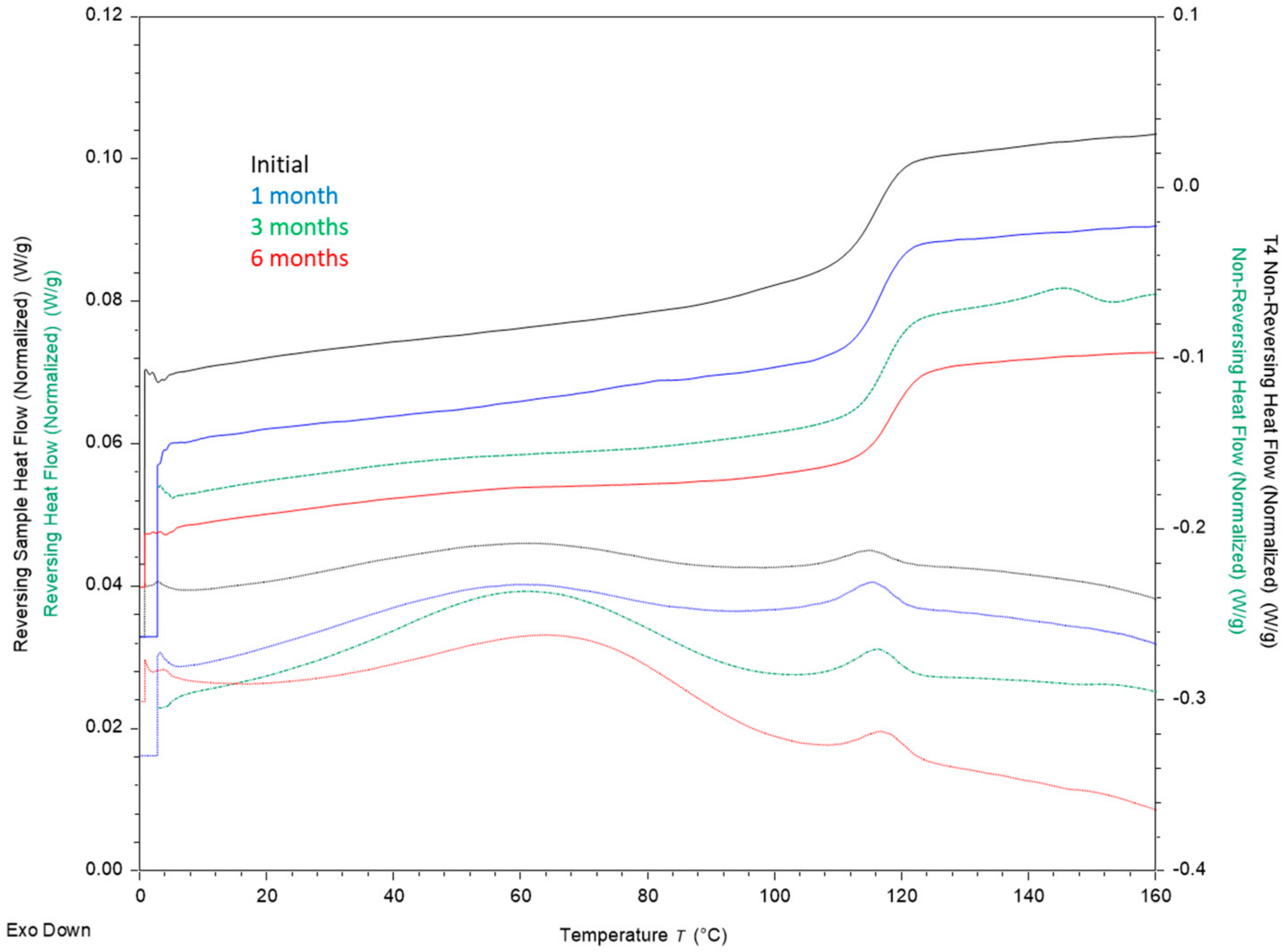
3.2. Stability
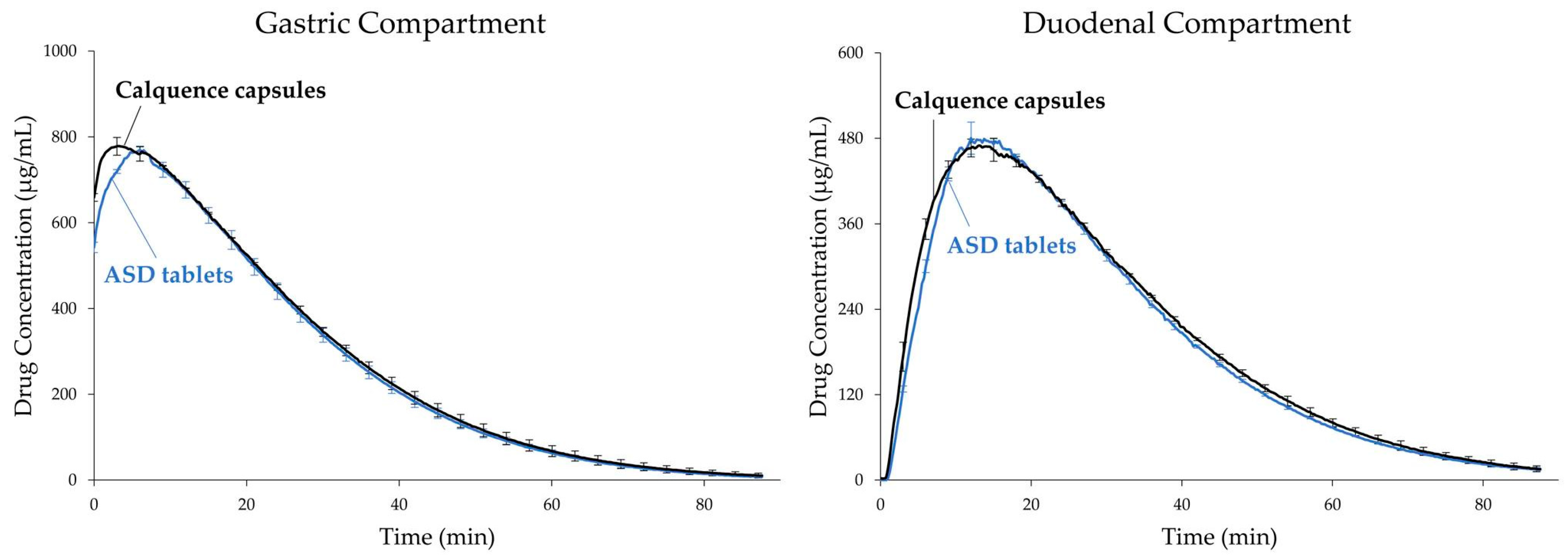
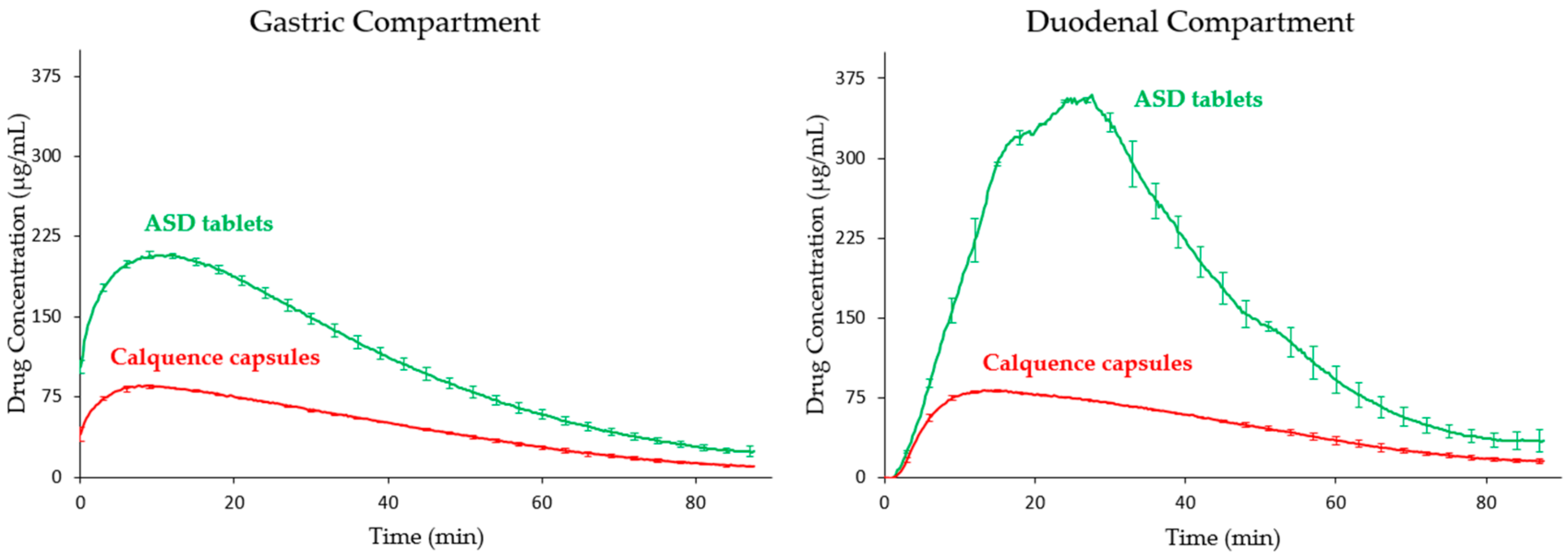
3.3. Tablet In Vitro Dissolution Performance
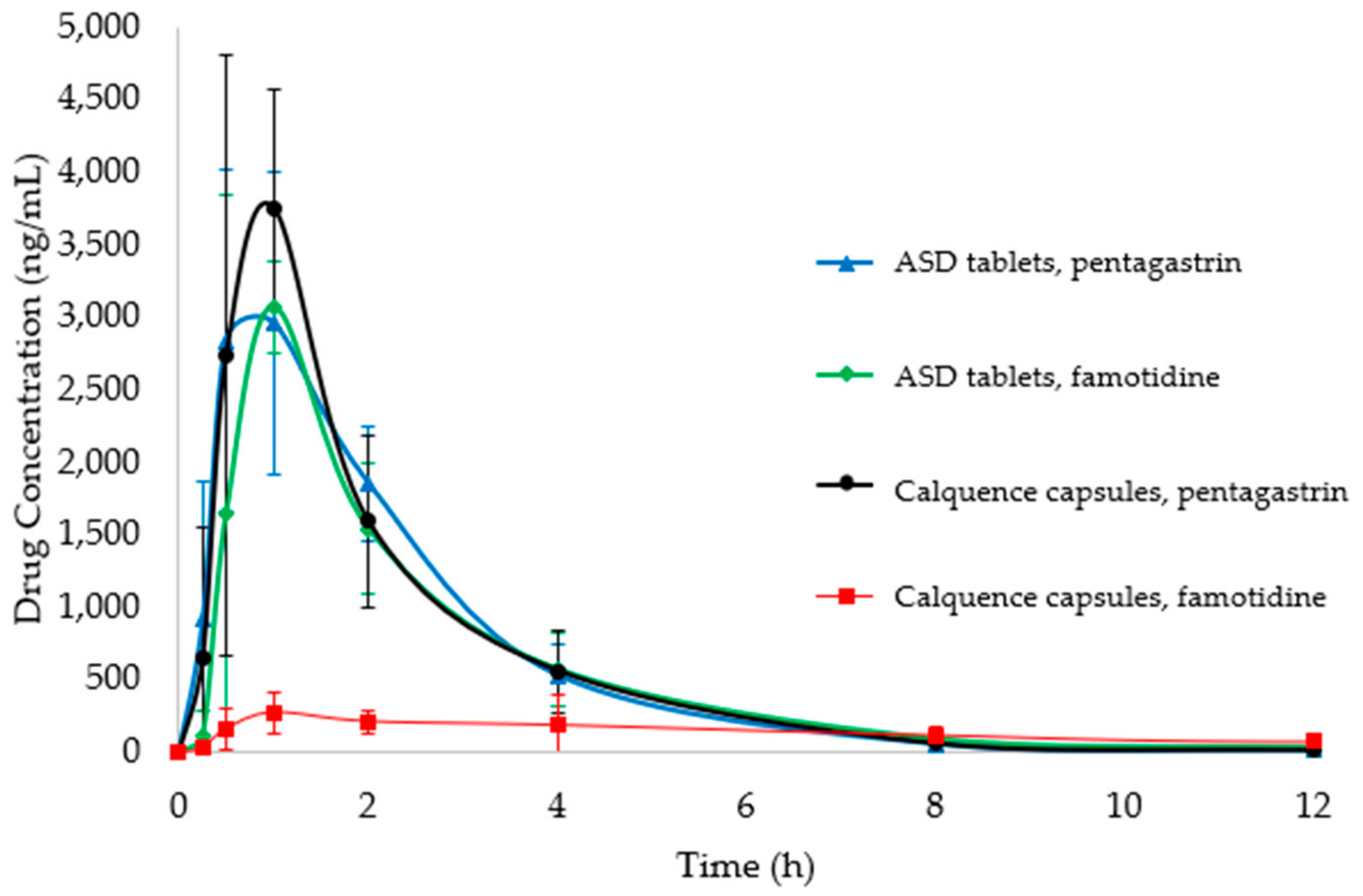
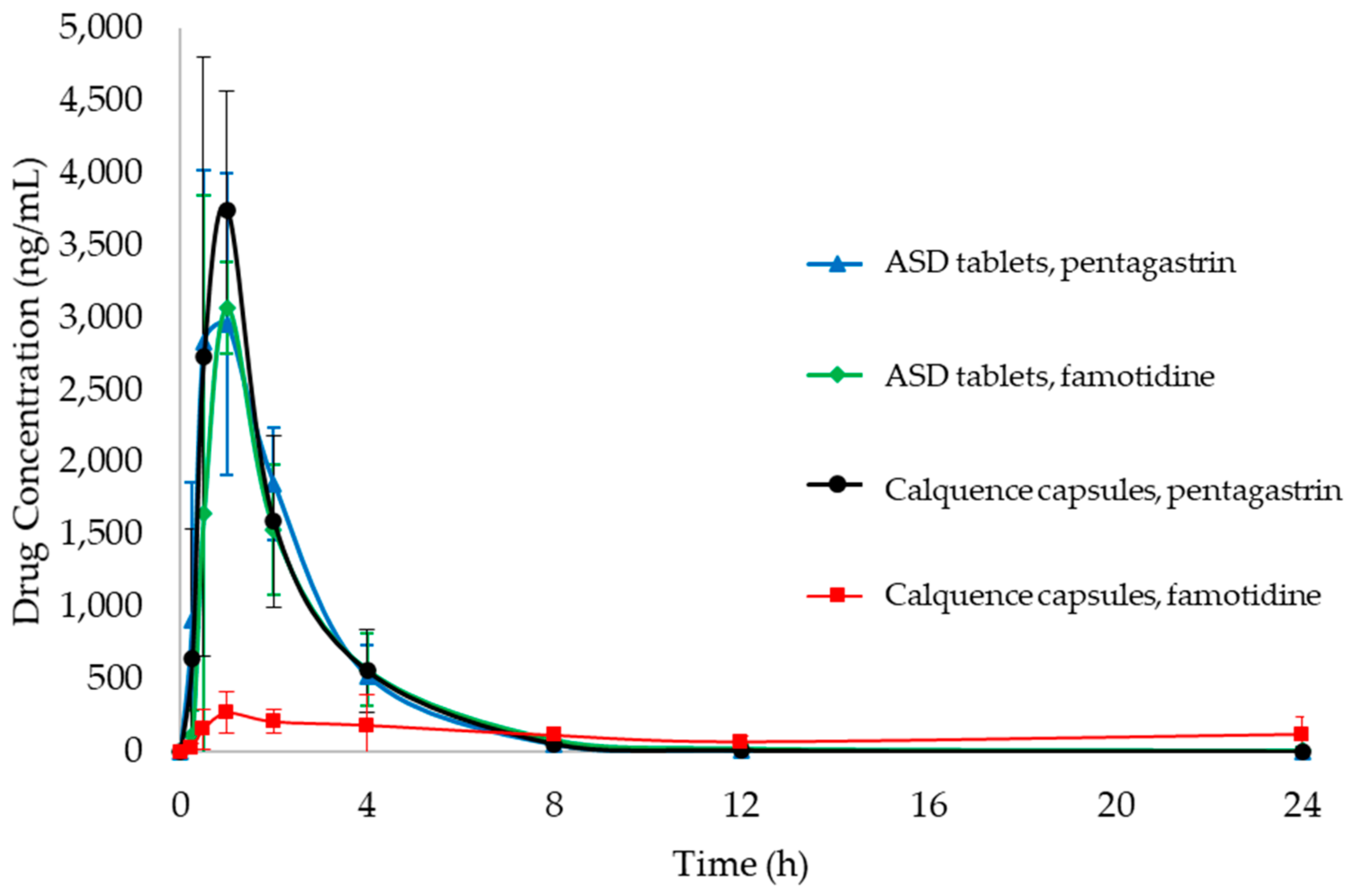
3.4. PK Study
4. Discussion
4.1. ASDs for Improving Low-Solubility, Weakly Basic Drugs
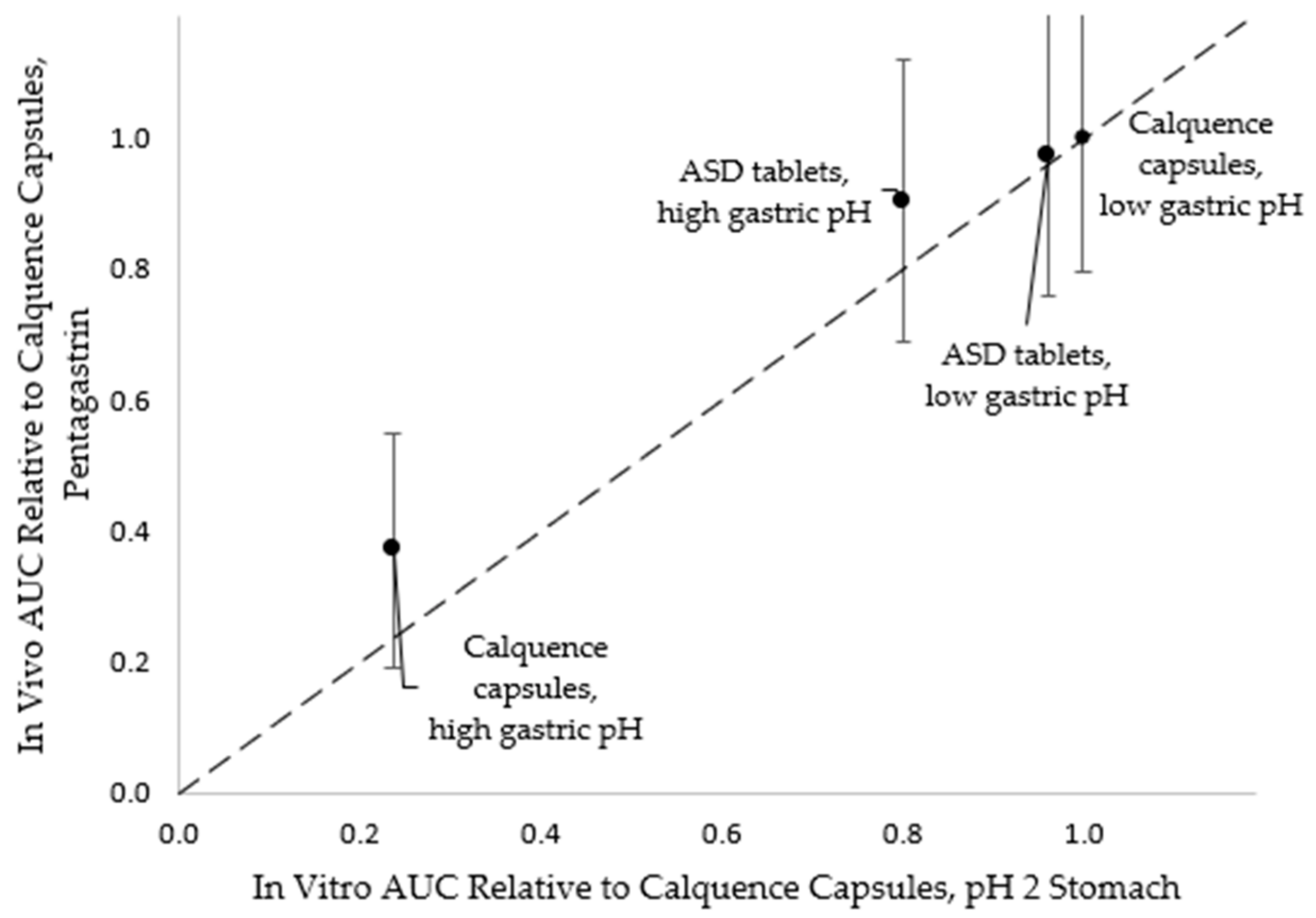
4.2. In Vitro Versus In Vivo Performance
4.3. Dog Model for Studying ARA Effect
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Active Pharmaceutical Ingredient (API), Amorphous Solid Dispersion (ASD) and ASD Tablet Characterization Methods
Appendix A.1.1. Modulated Differential Scanning Calorimetry (mDSC)
Appendix A.1.2. Powder X-ray Diffraction (PXRD)
Appendix A.1.3. Scanning Electron Micrography (SEM)
Appendix A.1.4. Reverse-Phase High-Performance Liquid Chromatography (RP-HPLC)
Appendix A.1.5. RP-HPLC–Mass Spectrometry (MS)
Appendix A.2. ASD Tablet Manufacturing Methods
Appendix A.3. Physical and Chemical Stability Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Storage Conditions | ASD Physical Stability | ASD Chemical Stability | ASD Tablet Chemical Stability |
|---|---|---|---|---|
| 1 | 5 °C/sealed with desiccant | ✔ | ✔ | |
| 2 | 5 °C/sealed | ✔ | ✔ | |
| 3 | 25 °C/60% RH/sealed with desiccant | ✔ | ✔ | |
| 4 | 25 °C/60% RH/open, perforated seal | ✔ | ✔ | |
| 5 | 40 °C/75% RH/open, perforated seal | ✔ |
Appendix A.4. ASD and API Characterization Results



Appendix A.5. Physical and Chemical Stability Results and Discussion



| Storage Condition | ASD | ASD Tablet | ||||
|---|---|---|---|---|---|---|
| Initial Impurities (area%) | Impurities after 12 wk (area%) | Change after 12 wk (%) a | Initial Impurities (area%) | Impurities after 12 wk (area%) | Change after 12 wk (%) a | |
| 5 °C/sealed with desiccant | 0.89 ± 0.01 | 0.85 ± 0.00 | −4.5 | 0.65 ± 0.01 | 0.63 ± 0.01 | −3.1 |
| 5 °C/sealed | 0.89 ± 0.01 | 0.89 ± 0.01 | 0.0 | 0.65 ± 0.01 | 0.63 ± 0.01 | −3.1 |
| 25 °C/60% RH/ sealed with desiccant | 0.89 ± 0.01 | 0.89 ± 0.01 | 0.0 | 0.65 ± 0.01 | 0.69 ± 0.00 | 6.2 |
| 25 °C/60% RH/open, perforated seal | 0.89 ± 0.01 | 1.15 ± 0.07 | 29.2 | 0.65 ± 0.01 | 0.82 ± 0.05 | 26.2 |
| 40 °C/75% RH/ open, perforated seal | 0.60 b ± 0.01 | 5.3 ± 0.08 | 783.3 | - | - | - |
| Total Impurities (area%) | |||||
|---|---|---|---|---|---|
| Storage Time | Initial | 2 wk | 4 wk | 5 wk | 12 wk |
| ASD | |||||
| 5 °C/sealed with desiccant | 0.89 ± 0.01 | 0.87 ± 0.10 | -- | 0.87 ± 0.07 | 0.85 ± 0.00 |
| 5 °C/sealed | 0.89 ± 0.01 | 0.82 ± 0.08 | -- | 0.89 ± 0.00 | 0.89 ± 0.01 |
| 25 °C/60% RH/sealed with desiccant | 0.89 ± 0.01 | 0.84 ± 0.06 | -- | 0.81 ± 0.05 | 0.89 ± 0.01 |
| 25 °C/60% RH/open, perforated seal | 0.89 ± 0.01 | 0.90 ± 0.07 | -- | 0.95 ± 0.08 | 1.15 ± 0.07 |
| 40 °C/75% RH/open, perforated seal | 0.60 ± 0.01 | -- | 1.25 ± 0.03 | -- | 5.27 ± 0.08 |
| ASD Tablets | |||||
| 5 °C/sealed with desiccant | 0.65 ± 0.01 | 0.65 ± 0.02 | -- | 0.65 ± 0.06 | 0.63 ± 0.01 |
| 5 °C/sealed | 0.65 ± 0.01 | 0.66 ± 0.06 | -- | 0.70 ± 0.15 | 0.63 ± 0.01 |
| 25 °C/60% RH/sealed with desiccant | 0.65 ± 0.01 | 0.70 ± 0.02 | -- | 0.62 ± 0.00 | 0.69 ± 0.00 |
| 25 °C/60% RH/open, perforated seal | 0.65 ± 0.01 | 0.70 ± 0.05 | -- | 0.74 ± 0.08 | 0.82 ± 0.05 |
| Crystalline Drug | |||||
| 40 °C/75% RH/open, perforated seal | 0.59 ± 0.00 | -- | 0.70 ± 0.04 | -- | 0.94 ± 0.05 |
| Peak Retention Time (min) | Relative Retention Time (min) | Degradant | Characteristic Ion Mass-to-Charge Ratio (m/z) | Proposed Degradation Pathway | Formula Weight of Proposed Product (Da) a |
|---|---|---|---|---|---|
| 11.05 | 0.65 | Ad1 | 442.6 | Transamidation with acetate at pyrrolidine ring | 441.4 |
| 14.79 | 0.87 | Ad2 | 400.6 | Amide cleavage at pyrrolidine ring | 399.4 |
| 15.09 | 0.89 | Ad3 | 550.8 | Putative addition product | |
| 16.87 | 0.99 | Ad4 | 484.6 | Alkyne hydration | 483.5 |
| 17.45 | 1.03 | Ad5 | 532.7 | Amidation of free amine with 2-butyne-4-one fragment | 532 |
| 20.23 | 1.19 | Ad6 | 546/548 | Succinamide formation at free amine | 548 |
| 23.07 | 1.36 | Ad7 | 457.7 | Unknown | |
| 25.8 | 1.52 | Ad8 | 932.2/466.7 | Addition and dimerization | |
| 27.9 | 1.64 | Ad9 | 888/444 | Dimerization |
Appendix A.6. Pharmacokinetic (PK) Study Results

| Test Article, Pretreatment | ASD Tablet, Pentagastrin | ASD Tablet, Famotidine | Calquence Capsules, Pentagastrin | Calquence Capsules, Famotidine |
|---|---|---|---|---|
| AUC0-24 (ng h/mL) | 8157 (1363) | 7558 (1429) | 8363 (1199) | 2875 (1284) |
| AUC0-inf (ng h/mL) | 8161 (1364) | 7579 (1423) | 8365 (1201) | 3112 (1415) |
| AUC0-inf p-value (compared to Calquence, famotidine) | <0.05 | <0.05 | <0.05 | n/a a |
| AUC0-inf p-value (compared to Calquence capsules, pentagastrin) | 0.81 | 0.37 | n/a | <0.05 |
References
- Deisseroth, A.B. Calquence FDA Label; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2019; Volume 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/210259s006s007lbl.pdf (accessed on 15 March 2021).
