
Phosphatidylinositol Stabilizes Fluid-Phase Liposomes Loaded with a Melphalan Lipophilic Prodrug

 , , , ,
, , , ,  and
and
Abstract
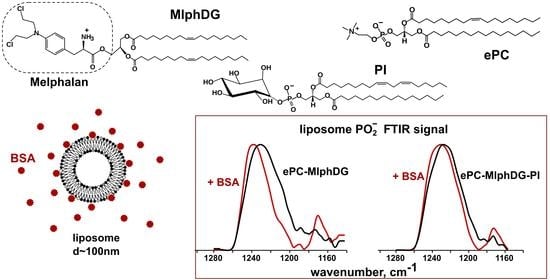
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Preparation of Liposomes
2.3. Hydrodynamic Diameter and Zeta Potential Measurements
2.4. ATR-FTIR Spectroscopy
2.5. Fluorescence Measurements
2.6. Asymmetrical Flow Field-Flow Fractionation (AF4)
2.6.1. Size Determination
2.6.2. Batch Analysis (Determination of Extinction Coefficient)
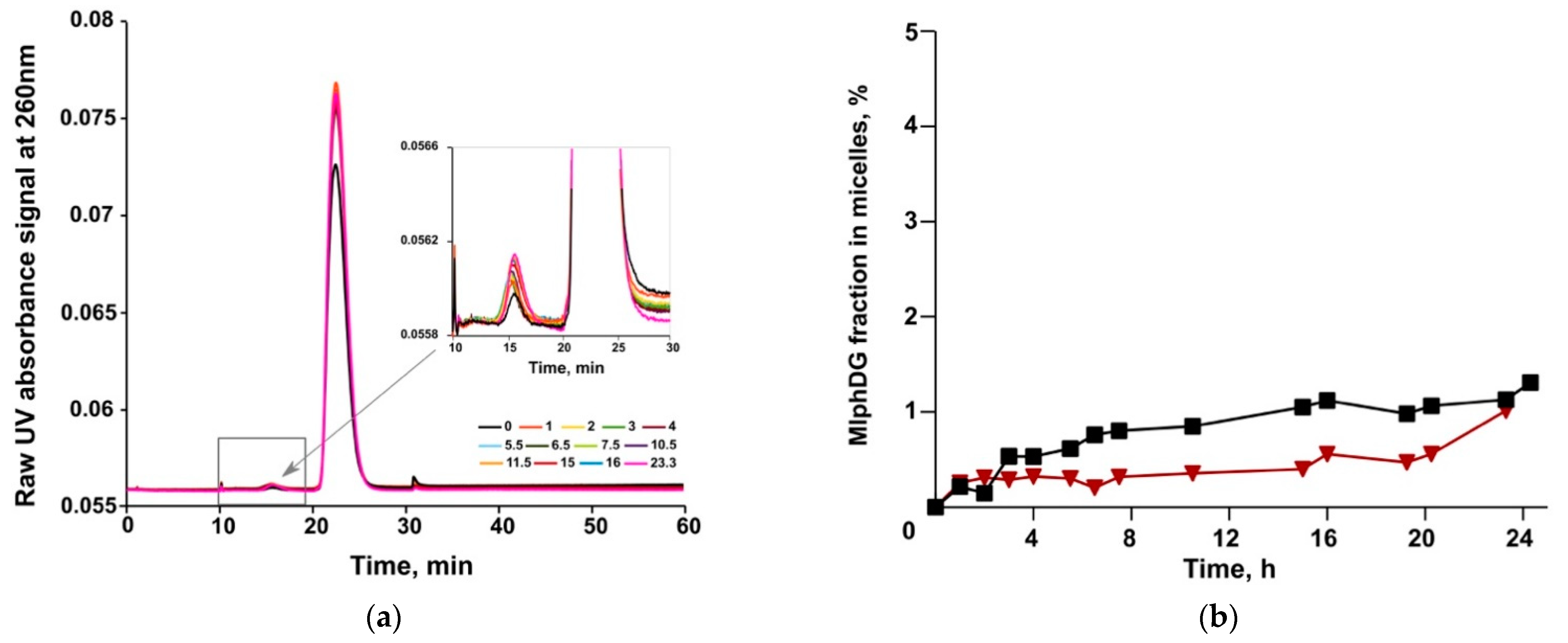
2.6.3. MlphDG Release
3. Results and Discussion
3.1. Characteristics of the Liposomes
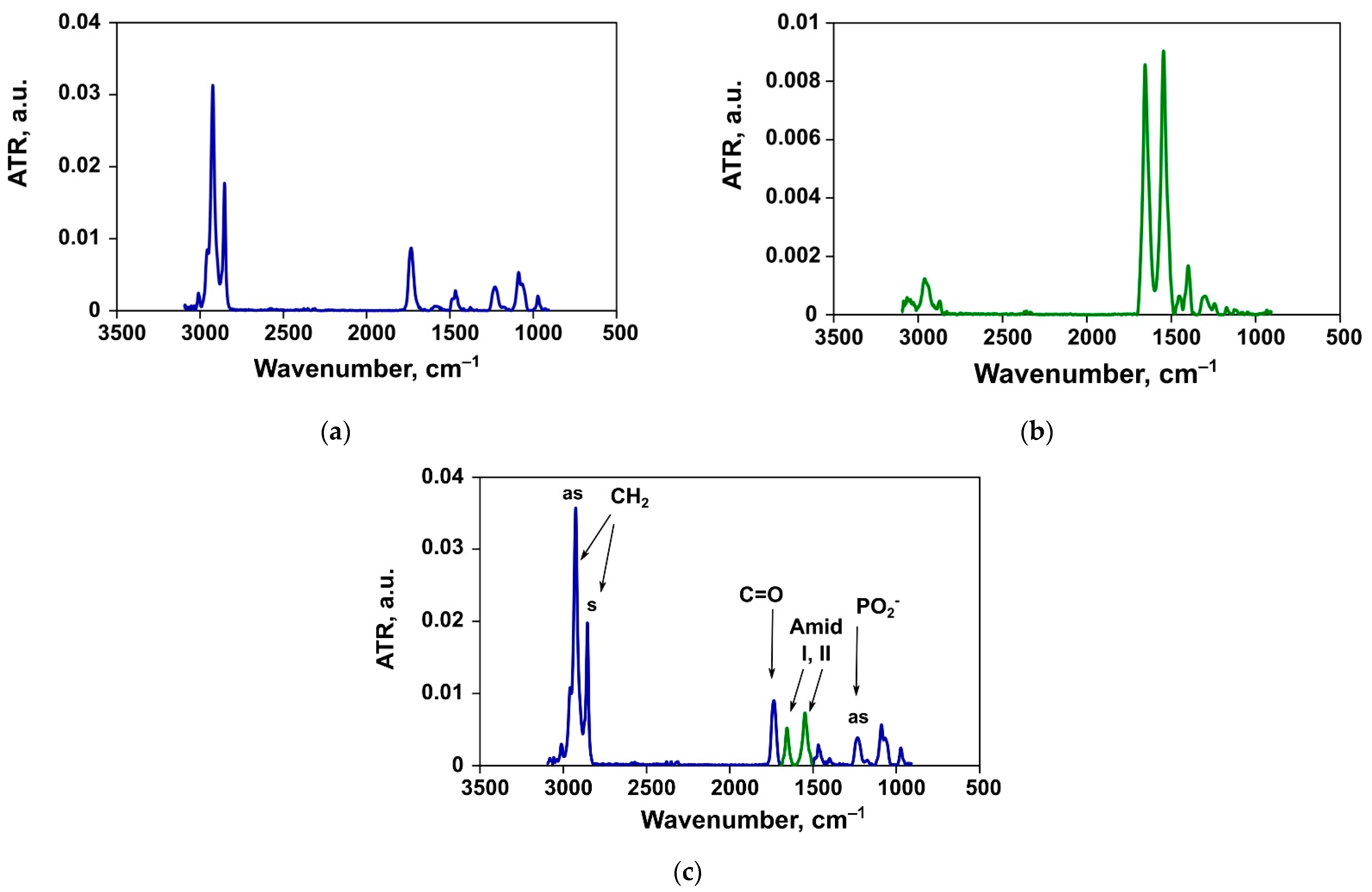
3.2. ATR-FTIR Spectroscopy
3.2.1. Changes in the Structure of Lipid Bilayer
Asymmetric Stretching Vibrations of Phosphate Groups
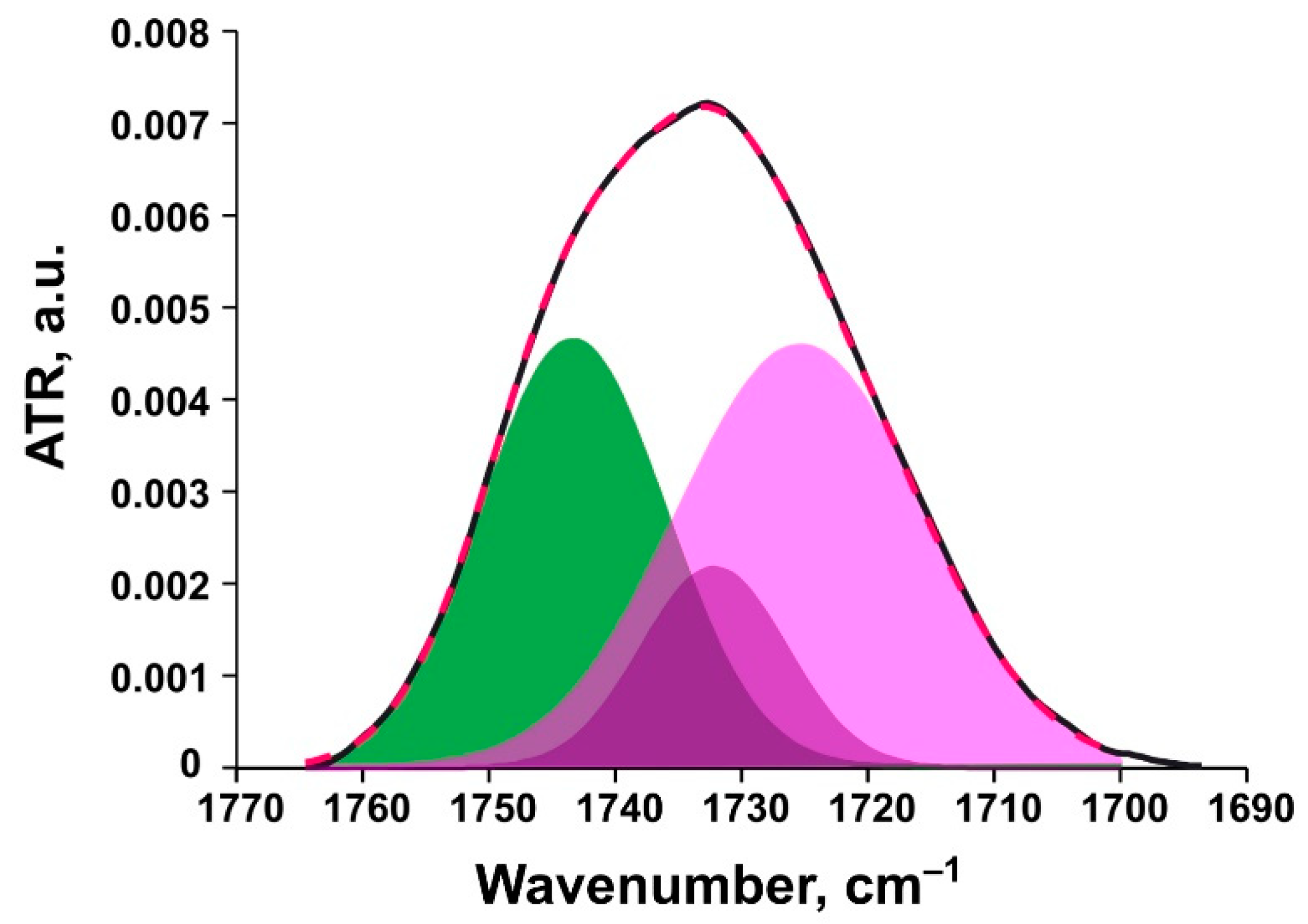
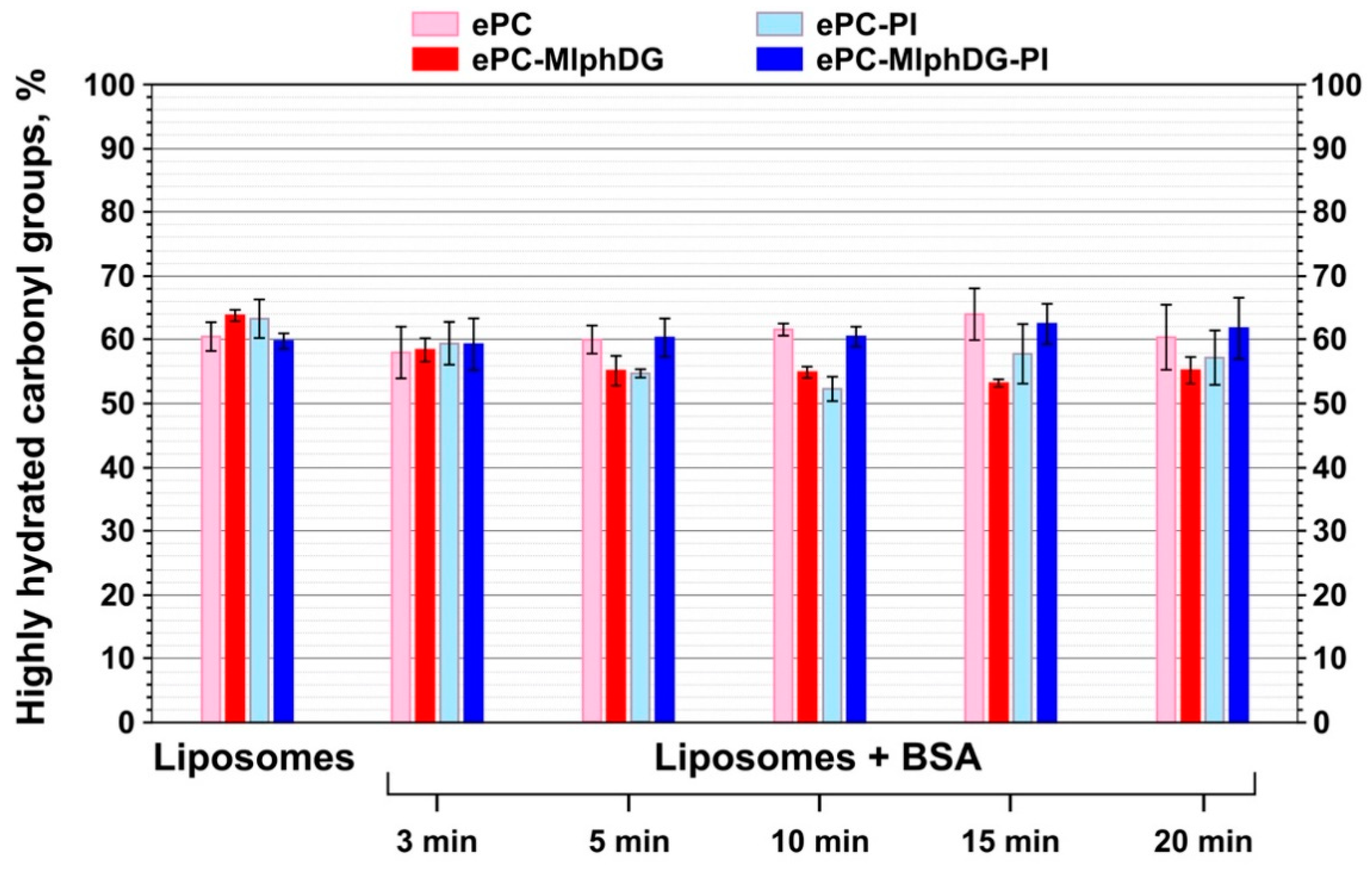
Ester Carbonyl Stretching Bands
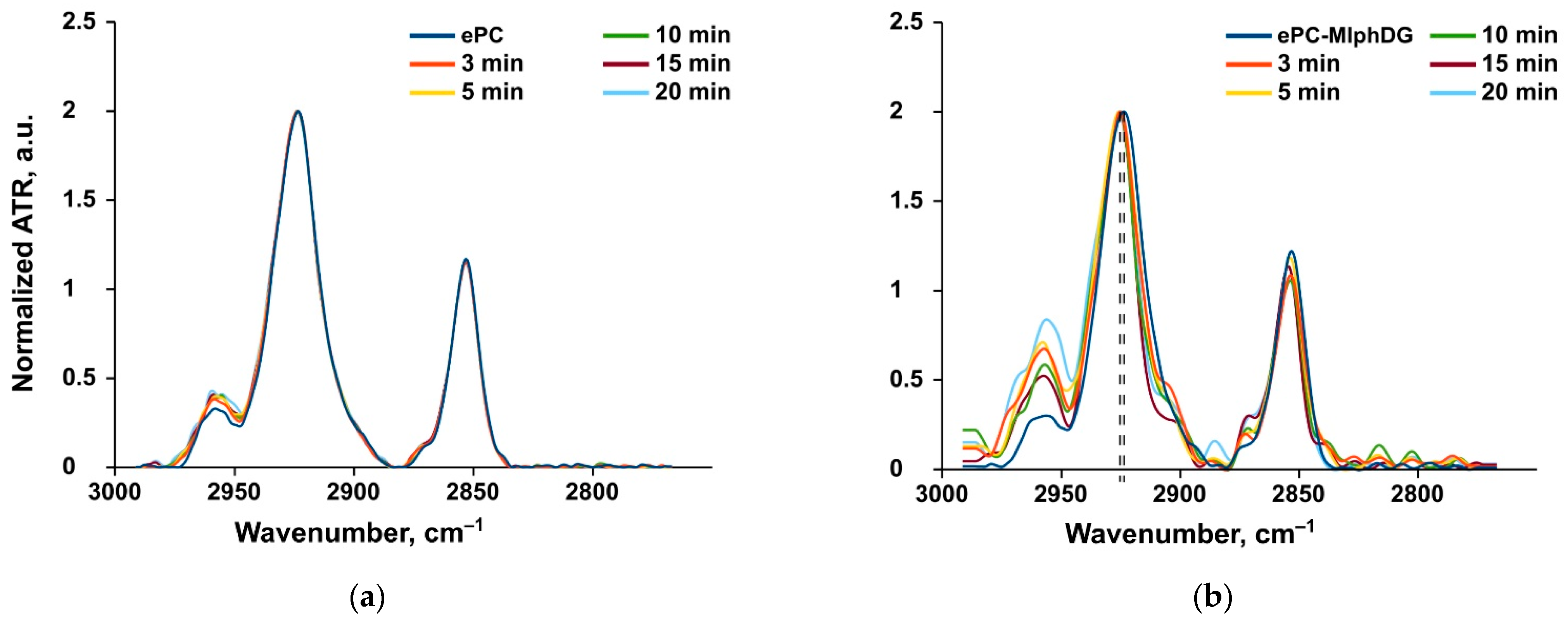
Methylene Stretching Vibrations
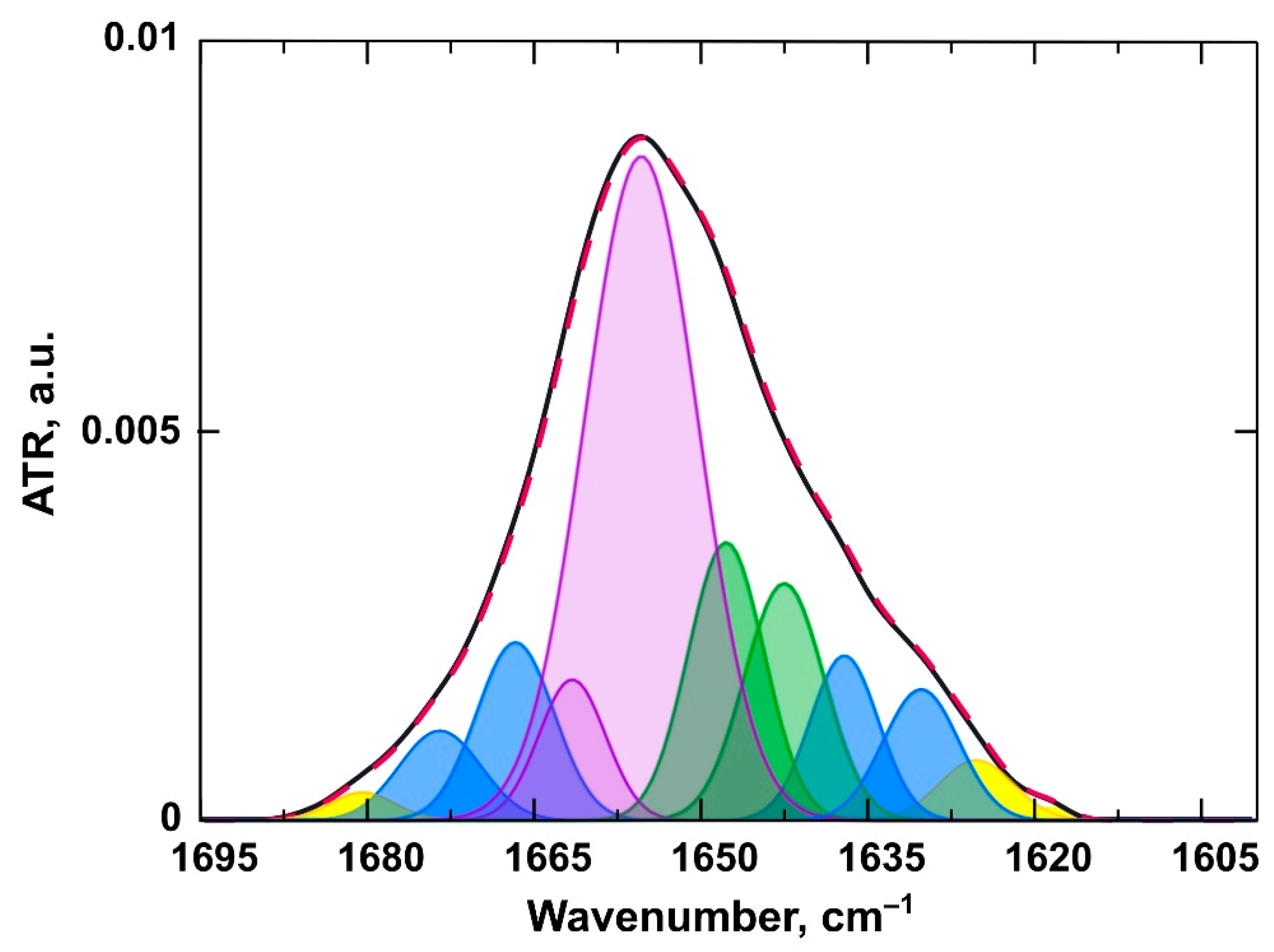
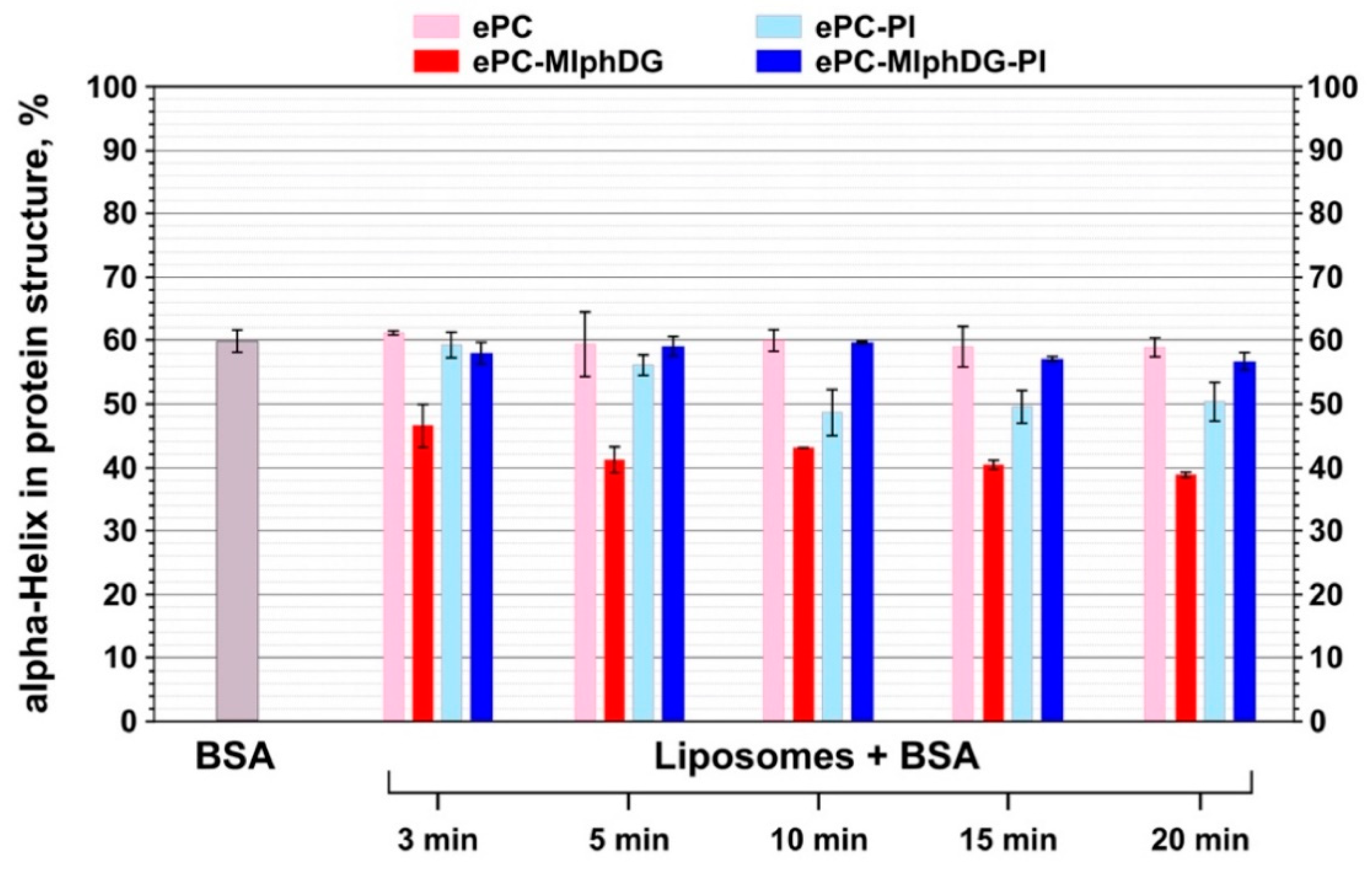
3.2.2. Changes in the Structure of the Protein
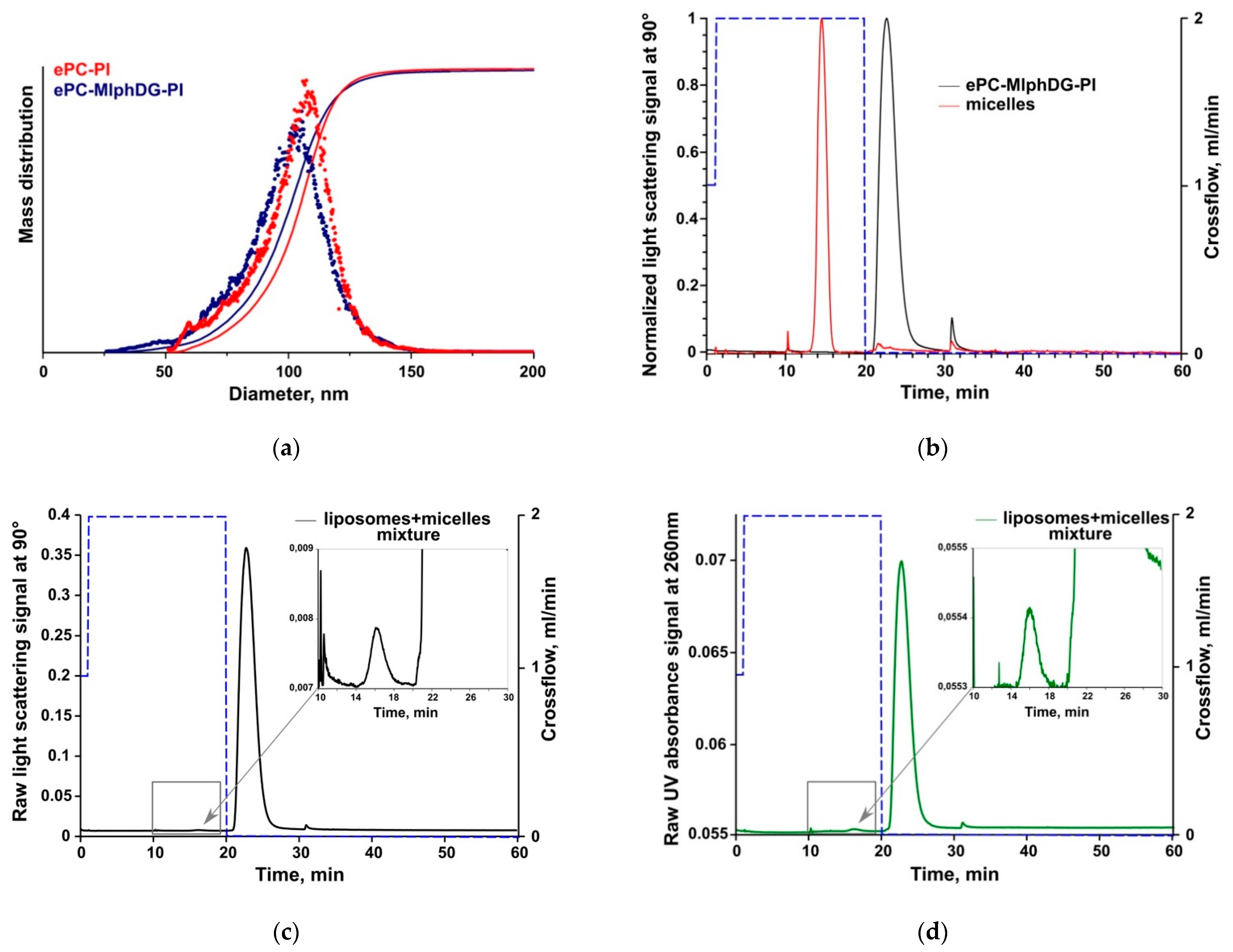
3.3. Asymmetrical Flow-Field-Flow Fractionation (AF4)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Barenholz, Y. Doxil—The first FDA approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat. Nanotechn. 2013, 8, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Bros, M.; Nuhn, L.; Simon, J.; Moll, L.; Mailänder, V.; Landfester, K.; Grabbe, S. The protein corona as a confounding variable of nanoparticle-mediated targeted vaccine delivery. Front. Immunol. 2018, 9, 1760. [Google Scholar] [CrossRef]
- Caracciolo, G. Liposome-protein corona in a physiological environment: Challenges and opportunities for targeted delivery of nanomedicines. Nanomedicine 2015, 11, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Digiacomo, L.; Pozzi, D.; Palchetti, S.; Zingoni, A.; Caracciolo, G. Impact of the protein corona on nanomaterial immune response and targeting ability. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Betker, J.L.; Jones, D.; Childs, C.R.; Helm, K.M.; Terrell, K.; Nagel, M.A.; Anchordoquy, T.J. Nanoparticle uptake by circulating leukocytes: A major barrier to tumor delivery. J. Control. Release 2018, 286, 85–93. [Google Scholar] [CrossRef]
- Milla, P.; Dosio, F.; Cattel, L. PEGylation of proteins and liposomes: A powerful and flexible strategy to improve the drug delivery. Curr. Drug. Metab. 2012, 13, 105–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trucillo, P.; Reverchon, E. Production of PEG-coated liposomes using a continuous supercritical assisted process. J. Supercrit. Fluids 2021, 167, 105048. [Google Scholar] [CrossRef]
- Szebeni, J.; Muggia, F.; Gabizon, A.; Barenholz, Y. Activation of complement by therapeutic liposomes and other lipid excipient-based therapeutic products: Prediction and prevention. Adv. Drug Deliv. Rev. 2011, 63, 1020–1030. [Google Scholar] [CrossRef]
- Ilinskaya, A.N.; Dobrovolskaia, M.A. Understanding the immunogenicity and antigenicity of nanomaterials: Past, present and future. Toxicol. Appl. Pharmacol. 2016, 299, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Anchordoquy, T.J.; Simberg, D. Watching the gorilla and questioning delivery dogma. J. Control. Release 2017, 262, 87–90. [Google Scholar] [CrossRef]
- Kuznetsova, N.; Kandyba, A.; Vostrov, I.; Kadykov, V.; Gaenko, G.; Molotkovsky, J.; Vodovozova, E. Liposomes loaded with lipophilic prodrugs of methotrexate and melphalan as convenient drug delivery vehicles. J. Drug Deliv. Sci. Technol. 2009, 19, 51–59. [Google Scholar] [CrossRef]
- Musto, P.; D’Auria, F. Melphalan: Old and new uses of a still master drug for multiple myeloma. Expert Opin. Investig. Drugs 2007, 16, 1467–1487. [Google Scholar] [CrossRef]
- Chemocare. Available online: Chemocare.com/chemotherapy/drug-info/melphalan.aspx (accessed on 24 February 2021).
- Sims, L.B.; Tyo, K.M.; Stocke, S.; Mahmoud, M.Y.; Ramasubramanian, A.; Steinbach-Rankins, J.M. Surface-modified melphalan nanoparticles for intravitreal chemotherapy of retinoblastoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1696–1705. [Google Scholar] [CrossRef] [PubMed]
- Wen, A.; Mei, X.; Feng, C.; Shen, C.; Wange, B.; Zhang, X. Electrosprayed nanoparticles of poly(p-dioxanone-co-melphalan) macromolecular prodrugs for treatment of xenograft ovarian carcinoma. Mater. Sci. Eng. C 2020, 111, 110759. [Google Scholar] [CrossRef] [PubMed]
- Tabatabaei, S.N.; Derbalic, R.M.; Yang, C.; Superstein, R.; Hamel, P.; Chain, J.L.; Hardy, P. Co-delivery of miR-181a and melphalan by lipid nanoparticles for treatment of seeded retinoblastoma. J. Control. Release 2019, 298, 177–185. [Google Scholar] [CrossRef]
- Rudhrabatla, V.P.; Sudhakar, B.; Reddy, K.S. In vitro and in vivo assessment of designed melphalan loaded stealth solid lipid nanoparticles for parenteral delivery. BioNano Sci. 2020, 10, 168–190. [Google Scholar] [CrossRef]
- Zucker, D.; Marcus, D.; Barenholz, Y.; Goldblum, A. Liposome drugs’ loading efficiency: A working model based on loading conditions and drug’s physicochemical properties. J. Control. Release 2009, 139, 73–80. [Google Scholar] [CrossRef]
- Mura, S.; Bui, D.T.; Couvreur, P.; Nicolas, J. Lipid prodrug nanocarriers in cancer therapy. J. Control. Release 2015, 208, 25–41. [Google Scholar] [CrossRef]
- Signorell, R.D.; Luciani, P.; Brambilla, D.; Leroux, J.-C. Pharmacokinetics of lipid-drug conjugates loaded into liposomes. Eur. J. Pharm. Biopharm. 2018, 128, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, P.J.; Christensen, M.S.; Ruysschaert, T.; Linderoth, L.; Andresen, T.L.; Melander, F.; Mouritsen, O.G.; Madsen, R.; Clausen, M.H. Synthesis and biophysical characterization of chlorambucil anticancer ether lipid prodrugs. J. Med. Chem. 2009, 52, 3408–3415. [Google Scholar] [CrossRef]
- Gabizon, A.; Papahadjopoulos, D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc. Natl. Acad. Sci. USA 1988, 85, 6949–6953. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.M.; Hansen, C.; Rutledge, J. Liposomes with prolonged circulation times: Factors affecting uptake by reticuloendothelial and other tissues. Biochim. Biophys. Acta 1989, 981, 27–35. [Google Scholar] [CrossRef]
- Kuznetsova, N.R.; Sevrin, C.; Lespineux, D.; Bovin, N.V.; Vodovozova, E.L.; Mészáros, T.; Szebeni, J.; Grandfils, C. Hemocompatibility of liposomes loaded with lipophilic prodrugs of methotrexate and melphalan in the lipid bilayer. J. Control. Release 2012, 160, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, N.R.; Stepanova, E.V.; Peretolchina, N.M.; Khochenkov, D.A.; Boldyrev, I.A.; Bovin, N.V.; Vodovozova, E.L. Targeting liposomes loaded with melphalan prodrug to tumour vasculature via the sialyl Lewis X selectin ligand. J. Drug Target. 2014, 22, 242–250. [Google Scholar] [CrossRef]
- Tretiakova, D.; Svirshchevskaya, E.; Onishchenko, N.; Alekseeva, A.; Boldyrev, I.; Kamyshinsky, R.; Natykan, A.; Lokhmotov, A.; Arantseva, D.; Shobolov, D.; et al. Liposomal formulation of a melphalan lipophilic prodrug: Studies of acute toxicity, tolerability, and antitumor efficacy. Curr. Drug Deliv. 2020, 17, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Tretiakova, D.; Onishchenko, N.; Boldyrev, I.; Mikhalyov, I.; Tuzikov, A.; Bovin, N.; Evtushenko, E.; Vodovozova, E. Influence of stabilizing components on the integrity of antitumor liposomes loaded with lipophilic prodrug in the bilayer. Colloids Surf. B Biointerfaces 2018, 166, 45–53. [Google Scholar] [CrossRef]
- Garbuzenko, O.; Barenholz, Y.; Priev, A. Effect of grafted PEG on liposome size and on compressibility and packing of lipid bilayer. Chem. Phys. Lipids 2005, 135, 117–129. [Google Scholar] [CrossRef]
- Charbonneau, D.; Beauregard, M.; Tajmir-Riahi, H.A. Structural analysis of human serum albumin complexes with cationic lipids. J. Phys. Chem. B 2009, 113, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Sabín, J.; Prieto, G.; Ruso, J.M.; Messina, P.V.; Salgado, F.J.; Nogueira, M.; Costas, M.; Sarmiento, F. Interactions between DMPC liposomes and the serum blood proteins HSA and IgG. J. Phys. Chem. B 2009, 113, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Galántai, R.; Bárdos-Nagy, I.; Módos, K.; Kardos, J.; Závodszky, P.; Fidy, J. Serum albumin-lipid membrane interaction influencing the uptake of porphyrins. Arch. Biochem. Biophys. 2000, 373, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.; Das, A.; Chakraborty, A. Interaction of human serum albumin with liposomes of saturated and unsaturated lipids with different phase transition temperatures: A spectroscopic investigation by membrane probe PRODAN. RSC Adv. 2014, 4, 14335–14347. [Google Scholar] [CrossRef]
- Tatulian, S.A. Structural characterization of membrane proteins and peptides by FTIR and ATR-FTIR spectroscopy. In Lipid-Protein Interactions: Methods and Protocols, Methods in Molecular Biology; Kleinschmidt, J.H., Ed.; Humana Press: Totowa, NJ, USA, 2013; Volume 974, pp. 177–216. [Google Scholar] [CrossRef]
- Yohannes, G.; Jussila, M.; Hartonen, K.; Riekkola, M.L. Asymmetrical flow field-flow fractionation technique for separation and characterization of biopolymers and bioparticles. J. Chromatogr. A 2011, 1218, 4104–4116. [Google Scholar] [CrossRef]
- Hinna, A.H.; Hupfeld, S.; Kuntsche, J.; Bauer-Brandl, A.; Brandl, M. Mechanism and kinetics of the loss of poorly soluble drugs from liposomal carriers studied by a novel flow field-flow fractionation-based drug release-/transfer-assay. J. Control. Release 2016, 232, 228–237. [Google Scholar] [CrossRef]
- Alekseeva, A.S.; Tretiakova, D.S.; Melnikova, D.N.; Molotkovsky, U.G.; Boldyrev, I.A. Novel fluorescent membrane probe 2,3,5,6-bis(cyclohexyl)-bodipy-labeled phosphatidylcholine. Russ. J. Bioorg. Chem. 2016, 42, 305–309. [Google Scholar] [CrossRef]
- Stewart, J.C.M. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem. 1980, 104, 10–14. [Google Scholar] [CrossRef]
- Barbero, N.; Barolo, C.; Viscardi, G. Bovine serum albumin conjugation with FITC. World J. Chem. Educ. 2016, 4, 80–85. [Google Scholar] [CrossRef]
- Kuntsche, J.; Decker, C.; Fahr, A. Analysis of liposomes using asymmetrical flow field-flow fractionation: Separation conditions and drug/lipid recovery. J. Sep. Sci. 2012, 35, 1993–2001. [Google Scholar] [CrossRef] [PubMed]
- Hinna, A.; Steiniger, F.; Hupfeld, S.; Brandl, M.; Kuntsche, J. Asymmetrical flow field-flow fractionation with on-line detection for drug transfer studies: A feasibility study. Anal. Bioanal. Chem. 2014, 406, 7827–7839. [Google Scholar] [CrossRef]
- Hinna, A.H.; Hupfeld, S.; Kuntsche, J.; Brandl, M. The use of asymmetrical flow field-flow fractionation with on-line detection in the study of drug retention within liposomal nanocarriers and drug transfer kinetics. J. Pharm. Biomed. Anal. 2016, 124, 157–163. [Google Scholar] [CrossRef]
- Muller, M.; Zschornig, O.; Ohki, S.; Arnold, K. Fusion, leakage and surface hydrophobicity of vesicles containing phosphoinositides: Influence of steric and electrostatic effects. J. Membr. Biol. 2003, 192, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Tretiakova, D.S.; Alekseeva, A.S.; Galimzyanov, T.R.; Boldyrev, A.M.; Chernyadyev, A.Y.; Ermakov, Y.A.; Batishchev, O.V.; Vodovozova, E.L.; Boldyrev, I.A. Lateral stress profile and fluorescent lipid probes. FRET pair of probes that introduces minimal distortions into lipid packing. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Deygen, I.M.; Seidl, C.; Kölmel, D.; Bednarek, C.; Heissler, S.; Kudryashova, E.V.; Bräse, S.; Schepers, U. Novel prodrug of doxorubicin modified by stearoylspermine encapsulated into PEG-chitosan stabilized liposomes. Langmuir 2016, 32, 10861–10869. [Google Scholar] [CrossRef]
- Le-Deygen, I.M.; Skuredina, A.A.; Safronova, A.S.; Yakimov, I.D.; Kolmogorov, I.M.; Deygen, D.M.; Burova, T.V.; Grinberg, N.V.; Grinberg, V.Y.; Kudryashova, E.V. Moxifloxacin interacts with lipid bilayer, causing dramatic changes in its structure and phase transitions. Chem. Phys. Lipids 2020, 228, 104891. [Google Scholar] [CrossRef]
- Deygen, I.M.; Kudryashova, E.V. New versatile approach for analysis of PEG content in conjugates and complexes with biomacromolecules based on FTIR spectroscopy. Colloids Surf. B Biointerfaces 2016, 141, 36–43. [Google Scholar] [CrossRef]
- Lewis, R.N.; McElhaney, R.N. Studies of mixed chain diacyl phosphatidylcholines with highly asymmetric acyl chains: A Fourier transform infrared spectroscopic study of interfacial hydration and hydrocarbon chain packing in the mixed interdigitated gel phase. Biophys. J. 1993, 65, 1866–1877. [Google Scholar] [CrossRef] [Green Version]
- Blume, A.; Hübner, W.; Messner, G. Fourier transform infrared spectroscopy of 13C=O-labeled phospholipids hydrogen bonding to carbonyl groups. Biochemistry 1988, 27, 8239–8249. [Google Scholar] [CrossRef] [PubMed]
- Mantsch, H.H.; McElhaney, R.N. Phospholipid phase transitions in model and biological membranes as studied by infrared spectroscopy. Chem. Phys. Lipids 1991, 57, 213–226. [Google Scholar] [CrossRef]
- Tatulian, S.A. Attenuated total reflection Fourier transform infrared spectroscopy: A method of choice for studying membrane proteins and lipids. Biochemistry 2003, 42, 11898–11907. [Google Scholar] [CrossRef] [PubMed]
- Cieślik-Boczula, K.; Koll, A. The effect of 3-pentadecylphenol on DPPC Bilayers ATR-IR and 31P NMR Studies. Biophys. Chem. 2009, 140, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Yang, S.; Kong, J.; Dong, A.; Yu, S. Obtaining information about protein secondary structures in aqueous solution using Fourier transform IR spectroscopy. Nat. Protoc. 2015, 10, 382–396. [Google Scholar] [CrossRef]
- Usoltsev, D.; Sitnikova, V.; Kajava, A.; Uspenskaya, M. Systematic FTIR spectroscopy study of the secondary structure changes in human serum albumin under various denaturation conditions. Biomolecules 2019, 9, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Schubert, D.; Sawaya, M.R.; Eisenberg, D.; Riek, R. Multidimensional structure-activity relationship of a protein in its aggregated states. Angew. Chem. Int. Ed. Engl. 2010, 49, 3904–3908. [Google Scholar] [CrossRef]
- Abrosimova, K.V.; Shulenina, O.V.; Paston, S.V. FTIR study of secondary structure of bovine serum albumin and ovalbumin. J. Phys. Conf. Ser. 2016, 769, 012016. Available online: https://iopscience.iop.org/article/10.1088/1742-6596/769/1/012016 (accessed on 24 February 2021). [CrossRef]
- Fleischer, C.C.; Payne, C.K. Secondary structure of corona proteins determines the cell surface receptors used by nanoparticles. J. Phys. Chem. B 2014, 118, 14017–14026. [Google Scholar] [CrossRef]
- Schnitzer, J.E. High affinity binding, endocytosis, and degradation of conformationally modified albumins. Potential role of gp30 and gp18 as novel scavenger receptors. J. Biol. Chem. 1993, 268, 7562–7570. [Google Scholar] [CrossRef]
- Johnsson, M.; Hansson, P.; Edwards, K. Spherical micelles and other self-assembled structures in dilute aqueous mixtures of poly(ethylene glycol) lipids. J. Phys. Chem. B 2001, 105, 8420–8430. [Google Scholar] [CrossRef]
- Washington, C. Drug release from microdisperse systems: A critical review. Int. J. Pharm. 1990, 58, 1–12. [Google Scholar] [CrossRef]
- Loew, S.; Fahr, A.; May, S. Modeling the release kinetics of poorly water-soluble drug molecules from liposomal nanocarriers. J. Drug Del. 2011, 2011, 376548. [Google Scholar] [CrossRef] [Green Version]
- Kenworthy, A.K.; Hristova, K.; Needham, D.; McIntosh, T.J. Range and magnitude of the steric pressure between bilayers containing phospholipids with covalently attached poly(ethylene glycol). Biophys. J. 1995, 68, 1921–1936. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode | Duration, min | Cross/Focus Flow Start, mL/min | Cross/Focus Flow End, mL/min |
|---|---|---|---|
| Elution | 1 | 1 | 1 |
| Focus | 1 | 2 | 2 |
| Focus and inject | 5 | 2 | 2 |
| Focus | 3 | 2 | 2 |
| Elution | 10 | 2 | 2 |
| Elution | 35 | 0 | 0 |
| Elution and inject | 5 | 0 | 0 |
| Sample Composition, by mol | Z-Average Size, nm 1 | PdI Width, nm (Average) | PdI | Zeta-Potential, mV 2 |
|---|---|---|---|---|
| ePC | 121.3 ± 2.4 | 35.2 ± 1.1 | 0.084 ± 0.003 | −1.4 ± 1.6 |
| ePC–MlphDG, 9:1 | 117.3 ± 2.2 | 33.1 ± 1.1 | 0.079 ± 0.003 | +18.4 ± 1.9 |
| ePC–PI, 9:1 | 104.4 ± 1.8 | 28.5 ± 2.6 | 0.068 ± 0.016 | −48.8 ± 2.6 |
| ePC–MlphDG–PI, 8:1:1 | 121.3 ± 2.3 | 34.8 ± 1.1 | 0.082 ± 0.003 | −25.3 ± 1.9 |
| Liposome Composition | Before Incubation | 5-Min Incubation |
|---|---|---|
| ePC | 1230.4 | 1230.9 |
| ePC–MlphDG, 9:1 | 1231.3 | 1238.6 |
| ePC–PI, 9:1 | 1229.9 | 1233.3 |
| ePC–MlphDG–PI, 8:1:1 | 1228.4 | 1230.9 |
| Liposome Composition | Before Incubation | 5-Min Incubation | ||
|---|---|---|---|---|
| νas | νsim | νas | νsim | |
| ePC | 2923.6 | 2853.2 | 2923.6 | 2853.2 |
| ePC–MlphDG, 9:1 | 2923.7 | 2853.2 | 2925.5 | 2853.7 |
| ePC–PI, 9:1 | 2924.1 | 2853.2 | 2924.5 | 2853.2 |
| ePC–MlphDG–PI, 8:1:1 | 2924.1 | 2853.2 | 2924.1 | 2853.2 |
| Secondary Structure | Range, cm−1 |
|---|---|
| α-helix | 1650–1660 |
| β-sheet | 1628–1639 |
| β-turn | 1664–1687 |
| random coil | 1640–1649 |
| Intermolecular β-sheet (aggregates) | 1618–1626, 1688–1696 |
| Sample | DLS | AF4 | ||||
|---|---|---|---|---|---|---|
| Dh, nm | PdI | D10, nm | D50, nm | D90, nm | Dmean, nm | |
| ePC–PI, 9:1 | 104 ± 2 | 0.091 ± 0.025 | 78.6 * | 104.4 * | 120.6 * | 106.4 * |
| ePC–MlphDG–PI, 8:1:1 | 95 ± 2 | 0.132 ± 0.011 | 75.2 ± 1.3 | 100.5 ± 0.3 | 120.7 ± 0.5 | 104.1 ± 0.5 |
| DSPE-PEG (micelles) | 15 ± 1 | 0.310 ± 0.148 | n.a. | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tretiakova, D.; Le-Deigen, I.; Onishchenko, N.; Kuntsche, J.; Kudryashova, E.; Vodovozova, E. Phosphatidylinositol Stabilizes Fluid-Phase Liposomes Loaded with a Melphalan Lipophilic Prodrug. Pharmaceutics 2021, 13, 473. https://doi.org/10.3390/pharmaceutics13040473
Tretiakova D, Le-Deigen I, Onishchenko N, Kuntsche J, Kudryashova E, Vodovozova E. Phosphatidylinositol Stabilizes Fluid-Phase Liposomes Loaded with a Melphalan Lipophilic Prodrug. Pharmaceutics. 2021; 13(4):473. https://doi.org/10.3390/pharmaceutics13040473
Chicago/Turabian StyleTretiakova, Daria, Irina Le-Deigen, Natalia Onishchenko, Judith Kuntsche, Elena Kudryashova, and Elena Vodovozova. 2021. "Phosphatidylinositol Stabilizes Fluid-Phase Liposomes Loaded with a Melphalan Lipophilic Prodrug" Pharmaceutics 13, no. 4: 473. https://doi.org/10.3390/pharmaceutics13040473