
Rapamycin-Loaded Polymeric Nanoparticles as an Advanced Formulation for Macrophage Targeting in Atherosclerosis

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis and Characterization of Poly-ε-Caprolactone-Succinate (PCL-SUCC)
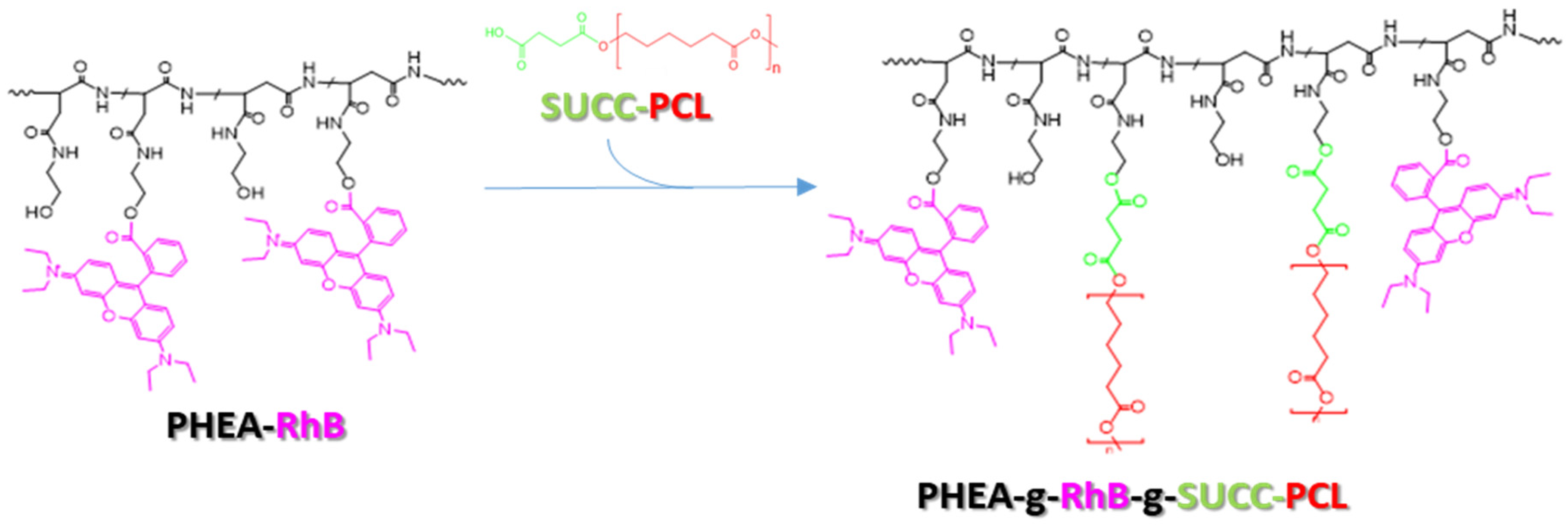
2.3. Synthesis and Characterization of PHEA-g-RhB-g-SUCC-PCL
2.4. Nano-Precipitation
2.5. Nanoparticle Drying
2.6. Nanoparticle Characterization
2.6.1. Size and ζ Potential Measurements
2.6.2. X-ray Photoelectron Spectroscopy (XPS) Analysis
2.6.3. Kodia-PC Quantification
2.6.4. Differential Scanning Calorimetry (DSC) Analysis
2.7. Rapa Entrapment
2.7.1. Determination of Drug Loading (DL%)
2.7.2. Drug Stability
2.7.3. Drug Release
2.8. Evaluation of Storage Stability
2.9. Biological Characterization
2.9.1. Cell Culture
2.9.2. Cell Viability
2.9.3. Nanoparticle Uptake
2.9.4. Biological Evaluation of Rapa Activity [ALICE: DA CAMBIARE TESTO]
3. Results and Discussion
3.1. Copolymer Synthesis
3.2. Production of Nanoparticles
3.3. Quali-Quantitative Analysis of Kodia-PC
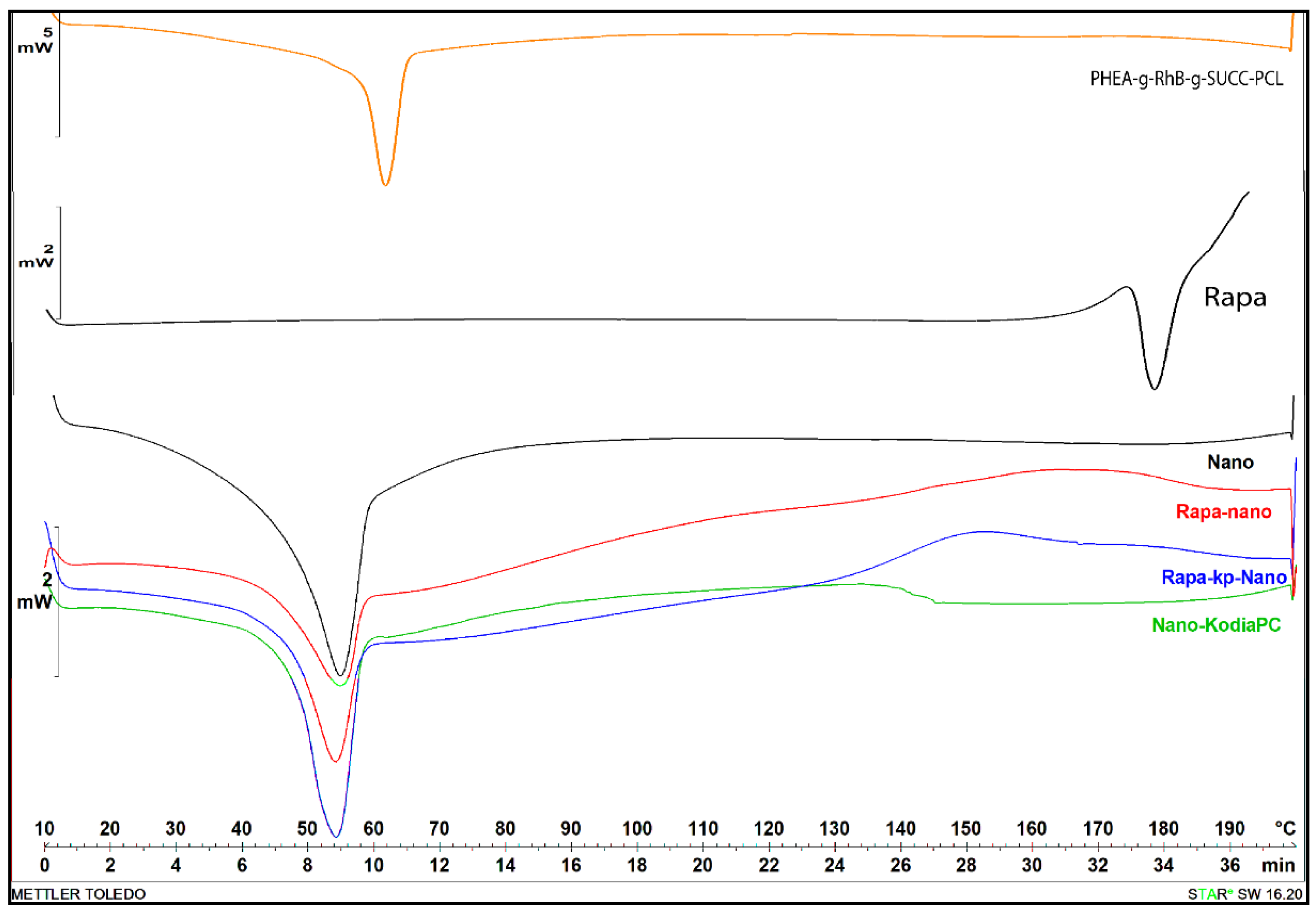
3.4. Thermal Analysis
3.5. Optimization of the Drying Process
3.6. Drug Stability and Release from KP-Nano
3.7. Long Term Storage
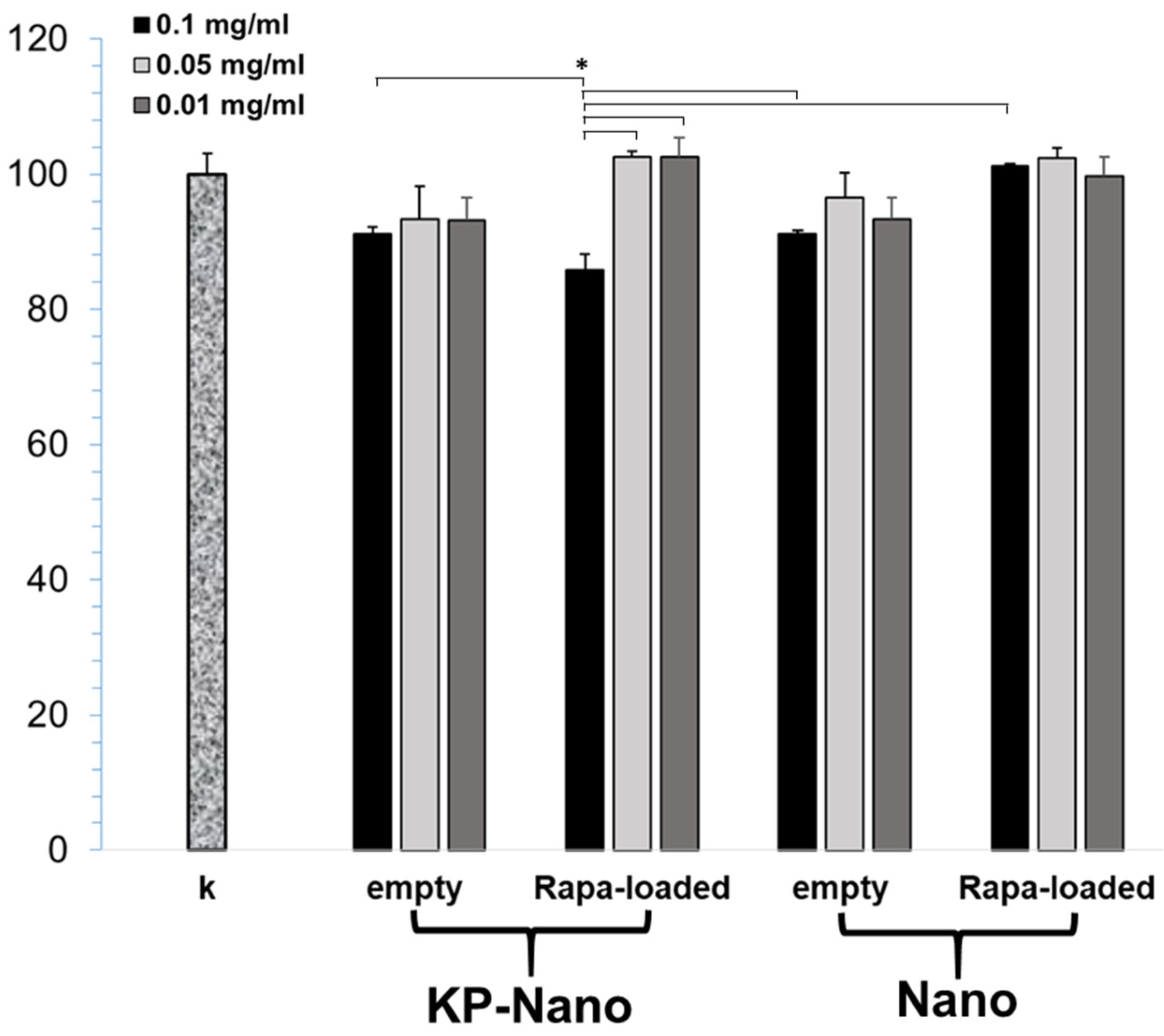
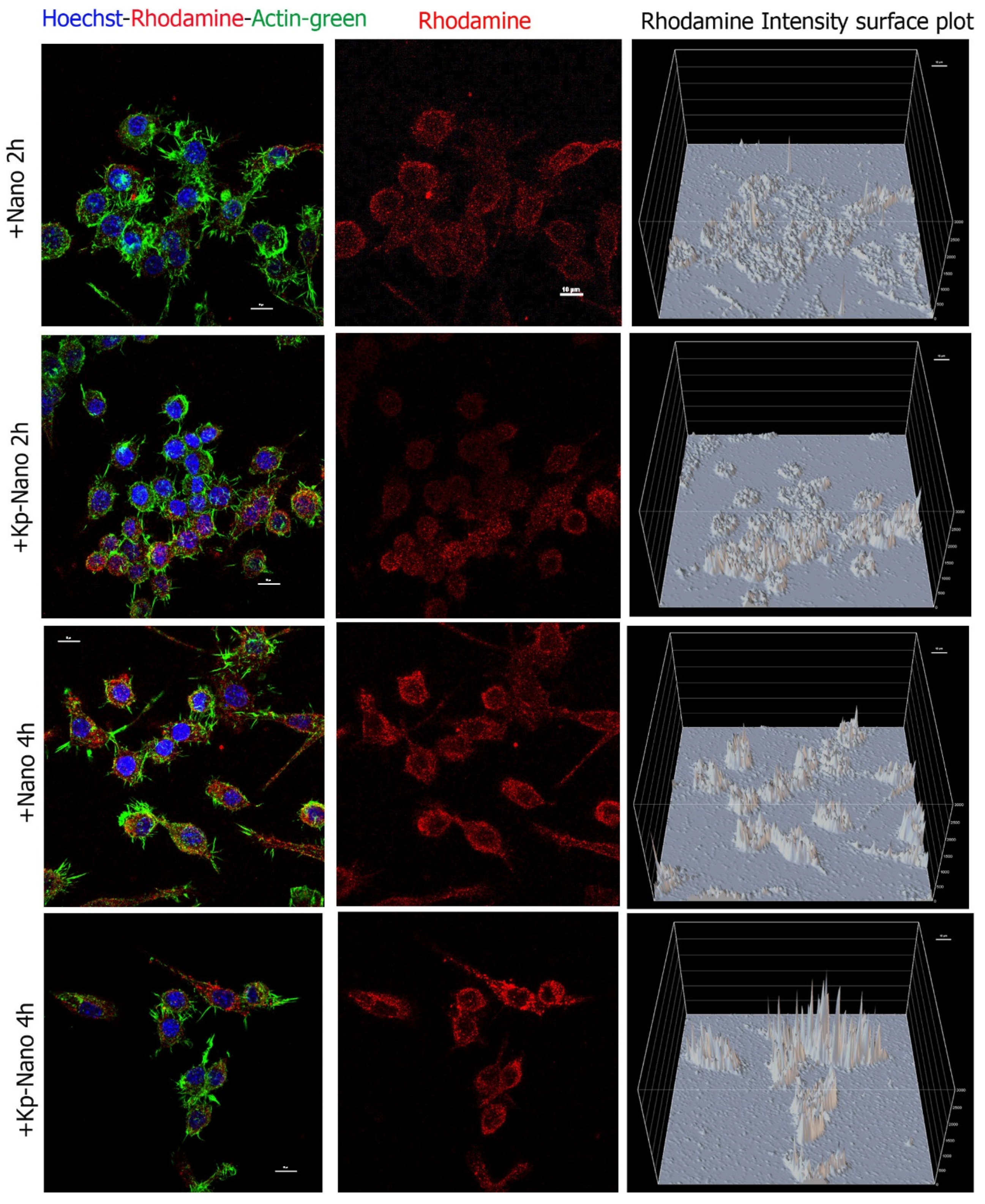
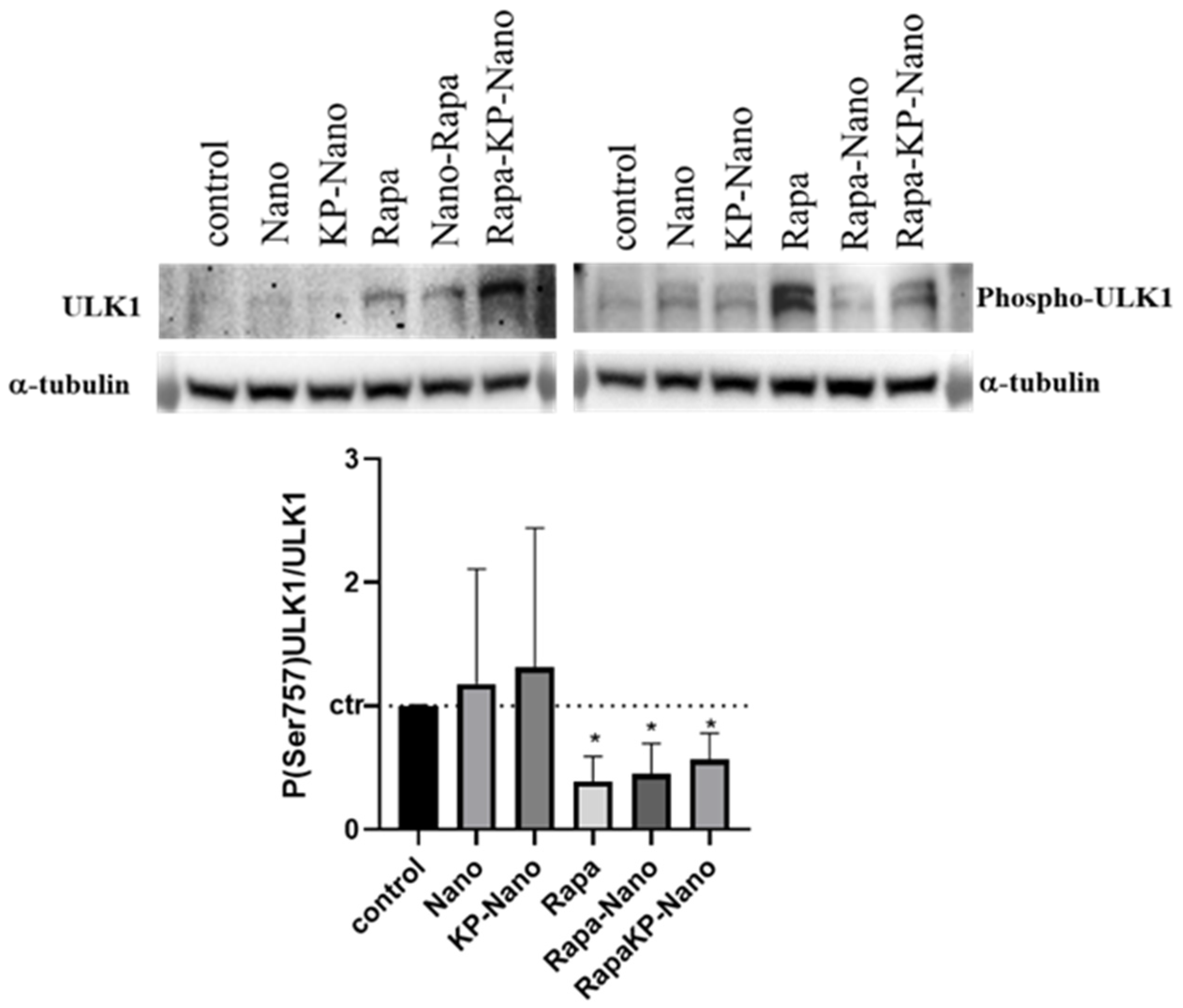
3.8. Cellular Viability, Internalization and Validation of Rapa-Nano Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geovanini, G.R.; Libby, P. Atherosclerosis and inflammation: Overview and updates. Clin. Sci. 2018, 132, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.K.W.; Zhang, L.; Cheng, C.K.; Yang, H.; Huang, Y.; Tian, X.Y.; Choi, C.H.J. Recent advances in managing atherosclerosis via nanomedicine. Small 2018, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Martinet, W.; De Loof, H.; De Meyer, G.R.Y. MTOR inhibition: A promising strategy for stabilization of atherosclerotic plaques. Atherosclerosis 2014, 233, 601–607. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, F.; Zou, S.; Qu, L. Rapamycin: A bacteria-derived immunosuppressant that has anti-atherosclerotic effects and its clinical application. Front. Pharmacol. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Haeri, A.; Osouli, M.; Bayat, F.; Alavi, S.; Dadashzadeh, S. Nanomedicine approaches for sirolimus delivery: A review of pharmaceutical properties and preclinical studies. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. MTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Guan, X. Autophagy: A new target for the treatment of atherosclerosis. Front. Lab. Med. 2018, 2, 68–71. [Google Scholar] [CrossRef]
- He, Y.; Zuo, C.; Jia, D.; Bai, P.; Kong, D.; Chen, D.; Liu, G.; Li, J.; Wang, Y.; Chen, G.; et al. Loss of DP1 aggravates vascular remodeling in pulmonary arterial hypertension via mTORC1 signaling. Am. J. Respir. Crit. Care Med. 2020, 201, 1263–1276. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Zhou, J.-S.; Huang, H.-Q.; Li, Z.-Y.; Xu, X.-C.; Lai, T.-W.; Hu, Y.; Zhou, H.-B.; Chen, H.-P.; et al. MTOR suppresses cigarette smoke–induced epithelial cell death and airway inflammation in chronic obstructive pulmonary disease. J. Immunol. 2018, 200, 2571–2580. [Google Scholar] [CrossRef] [PubMed]
- Pelaz, B.; Alexiou, C.; Alvarez-Puebla, R.A.; Alves, F.; Andrews, A.M.; Ashraf, S.; Balogh, L.P.; Ballerini, L.; Bestetti, A.; Brendel, C.; et al. Diverse applications of nanomedicine. ACS Nano 2017, 11, 2313–2381. [Google Scholar] [CrossRef] [Green Version]
- Cassano, R.; Cuconato, M.; Calviello, G.; Serini, S.; Trombino, S. Recent advances in nanotechnology for the treatment of melanoma. Molecules 2021, 26, 785. [Google Scholar] [CrossRef]
- Atukorale, P.U.; Covarrubias, G.; Bauer, L.; Karathanasis, E. Vascular targeting of nanoparticles for molecular imaging of diseased endothelium. Adv. Drug Deliv. Rev. 2017, 113, 141–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.H.; Muktabar, A.; Tang, J.; Dravid, V.P.; Thaxton, C.S.; Venkatraman, S.; Ng, K.W. Engineered nanoparticles for the detection, treatment and prevention of atherosclerosis: How close are we? Drug Discov. Today 2017, 22, 1438–1446. [Google Scholar] [CrossRef]
- Zhang, J.; Zu, Y.; Dhanasekara, C.S.; Li, J.; Wu, D.; Fan, Z.; Wang, S. Detection and treatment of atherosclerosis using nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Flores, A.M.; Ye, J.; Jarr, K.U.; Hosseini-Nassab, N.; Smith, B.R.; Leeper, N.J. Nanoparticle therapy for vascular diseases. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 635–646. [Google Scholar] [CrossRef]
- Boada, C.; Zinger, A.; Tsao, C.; Zhao, P.; Martinez, J.O.; Hartman, K.; Naoi, T.; Sukhoveshin, R.; Sushnitha, M.; Molinaro, R.; et al. Rapamycin-loaded biomimetic nanoparticles reverse vascular inflammation. Circ. Res. 2020, 25–37. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, K.; Qin, X.; Li, T.; Qiu, J.; Yin, T.; Huang, J.; McGinty, S.; Pontrelli, G.; Ren, J.; et al. Biomimetic nanotherapies: Red blood cell based core–shell structured nanocomplexes for atherosclerosis management. Adv. Sci. 2019, 6. [Google Scholar] [CrossRef]
