
Cyanocobalamin Ultraflexible Lipid Vesicles: Characterization and In Vitro Evaluation of Drug-Skin Depth Profiles

 , , , and
, , , and
Abstract
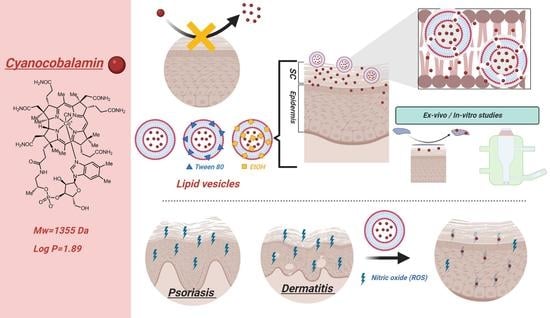
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cyanocobalamin Quantification. Development and Validation of a HPLC Method
2.3. Preparation of Liposomes, Transferosomes and Ethosomes
2.4. Characterization of the Lipid Vesicles
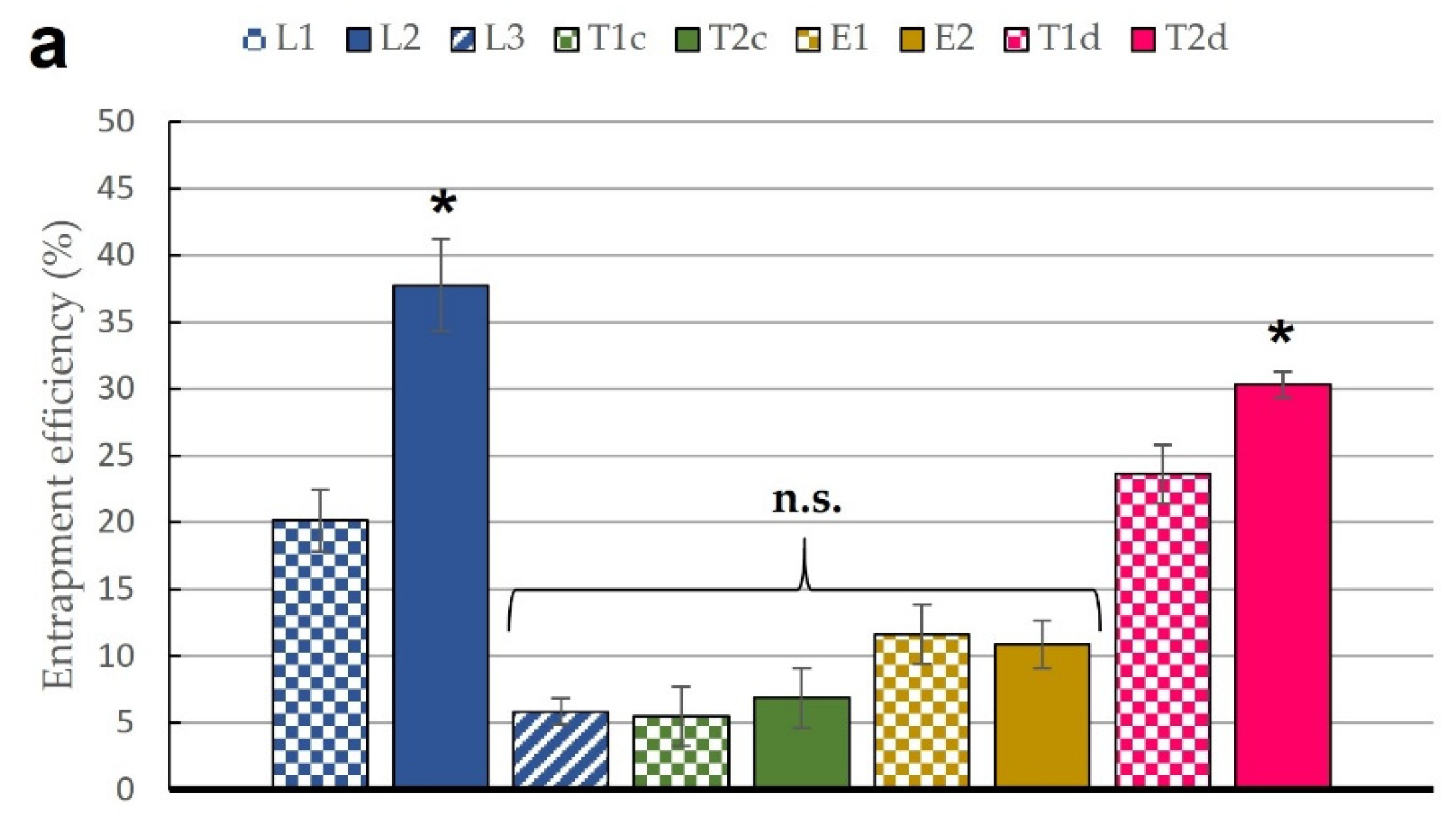
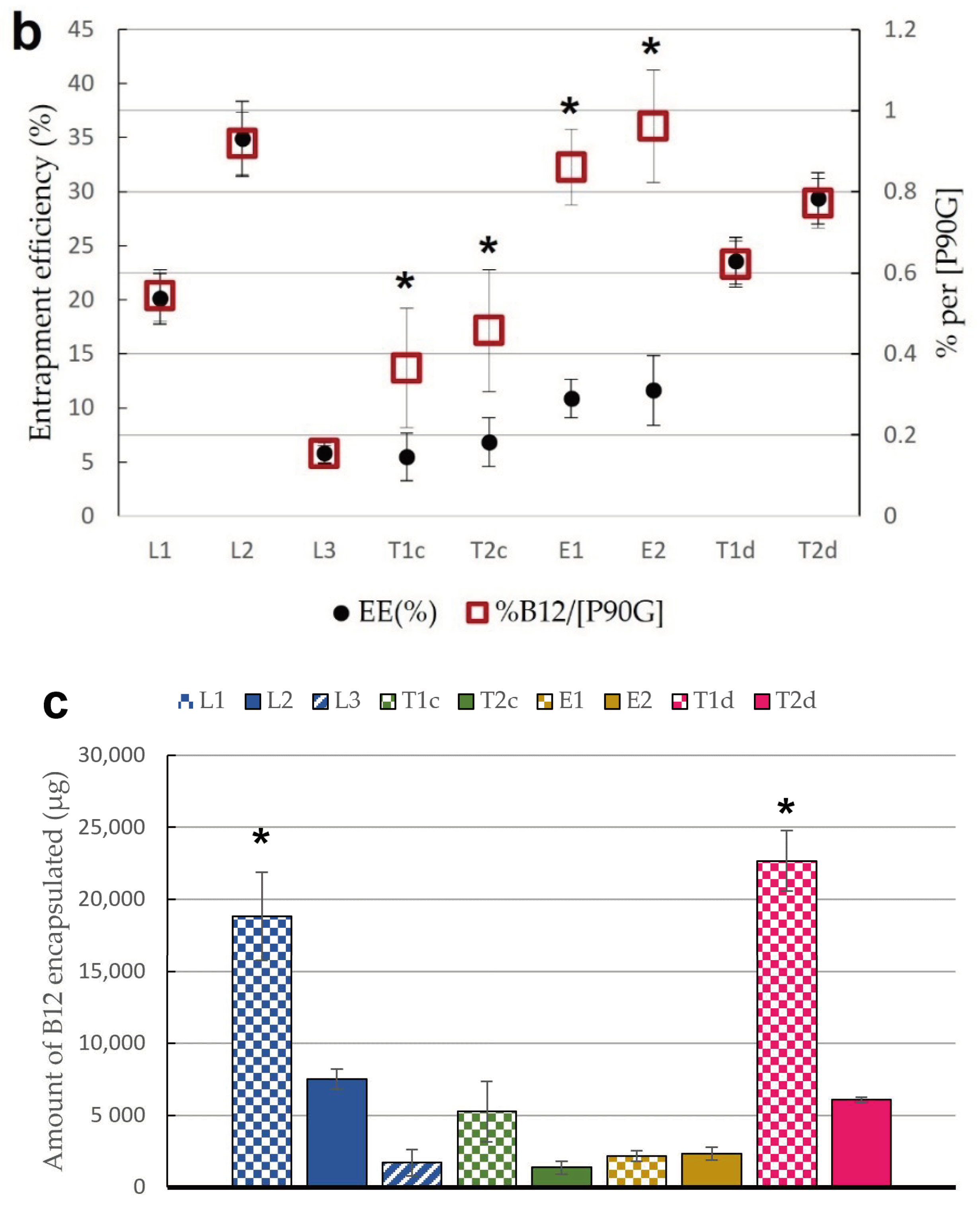
2.4.1. Determination of Entrapment Efficiency
2.4.2. Determination of Particle Size, Polidispersity Index (PDI) and Zeta-Potential
2.4.3. Determination of Phospholipid Content of Lipid Vesicles
2.4.4. Evaluation of Lipid Vesicle Flexibility
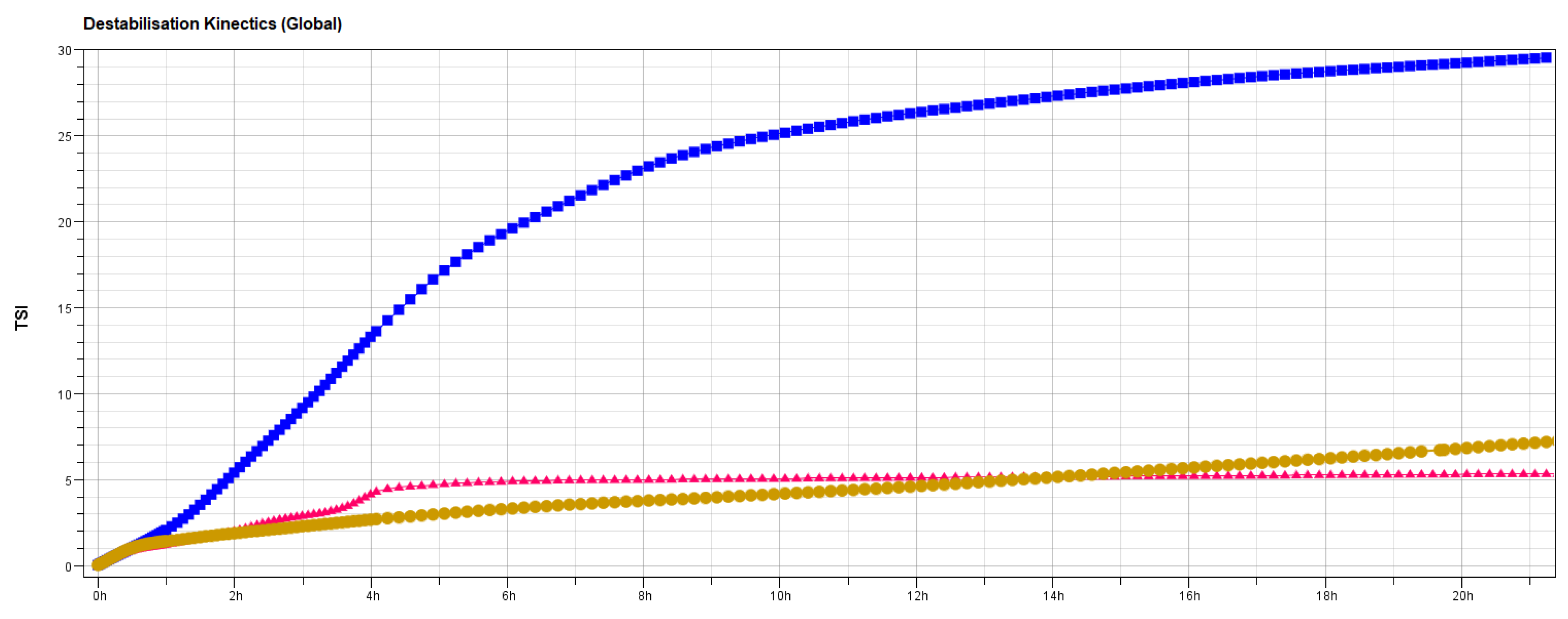
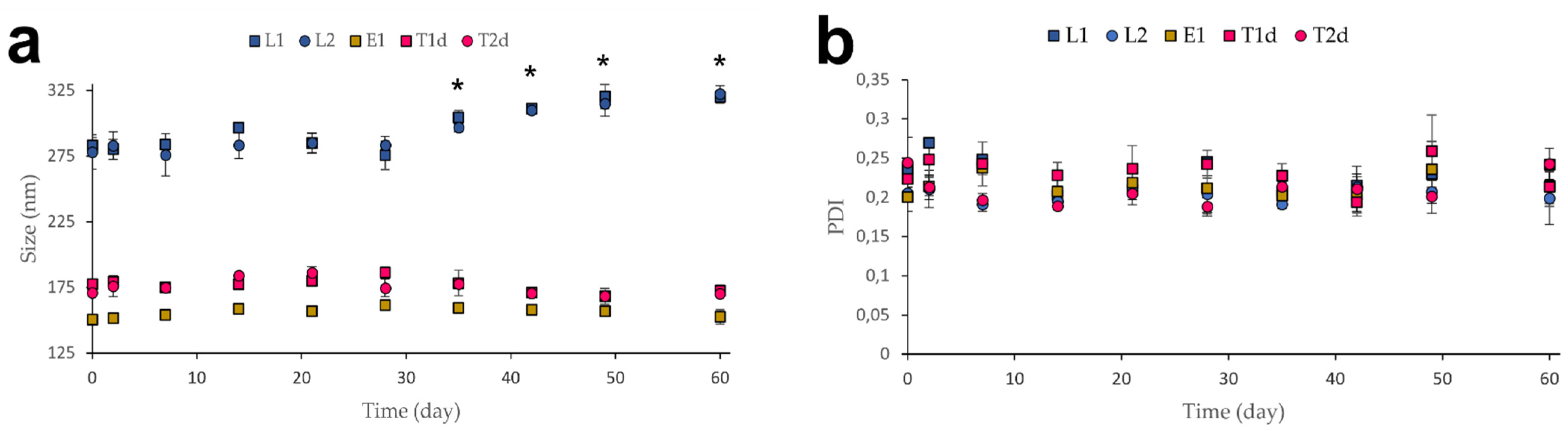
2.4.5. Stability Studies
2.5. Release Studies
2.6. Tape-Stripping Studies. Drug Penetration through the Skin and Stratum Corneum Depth
2.7. Data Analysis and Statistical Analysis
3. Results and Discussion
3.1. Characterization of Lipid Vesicles
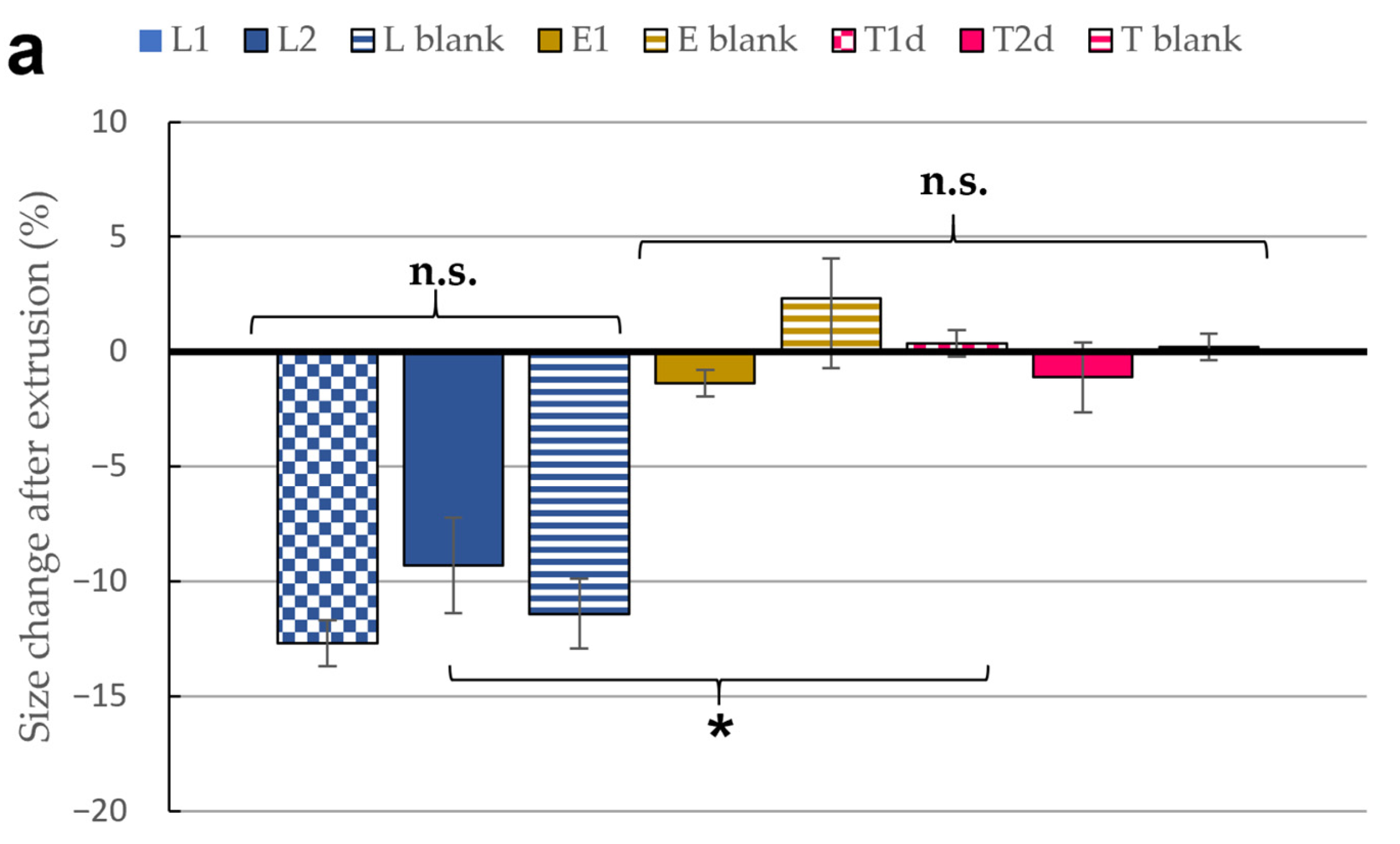
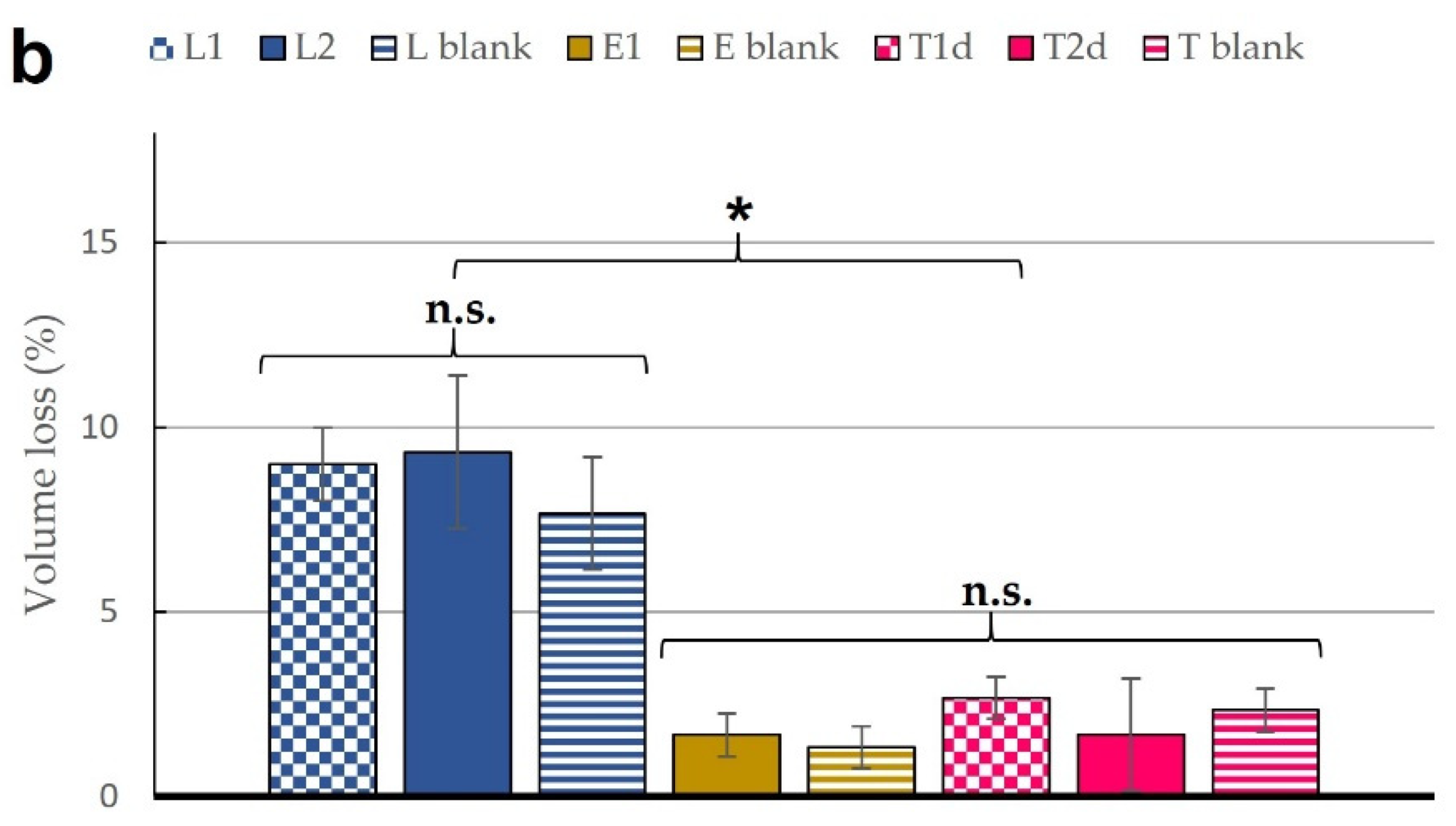
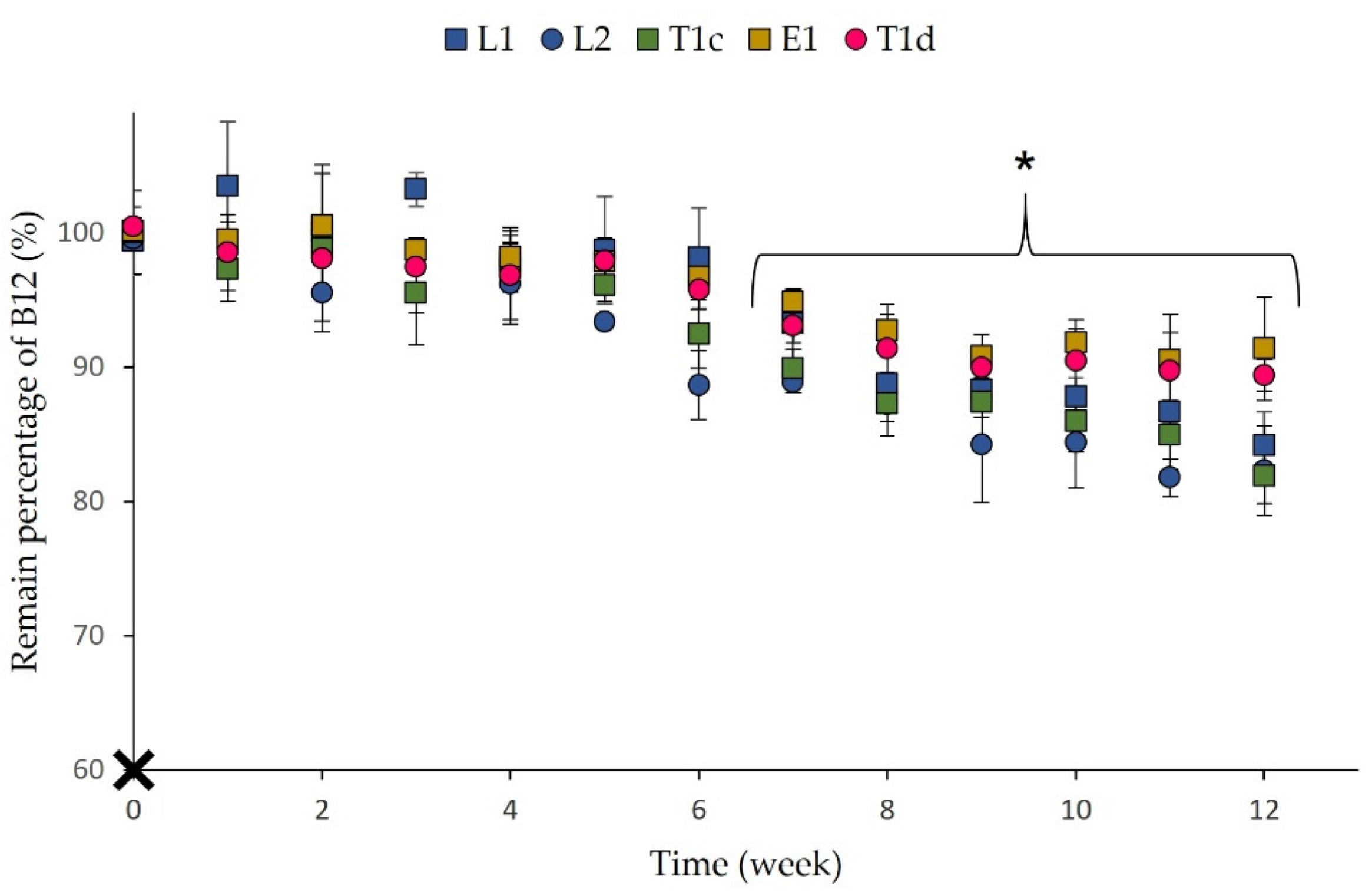
3.2. Stability Studies
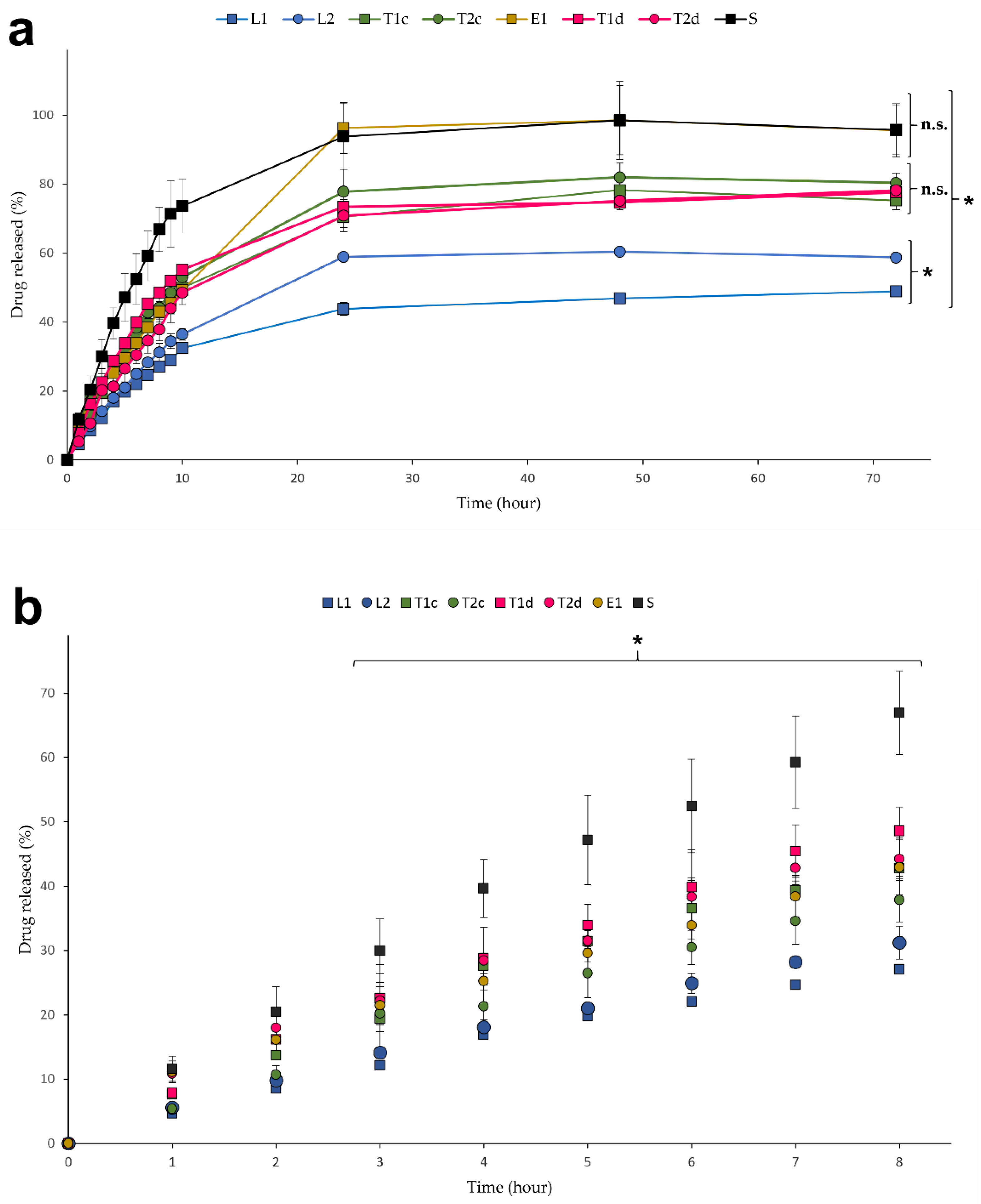
3.3. In Vitro Drug Release Studies
3.4. Model Fitting: Kinetic Drug Release Mechanisms
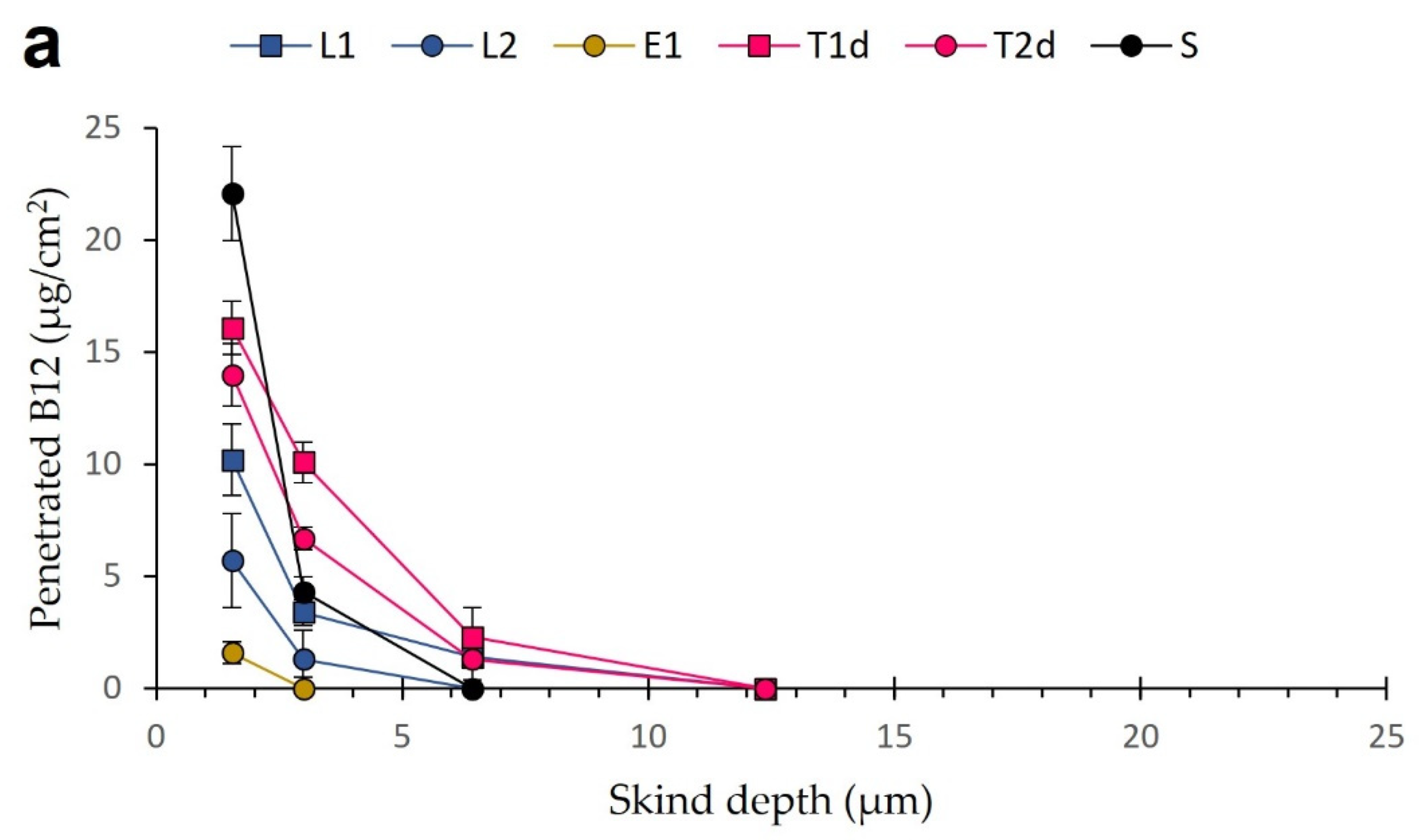
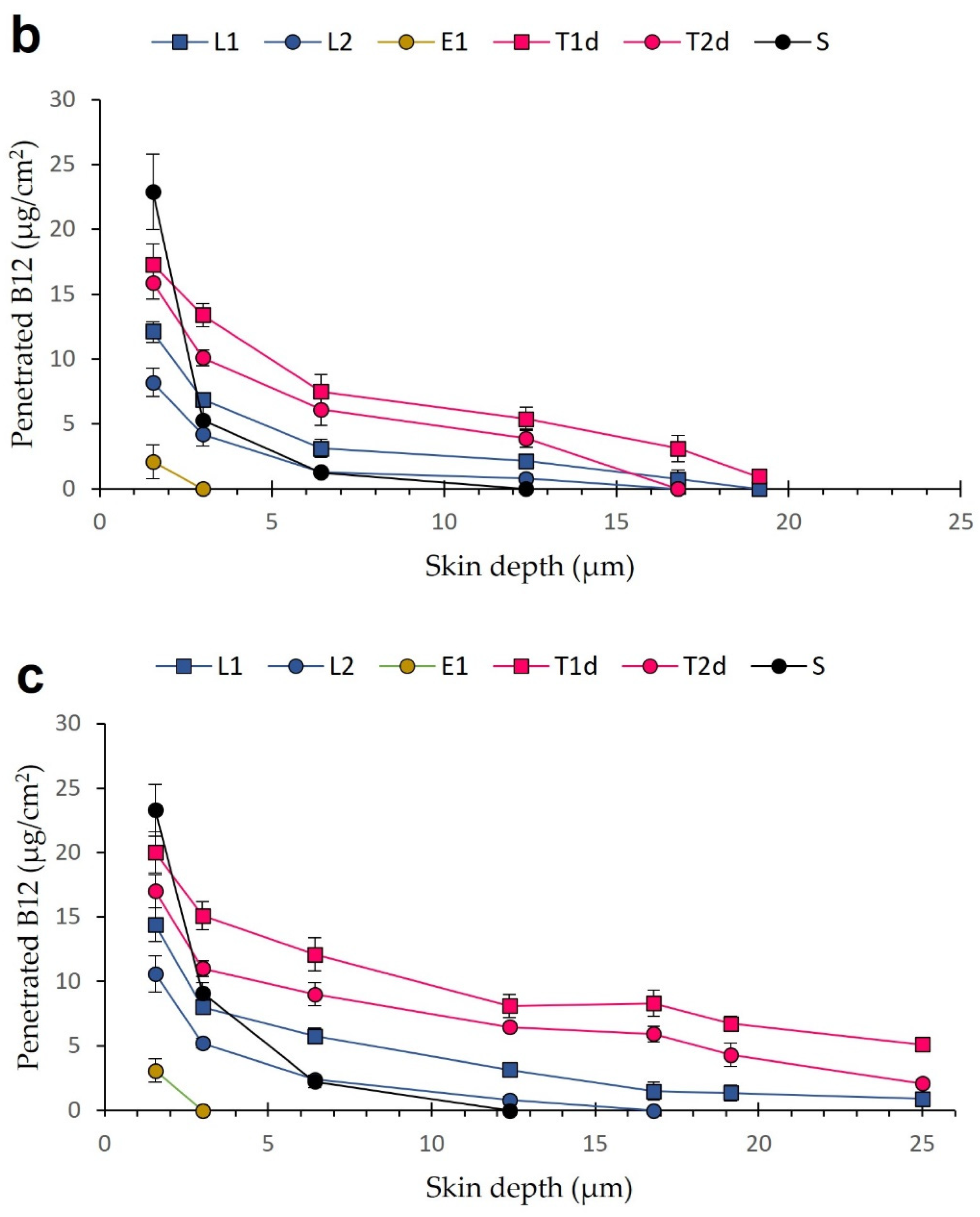
3.5. Tape-Stripping Drug Penetration Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adler-Neal, A.L.; Cline, A.; Frantz, T.; Strowd, L.; Feldman, S.R.; Taylor, S. Complementary and Integrative Therapies for Childhood Atopic Dermatitis. Children 2019, 6, 121. [Google Scholar] [CrossRef] [Green Version]
- Kapur, S.; Watson, W.; Carr, S. Atopic Dermatitis. Allergy Asthma Clin. Immunol. 2018, 14 (Suppl. 2). [Google Scholar] [CrossRef] [Green Version]
- Hirabayashi, T.; Anjo, T.; Kaneko, A.; Senoo, Y.; Shibata, A.; Takama, H.; Yokoyama, K.; Nishito, Y.; Ono, T.; Taya, C.; et al. PNPLA1 Has a Crucial Role in Skin Barrier Function by Directing Acylceramide Biosynthesis. Nat. Commun. 2017, 8, 14609. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Woodfolk, J.A. Skin Barrier Defects in Atopic Dermatitis. Curr. Allergy Asthma Rep. 2014, 14, 433. [Google Scholar] [CrossRef]
- Zaniboni, M.C.; Samorano, L.P.; Orfali, R.L.; Aoki, V. Skin Barrier in Atopic Dermatitis: Beyond Filaggrin. An. Bras. Dermatol. 2016, 91, 472–478. [Google Scholar] [CrossRef]
- Kolb, L.; Ferrer-Bruker, S.J. Atopic Dermatitis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Parisi, R.; Symmons, D.P.M.; Griffiths, C.E.M.; Ashcroft, D.M.; Identification and Management of Psoriasis and Associated ComorbidiTy (IMPACT) Project Team. Global Epidemiology of Psoriasis: A Systematic Review of Incidence and Prevalence. J. Investig. Dermatol. 2013, 133, 377–385. [Google Scholar] [CrossRef] [Green Version]
- Arima, K.; Ohta, S.; Takagi, A.; Shiraishi, H.; Masuoka, M.; Ontsuka, K.; Suto, H.; Suzuki, S.; Yamamoto, K.-I.; Ogawa, M.; et al. Periostin Contributes to Epidermal Hyperplasia in Psoriasis Common to Atopic Dermatitis. Allergol. Int. 2015, 64, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Heidenreich, R.; Röcken, M.; Ghoreschi, K. Angiogenesis Drives Psoriasis Pathogenesis. Int. J. Exp. Pathol. 2009, 90, 232–248. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, L.; Chen, Y.L.; Ogg, G.S. Role of Regulatory T Cells in Psoriasis Pathogenesis and Treatment. Br. J. Dermatol. 2021, 184, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Badri, T.; Kumar, P.; Oakley, A.M. Plaque Psoriasis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Thomsen, S.F. Atopic Dermatitis: Natural History, Diagnosis, and Treatment. Int. Sch. Res. Not. 2014, 2014, 354250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiore, M.; Leone, S.; Maraolo, A.E.; Berti, E.; Damiani, G. Liver Illness and Psoriatic Patients. BioMed Res. Int. 2018, 2018, 3140983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandinetti, V.; Baraldi, O.; Comai, G.; Corradetti, V.; Aiello, V.; Bini, C.; Minerva, V.; Barbuto, S.; Fabbrizio, B.; Donati, G.; et al. Renal Dysfunction in Psoriatic Patients. G. Ital. Nefrol. 2020, 37, 1–12. [Google Scholar]
- Jindal, N.; Arora, K.; Jindal, P.; Jain, V.K.; Ghosh, S. Inflamed Psoriatic Plaques: Drug Toxicity or Disease Exacerbation? Indian J. Pharmacol. 2013, 45, 410–411. [Google Scholar] [CrossRef]
- Oray, M.; Abu Samra, K.; Ebrahimiadib, N.; Meese, H.; Foster, C.S. Long-Term Side Effects of Glucocorticoids. Expert Opin. Drug Saf. 2016, 15, 457–465. [Google Scholar] [CrossRef]
- Siegfried, E.C.; Jaworski, J.C.; Hebert, A.A. Topical Calcineurin Inhibitors and Lymphoma Risk: Evidence Update with Implications for Daily Practice. Am. J. Clin. Dermatol. 2013, 14, 163–178. [Google Scholar] [CrossRef] [Green Version]
- Sirsjö, A.; Karlsson, M.; Gidlöf, A.; Rollman, O.; Törmä, H. Increased Expression of Inducible Nitric Oxide Synthase in Psoriatic Skin and Cytokine-Stimulated Cultured Keratinocytes. Br. J. Dermatol. 1996, 134, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Brescoll, J.; Daveluy, S. A Review of Vitamin B12 in Dermatology. Am. J. Clin. Dermatol. 2015, 16, 27–33. [Google Scholar] [CrossRef]
- Baker, H.; Comaish, J.S. Is Vitamin B12 of Value in Psoriasis? Br. Med. J. 1962, 2, 1729–1730. [Google Scholar] [CrossRef] [Green Version]
- Januchowski, R. Evaluation of Topical Vitamin B(12) for the Treatment of Childhood Eczema. J. Altern Complement. Med. 2009, 15, 387–389. [Google Scholar] [CrossRef]
- Stücker, M.; Memmel, U.; Hoffmann, M.; Hartung, J.; Altmeyer, P. Vitamin B(12) Cream Containing Avocado Oil in the Therapy of Plaque Psoriasis. Dermatology 2001, 203, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Del Duca, E.; Farnetani, F.; De Carvalho, N.; Bottoni, U.; Pellacani, G.; Nisticò, S.P. Superiority of a Vitamin B12-Containing Emollient Compared to a Standard Emollient in the Maintenance Treatment of Mild-to-Moderate Plaque Psoriasis. Int. J. Immunopathol. Pharmacol. 2017, 30, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Moore, R.; Harry, R. Harry’s Cosmeticology, 7th ed.; Chemical Publishing: New York, NY, USA, 1982; pp. 1–41. [Google Scholar]
- Paudel, K.S.; Milewski, M.; Swadley, C.L.; Brogden, N.K.; Ghosh, P.; Stinchcomb, A.L. Challenges and Opportunities in Dermal/Transdermal Delivery. Ther. Deliv. 2010, 1, 109–131. [Google Scholar] [CrossRef] [Green Version]
- Howe, E.E.; Dooley, C.L.; Geoffroy, R.F.; Rosenblum, C. Percutaneous Absorption of Vitamin B12 in the Rat and Guinea Pig. J. Nutr. 1967, 92, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kalluri, H.; Banga, A.K. Effects of Chemical and Physical Enhancement Techniques on Transdermal Delivery of Cyanocobalamin (Vitamin B12) In Vitro. Pharmaceutics 2011, 3, 474–484. [Google Scholar] [CrossRef] [Green Version]
- Ramöller, I.K.; Tekko, I.A.; McCarthy, H.O.; Donnelly, R.F. Rapidly Dissolving Bilayer Microneedle Arrays—A Minimally Invasive Transdermal Drug Delivery System for Vitamin B12. Int. J. Pharm. 2019, 566, 299–306. [Google Scholar] [CrossRef]
- Kim, M.-H.; Jeon, Y.-E.; Kang, S.; