
Delivery Systems for Nucleic Acids and Proteins: Barriers, Cell Capture Pathways and Nanocarriers



Abstract
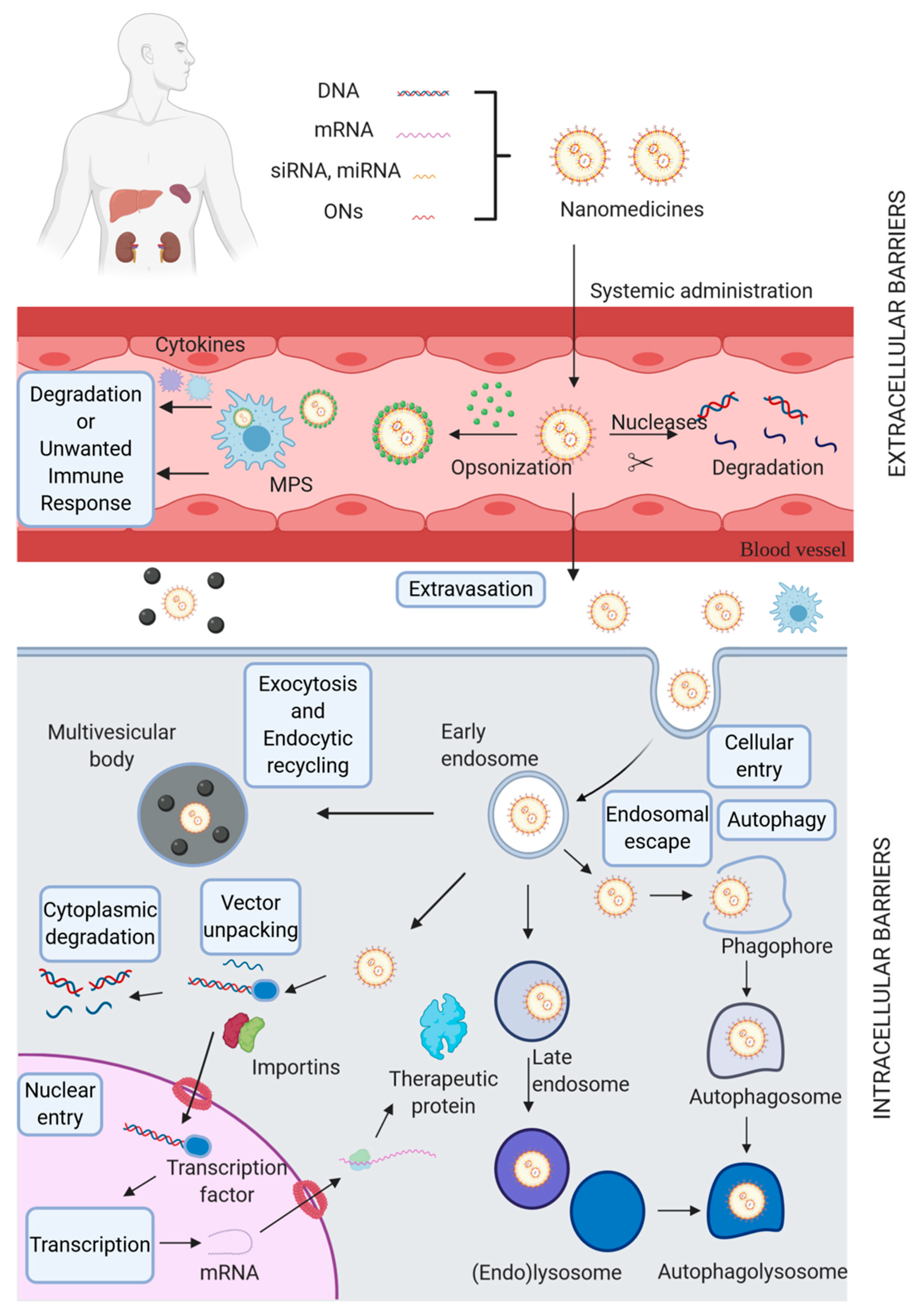
:1. Introduction
2. Biological Barriers
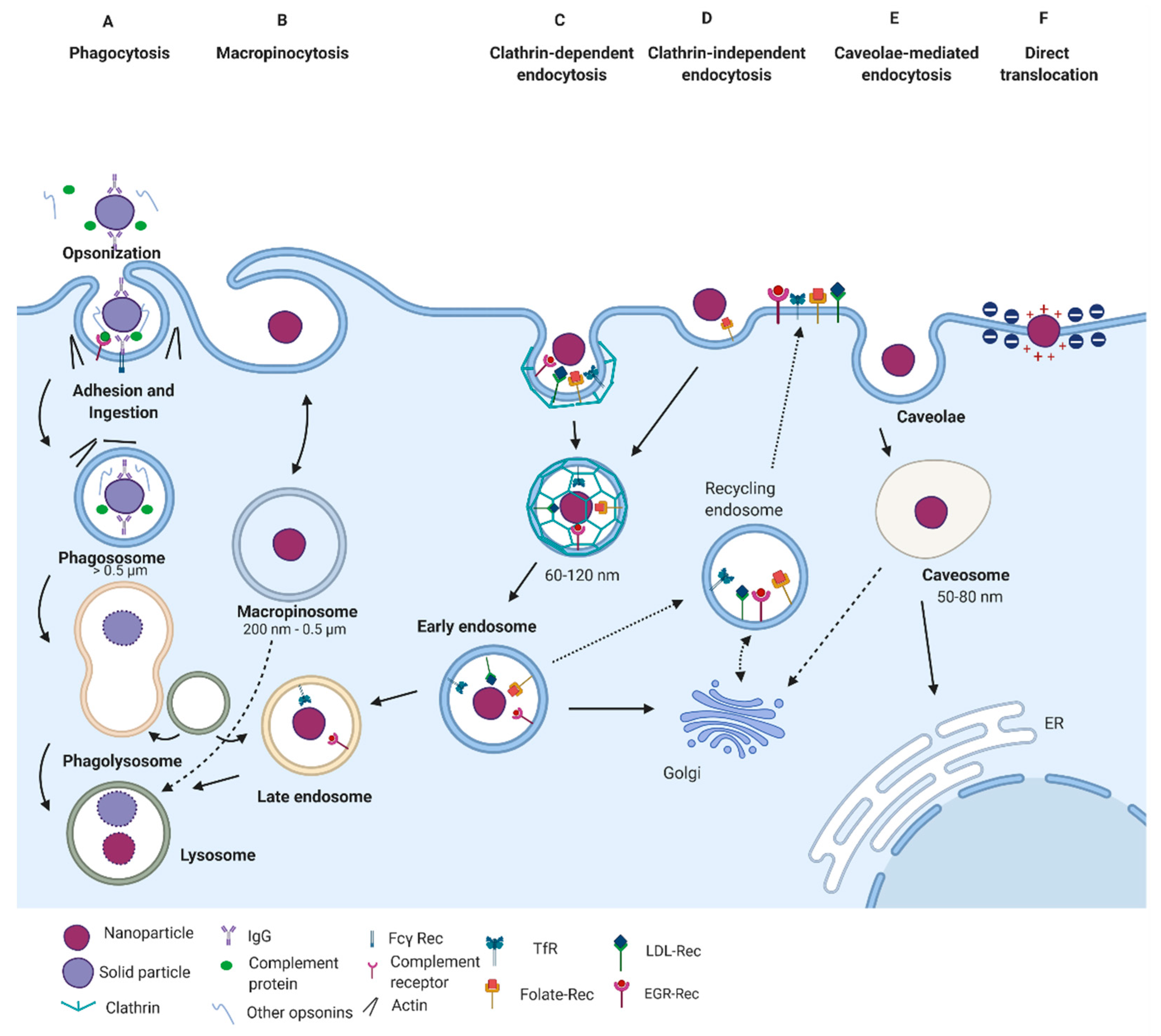
3. Internalization Pathways
3.1. Phagocytosis
3.2. Endocytic Pathways
3.2.1. Clathrin-Mediated Endocytosis
3.2.2. Caveolae-Mediated Endocytosis
3.2.3. Clathrin- and Caveolin-Independent Endocytosis
3.3. Macropinocytosis
3.4. Direct Translocation
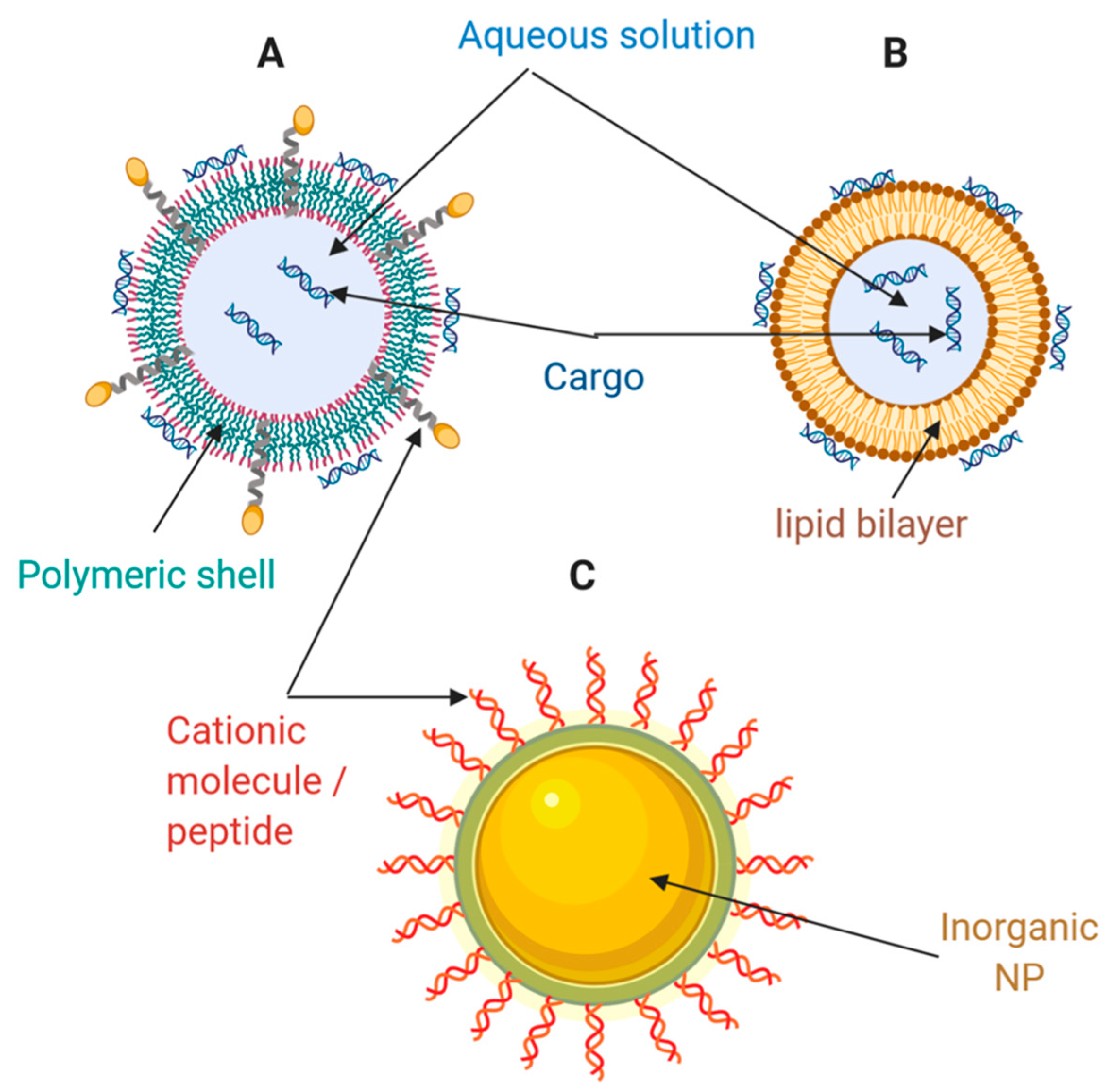
4. Nanocarriers for the Delivery of Nucleic Acids and Proteins
4.1. Lipid-Based Nanocarriers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Chemical Name | Findings/Relevant Data | Reference |
|---|---|---|---|
| DLin-MC3-DMA | 6Z,9Z,28Z,31Z-heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino)-butanoate | Used for the first time in Patisiran (liposome formulation). | [90] |
| DLin-KC2-DMA | 1,2-dilinoleyl-4-(2-dimethylaminoethyl)-1,3-dioxolane | Demonstrated to have in vivo activity at siRNA doses as low as 0.01 mg/kg in rodents and 0.1 mg/kg in nonhuman primates. | [99] |
| L319 | di((Z)-non-2-en-1-yl)-9-((4-(dimethylamino)butanoyl)oxy)heptadecanedioate)-9-((4-(dimethylamino)butanoyl)oxy)heptadecanedioate | Biodegradable lipid displaying rapid elimination from plasma and tissues, substantially improved tolerability in preclinical studies. | [100] |
| C12-200 | - | Over 95% silencing at a dose of 0.03 mg/kg in non-human primates and 0.01 mg/kg in mice. | [101] |
| cKK-E12 | - | Over 95% silencing at a dose of 0.3 mg/kg in nonhuman primates. Toxicity studies showed that cKK-E12 was well tolerated in rats at a dose of 1 mg/kg. | [102] |
4.2. Polymeric-Based Nanocarriers
| Abbreviation | Chemical Name | Findings/Relevant Properties | Reference |
|---|---|---|---|
| PEI | Polyethylenimine | The most widely used. It is the organic macromolecule with the highest cationic-charge-density potential. | [90,111] |
| pDMAEMA | Poly(2-dimethylamino)ethyl methacrylate | Extensively studied and widely used for the delivery of DNA, siRNA, mRNA and miRNA. It had tertiary amines in its structure. | [126] |
| hDD90-118 | - | An hyperbranched poly(beta amino ester) capable of save and effective delivering of mRNA to lung epithelium. | [127] |
| N5 | - | An assembly of poly A binding proteins and cationic polypeptides for enhanced mRNA delivery. | [128] |
| PAA8k-(2-3-2) | - | A poly(acrylic acid) scaffold grafted with oligoalkylamines promoting enhanced mRNA delivery. | [129] |
4.3. Inorganic Nanomaterials
4.3.1. Mesoporous Silica
4.3.2. Hydroxyapatite and Other Calcium Phosphates
4.3.3. Layered Double Hydroxides
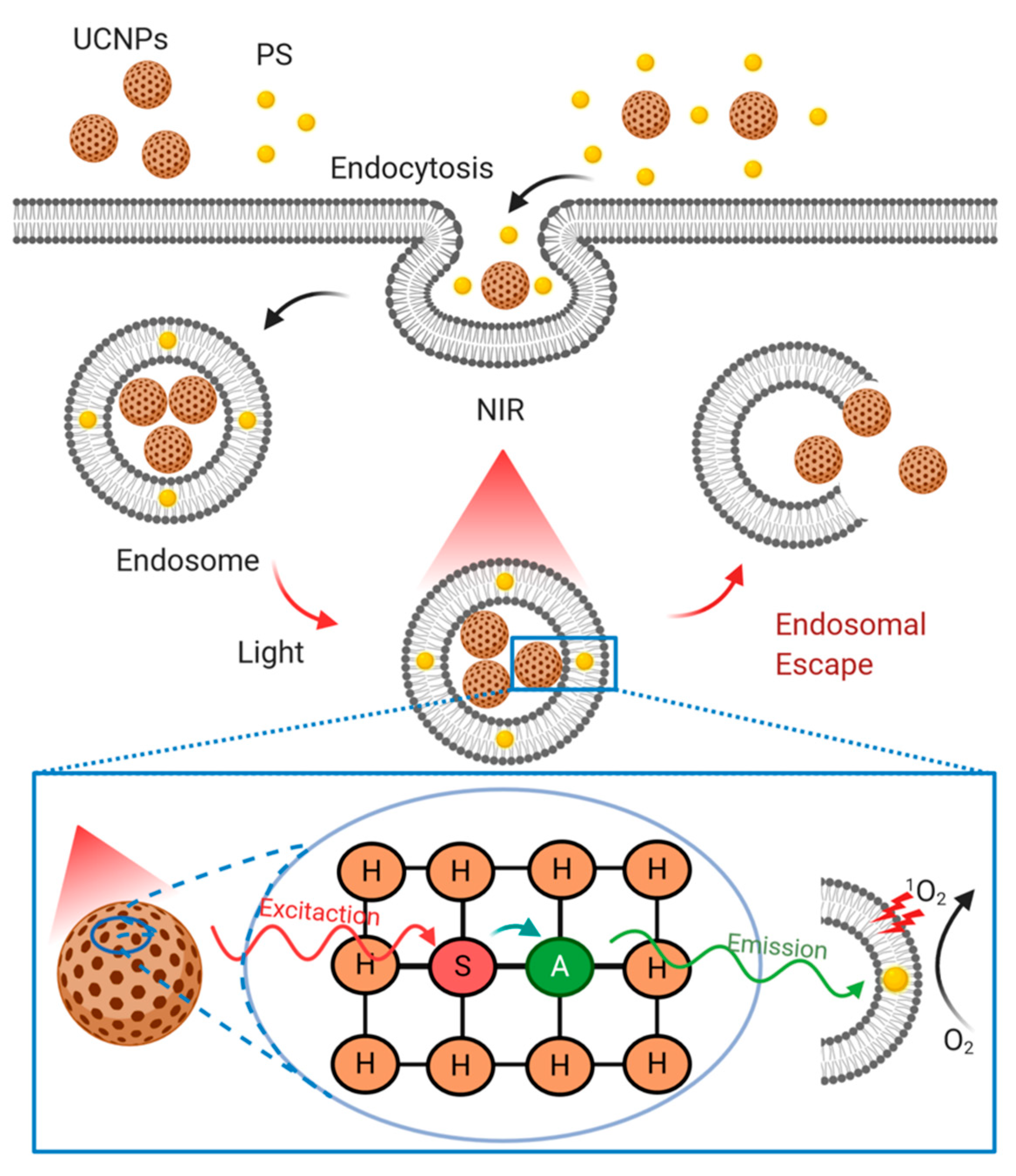
4.3.4. Lanthanide Upconversion Particles
4.3.5. Gold Nanoparticles
4.3.6. Magnetic Nanoparticles
4.3.7. Carbon-Based Nanostructures
4.3.8. Quantum Dots
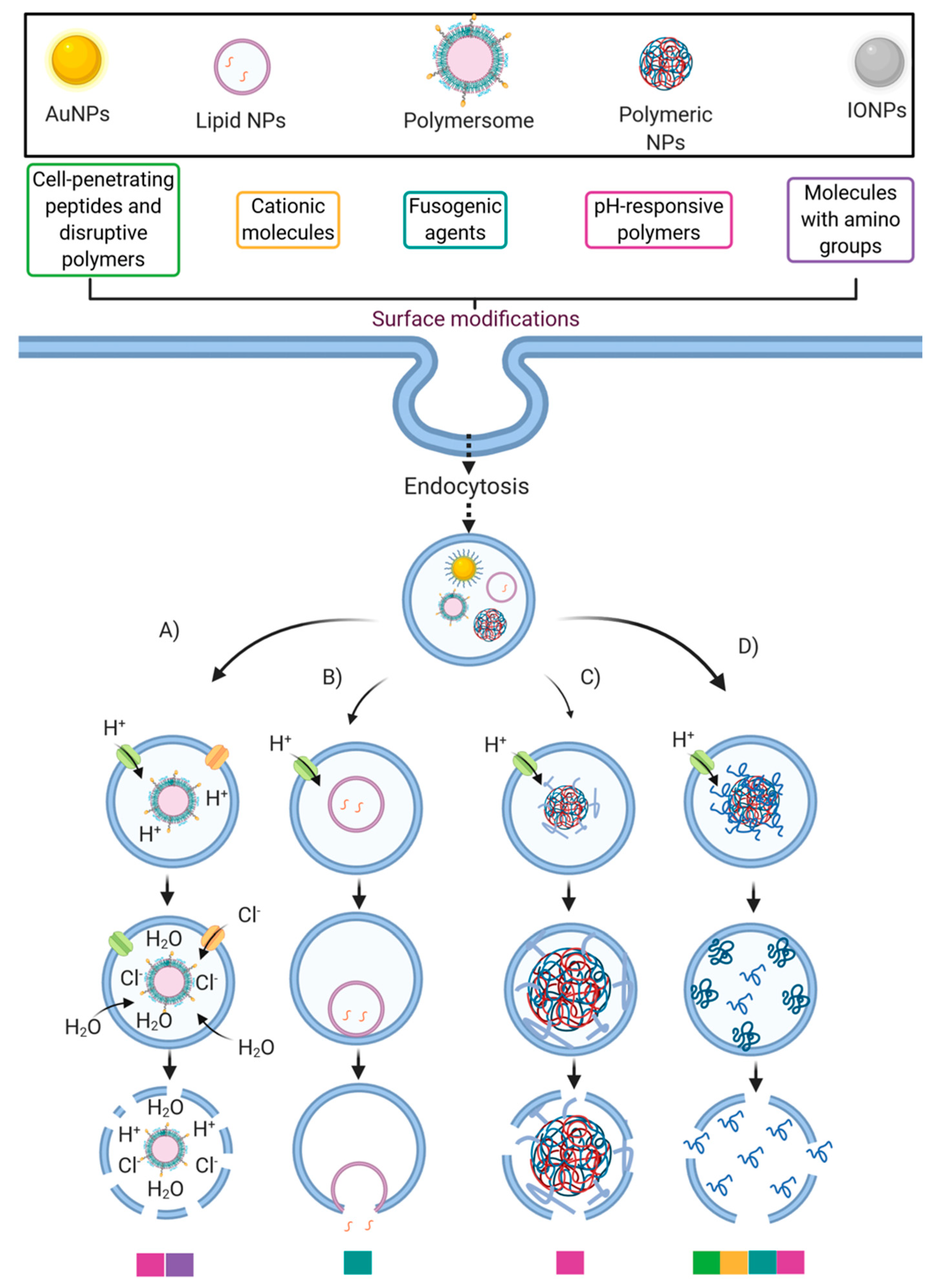
4.4. Cell-Penetrating Peptides
5. Enhancing Endosomal Escape of Nanocarriers by Conjugating Cell-Penetrating Peptides
5.1. Nanoparticles
5.2. Micelles
5.3. Liposomes
6. Conclusions and Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ibraheem, D.; Elaissari, A.; Fessi, H. Gene therapy and DNA delivery systems. Int. J. Pharm. 2014, 459, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Nayerossadat, N.; Ali, P.A.; Maedeh, T. Viral and nonviral delivery systems for gene delivery. Adv. Biomed. Res. 2012, 1, 27. [Google Scholar] [CrossRef]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- Santos, S.A.; Vidigal, P.M.; Thrimawithana, A.; Betancourth, B.M.; Guimarães, L.M.; Templeton, M.D.; Alfenas, A.C. Comparative genomic and transcriptomic analyses reveal different pathogenicity-related genes among three eucalyptus fungal pathogens. Fungal Genet. Biol. 2020, 137, 103332. [Google Scholar] [CrossRef]
- Nagasaki, T.; Shinkai, S. The concept of molecular machinery is useful for design of stimuli-responsive gene delivery systems in the mammalian cell. J. Incl. Phenom. Macrocycl. Chem. 2007, 58, 205–219. [Google Scholar] [CrossRef]
- Gao, X.; Kim, K.-S.; Liu, D. Nonviral gene delivery: What we know and what is next. AAPS J. 2007, 9, E92–E104. [Google Scholar] [CrossRef]
- Lin, G.; Li, L.; Panwar, N.; Wang, J.; Tjin, S.C.; Wang, X.; Yong, K.T. Non-viral gene therapy using multifunctional nanoparticles: Status, challenges, and opportu-nities. Coord. Chem. Rev. 2018, 374, 133–152. [Google Scholar] [CrossRef]
- Malmsten, M. Inorganic nanomaterials as delivery systems for proteins, peptides, DNA, and siRNA. Curr. Opin. Colloid Interface Sci. 2013, 18, 468–480. [Google Scholar] [CrossRef]
- Botto, C.; Augello, G.; Amore, E.; Emma, M.R.; Azzolina, A.; Cavallaro, G.; Cervello, M.; Bondì, M.L. Cationic Solid Lipid Nanoparticles as Non Viral Vectors for the Inhibition of Hepatocellular Carcinoma Growth by RNA Interference. J. Biomed. Nanotechnol. 2018, 14, 1009–1016. [Google Scholar] [CrossRef]
- Gardlík, R.; Pálffy, R.; Hodosy, J.; Lukács, J.; Turna, J.; Celec, P. Vectors and delivery sys-tems in gene therapy. Med. Sci. Monit. 2005, 11, RA110–RA121. [Google Scholar] [PubMed]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Vermeulen, L.M.; Brans, T.; De Smedt, S.C.; Remaut, K.; Braeckmans, K. Methodologies to investigate intracellular barriers for nucleic acid delivery in non-viral gene therapy. Nano Today 2018, 21, 74–90. [Google Scholar] [CrossRef] [Green Version]
- Okholm, A.H.; Kjems, J. DNA nanovehicles and the biological barriers. Adv. Drug Deliv. Rev. 2016, 106, 183–191. [Google Scholar] [CrossRef]
- Liu, H.; Kang, R.S.; Bagnowski, K.; Yu, J.M.; Radecki, S.; Daniel, W.L.; Anderson, B.R.; Nallagatla, S.; Schook, A.; Agarwal, R.; et al. Targeting the IL-17 Receptor Using Liposomal Spherical Nucleic Acids as Topical Therapy for Psoriasis. J. Investig. Dermatol. 2020, 140, 435–444.e4. [Google Scholar] [CrossRef]
- Desravines, N.; Miele, K.; Carlson, R.; Chibwesha, C.; Rahangdale, L. Topical therapies for the treatment of cervical intraepithelial neoplasia (CIN) 2–3: A narrative review. Gynecol. Oncol. Rep. 2020, 33, 100608. [Google Scholar] [CrossRef]
- Alqawlaq, S.; Sivak, J.M.; Huzil, J.T.; Ivanova, M.V.; Flanagan, J.G.; Beazely, M.A.; Foldvari, M. Preclinical development and ocular biodistribution of gemini-DNA nanoparticles after intravitreal and topical administration: Towards non-invasive glaucoma gene therapy. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Han, P.; Chen, R.; Lin, P.; Luo, M.; Cai, Q.; Huang, X. Topical 5-aminolevulinic acid photodynamic therapy for laryngeal papillomatosistosis treatment. Photodiagn. Photodyn. Ther. 2019, 28, 136–141. [Google Scholar] [CrossRef]
- Guo, X.; Huang, L. Recent Advances in Nonviral Vectors for Gene Delivery. Acc. Chem. Res. 2012, 45, 971–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Khan, A.I.; Cai, X.; Song, Y.; Lyu, Z.; Du, D.; Dutta, P.; Lin, Y. Overcoming blood–brain barrier transport: Advances in nanoparticle-based drug delivery strategies. Mater. Today 2020, 37, 112–125. [Google Scholar] [CrossRef]
- Gottfried, L.F.; Dean, D.A. Extracellular and Intracellular Barriers to Non-Viral Gene Transfer. In Novel Gene Therapy Approaches; IntechOpen: London, UK, 2013. [Google Scholar]
- Li, L.; Wei, Y.; Gong, C. Polymeric Nanocarriers for Non-Viral Gene Delivery. J. Biomed. Nanotechnol. 2015, 11, 739–770. [Google Scholar] [CrossRef]
- Ramamoorth, M.; Narvekar, A. Non viral vectors in gene therapy- An overview. J. Clin. Diagn. Res. 2015, 9, GE01–GE06. [Google Scholar] [CrossRef]
- Mellott, A.J.; Forrest, M.L.; Detamore, M.S. Physical non-viral gene delivery methods for tissue engineering. Ann. Biomed. Eng. 2013, 41, 446–468. [Google Scholar] [CrossRef] [Green Version]
- Xiong, R.; Samal, S.K.; Demeester, J.; Skirtach, A.G.; De Smedt, S.C.; Braeckmans, K. Laser-assisted pho-toporation: Fundamentals, technological advances and applications. Adv. Phys. X 2016, 1, 596–620. [Google Scholar]
- Khalil, I.A.; Kogure, K.; Akita, H.; Harashima, H. Uptake pathways and subsequent intracellular traf-ficking in nonviral gene delivery. Pharmacol. Rev. 2006, 58, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.H.; Chen, C.K.; Ravikrishnan, A.; Rane, S.; Pfeifer, B.A. Overcoming nonviral gene delivery bar-riers: Perspective and future. Mol. Pharm. 2013, 10, 4082–4098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahay, G.; Querbes, W.; Alabi, C.A.; Eltoukhy, A.A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.T.; Chen, D.; Zoncu, R.; et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaffer, D.V.; Fidelman, N.A.; Dan, N.; Lauffenburger, D.A. Vector unpacking as a potential barrier for receptor-mediated polyplex gene delivery. Biotechnol. Bioeng. 2000, 67, 598–606. [Google Scholar] [CrossRef]
- Capecchi, M.R. High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell 1980, 22, 479–488. [Google Scholar] [CrossRef]
- Vaughan, E.E.; DeGiulio, J.V.; Dean, D.A. Intracellular Trafficking of Plasmids for Gene Therapy: Mechanisms of Cytoplasmic Movement and Nuclear Import. Curr. Gene Ther. 2006, 6, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Graessmann, M.; Menne, J.; Liebler, M.; Graeber, I.; Graessmann, A. Helper activity for gene expression, a novel function of the SV40 enhancer. Nucleic Acids Res. 1989, 17, 6603–6612. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Aubin, R.A.; Paterson, M.C. Differential expression and stability of foreign genes intro-duced into human fibroblasts by nuclear versus cytoplasmic microinjection. Mutat. Res. Lett. 1992, 281, 115–122. [Google Scholar] [CrossRef]
- Fasbender, A.; Zabner, J.; Zeiher, B.; Welsh, M. A low rate of cell proliferation and reduced DNA uptake limit cationic lipid-mediated gene transfer to primary cultures of ciliated human airway epithelia. Gene Ther. 1997, 4, 1173–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, W.-C.; Haselton, F.R.; Giorgio, T.D. Mitosis enhances transgene expression of plasmid delivered by cationic liposomes. Biochim. Biophys. Acta BBA Gene Struct. Expr. 1999, 1445, 53–64. [Google Scholar] [CrossRef]
- Görlich, D. Nuclear protein import. Curr. Opin. Cell Biol. 1997, 9, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Dean, D.A.; Byrd, J.N.; Dean, B.S. Nuclear targeting of plasmid DNA in human corneal cells. Curr. Eye Res. 1999, 19, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Dynan, W.S.; Tjian, R. The promoter-specific transcription factor Sp1 binds to upstream sequences in the SV40 early promoter. Cell 1983, 35, 79–87. [Google Scholar] [CrossRef]
- Wildeman, A.G. Regulation of SV40 early gene expression. Biochem. Cell Biol. 1988, 66, 567–577. [Google Scholar] [CrossRef]
- Dynan, W.S.; Chervitz, S.A. Characterization of a minimal simian virus 40 late promoter: Enhancer elements in the 72-base-pair repeat not required. J. Virol. 1989, 63, 1420–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacik, J.; Dean, B.S.; Zimmer, W.E.; Dean, D.A. Cell-specific nuclear import of plasmid DNA. Gene Ther. 1999, 6, 1006–1014. [Google Scholar] [CrossRef]
- Hayden, M.S. Signaling to NF-B. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Längle-Rouault, F.; Patzel, V.; Benavente, A.; Taillez, M.; Silvestre, N.; Bompard, A.; Sczakiel, G.; Jacobs, E.; Rittner, K. Up to 100-Fold Increase of Apparent Gene Expression in the Presence of Ep-stein-Barr Virus oriP Sequences and EBNA1: Implications of the Nuclear Import of Plasmids. J. Virol. 1998, 72, 6181–6185. [Google Scholar] [CrossRef] [Green Version]
- Vaysse, L.; Gregory, L.G.; Harbottle, R.P.; Perouzel, E.; Tolmachov, O.; Coutelle, C. Nuclear-targeted minicircle to enhance gene transfer with non-viral vectorsin vitro andin vivo. J. Gene Med. 2006, 8, 754–763. [Google Scholar] [CrossRef]
- Donahue, N.D.; Acar, H.; Wilhelm, S. Concepts of nanoparticle cellular uptake, intracellular trafficking, and kinetics in nanomedicine. Adv. Drug Deliv. Rev. 2019, 143, 68–96. [Google Scholar] [CrossRef]
- Rivolta, I.; Panariti, A.; Miserocchi, G. The effect of nanoparticle uptake on cellular behavior: Disrupting or enabling functions? Nanotechnol. Sci. Appl. 2012, 5, 87–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clift, M.J.; Brandenberger, C.; Rothen-Rutishauser, B.; Brown, D.M.; Stone, V. The uptake and intra-cellular fate of a series of different surface coated quantum dots in vitro. Toxicology 2011, 286, 58–68. [Google Scholar] [CrossRef]
- Chen, J.; Yu, Z.; Chen, H.; Gao, J.; Liang, W. Transfection efficiency and intracellular fate of polycation liposomes combined with protamine. Biomaterials 2011, 32, 1412–1418. [Google Scholar] [CrossRef] [PubMed]
- Hillaireau, H.; Couvreur, P. Nanocarriers’ entry into the cell: Relevance to drug delivery. Cell. Mol. Life Sci. 2009, 66, 2873–2896. [Google Scholar] [CrossRef] [PubMed]
- Aderem, A.; Underhill, D.M. MECHANISMS OF PHAGOCYTOSIS IN MACROPHAGES. Annu. Rev. Immunol. 1999, 17, 593–623. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M. Professional and non-professional phagocytes: An introduction. Trends Cell Biol. 1995, 5, 85–87. [Google Scholar] [CrossRef]
- Chen, F.; Wang, G.; Griffin, J.I.; Brenneman, B.; Banda, N.K.; Holers, N.K.B.V.M.; Backos, D.S.; Wu, L.; Moghimi, L.W.S.M.; Simberg, D. Complement proteins bind to nanoparticle protein corona and undergo dynamic exchange in vivo. Nat. Nanotechnol. 2017, 12, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Tavano, R.; Gabrielli, L.; Lubian, E.; Fedeli, C.; Visentin, S.; De Laureto, P.P.; Arrigoni, G.; Geffner-Smith, A.; Chen, F.; Simberg, D.; et al. C1q-Mediated Complement Activation and C3 Opsonization Trigger Recognition of Stealth Poly(2-methyl-2-oxazoline)-Coated Silica Nanoparticles by Human Phagocytes. ACS Nano 2018, 12, 5834–5847. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.L.; Ezekowitz, R.A.B. Phagocytosis: Elegant complexity. Immunity 2005, 22, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-N.; Poon, W.; Tavares, A.J.; McGilvray, I.D.; Chan, W.C. Nanoparticle–liver interactions: Cellular uptake and hepatobiliary elimination. J. Control. Release 2016, 240, 332–348. [Google Scholar] [CrossRef]
- Daneman, R. The blood-brain barrier in health and disease. Ann. Neurol. 2012, 72, 648–672. [Google Scholar] [CrossRef] [PubMed]
- Walkey, C.D.; Olsen, J.B.; Guo, H.; Emili, A.; Chan, W.C.W. Nanoparticle Size and Surface Chemistry Determine Serum Protein Adsorption and Macrophage Uptake. J. Am. Chem. Soc. 2012, 134, 2139–2147. [Google Scholar] [CrossRef]
- Dai, Q.; Walkey, C.; Chan, W.C.W. Polyethylene Glycol Backfilling Mitigates the Negative Impact of the Protein Corona on Nanoparticle Cell Targeting. Angew. Chem. Int. Ed. 2014, 53, 5093–5096. [Google Scholar] [CrossRef]
- McMahon, H.T.; Boucrot, E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2011, 12, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, A.; Costa, M. Elucidating the mechanisms of nickel compound uptake: A review of particulate and nano-nickel endocytosis and toxicity. Toxicol. Appl. Pharmacol. 2012, 260, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- Mattila, J.P.; Shnyrova, A.V.; Sundborger, A.C.; Hortelano, E.R.; Fuhrmans, M.; Neumann, S.; Müller, M.; Hinshaw, J.E.; Schmid, S.L.; Frolov, V.A. A hemi-fission intermediate links two mechanistically distinct stages of membrane fission. Nature 2015, 524, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef]
- Bareford, L.M.; Swaan, P.W. Endocytic mechanisms for targeted drug delivery. Adv. Drug Deliv. Rev. 2007, 59, 748–758. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Ghosh, R.N.; Maxfield, F.R. Endocytosis. Physiol. Rev. 1997, 77, 759–803. [Google Scholar] [CrossRef]
- Elkin, S.R.; Lakoduk, A.M.; Schmid, S.L. Endocytic pathways and endosomal trafficking: A primer. Wien. Med. Wochenschr. 2016, 166, 196–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parton, R.G.; Del Pozo, M.A. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 2013, 14, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Kiss, A.L. Caveolae and the Regulation of Endocytosis. In Caveolins and Caveolae; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2012; Volume 729, pp. 14–28. [Google Scholar] [CrossRef]
- Yameen, B.; Choi, W.I.; Vilos, C.; Swami, A.; Shi, J.; Farokhzad, O.C. Insight into nanoparticle cellular uptake and intracellular targeting. J. Control. Release 2014, 190, 485–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Wang, Z.; Tiruppathi, C.; Cho, J.; Minshall, R.D.; Malik, A.B. Delivery of nanoparticle-complexed drugs across the vascular endothelial barrier via caveolae. IUBMB Life 2011, 63, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Oh, P.; Testa, J.E.; Borgstrom, P.; Witkiewicz, H.; Li, Y.; Schnitzer, J.E. In vivo proteomic imaging analysis of caveolae reveals pumping system to penetrate solid tumors. Nat. Med. 2014, 20, 1062–1068. [Google Scholar] [CrossRef]
- Rueda-Gensini, L.; Cifuentes, J.; Castellanos, M.C.; Puentes, P.R.; Serna, J.A.; Muñoz-Camargo, C.; Cruz, J.C. Tailoring iron oxide nanoparticles for efficient cellular internalization and endoso-mal escape. Nanomaterials 2020, 10, 1816. [Google Scholar] [CrossRef]
- Goulatis, L.I.; Shusta, E.V. Protein engineering approaches for regulating blood–brain barrier transcytosis. Curr. Opin. Struct. Biol. 2017, 45, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, G.J.; McMahon, H.T. Mechanisms of Endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, D.R.; Gosse, J.A.; Holowka, D.A.; Baird, B.A.; Webb, W.W. Temporally resolved interactions between antigen-stimulated IgE receptors and Lyn kinase on living cells. J. Cell Biol. 2005, 171, 527–536. [Google Scholar] [CrossRef]
- Tuli, A.; Sharma, M.; McIlhaney, M.M.; Talmadge, J.E.; Naslavsky, N.; Caplan, S.; Solheim, J.C. Amyloid Precursor-Like Protein 2 Increases the Endocytosis, Instability, and Turnover of the H2-K d MHC Class I Molecule. J. Immunol. 2008, 181, 1978–1987. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Low, P.S. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv. Drug Deliv. Rev. 2002, 54, 675–693. [Google Scholar] [CrossRef]
- Rijnboutt, S.; Jansen, G.; Posthuma, G.; Hynes, J.B.; Schornagel, J.H.; Strous, G.J. Endocytosis of GPI-linked membrane folate receptor-α. J. Cell Biol. 1996, 132, 35–47. [Google Scholar] [CrossRef]
- Fiorentini, C.; Falzano, L.; Fabbri, A.; Stringaro, A.; Logozzi, M.; Travaglione, S.; Contamin, S.; Arancia, G.; Malorni, W.; Fais, S. Activation of Rho GTPases by Cytotoxic Necrotizing Factor 1 Induces Macropinocytosis and Scavenging Activity in Epithelial Cells. Mol. Biol. Cell 2001, 12, 2061–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, J.; Helenius, A. Virus entry by macropinocytosis. Nat. Cell Biol. 2009, 11, 510–520. [Google Scholar] [CrossRef]
- Kolb-Mäurer, A.; Wilhelm, M.; Weissinger, F.; Bröcker, E.B.; Goebel, W. Interaction of human hematopoietic stem cells with bacterial pathogens. Blood 2002, 100, 3703–3709. [Google Scholar] [CrossRef] [Green Version]
- Steinman, R.M.; Swanson, J.J. The endoeytie activity of dendritic cells. J. Exp. Med. 1995, 182, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Cella, M.; Danieli, C.; Lanzavecchia, A. Dendritic cells use macropinocytosis and the man-nose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: Downregulation by cytokines and bacterial products. J. Exp. Med. 1995, 182, 389–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcone, S.; Cocucci, E.; Podini, P.; Kirchhausen, T.; Clementi, E.; Meldolesi, J. Macropinocytosis: Regulated coordination of endocytic and exocytic membrane traffic events. J. Cell Sci. 2006, 119, 4758–4769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, M.C.; Teasdale, R.D. Defining Macropinocytosis. Traffic 2009, 10, 364–371. [Google Scholar] [CrossRef]
- Schafer, D.A.; D’Souza-Schorey, C.; Cooper, J.A. Actin assembly at membranes controlled by ARF6. Traffic 2000, 1, 896–907. [Google Scholar] [CrossRef]
- Racoosin, E.L.; Swanson, J.A. Macropinosome maturation and fusion with tubular lysosomes in macrophages. J. Cell Biol. 1993, 121, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Alexander-Katz, A. Cell membranes open ‘doors’ for cationic nanoparticles/ biomolecules: In-sights into uptake kinetics. ACS Nano 2013, 7, 10799–10808. [Google Scholar] [CrossRef]
- Weng, Y.; Huang, Q.; Li, C.; Yang, Y.; Wang, X.; Yu, J.; Huang, Y.; Liang, X.-J. Improved Nucleic Acid Therapy with Advanced Nanoscale Biotechnology. Mol. Ther. Nucleic Acids 2020, 19, 581–601. [Google Scholar] [CrossRef]
- Fraley, R.; Subramani, S.; Berg, P.; Papahadjopoulos, D. Introduction of lipo-some-encapsulated SV40 DNA into cells. J. Biol. Chem. 1980, 255, 10431–10435. [Google Scholar] [CrossRef]
- Ozpolat, B.; Sood, A.K.; Lopez-Berestein, G. Liposomal siRNA nanocarriers for cancer therapy. Adv. Drug Deliv. Rev. 2014, 66, 110–116. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, P.J.; Elahipanah, S.; Rogozhnikov, D.; Yousaf, M.N. Bio-Orthogonal Mediated Nucleic Acid Transfection of Cells via Cell Surface Engineering. ACS Cent. Sci. 2017, 3, 489–500. [Google Scholar] [CrossRef]
- Li, L.; Zahner, D.; Su, Y.; Gruen, C.; Davidson, G.; Levkin, P.A. A biomimetic lipid library for gene delivery through thiol-yne click chemistry. Biomaterials 2012, 33, 8160–8166. [Google Scholar] [CrossRef]
- Li, Y.; Liu, R.; Shi, Y.; Zhang, Z.; Zhang, X. Zwitterionic Poly(carboxybetaine)-based Cationic Liposomes for Effective Delivery of Small Interfering RNA Therapeutics without Accelerated Blood Clearance Phenomenon. Theranostics 2015, 5, 583–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.A.; Selby, L.I.; Johnston, A.P.R.; Such, G.K. The Endosomal Escape of Nanoparticles: Toward More Efficient Cellular Delivery. Bioconjug. Chem. 2019, 30, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Selby, L.I.; Cortez-Jugo, C.M.; Such, G.K.; Johnston, A.P. Nanoescapology: Progress toward under-standing the endosomal escape of polymeric nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1452. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, D.; Marchini, C.; Cardarelli, F.; Amenitsch, H.; Garulli, C.; Bifone, A.; Caracciolo, G. Transfection efficiency boost of cholesterol-containing lipoplexes. Biochim. Biophys. Acta BBA Biomembr. 2012, 1818, 2335–2343. [Google Scholar] [CrossRef] [Green Version]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef]
- Maier, M.A.; Jayaraman, M.; Matsuda, S.; Liu, J.; Barros, S.; Querbes, W.; Tam, Y.K.; Ansell, S.M.; Kumar, V.; Qin, J.; et al. Biodegradable Lipids Enabling Rapidly Eliminated Lipid Nanoparticles for Systemic Delivery of RNAi Therapeutics. Mol. Ther. 2013, 21, 1570–1578. [Google Scholar] [CrossRef] [Green Version]
- Love, K.T.; Mahon, K.P.; Levins, C.G.; Whitehead, K.A.; Querbes, W.; Dorkin, J.R.; Qin, J.; Cantley, W.; Qin, L.L.; Racie, T.; et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA 2010, 107, 1864–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Love, K.T.; Dorkin, J.R.; Sirirungruang, S.; Zhang, Y.; Chen, D.; Bogorad, R.L.; Yin, H.; Chen, Y.; Vegas, A.J.; et al. Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proc. Natl. Acad. Sci. USA 2014, 111, 3955–3960. [Google Scholar] [CrossRef] [Green Version]
- Mellet, C.O.; Fernández, J.M.G.; Benito, J.M. Cyclodextrin-based gene delivery systems. Chem. Soc. Rev. 2011, 40, 1586–1608. [Google Scholar] [CrossRef]
- Davis, M.E. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: From concept to clinic. Mol. Pharm. 2009, 6, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Barata, P.; Sood, A.K.; Hong, D.S. RNA-targeted therapeutics in cancer clinical trials: Current status and future directions. Cancer Treat. Rev. 2016, 50, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Huang, W.; Jin, M.; Wang, Q.; Fan, B.; Kang, L.; Gao, Z. Chitosan-based nanoparticles for survivin targeted siRNA delivery in breast tumor therapy and preventing its metastasis. Int. J. Nanomed. 2016, 11, 4931–4945. [Google Scholar] [CrossRef] [Green Version]
- Siahmansouri, H.; Somi, M.H.; Babaloo, Z.; Baradaran, B.; Jadidi-Niaragh, F.; Atyabi, F.; Mohammadi, H.; Ahmadi, M.; Yousefi, M. Effects of HMGA2 siRNA and doxorubicin dual delivery by chitosan nanoparticles on cytotoxicity and gene expression of HT-29 colorectal cancer cell line. J. Pharm. Pharmacol. 2016, 68, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Yang, J.A.; Lee, M.Y.; Lee, H.; Hahn, S.K. Reducible hyaluronic acid-siRNA conjugate for target specific gene silencing. Bioconjug. Chem. 2013, 24, 1201–1209. [Google Scholar] [CrossRef]
- Kim, J.S.; Oh, M.H.; Park, J.Y.; Park, T.G.; Nam, Y.S. Protein-resistant, reductively dissociable polyplex-es for in vivo systemic delivery and tumor-targeting of siRNA. Biomaterials 2013, 34, 2370–2379. [Google Scholar] [CrossRef]
- Xiao, Y.; Shi, K.; Qu, Y.; Chu, B.; Qian, Z. Engineering Nanoparticles for Targeted Delivery of Nucleic Acid Therapeutics in Tumor. Mol. Ther. Methods Clin. Dev. 2019, 12, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Boussif, O.; Lezoualc’H, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Wang, M.; Wang, T.; Zhang, B.; Liu, C.; Zhang, N. Multifunctionalized polyethyleneimine-based nanocarriers for gene and chemotherapeutic drug combination therapy through one-step assembly strategy. Int. J. Nanomed. 2017, 12, 8681–8698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Li, Z.; Sun, J.; Luo, C.; Li, L.; Liu, Y.; Du, Y.; Qiu, S.; Ai, X.; Wu, C.; et al. Stealth CD44-targeted hyaluronic acid supramolecular nanoassemblies for doxorubicin deliv-ery: Probing the effect of uncovalent pegylation degree on cellular uptake and blood long circulation. J. Control. Release 2015, 197, 29–40. [Google Scholar] [CrossRef]
- Lu, H.H.; Huang, C.H.; Shiue, T.Y.; Wang, F.S.; Chang, K.K.; Chen, Y.; Peng, C.H. Erratum: Highly efficient gene release in spatiotemporal precision approached by light and pH dual responsive copolymers. Chem. Sci. 2019, 10, 284–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Wu, Z.; Wu, C.; Wang, X.; Liow, S.S.; Li, Z.; Wu, Y.-L. Overcoming STC2 mediated drug resistance through drug and gene co -delivery by PHB-PDMAEMA cationic polyester in liver cancer cells. Mater. Sci. Eng. C 2018, 83, 210–217. [Google Scholar] [CrossRef]
- Ramírez-Acosta, C.M.; Cifuentes, J.; Castellanos, M.C.; Moreno, R.J.; Muñoz-Camargo, C.; Cruz, J.C.; Reyes, L.H. PH-Responsive, Cell-Penetrating, Core/Shell Magnetite/Silver Nanoparticles for the Delivery of Plasmids: Preparation, Characterization, and Preliminary In Vitro Evaluation. Pharmaceutics 2020, 12, 561. [Google Scholar] [CrossRef]
- Ramírez-Acosta, C.M.; Cifuentes, J.; Cruz, J.C.; Reyes, L.H. Patchy Core/Shell, Magnetite/Silver Na-noparticles via Green and Facile Synthesis: Routes to Assure Biocompatibility. Nanomatererials 2020, 10, 1857. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, W.; Guo, S.; Wang, Y.; Miao, L.; Xiong, Y.; Huang, L. PolyMetformin combines carrier and anticancer activities for in vivo siRNA delivery. Nat. Commun. 2016, 7, 11822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernicova, I.; Korbonits, M. Metformin—Mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP ki-nase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [Green Version]
- Dowling, R.J.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin Inhibits Mammalian Target of Rapamycin–Dependent Translation Initiation in Breast Cancer Cells. Cancer Res. 2007, 67, 10804–10812. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Martín, A.; Oliveras-Ferraros, C.; del Barco, S.; Martín-Castillo, B.; Menéndez, J.A. MTOR in-hibitors and the anti-diabetic biguanide metformin: New insights into the molecular management of breast cancer resistance to the HER2 tyrosine kinase inhibitor lapatinib (Tykerb®). Clin. Transl. Oncol. 2009, 11, 455–459. [Google Scholar] [CrossRef]
- Stanzl, E.G.; Trantow, B.M.; Vargas, J.R.; Wender, P.A. Fifteen years of cell-penetrating, guanidini-um-rich molecular transporters: Basic science, research tools, and clinical applications. Acc. Chem. Res. 2013, 46, 2944–2954. [Google Scholar] [CrossRef] [Green Version]
- Bechara, C.; Sagan, S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef]
- Lyu, Y.; Cui, D.; Sun, H.; Miao, Y.; Duan, H.; Pu, K. Dendronized Semiconducting Polymer as Photothermal Nanocarrier for Remote Activation of Gene Expression. Angew. Chem. Int. Ed. 2017, 56, 9155–9159. [Google Scholar] [CrossRef]
- Olden, B.R.; Cheng, Y.; Yu, J.L.; Pun, S.H. Cationic polymers for non-viral gene delivery to human T cells. J. Control. Release 2018, 282, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.K.; Kaczmarek, J.C.; Bose, S.; Kauffman, K.J.; Mir, F.; Heartlein, M.W.; DeRosa, F.; Langer, R.; Anderson, D.G. Inhaled Nanoformulated mRNA Polyplexes for Protein Production in Lung Epithelium. Adv. Mater. 2019, 31, e1805116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; He, Y.; Wang, W.; Wu, C.; Hong, C.; Hammond, P.T. Polyamine-Mediated Stoichiometric Assembly of Ribonucleoproteins for Enhanced mRNA Delivery. Angew. Chem. Int. Ed. 2017, 56, 13709–13712. [Google Scholar] [CrossRef] [Green Version]
- Jarzębińska, A.; Pasewald, T.; Lambrecht, J.; Mykhaylyk, O.; Kümmerling, L.; Beck, P.; Hasenpusch, G.; Rudolph, C.; Plank, C.; Dohmen, C. A Single Methylene Group in Oligoalkylamine-Based Cationic Polymers and Lipids Promotes Enhanced mRNA Delivery. Angew. Chem. Int. Ed. 2016, 55, 9591–9595. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Wei, T.; Jia, Y.; Farbiak, L.; Zhou, K.; Zhang, S.; Wei, Y.; Zhu, H.; Siegwart, D.J. Dendrimer-Based Lipid Nanoparticles Deliver Therapeutic FAH mRNA to Normalize Liver Function and Extend Survival in a Mouse Model of Hepatorenal Tyrosinemia Type I. Adv. Mater. 2018, 30, e1805308. [Google Scholar] [CrossRef]
- Sharfstein, S.T. Non-protein biologic therapeutics. Curr. Opin. Biotechnol. 2018, 53, 65–75. [Google Scholar] [CrossRef]
- Li, X.; Sun, A.-N.; Liu, Y.-J.; Zhang, W.-J.; Pang, N.; Cheng, S.-X.; Qi, X.-R. Amphiphilic dendrimer engineered nanocarrier systems for co-delivery of siRNA and paclitaxel to matrix metalloproteinase-rich tumors for synergistic therapy. NPG Asia Mater. 2018, 10, 238–254. [Google Scholar] [CrossRef] [Green Version]
- Navarro, G.; Pan, J.; Torchilin, V.P. Micelle-like Nanoparticles as Carriers for DNA and siRNA. Mol. Pharm. 2015, 12, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Lao, Y.H.; Li, M.; Gao, M.A.; Shao, D.; Chi, C.W.; Huang, D.; Chakraborty, S.; Ho, T.C.; Jiang, W.; Wang, H.X.; et al. HPV Oncogene Manipulation Using Nonvirally Delivered CRISPR/Cas9 or Natronobacte-rium gregoryi Argonaute. Adv. Sci. 2018, 5, 1700540. [Google Scholar] [CrossRef]
- Davis, M.E. Design and development of IT-101, a cyclodextrin-containing polymer conjugate of camptoth-ecin. Adv. Drug Deliv. Rev. 2009, 61, 1189–1192. [Google Scholar] [CrossRef]
- Freeman, E.C.; Weiland, L.M.; Meng, W.S. Modeling the proton sponge hypothesis: Examining proton sponge effectiveness for enhancing intracellular gene delivery through multiscale modeling. J. Biomater. Sci. Polym. Ed. 2013, 24, 398–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeulen, L.M.; De Smedt, S.C.; Remaut, K.; Braeckmans, K. The proton sponge hypothesis: Fable or fact? Eur. J. Pharm. Biopharm. 2018, 129, 184–190. [Google Scholar] [CrossRef]
- Hu, Y.; Litwin, T.; Nagaraja, A.R.; Kwong, B.; Katz, J.; Watson, N.; Irvine, D.J. Cytosolic delivery of membrane-impermeable molecules in dendritic cells using pH-responsive core-shell nanoparticles. Nano Lett. 2007, 7, 3056–3064. [Google Scholar] [CrossRef]
- Arcos, D.; Vallet-Regí, M. Bioceramics for drug delivery. Acta Mater. 2013, 61, 890–911. [Google Scholar] [CrossRef]
- Gultepe, E.; Nagesha, D.; Sridhar, S.; Amiji, M. Nanoporous inorganic membranes or coatings for sus-tained drug delivery in implantable devices. Adv. Drug Deliv. Rev. 2010, 62, 305–315. [Google Scholar] [CrossRef]
- Vivero-Escoto, J.L.; Slowing, I.I.; Trewyn, B.G.; Lin, V.S. Mesoporous Silica Nanoparticles for Intra-cellular Controlled Drug Delivery. Small 2010, 6, 1952–1967. [Google Scholar] [CrossRef]
- Kim, M.-H.; Na, H.-K.; Kim, Y.-K.; Ryoo, S.-R.; Cho, H.S.; Lee, K.E.; Jeon, H.; Ryoo, R.; Min, D.-H. Facile Synthesis of Monodispersed Mesoporous Silica Nanoparticles with Ultralarge Pores and Their Application in Gene Delivery. ACS Nano 2011, 5, 3568–3576. [Google Scholar] [CrossRef]
- Li, X.; Xie, Q.R.; Zhang, J.; Xia, W.; Gu, H. The packaging of siRNA within the mesoporous structure of silica nanoparticles. Biomaterials 2011, 32, 9546–9556. [Google Scholar] [CrossRef]
- Na, H.K.; Kim, M.H.; Park, K.; Ryoo, S.R.; Lee, K.E.; Jeon, H.; Ryoo, R.; Hyeon, C.; Min, D.H. Efficient Functional Delivery of siRNA using Mesoporous Silica Nanoparticles with Ultra-large Pores. Small 2012, 8, 1752–1761. [Google Scholar] [CrossRef]
- Meng, H.; Liong, M.; Xia, T.; Li, Z.; Ji, Z.; Zink, J.I.; Nel, A.E. Engineered Design of Mesoporous Silica Nanoparticles to Deliver Doxorubicin and P-Glycoprotein siRNA to Overcome Drug Resistance in a Cancer Cell Line. ACS Nano 2010, 4, 4539–4550. [Google Scholar] [CrossRef]
- Izquierdo-Barba, I.; Vallet-Regí, M.; Kupferschmidt, N.; Terasaki, O.; Schmidtchen, A.; Malmsten, M. Incorporation of antimicrobial compounds in mesoporous silica film monolith. Biomaterials 2009, 30, 5729–5736. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Jiang, H.; Zhao, Q.; Wang, S.; Zou, M.; Cheng, G. Enhanced mucosal and systemic immune responses obtained by porous silica nanoparticles used as an oral vaccine adjuvant: Effect of silica architecture on immunological properties. Int. J. Pharm. 2012, 436, 351–358. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.; Wang, M.; Ma, Y.; Xia, W.; Gu, H. A mesoporous silica nanoparticle—PEI—Fusogenic pep-tide system for siRNA delivery in cancer therapy. Biomaterials 2013, 34, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; He, Q.; Liu, J.; Chen, Y.; Ma, M.; Zhang, L.; Shi, J. Nuclear-targeted drug delivery of tat peptide-conjugated monodisperse mesoporous silica nanoparticles. J. Am. Chem. Soc. 2012, 134, 5722–5725. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Tarafder, S. Calcium phosphate ceramic systems in growth factor and drug delivery for bone tissue engineering: A review. Acta Biomater. 2012, 8, 1401–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, T.N.T.; Lee, W.-H.; Loo, C.-Y.; Zavgorodniy, A.V.; Rohanizadeh, R. Hydroxyapatite nanoparticles as vectors for gene delivery. Ther. Deliv. 2012, 3, 623–632. [Google Scholar] [CrossRef]
- Sokolova, V.V.; Radtke, I.; Heumann, R.; Epple, M. Effective transfection of cells with multi-shell calcium phosphate-DNA nanoparticles. Biomatererials 2006, 27, 3147–3153. [Google Scholar] [CrossRef]
- Chen, F.; Tang, Q.-L.; Zhu, Y.-J.; Wang, K.-W.; Zhang, M.-L.; Zhai, W.-Y.; Chang, J. Hydroxyapatite nanorods/poly(vinyl pyrolidone) composite nanofibers, arrays and three-dimensional fabrics: Electrospun preparation and transformation to hydroxyapatite nanostructures. Acta Biomater. 2010, 6, 3013–3020. [Google Scholar] [CrossRef]
- Wang, J.W.; Chen, C.Y.; Kuo, Y.M. Preparation and characterization of chitosan-coated hydroxyap-atite nanoparticles as a promising non-viral vector for gene delivery. J. Appl. Polym. Sci. 2011, 121, 3531–3540. [Google Scholar] [CrossRef]
- Wu, X.; Ding, D.; Jiang, H.; Xing, X.; Huang, S.; Liu, H.; Chen, Z.; Sun, H. Transfection using hydroxyapatite nanoparticles in the inner ear via an intact round window membrane in chinchilla. J. Nanopart. Res. 2012, 14, 708. [Google Scholar] [CrossRef]
- Yazaki, Y.; Oyane, A.; Araki, H.; Sogo, Y.; Ito, A.; Yamazaki, A.; Tsurushima, H. Fabrication of DNA-antibody–apatite composite layers for cell-targeted gene transfer. Sci. Technol. Adv. Mater. 2012, 13, 064204. [Google Scholar] [CrossRef] [Green Version]
- Ladewig, K.; Xu, Z.P.; Lu, G.Q. Layered double hydroxide nanoparticles in gene and drug delivery. Expert Opin. Drug Deliv. 2009, 6, 907–922. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.P.; Walker, T.L.; Liu, K.-L.; Cooper, H.M.; Lu, G.Q.M.; Bartlett, P.F. Layered double hydroxide nanoparticles as cellular delivery vectors of supercoiled plasmid DNA. Int. J. Nanomed. 2007, 2, 163–174. [Google Scholar]
- Wong, Y.; Markham, K.; Xu, Z.P.; Chen, M.; Lu, G.Q.; Bartlett, P.F.; Cooper, H.M. Efficient delivery of siRNA to cortical neurons using layered double hydroxide nanoparticles. Biomaterials 2010, 31, 8770–8779. [Google Scholar] [CrossRef]
- Chen, M.; Cooper, H.M.; Zhou, J.Z.; Bartlett, P.F.; Xu, Z.P. Reduction in the size of layered double hy-droxide nanoparticles enhances the efficiency of siRNA delivery. J. Colloid Interface Sci. 2013, 390, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Qin, L.; Wang, W.; Zhu, R.; Yu, Y.; Liu, H.; Wang, S. The use of layered double hydroxides as DNA vaccine delivery vector for enhancement of an-ti-melanoma immune response. Biomaterials 2011, 32, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Zhao, Y.; Wang, S.; Liu, M.; Duan, Z.; Chen, Y.; Li, F.; Xu, F.; Lu, T. Recent advances in synthesis and surface modification of lanthanide-doped upconversion na-noparticles for biomedical applications. Biotechnol. Adv. 2012, 30, 1551–1561. [Google Scholar] [CrossRef]
- Gai, S.; Yang, P.; Li, C.; Wang, W.; Dai, Y.; Niu, N.; Lin, J. Synthesis of Magnetic, Up-Conversion Luminescent, and Mesoporous Core-Shell-Structured Nanocomposites as Drug Carriers. Adv. Funct. Mater. 2010, 20, 1166–1172. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, Y.; Lim, K.M.; Sim, E.K.; Ye, L. NIR-to-visible upconversion nanoparticles for fluo-rescent labeling and targeted delivery of siRNA. Nanotechnology 2009, 20, 155101. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, F.; Liu, X.; Xing, B. NIR light controlled photorelease of siRNA and its targeted intracellular delivery based on upconversion nanoparticles. Nanoscale 2013, 5, 231–238. [Google Scholar] [CrossRef]
- Guo, H.; Hao, R.; Qian, H.; Sun, S.; Sun, D.; Yin, H.; Liu, Z.; Liu, X. Upconversion na-noparticles modified with aminosilanes as carriers of DNA vaccine for foot-and-mouth disease. Appl. Microbiol. Biotechnol. 2012, 95, 1253–1263. [Google Scholar] [CrossRef]
- Lytton-Jean, A.K.; Langer, R.; Anderson, D.G. Five Years of siRNA Delivery: Spotlight on Gold Nanoparticles. Small 2011, 7, 1932–1937. [Google Scholar] [CrossRef]
- Oishi, M.; Nakaogami, J.; Ishii, T.; Nagasaki, Y. Smart PEGylated Gold Nanoparticles for the Cytoplasmic Delivery of siRNA to Induce Enhanced Gene Silencing. Chem. Lett. 2006, 35, 1046–1047. [Google Scholar] [CrossRef]
- Giljohann, D.A.; Seferos, D.S.; Prigodich, A.E.; Patel, P.C.; Mirkin, C.A. Gene regulation with polyvalent siRNA-nanoparticle conjugates. J. Am. Chem. Soc. 2009, 131, 2072–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akinc, A.; Lynn, D.M.; Anderson, A.D.G.; Langer, R. Parallel Synthesis and Biophysical Characterization of a Degradable Polymer Library for Gene Delivery. J. Am. Chem. Soc. 2003, 125, 5316–5323. [Google Scholar] [CrossRef] [PubMed]
- Elbakry, A.; Zaky, A.; Liebl, R.; Rachel, R.; Goepferich, A.; Breunig, M. Layer-by-layer assembled gold nanoparticles for sirna delivery. Nano Lett. 2009, 9, 2059–2064. [Google Scholar] [CrossRef] [PubMed]
- Elbakry, A.M.; Wurster, E.-C.; Zaky, A.; Liebl, R.; Schindler, E.; Bauer-Kreisel, P.; Blunk, T.; Rachel, R.; Goepferich, A.; Breunig, M. Layer-by-Layer Coated Gold Nanoparticles: Size-Dependent Delivery of DNA into Cells. Small 2012, 8, 3847–3856. [Google Scholar] [CrossRef]
- Guo, S.; Huang, Y.; Jiang, Q.; Sun, Y.; Deng, L.; Liang, Z.; Du, Q.; Xing, J.; Zhao, Y.; Wang, P.C.; et al. Enhanced Gene Delivery and siRNA Silencing by Gold Nanoparticles Coated with Charge-Reversal Polyelectrolyte. ACS Nano 2010, 4, 5505–5511. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Zhao, J.; Zhang, X.; Cao, W.; Hu, X.; Zou, G.; Duan, X.; Liang, X.-J. Enhanced siRNA Delivery and Silencing Gold–Chitosan Nanosystem with Surface Charge-Reversal Polymer Assembly and Good Biocompatibility. ACS Nano 2012, 6, 7340–7351. [Google Scholar] [CrossRef]
- Lee, M.Y.; Park, S.J.; Park, K.; Kim, K.S.; Lee, H.; Hahn, S.K. Target-specific gene silencing of lay-er-by-layer assembled gold-cysteamine/siRNA/PEI/HA nanocomplex. ACS Nano 2011, 5, 6138–6147. [Google Scholar] [CrossRef]
- Lu, W.; Zhang, G.; Zhang, R.; Flores, L.G.; Huang, Q.; Gelovani, J.G.; Li, C. Tumor site-specific silencing of NF-kappaB p65 by targeted hollow gold nanosphere-mediated photothermal trans-fection. Cancer Res. 2010, 70, 3177–3188. [Google Scholar] [CrossRef] [Green Version]
- Conde, J.; Ambrosone, A.; Sanz, V.; Hernandez, Y.; Marchesano, V.; Tian, F.; Child, B.H.; Berry, C.C.; Ibarra, M.R.; Baptista, P.V.; et al. Design of Multifunctional Gold Nanoparticles for In Vitro and In Vivo Gene Silencing. ACS Nano 2012, 6, 8316–8324. [Google Scholar] [CrossRef]
- Braun, G.B.; Pallaoro, A.; Wu, G.; Missirlis, D.; Zasadzinski, J.A.; Tirrell, M.; Reich, N.O. Laser-activated gene silencing via gold nanoshell-siRNA conjugates. ACS Nano 2009, 3, 2007–2015. [Google Scholar] [CrossRef]
- Huschka, R.; Barhoumi, A.; Liu, Q.; Roth, J.A.; Ji, L.; Halas, N.J. Gene Silencing by Gold Nanoshell-Mediated Delivery and Laser-Triggered Release of Antisense Oligonucleotide and siRNA. ACS Nano 2012, 6, 7681–7691. [Google Scholar] [CrossRef] [Green Version]
- Oyelere, A.K.; Chen, P.C.; Huang, X.; El-Sayed, A.I.H.; El-Sayed, M.A. Peptide-Conjugated Gold Nanorods for Nuclear Targeting. Bioconjug. Chem. 2007, 18, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-Y.; Yang, J.-A.; Jung, H.S.; Beack, S.; Choi, J.E.; Hur, W.; Koo, H.; Kim, K.; Yoon, S.K.; Hahn, S.K. Hyaluronic Acid–Gold Nanoparticle/Interferon α Complex for Targeted Treatment of Hepatitis C Virus Infection. ACS Nano 2012, 6, 9522–9531. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Simard, J.M.; Worrall, A.J.W.E.; Rotello, V.M. Tunable Reactivation of Nanoparticle-Inhibited β-Galactosidase by Glutathione at Intracellular Concentrations. J. Am. Chem. Soc. 2004, 126, 13987–13991. [Google Scholar] [CrossRef]
- Liu, C.L.; Wu, H.T.; Hsiao, Y.H.; Lai, C.W.; Shih, C.W.; Peng, Y.K.; Tang, K.C.; Chang, H.W.; Chien, Y.C.; Hsiao, J.K.; et al. Insulin-Directed Synthesis of Fluorescent Gold Nanoclusters: Preservation of Insulin Bioactivity and Versatility in Cell Imaging. Angew. Chem. Int. Ed. 2011, 50, 7056–7060. [Google Scholar] [CrossRef]
- Paciotti, G.F.; Myer, L.; Weinreich, D.; Goia, D.; Pavel, N.; McLaughlin, R.E.; Tamarkin, L. Colloidal Gold: A Novel Nanoparticle Vector for Tumor Directed Drug Delivery. Drug Deliv. 2004, 11, 169–183. [Google Scholar] [CrossRef]
- Mok, H.; Zhang, M. Superparamagnetic iron oxide nanoparticle-based delivery systems for biotherapeutics. Expert Opin. Drug Deliv. 2013, 10, 73–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Deen, F.N.; Selomulya, C.; Williams, T. On designing stable magnetic vectors as carriers for malaria DNA vaccine. Colloids Surfaces B Biointerfaces 2013, 102, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Asdi, M.; Hashmi, M.; Umar, M.; Awan, S.-U. Thermo-responsive copolymer coated MnFe2O4 magnetic nanoparticles for hyperthermia therapy and controlled drug delivery. Mater. Chem. Phys. 2012, 137, 365–371. [Google Scholar] [CrossRef]
- Cormode, D.P.; Skajaa, G.O.; Delshad, A.; Parker, N.; Jarzyna, P.A.; Calcagno, C.; Galper, M.W.; Skajaa, T.; Briley-Saebo, K.C.; Bell, H.M.; et al. A Versatile and Tunable Coating Strategy Allows Control of Nanocrystal Delivery to Cell Types in the Liver. Bioconjug. Chem. 2011, 22, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Kenny, G.D.; Kamaly, N.; Kalber, T.L.; Brody, L.P.; Sahuri, M.; Shamsaei, E.; Miller, A.D.; Bell, J.D. Novel multifunctional nanoparticle mediates siRNA tumour delivery, visualisation and therapeutic tumour reduction in vivo. J. Control. Release 2011, 149, 111–116. [Google Scholar] [CrossRef]
- Al-Deen, F.N.; Ho, J.; Selomulya, C.; Ma, C.; Coppel, R. Superparamagnetic Nanoparticles for Effective Delivery of Malaria DNA Vaccine. Langmuir 2011, 27, 3703–3712. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, T.; Li, L.; Wang, C.; Su, Z.; Li, J. Multifunctional fluorescent-magnetic polyethyleneimine functionalized Fe3O4–mesoporous silica yolk–shell nanocapsules for siRNA delivery. Chem. Commun. 2012, 48, 8706–8708. [Google Scholar] [CrossRef]
- Liu, W.-M.; Xue, Y.-N.; Peng, N.; He, W.-T.; Zhuo, R.-X.; Huang, S.-W. Dendrimer modified magnetic iron oxide nanoparticle/DNA/PEI ternary magnetoplexes: A novel strategy for magnetofection. J. Mater. Chem. 2011, 21, 13306–13315. [Google Scholar] [CrossRef]
- Veiseh, O.; Gunn, J.W.; Kievit, F.M.; Sun, C.; Fang, C.; Lee, J.S.; Zhang, M. Inhibition of Tumor-Cell Invasion with Chlorotoxin-Bound Superparamagnetic Nanoparticles. Small 2008, 5, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Chertok, B.; David, A.E.; Yang, V.C. Magnetically-enabled and MR-monitored selective brain tumor protein delivery in rats via magnetic nanocarriers. Biomaterials 2011, 32, 6245–6253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkpatrick, D.L.; Weiss, M.; Naumov, A.; Bartholomeusz, G.; Weisman, R.B.; Gliko, O. Carbon Nanotubes: Solution for the Therapeutic Delivery of siRNA? Materials 2012, 5, 278–301. [Google Scholar] [CrossRef] [Green Version]
- Rotoli, B.M.; Gatti, R.; Movia, D.; Bianchi, M.G.; Di Cristo, L.; Fenoglio, I.; Sonvico, F.; Bergamaschi, E.; Prina-Mello, A.; Bussolati, O. Identifying contact-mediated, localized toxic effects of MWCNT aggregates on epithelial monolayers: A single-cell monitoring toxicity assay. Nanotoxicology 2014, 9, 230–241. [Google Scholar] [CrossRef]
- Kang, B.; Chang, S.; Dai, Y.; Yu, D.; Chen, D. Cell Response to Carbon Nanotubes: Size-Dependent Intra-cellular Uptake Mechanism and Subcellular Fate. Small 2010, 6, 2362–2366. [Google Scholar] [CrossRef]
- Park, J.; Kim, W.J. Current status of gene delivery: Spotlight on nanomaterial-polymer hybrids. J. Drug Target. 2012, 20, 648–666. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, D.C.; Zhang, W.D.; Jiang, X.; He, C.B.; Chung, T.S.; Goh, S.H.; Leong, K.W. Polyethylenimine-Grafted Multiwalled Carbon Nanotubes for Secure Noncovalent Immobili-zation and Efficient Delivery of DNA. Angew. Chem. Int. Ed. 2005, 44, 4782–4785. [Google Scholar] [CrossRef]
- Liu, Y.-P.; Yu, Z.-L.; Zhang, Y.-M.; Guo, D.-S. Supramolecular Architectures of β-Cyclodextrin-Modified Chitosan and Pyrene Derivatives Mediated by Carbon Nanotubes and Their DNA Condensation. J. Am. Chem. Soc. 2008, 130, 10431–10439. [Google Scholar] [CrossRef]
- Kam, N.W.S.; Liu, Z.; Dai, H. Functionalization of Carbon Nanotubes via Cleavable Disulfide Bonds for Efficient Intracellular Delivery of siRNA and Potent Gene Silencing. J. Am. Chem. Soc. 2005, 127, 12492–12493. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, J.; Baigude, H.; Yang, C.-S.; Rana, T.M. Nanotubes Functionalized with Lipids and Natural Amino Acid Dendrimers: A New Strategy to Create Nanomaterials for Delivering Systemic RNAi. Bioconjug. Chem. 2009, 21, 56–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, C.H.; Dao, T.; Ahearn, I.; Fehrenbacher, N.; Casey, E.; Rey, D.A.; Korontsvit, T.; Zakhaleva, V.; Batt, C.A.; Philips, M.R.; et al. Single-Walled Carbon Nanotubes Deliver Peptide Antigen into Dendritic Cells and Enhance IgG Responses to Tumor-Associated Antigens. ACS Nano 2011, 5, 5300–5311. [Google Scholar] [CrossRef]
- Mu, Q.; Su, G.; Li, L.; Gilbertson, B.O.; Yu, L.H.; Zhang, Q.; Sun, Y.-P.; Yan, B. Size-Dependent Cell Uptake of Protein-Coated Graphene Oxide Nanosheets. ACS Appl. Mater. Interfaces 2012, 4, 2259–2266. [Google Scholar] [CrossRef]
- Liu, J.-H.; Yang, S.-T.; Wang, H.; Chang, Y.; Cao, A.; Liu, Y. Effect of size and dose on the biodistribution of graphene oxide in mice. Nanomedicine 2012, 7, 1801–1812. [Google Scholar] [CrossRef]
- Feng, L.; Zhang, S.; Liu, Z. Graphene based gene transfection. Nanoscale 2011, 3, 1252–1257. [Google Scholar] [CrossRef]
- Ren, T.; Li, L.; Cai, X.; Dong, H.; Liu, S.; Li, Y. Engineered polyethylenimine/graphene oxide nanocomposite for nuclear localized gene delivery. Polym. Chem. 2012, 3, 2561–2569. [Google Scholar] [CrossRef]
- Yang, X.; Niu, G.; Cao, X.; Wen, Y.; Xiang, R.; Duan, H.; Chen, Y. The preparation of functionalized graphene oxide for targeted intracellular delivery of siRNA. J. Mater. Chem. 2012, 22, 6649–6654. [Google Scholar] [CrossRef]
- Shen, H.; Liu, M.; He, H.; Zhang, L.; Huang, J.; Chong, Y.; Dai, J.; Zhang, Z. PEGylated Graphene Oxide-Mediated Protein Delivery for Cell Function Regulation. ACS Appl. Mater. Interfaces 2012, 4, 6317–6323. [Google Scholar] [CrossRef]
- Kargozar, S.; Mozafari, M. Nanotechnology and Nanomedicine: Start small, think big. Mater. Today Proc. 2018, 5, 15492–15500. [Google Scholar] [CrossRef]
- Roursgaard, M.; Poulsen, S.S.; Kepley, C.L.; Hammer, M.; Nielsen, G.D.; Larsen, S.T. Polyhydroxylated C60Fullerene (Fullerenol) Attenuates Neutrophilic Lung Inflammation in Mice. Basic Clin. Pharmacol. Toxicol. 2008, 103, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Sergio, M.; Behzadi, H.; Otto, A.; Van Der Spoel, D. Fullerenes toxicity and electronic properties. Environ. Chem. Lett. 2012, 11, 105–118. [Google Scholar] [CrossRef]
- Kazemzadeh, H.; Mozafari, M. Fullerene-based delivery systems. Drug Discov. Today 2019, 24, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, S.; Da Ros, T.; Conde, J.; Sefat, F.; Mozafari, M. Fullerene: Biomedical engineers get to revisit an old friend. Mater. Today 2017, 20, 460–480. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Gao, N.; Wang, X.; Ren, J.; Qu, X. Near-Infrared Switchable Fullerene-Based Synergy Therapy for Alzheimer’s Disease. Small 2018, 14, e1801852. [Google Scholar] [CrossRef]
- Wang, J.; Xie, L.; Wang, T.; Wu, F.; Meng, J.; Liu, J.; Xu, H. Visible light-switched cytosol release of siRNA by amphiphilic fullerene derivative to en-hance RNAi efficacy in vitro and in vivo. Acta Biomater. 2017, 59, 158–169. [Google Scholar] [CrossRef]
- Obonyo, O.; Fisher, E.; Edwards, M.; Douroumis, D. Quantum dots synthesis and biological applications as imaging and drug delivery systems. Crit. Rev. Biotechnol. 2010, 30, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Long, J.; Zhang, B.; Liu, C.; Ji, S.; Xu, J.; Yu, X.; Ni, Q. Quantum dots in cancer therapy. Expert Opin. Drug Deliv. 2011, 9, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Zhou, X.; Yang, J. TAT modified and lipid—PEI hybrid nanoparticles for co-delivery of docetaxel and Pdna. Biomed. Pharmacother. 2016, 84, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Yezhelyev, M.V.; Qi, L.; O’Regan, R.M.; Nie, S.; Gao, X. Proton-sponge coated quantum dots for siRNA delivery and intracellular imaging. J. Am. Chem. Soc. 2008, 130, 9006–9012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.; Gao, X. Quantum Dot−Amphipol Nanocomplex for Intracellular Delivery and Real-Time Imaging of siRNA. ACS Nano 2008, 2, 1403–1410. [Google Scholar] [CrossRef]
- Ramsey, J.D.; Flynn, N.H. Cell-penetrating peptides transport therapeutics into cells. Pharmacol. Ther. 2015, 154, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Kardani, K.; Milani, A.; Shabani, S.H.; Bolhassani, A. Cell penetrating peptides: The potent multi-cargo intracellular carriers. Expert Opin. Drug Deliv. 2019, 16, 1227–1258. [Google Scholar] [CrossRef]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.-E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, A.; Dowdy, S.F. Efficient siRNA delivery by novel PTD-DRBD fusion proteins. Cell Cycle 2010, 9, 424–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, A.; Meade, B.R.; Chang, Y.C.; Fredrickson, C.T.; Willert, K.; Puri, N.; Dowdy, S.F. Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein. Nat. Biotechnol. 2009, 27, 567–571. [Google Scholar] [CrossRef]
- Simeoni, F.; Morris, M.C.; Heitz, F.; Divita, G. Insight into the mechanism of the peptide-based gene de-livery system MPG: Implications for delivery of siRNA into mammalian cells. Nucleic Acids Res. 2003, 31, 2717–2724. [Google Scholar] [CrossRef] [Green Version]
- Trabulo, S.; Cardoso, A.L.; Mano, M.; De Lima, M.C.P. Cell-Penetrating Peptides—Mechanisms of Cellular Uptake and Generation of Delivery Systems. Pharmaceuticals 2010, 3, 961–993. [Google Scholar] [CrossRef] [Green Version]
- Lehto, T.; Kurrikoff, K.; Langel, Ü. Cell-penetrating peptides for the delivery of nucleic acids. Expert Opin. Drug Deliv. 2012, 9, 823–836. [Google Scholar] [CrossRef]
- Nakase, I.; Tanaka, G.; Futaki, S. Cell-penetrating peptides (CPPs) as a vector for the delivery of siRNAs into cells. Mol. BioSyst. 2013, 9, 855. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Xu, W.; Ding, Y.; Lu, S.; Chen, P. Uptake Mechanism and Direct Translocation of a New CPP for siRNA Delivery. Mol. Pharm. 2016, 13, 1366–1374. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a bio-logically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Pei, W.; Ge, H.; Liang, Q.; Luo, Y.; Sharp, F.R.; Lu, A.; Ran, R.; Graham, S.H.; Chen, J. In vivo delivery of a Bcl-xL fusion protein containing the TAT protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J. Neurosci. 2002, 22, 5423–5431. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, S.B.; Pereira, M.P.; Kelley, S.O. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv. Drug Deliv. Rev. 2009, 61, 953–964. [Google Scholar] [CrossRef]
- Kristensen, M.; Birch, D.; Nielsen, H.M. Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. Int. J. Mol. Sci. 2016, 17, 185. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Yamashita, H.; Misawa, T.; Nishida, K.; Kurihara, M.; Tanaka, M.; Demizu, Y.; Oba, M. Plasmid DNA delivery by arginine-rich cell-penetrating peptides containing unnatural amino acids. Bioorg. Med. Chem. 2016, 24, 2681–2687. [Google Scholar] [CrossRef]
- Gomes dos Reis, L.; Lee, W.H.; Svolos, M.; Moir, L.M.; Jaber, R.; Engel, A.; Windhab, N.; Young, P.M.; Traini, D. Delivery of pDNA to lung epithelial cells using PLGA nanoparticles formulated with a cell-penetrating peptide: Understanding the intracellular fate. Drug Dev. Ind. Pharm. 2020, 46, 427–442. [Google Scholar] [CrossRef]
- Suresh, B.; Ramakrishna, S.; Kim, H. Cell-Penetrating Peptide-Mediated Delivery of Cas9 Protein and Guide RNA for Genome Editing. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2016; Volume 1507, pp. 81–94. [Google Scholar]
- Uhl, P.; Grundmann, C.; Sauter, M.; Storck, P.; Tursch, A.; Özbek, S.; Leotta, K.; Roth, R.; Witzigmann, D.; Kulkarni, J.; et al. Coating of PLA-nanoparticles with cyclic, arginine-rich cell penetrating peptides enables oral delivery of liraglutide. Nanomed. Nanotechnol. Biol. Med. 2020, 24, 102132. [Google Scholar] [CrossRef]
- He, L.; Sayers, E.J.; Watson, P.; Jones, A.T. Contrasting roles for actin in the cellular uptake of cell pene-trating peptide conjugates. Sci. Rep. 2018, 8, 7318. [Google Scholar] [CrossRef]
- Khan, M.M.; Filipczak, N.; Torchilin, V.P. Cell penetrating peptides: A versatile vector for co-delivery of drug and genes in cancer. J. Control. Release 2021, 330, 1220–1228. [Google Scholar] [CrossRef]
- Gessner, I.; Klimpel, A.; Klußmann, M.; Neundorf, I.; Mathur, S. Interdependence of charge and secondary structure on cellular uptake of cell penetrating peptide functionalized silica nanoparticles. Nanoscale Adv. 2019, 2, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Allolio, C.; Magarkar, A.; Jurkiewicz, P.; Baxová, K.; Javanainen, M.; Mason, P.E.; Šachl, R.; Cebecauer, M.; Hof, M.; Horinek, D.; et al. Arginine-rich cell-penetrating peptides induce membrane multilamellarity and subse-quently enter via formation of a fusion pore. Proc. Natl. Acad. Sci. USA 2018, 115, 11923–11928. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.E.; Zahid, M. Cell Penetrating Peptides, Novel Vectors for Gene Therapy. Pharmaceutics 2020, 12, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuellar, M.; Cifuentes, J.; Perez, J.; Suarez-Arnedo, A.; Serna, J.A.; Groot, H.; Muñoz-Camargo, C.; Cruz, J.C. Novel BUF2-magnetite nanobioconjugates with cell-penetrating abilities. Int. J. Nanomed. 2018, 13, 8087–8094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Barbosa, N.; Suárez-Arnedo, A.; Cifuentes, J.; Gonzalez Barrios, A.F.; Silvera Batista, C.A.; Osma, J.F.; Muñoz-Camargo, C.; Cruz, J.C. Magnetite-OmpA nanobioconjugates as cell-penetrating vehicles with endosomal escape abilities. ACS Biomater. Sci. Eng. 2019, 6, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Pan, W.; Li, N.; Tang, B. A nuclear targeted dual-photosensitizer for drug-resistant cancer therapy with NIR activated multiple ROS. Chem. Sci. 2016, 7, 4237–4244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.-T.; Liu, Z.-K.; Zhu, Q.-L.; Rong, X.-H.; Liang, C.-L.; Wang, J.; Ma, D.; Sun, J.; Wang, G.-H. Redox-responsive nanocarriers for drug and gene co-delivery based on chitosan derivatives modified mesoporous silica nanoparticles. Colloids Surfaces B Biointerfaces 2017, 155, 41–50. [Google Scholar] [CrossRef]
- Ding, J.; Liang, T.; Zhou, Y.; He, Z.; Min, Q.; Jiang, L.; Zhu, J. Hyaluronidase-triggered anticancer drug and siRNA delivery from cascaded targeting nano-particles for drug-resistant breast cancer therapy. Nano Res. 2017, 10, 690–703. [Google Scholar] [CrossRef]
- Han, L.; Tang, C.; Yin, C. Dual-targeting and pH/redox-responsive multi-layered nanocomplexes for smart co-delivery of doxorubicin and siRNA. Biomaterials 2015, 60, 42–52. [Google Scholar] [CrossRef]
- Costa, D.; Albuquerque, T.; Queiroz, J.A.; Valente, A.J. A co-delivery platform based on plasmid DNA peptide-surfactant complexes: Formation, characterization and release behavior. Colloids Surfaces B Biointerfaces 2019, 178, 430–438. [Google Scholar] [CrossRef]
- Tang, Y.; Liang, J.; Wu, A.; Chen, Y.; Zhao, P.; Lin, T.; Zhang, M.; Xu, Q.; Wang, J.; Huang, Y. Co-Delivery of Trichosanthin and Albendazole by Nano-Self-Assembly for Overcoming Tu-mor Multidrug-Resistance and Metastasis. ACS Appl. Mater. Interfaces 2017, 9, 26648–26664. [Google Scholar] [CrossRef]
- Yang, Y.; Meng, Y.; Ye, J.; Xia, X.; Wang, H.; Li, L.; Dong, W.; Jin, D.; Liu, Y. Sequential delivery of VEGF siRNA and paclitaxel for PVN destruction, anti-angiogenesis, and tumor cell apoptosis procedurally via a multi-functional polymer micelle. J. Control. Release 2018, 287, 103–120. [Google Scholar] [CrossRef]
- Wang, X.; Wu, F.; Li, G.; Zhang, N.; Song, X.; Zheng, Y.; Gong, C.; Han, B.; He, G. Lipid-modified cell-penetrating peptide-based self-assembly micelles for co-delivery of nar-ciclasine and siULK1 in hepatocellular carcinoma therapy. Acta Biomater. 2018, 74, 414–429. [Google Scholar] [CrossRef]
- Wang, G.-H.; Cai, Y.-Y.; Du, J.-K.; Li, L.; Li, Q.; Yang, H.-K.; Lin, J.-T. TAT-conjugated chitosan cationic micelle for nuclear-targeted drug and gene co-delivery. Colloids Surfaces B Biointerfaces 2018, 162, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-T.; Chen, H.; Wang, D.; Xiong, L.; Li, J.-Z.; Chen, G.-H.; Chen, G.-B. Nuclear-targeted p53 and DOX co-delivery of chitosan derivatives for cancer therapy in vitro and in vivo. Colloids Surfaces B Biointerfaces 2019, 183, 110440. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, P.P.; Biswas, S.; Torchilin, V.P. Current trends in the use of liposomes for tumor targeting. Nanomedicine 2013, 8, 1509–1528. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Ran, R.; Zhang, L.; Liu, Y.; Mei, L.; Zhang, Z.; Gao, H.; He, Q. Simultaneous delivery of therapeutic antagomirs with paclitaxel for the management of metastatic tumors by a pH-responsive anti-microbial peptide-mediated liposomal delivery system. J. Control. Release 2015, 197, 208–218. [Google Scholar] [CrossRef]
- Yuan, M.; Qiu, Y.; Zhang, L.; Gao, H.; He, Q. Targeted delivery of transferrin and TAT co-modified lipo-somes encapsulating both paclitaxel and doxorubicin for melanoma. Drug Deliv. 2016, 23, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Lakkadwala, S.; Rodrigues, B.D.S.; Sun, C.; Singh, J. Biodistribution of TAT or QLPVM coupled to receptor targeted liposomes for delivery of anticancer therapeutics to brain in vitro and in vivo. Nanomed. Nanotechnol. Biol. Med. 2020, 23, 102112. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Vanegas, J.D.; Cruz, J.C.; Reyes, L.H. Delivery Systems for Nucleic Acids and Proteins: Barriers, Cell Capture Pathways and Nanocarriers. Pharmaceutics 2021, 13, 428. https://doi.org/10.3390/pharmaceutics13030428
Torres-Vanegas JD, Cruz JC, Reyes LH. Delivery Systems for Nucleic Acids and Proteins: Barriers, Cell Capture Pathways and Nanocarriers. Pharmaceutics. 2021; 13(3):428. https://doi.org/10.3390/pharmaceutics13030428
Chicago/Turabian StyleTorres-Vanegas, Julian D., Juan C. Cruz, and Luis H. Reyes. 2021. "Delivery Systems for Nucleic Acids and Proteins: Barriers, Cell Capture Pathways and Nanocarriers" Pharmaceutics 13, no. 3: 428. https://doi.org/10.3390/pharmaceutics13030428