- Song, Y.; Huang, Z.; Liu, X.; Pang, Z.; Chen, J.; Yang, H.; Zhang, N.; Cao, Z.; Liu, M.; Cao, J.; et al. Platelet membrane-coated nanoparticle-mediated targeting delivery of Rapamycin blocks atherosclerotic plaque development and stabilizes plaque in apolipoprotein E-deficient (ApoE−/−) mice. Nanomed. Nanotechnol. Biol. Med. 2019, 15, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Guo, J.; Chen, Y.; Han, S.; Xu, X.; Shi, Q.; Jia, Y.; Liu, Y.; Deng, Y.; Wang, R.; et al. Sustained delivery by a cyclodextrin material-based nanocarrier potentiates antiatherosclerotic activity of rapamycin via selectively inhibiting mTORC1 in mice. J. Control. Release 2016, 235, 48–62. [Google Scholar] [CrossRef]
- Nie, S.; Zhang, J.; Martinez-Zaguilan, R.; Sennoune, S.; Hossen, M.N.; Lichtenstein, A.H.; Cao, J.; Meyerrose, G.E.; Paone, R.; Soontrapa, S.; et al. Detection of atherosclerotic lesions and intimal macrophages using CD36-targeted nanovesicles. J. Control. Release 2015, 220, 61–70. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Kapate, N.; Shields, C.W.; Mitragotri, S. Drug delivery to macrophages: A review of targeting drugs and drug carriers to macrophages for inflammatory diseases. Adv. Drug Deliv. Rev. 2019. [Google Scholar] [CrossRef]
- Martinet, W.; Coornaert, I.; Puylaert, P.; De Meyer, G.R.Y. Macrophage death as a pharmacological target in atherosclerosis. Front. Pharmacol. 2019, 10, 1–18. [Google Scholar] [CrossRef]
- Podrez, E.A.; Poliakov, E.; Shen, Z.; Zhang, R.; Deng, Y.; Sun, M.; Finton, P.J.; Shan, L.; Gugiu, B.; Fox, P.L.; et al. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J. Biol. Chem. 2002, 277, 38503–38516. [Google Scholar] [CrossRef] [Green Version]
- Giammona, G.; Pitarresi, G.; Craparo, E.F.; Cavallaro, G.; Buscemi, S. New biodegradable hydrogels based on a photo-cross-linkable polyaspartamide and poly(ethylene glycol) derivatives. Release studies of an anticancer drug. Colloid Polym. Sci. 2001, 279, 771–783. [Google Scholar] [CrossRef]
- Craparo, E.F.; Licciardi, M.; Conigliaro, A.; Palumbo, F.S.; Giammona, G.; Alessandro, R.; De Leo, G.; Cavallaro, G. Hepatocyte-targeted fluorescent nanoparticles based on a polyaspartamide for potential theranostic applications. Polymer (Guildf.) 2015, 70, 257–270. [Google Scholar] [CrossRef]
- Craparo, E.F.; Drago, S.E.; Giammona, G.; Cavallaro, G. Production of polymeric micro- and nanostructures with tunable properties as pharmaceutical delivery systems. Polymer (Guildf.) 2020, 200, 122596. [Google Scholar] [CrossRef]
- Stewart, J.C.M. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem. 1980, 104, 10–14. [Google Scholar] [CrossRef]
- Zhang, Y.; Schmid, Y.R.F.; Luginbühl, S.; Wang, Q.; Dittrich, P.S.; Walde, P. Spectrophotometric quantification of peroxidase with p-Phenylene-diamine for analyzing peroxidase-encapsulating lipid vesicles. Anal. Chem. 2017, 89, 5484–5493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ICH. Stability Testing for New Dosage Forms-Q1C; ICH: Geneva, Swziterland, 1996; pp. 4–6. [Google Scholar]
- ICH. Stability Testing of New Drug Substances and Products Q1A(R2); ICH: Geneva, Swziterland, 2003; p. 24. [Google Scholar] [CrossRef] [Green Version]
- Saieva, L.; Barreca, M.M.; Zichittella, C.; Prado, M.G.; Tripodi, M.; Alessandro, R.; Conigliaro, A. Hypoxia-induced MiR-675-5p supports β-catenin nuclear localization by regulating GSK3-β activity in colorectal cancer cell lines. Int. J. Mol. Sci. 2020, 21, 3832. [Google Scholar] [CrossRef]
- Craparo, E.F.; Porsio, B.; Mauro, N.; Giammona, G.; Cavallaro, G. Polyaspartamide-polylactide graft copolymers with tunable properties for the realization of fluorescent nanoparticles for imaging. Macromol. Rapid Commun. 2015, 36, 1409–1415. [Google Scholar] [CrossRef] [PubMed]
- Othman, R.; Vladisavljević, G.T.; Nagy, Z.K.; Holdich, R.G. Encapsulation and Controlled Release of Rapamycin from polycaprolactone nanoparticles prepared by membrane micromixing combined with antisolvent precipitation. Langmuir 2016, 32, 10685–10693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Li, X.; Le, Y. Amorphous nanoparticulate formulation of sirolimus and its tablets. Pharmaceutics 2018, 10, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musumeci, T.; Serapide, M.F.; Pellitteri, R.; Dalpiaz, A.; Ferraro, L.; Dal Magro, R.; Bonaccorso, A.; Carbone, C.; Veiga, F.; Sancini, G.; et al. Oxcarbazepine free or loaded PLGA nanoparticles as effective intranasal approach to control epileptic seizures in rodents. Eur. J. Pharm. Biopharm. 2018, 133, 309–320. [Google Scholar] [CrossRef]
- Bereczki, D.; Liu, M.; Fernandes do Prado, G.; Fekete, I. Mannitol for acute stroke. Cochr. Database Syst. Rev. 2007. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Campos, M.S.T.; Fialho, S.L.; Pereira, B.G.; Yoshida, M.I.; Oliveira, M.A. Kinetics studies of the degradation of sirolimus in solid state and in liquid medium. J. Therm. Anal. Calorim. 2017, 130, 1653–1661. [Google Scholar] [CrossRef]
- Sun, M.; Si, L.; Zhai, X.; Fan, Z.; Ma, Y.; Zhang, R.; Yang, X. The influence of co-solvents on the stability and bioavailability of rapamycin formulated in self-microemulsifying drug delivery systems. Drug Dev. Ind. Pharm. 2011, 37, 986–994. [Google Scholar] [CrossRef]
- Kabary, D.M.; Helmy, M.W.; Elkhodairy, K.A.; Fang, J.Y.; Elzoghby, A.O. Hyaluronate/lactoferrin layer-by-layer-coated lipid nanocarriers for targeted co-delivery of rapamycin and berberine to lung carcinoma. Colloids Surf. B Biointerfaces 2018, 169, 183–194. [Google Scholar] [CrossRef]
- Li, H.; Teng, Y.; Xu, X.; Liu, J. Enhanced rapamycin delivery to hemangiomas by lipid polymer nanoparticles coupled with anti-VEGFR antibody. Int. J. Mol. Med. 2018, 41, 3586–3596. [Google Scholar] [CrossRef] [PubMed]
- Linares-Alba, M.A.; Gómez-Guajardo, M.B.; Fonzar, J.F.; Brooks, D.E.; García-Sánchez, G.A.; Bernad-Bernad, M.J. Preformulation studies of a liposomal formulation containing sirolimus for the treatment of dry eye disease. J. Ocul. Pharmacol. Ther. 2016, 32, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Lamming, D.W. Inhibition of the mechanistic target of rapamycin (mTOR)–Rapamycin and beyond. Cold Spring Harb. Perspect. Med. 2016, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mean Size (nm ± S.D.) | PDI ± S.D. | ζ Potential (mV ± S.D.) |
|---|---|---|---|
| Nano | 56.6 ± 22.4 | 0.25 ± 0.03 | −17.2 ± 2.8 |
| Rapa-Nano | 57.3 ± 23.2 | 0.24 ± 0.07 | −15.2 ± 3.0 |
| KP-Nano | 50.9 ± 18.8 | 0.26 ± 0.10 | −10.3 ± 3.5 |
| Rapa-KP-Nano | 43.8 ± 27.4 | 0.25 ± 0.05 | −19.2 ± 1.6 |
| Sample | C 1s | O 1s | N 1s | P 2p |
|---|---|---|---|---|
| KP-Nano | 68.45 | 26.21 | 5.20 | 0.14 |
| Nano | 69.70 | 26.40 | 3.90 | --- |
| Samples | TM/°C | |
|---|---|---|
| Raw materials | Rapa | 178.36 |
| PHEA-g-RhB-g-SUCC-PCL | 69.66 | |
| Nano | 54.73 | |
| Rapa-Nano | 54.71 | |
| Rapa-KP-Nano | 54.09 | |
| KP-Nano | 54.08 | |
| Sample | Mean Size | PDI ± S.D. | ζ Potential |
|---|---|---|---|
| (nm ± S.D.) | (mV ± S.D.) | ||
| Pre-drying | |||
| Nano | 86.3 ± 16.2 | 0.30 ± 0.06 | −19.1 ± 2.5 |
| Rapa-Nano | 97.6 ± 24.7 | 0.32 ± 0.12 | −18.7 ± 3.2 |
| KP-Nano | 92.6 ± 16.7 | 0.37 ± 0.08 | −10.6 ± 2.1 |
| Rapa-KP-Nano | 98.1 ± 15.0 | 0.31± 0.05 | −21.2 ± 3.3 |
| Post-freeze drying | |||
| Nano | 170.7 ± 12.2 | 0.33 ± 0.07 | −10.4 ± 0.8 |
| Rapa-Nano | 198.6 ±17.5 | 0.23 ± 0.11 | −20.4 ± 6.1 |
| KP-Nano | 188.1 ± 20.7 | 0.37 ± 0.04 | −10.9 ± 1.4 |
| Rapa-KP-Nano | 194.5 ± 21.3 | 0.32 ± 0.05 | −21.7 ± 4.9 |
| Post-spray drying | |||
| Nano | 139.3 ± 24.5 | 0.48 ± 0.12 | −12.6 ± 0.9 |
| Rapa-Nano | 161.1 ± 20.8 | 0.35 ± 0.09 | −8.1 ± 4.9 |
| KP-Nano | 178.0 ± 20.1 | 0.36 ± 0.06 | −14.6 ± 0.9 |
| Rapa-KP-Nano | 213.8 ± 19.6 | 0.40 ± 0.07 | −11.4 ± 6.0 |
| Mean Size (nm ± S.D.) (PDI ± S.D.) | |||
| t0 | t12months, −20 °C | t12months, 5 °C | |
| After freeze-drying | 194.5 ± 21.3 (0.32 ± 0.05) | 201.5 ± 28.5 (0.34 ± 0.11) | 220.3 ± 20.2 (0.37 ± 0.08) |
| After spray-drying | 213.8 ± 19.6 (0.40± 0.07) | 209.7 ± 23.6 (0.40 ± 0.08) | 217.7 ± 22.5 (0.41 ± 0.09) |
| ζ potential (mV) (±S.D.) | |||
| t0 | t12months, −20 °C | t12months, 5 °C | |
| After freeze-drying | −21.7 ± 2.3 | −18.6 ± 4.4 | −17.6 ± 4.1 |
| After spray-drying | −11.4 ± 6.0 | −15.7 ± 6.9 | −16.0 ± 3.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Craparo, E.F.; Cabibbo, M.; Conigliaro, A.; Barreca, M.M.; Musumeci, T.; Giammona, G.; Cavallaro, G. Rapamycin-Loaded Polymeric Nanoparticles as an Advanced Formulation for Macrophage Targeting in Atherosclerosis. Pharmaceutics 2021, 13, 503. https://doi.org/10.3390/pharmaceutics13040503
Craparo EF, Cabibbo M, Conigliaro A, Barreca MM, Musumeci T, Giammona G, Cavallaro G. Rapamycin-Loaded Polymeric Nanoparticles as an Advanced Formulation for Macrophage Targeting in Atherosclerosis. Pharmaceutics. 2021; 13(4):503. https://doi.org/10.3390/pharmaceutics13040503
Chicago/Turabian StyleCraparo, Emanuela Fabiola, Marta Cabibbo, Alice Conigliaro, Maria Magdalena Barreca, Teresa Musumeci, Gaetano Giammona, and Gennara Cavallaro. 2021. "Rapamycin-Loaded Polymeric Nanoparticles as an Advanced Formulation for Macrophage Targeting in Atherosclerosis" Pharmaceutics 13, no. 4: 503. https://doi.org/10.3390/pharmaceutics13040503