- Pepin, X.J.H.; Moir, A.J.; Mann, J.C.; Sanderson, N.J.; Barker, R.; Meehan, E.; Plumb, A.P.; Bailey, G.R.; Murphy, D.S.; Krejsa, C.M.; et al. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part ii. A mechanistic pbpk model for ir formulation comparison, proton pump inhibitor drug interactions, and administration with acidic juices. Eur. J. Pharm. Biopharm. 2019, 142, 435–448. [Google Scholar] [PubMed]
- Center For Drug Evaluation and Research. Product Quality Review; Application Number: 210259orig1s000; Center For Drug Evaluation and Research: Silver Spring, MD, USA, 2017. [Google Scholar]
- Patel, D.; Bertz, R.; Ren, S.; Boulton, D.W.; Någård, M. A systematic review of gastric acid-reducing agent-mediated drug–Drug interactions with orally administered medications. Clin. Pharmacokinet. 2020, 59, 447–462. [Google Scholar] [PubMed] [Green Version]
- Smelick, G.S.; Heffron, T.P.; Chu, L.; Dean, B.; West, D.A.; DuVall, S.L.; Lum, B.L.; Budha, N.; Holden, S.N.; Benet, L.Z.; et al. Prevalence of acid-reducing agents (ara) in cancer populations and ara drug–Drug interaction potential for molecular targeted agents in clinical development. Mol. Pharm. 2013, 10, 4055–4062. [Google Scholar] [PubMed]
- Pepin, X.J.H.; Sanderson, N.J.; Blanazs, A.; Grover, S.; Ingallinera, T.G.; Mann, J.C. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part i. Mechanistic modelling of drug product dissolution to derive a p-psd for pbpk model input. Eur. J. Pharm. Biopharm. 2019, 142, 421–434. [Google Scholar]
- Zhang, L.; Wu, F.; Lee, S.C.; Zhao, H.; Zhang, L. Ph-dependent drug–Drug interactions for weak base drugs: Potential implications for new drug development. Clin. Pharmacol. Ther. 2014, 96, 266–277. [Google Scholar]
- Hens, B.; Tsume, Y.; Bermejo, M.; Paixao, P.; Koenigsknecht, M.J.; Baker, J.R.; Hasler, W.L.; Lionberger, R.; Fan, J.; Dickens, J.; et al. Low buffer capacity and alternating motility along the human gastrointestinal tract: Implications for in vivo dissolution and absorption of ionizable drugs. Mol. Pharm. 2017, 14, 4281–4294. [Google Scholar]
- Riethorst, D.; Mols, R.; Duchateau, G.; Tack, J.; Brouwers, J.; Augustijns, P. Characterization of human duodenal fluids in fasted and fed state conditions. J. Pharm. Sci. 2016, 105, 673–681. [Google Scholar]
- Shih, G.L.; Brensinger, C.; Katzka, D.A.; Metz, D.C. Influence of age and gender on gastric acid secretion as estimated by integrated acidity in patients referred for 24-h ambulatory ph monitoring. Am. J. Gastroenterol. 2003, 98, 1713–1718. [Google Scholar]
- Blatter, F.; Ingallinera, T.; Barf, T.; Aret, T.; Krejsa, C.; Evarts, J. Solid Forms and Formuations of (s)-4->-Amino-3-(l-(but-2-ynoyl)pyrrolidin-2-yl)imidazo[1,5-ajpyrazin- 1-yl)-n-(pyridin-2-yl)benzamide. 2016. Available online: https://patentimages.storage.googleapis.com/5e/4d/3f/0e3e4fb9e1a906/WO2017002095A1.pdf (accessed on 1 March 2021).
- Van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar]
- Almeida e Sousa, L.; Reutzel-Edens, S.M.; Stephenson, G.A.; Taylor, L.S. Assessment of the amorphous “solubility” of a group of diverse drugs using new experimental and theoretical approaches. Mol. Pharm. 2015, 12, 484–495. [Google Scholar]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class ii drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar]
- Mudie, D.M.; Buchanan, S.; Stewart, A.M.; Smith, A.; Shepard, K.B.; Biswas, N.; Marshall, D.; Ekdahl, A.; Pluntze, A.; Craig, C.D.; et al. A novel architecture for achieving high drug loading in amorphous spray dried dispersion tablets. Int. J. Pharm. X 2020, 2, 100042. [Google Scholar]
- Mudie, D.M.; Stewart, A.M.; Biswas, N.; Brodeur, T.J.; Shepard, K.B.; Smith, A.; Morgen, M.M.; Baumann, J.M.; Vodak, D.T. Novel high-drug-loaded amorphous dispersion tablets of posaconazole; in vivo and in vitro assessment. Mol. Pharm. 2020, 17, 4463–4472. [Google Scholar]
- Fancher, R.M.; Zhang, H.; Sleczka, B.; Derbin, G.; Rockar, R.; Marathe, P. Development of a canine model to enable the preclinical assessment of ph-dependent absorption of test compounds. J. Pharm. Sci. 2011, 100, 2979–2988. [Google Scholar]
- Koziolek, M.; Grimm, M.; Bollmann, T.; Schäfer, K.J.; Blattner, S.M.; Lotz, R.; Boeck, G.; Weitschies, W. Characterization of the gi transit conditions in beagle dogs with a telemetric motility capsule. Eur. J. Pharm. Biopharm. 2019, 136, 221–230. [Google Scholar]
- Meyer, J.H.; Dressman, J.; Fink, A.; Amidon, G. Effect of size and density on canine gastric emptying of nondigestible solids. Gastroenterology 1985, 89, 805–813. [Google Scholar]
- Dressman, J.B. Comparison of canine and human gastrointestinal physiology. Pharm. Res. 1986, 3, 123–131. [Google Scholar]
- Arndt, M.; Chokshi, H.; Tang, K.; Parrott, N.J.; Reppas, C.; Dressman, J.B. Dissolution media simulating the proximal canine gastrointestinal tract in the fasted state. Eur. J. Pharm. Biopharm. 2013, 84, 633–641. [Google Scholar]
- Wang, C.; Zhai, B.; Guo, H.; Wang, P.; Liu, Z.; Gu, H.; Ho, H.; Langguth, P.; Li, K.; Wang, C.; et al. In vivo measurement of gastric fluid volume in anesthetized dogs. J. Drug Deliv. Sci. Technol. 2020, 55, 101488. [Google Scholar]
- Smeets-Peeters, M.; Watson, T.; Minekus, M.; Havenaar, R. A review of the physiology of the canine digestive tract related to the development of in vitro systems. Nutr. Res. Rev. 1998, 11, 45–69. [Google Scholar]
- Drumond, N.; Stegemann, S. Better medicines for older patients: Considerations between patient characteristics and solid oral dosage form designs to improve swallowing experience. Pharmaceutics 2021, 13, 32. [Google Scholar]
- Raber-Durlacher, J.E.; Brennan, M.T.; Verdonck-de Leeuw, I.M.; Gibson, R.J.; Eilers, J.G.; Waltimo, T.; Bots, C.P.; Michelet, M.; Sollecito, T.P.; Rouleau, T.S.; et al. Swallowing dysfunction in cancer patients. Support. Care Cancer 2012, 20, 433–443. [Google Scholar]
- van Leeuwen, R.W.; van Gelder, T.; Mathijssen, R.H.; Jansman, F.G. Drug-drug interactions with tyrosine-kinase inhibitors: A clinical perspective. Lancet Oncol. 2014, 15, e315–e326. [Google Scholar]
- Uchiyama, A.A.T.; Silva, P.A.I.A.; Lopes, M.S.M.; Yen, C.T.; Ricardo, E.D.; Mutão, T.; Pimenta, J.R.; Machado, L.M.; Shimba, D.S.; Peixoto, R.D. Proton pump inhibitors and oncologic treatment efficacy: A practical review of the literature for oncologists. Curr. Oncol. 2021, 28, 783–799. [Google Scholar]
- Sawicki, E.; Schellens, J.H.; Beijnen, J.H.; Nuijen, B. Inventory of oral anticancer agents: Pharmaceutical formulation aspects with focus on the solid dispersion technique. Cancer Treat. Rev. 2016, 50, 247–263. [Google Scholar]
- Herbrink, M.; Nuijen, B.; Schellens, J.H.; Beijnen, J.H. Variability in bioavailability of small molecular tyrosine kinase inhibitors. Cancer Treat. Rev. 2015, 41, 412–422. [Google Scholar]
- Gala, U.H.; Miller, D.A.; Williams, R.O. Harnessing the therapeutic potential of anticancer drugs through amorphous solid dispersions. Biochim. Biophys. Acta (BBA) Rev. Cancer 2020, 1873, 188319. [Google Scholar]
- Roskoski, R. Properties of fda-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [PubMed]
- Veerman, G.D.M.; Hussaarts, K.; Jansman, F.G.A.; Koolen, S.W.L.; van Leeuwen, R.W.F.; Mathijssen, R.H.J. Clinical implications of food-drug interactions with small-molecule kinase inhibitors. Lancet Oncol. 2020, 21, e265–e279. [Google Scholar] [PubMed]
- Pathak, S.M.; Schaefer, K.J.; Jamei, M.; Turner, D.B. Biopharmaceutic ivive-mechanistic modeling of single- and two-phase in vitro experiments to obtain drug-specific parameters for incorporation into pbpk models. J. Pharm. Sci. 2019, 108, 1604–1618. [Google Scholar]
- Xu, H.; Vela, S.; Shi, Y.; Marroum, P.; Gao, P. In vitro characterization of ritonavir drug products and correlation to human in vivo performance. Mol. Pharm. 2017, 14, 3801–3814. [Google Scholar]
- Hens, B.; Talattof, A.; Paixão, P.; Bermejo, M.; Tsume, Y.; Löbenberg, R.; Amidon, G.L. Measuring the impact of gastrointestinal variables on the systemic outcome of two suspensions of posaconazole by a pbpk model. AAPS J. 2018, 20, 57. [Google Scholar]
- Jamei, M.; Abrahamsson, B.; Brown, J.; Bevernage, J.; Bolger, M.B.; Heimbach, T.; Karlsson, E.; Kotzagiorgis, E.; Lindahl, A.; McAllister, M.; et al. Current status and future opportunities for incorporation of dissolution data in pbpk modeling for pharmaceutical development and regulatory applications: Orbito consortium commentary. Eur. J. Pharm. Biopharm. 2020, 155, 55–68. [Google Scholar]
- Grignard, E.; Taylor, R.; McAllister, M.; Box, K.; Fotaki, N. Considerations for the development of in vitro dissolution tests to reduce or replace preclinical oral absorption studies. Eur. J. Pharm. Sci. 2017, 99, 193–201. [Google Scholar]
- Kararli, T.T. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm. Drug Dispos. 1995, 16, 351–380. [Google Scholar]
- Zane, P.; Guo, Z.; MacGerorge, D.; Vicat, P.; Ollier, C. Use of the pentagastrin dog model to explore the food effects on formulations in early drug development. Eur. J. Pharm. Sci. 2014, 57, 207–213. [Google Scholar]
- Mitra, A.; Kesisoglou, F. Impaired drug absorption due to high stomach ph: A review of strategies for mitigation of such effect to enable pharmaceutical product development. Mol. Pharm. 2013, 10, 3970–3979. [Google Scholar]
- Mitra, A.; Kesisoglou, F.; Beauchamp, M.; Zhu, W.; Chiti, F.; Wu, Y. Using absorption simulation and gastric ph modulated dog model for formulation development to overcome achlorhydria effect. Mol. Pharm. 2011, 8, 2216–2223. [Google Scholar]
- Litou, C.; Vertzoni, M.; Goumas, C.; Vasdekis, V.; Xu, W.; Kesisoglou, F.; Reppas, C. Characteristics of the human upper gastrointestinal contents in the fasted state under hypo- and a-chlorhydric gastric conditions under conditions of typical drug—Drug interaction studies. Pharm. Res. 2016, 33, 1399–1412. [Google Scholar]
- Tutuian, R.; Katz, P.O.; Bochenek, W.; Castell, D.O. Dose-dependent control of intragastric ph by pantoprazole, 10, 20 or 40 mg, in healthy volunteers. Aliment. Pharmacol. Ther. 2002, 16, 829–836. [Google Scholar]
- Prichard, P.J.; Yeomans, N.D.; Mihaly, G.W.; Jones, D.B.; Buckle, P.J.; Smallwood, R.A.; Louis, W.J. Omeprazole: A study of its inhibition of gastric ph and oral pharmacokinetics after morning or evening dosage. Gastroenterology 1985, 88, 64–69. [Google Scholar] [PubMed]
- Podoll, T.; Pearson, P.G.; Evarts, J.; Ingallinera, T.; Bibikova, E.; Sun, H.; Gohdes, M.; Cardinal, K.; Sanghvi, M.; Slatter, J.G. Bioavailability, biotransformation, and excretion of the covalent bruton tyrosine kinase inhibitor acalabrutinib in rats, dogs, and humans. Drug Metab. Dispos. 2019, 47, 145–154. [Google Scholar] [PubMed]





| Compound Property | Value |
|---|---|
| Molecular weight (g/mol) | 465.5 |
| pKas in physiological range | 3.5 (basic) and 5.8 (basic) a |
| log P | 2.0 a |
| Melting temperature (Tm) (°C) | 214 b |
| Glass-transition temperature (Tg) (°C) | 133 c |
| Crystalline intrinsic solubility (µg/mL) | 48 a |
| Function | Ingredient | Tablet Fraction (wt%) a |
|---|---|---|
| Intragranular | ||
| ASD | 50/50 acalabrutinib/Aqoat (HPMCAS-HF) | 50.0 |
| Ductile filler | Avicel® PH-101 | 14.3 |
| Brittle filler | Pearlitol® 25 | 28.7 |
| Disintegrant | Ac-Di-Sol® | 6.0 |
| Glidant | Cab-O-Sil® (M5P) | 0.25 |
| Lubricant | Magnesium stearate | 0.25 |
| Extragranular | ||
| Glidant | Cab-O-Sil | 0.25 |
| Lubricant | Magnesium stearate | 0.25 |
| Total tablet mass: | 400 mg | |
| Parameter | Value |
|---|---|
| Dose (mg) | 100 |
| Dosing volume (mL) | 50 |
| Dosing medium | Milli-Q water |
| Gastric resting mediumGastric secretion medium | HCl (pH 2) and 34 mM NaClHCl (pH 6) and 34 mM NaCl |
| Gastric resting volume (mL) | 50 |
| Gastric secretion rate (mL/min) | 2.4 |
| Gastric emptying half-life (monoexponential) (min) | 15 |
| Duodenal resting and secretion medium | Phosphate (pH 6.5) and FaSSIF powder a |
| Duodenal fluid volume (mL) | 50 |
| Duodenal secretion rate (mL/min) | 2.4 |
| Duodenal emptying rate | Gastric emptying and duodenal secretion |
| Jejunal medium | Gastric and duodenal composition |
| Jejunal volume | Starts at 0 and increases to 419 mL at 90 min |
| Phase a | Test Article | Pretreatment | Dosage Form per Animal |
|---|---|---|---|
| 1 | Acalabrutinib ASD | Pentagastrin b | 1 tablet |
| 2 | Acalabrutinib ASD | Famotidine c | 1 tablet |
| 3 | Calquence | Pentagastrin b | 1 capsule |
| 4 | Calquence | Famotidine c | 1 capsule |
| Test Article with Simulated Pretreatment | Average In Vitro Duodenal AUC (µg-min/mL) | In Vitro AUC Ratio (Famotidine/Pentagastrin) |
|---|---|---|
| ASD tablet, pentagastrin (pH 2 stomach) | 16,946 | 0.83 |
| ASD tablet, famotidine (pH 6 stomach) | 14,102 | - |
| Calquence, pentagastrin (pH 2 stomach) | 17,617 | 0.24 |
| Calquence, famotidine (pH 6 stomach) | 4173 | - |
| Test Article, Pretreatment | ASD Tablet, Pentagastrin | ASD Tablet, Famotidine | Calquence Capsule, Pentagastrin | Calquence Capsule, Famotidine |
|---|---|---|---|---|
| AUC0-inf (ng-h/mL) | 8161 (1364) a | 7579 (1423) a | 8365 (1201) | 3112 (1415) b |
| Cmax (ng/mL) | 3332 (769) | 3443 (996) | 4480 (516) | 355 (121) |
| Tmax (h) | 0.9 (0.5) | 0.9 (0.2) | 0.8 (0.2) | 1.6 (1.2) |
| AUC ratio compared to Calquence capsule, pentagastrin | 0.98 | 0.91 | 1.00 | 0.37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mudie, D.M.; Stewart, A.M.; Rosales, J.A.; Biswas, N.; Adam, M.S.; Smith, A.; Craig, C.D.; Morgen, M.M.; Vodak, D.T. Amorphous Solid Dispersion Tablets Overcome Acalabrutinib pH Effect in Dogs. Pharmaceutics 2021, 13, 557. https://doi.org/10.3390/pharmaceutics13040557
Mudie DM, Stewart AM, Rosales JA, Biswas N, Adam MS, Smith A, Craig CD, Morgen MM, Vodak DT. Amorphous Solid Dispersion Tablets Overcome Acalabrutinib pH Effect in Dogs. Pharmaceutics. 2021; 13(4):557. https://doi.org/10.3390/pharmaceutics13040557
Chicago/Turabian StyleMudie, Deanna M., Aaron M. Stewart, Jesus A. Rosales, Nishant Biswas, Molly S. Adam, Adam Smith, Christopher D. Craig, Michael M. Morgen, and David T. Vodak. 2021. "Amorphous Solid Dispersion Tablets Overcome Acalabrutinib pH Effect in Dogs" Pharmaceutics 13, no. 4: 557. https://doi.org/10.3390/pharmaceutics13040557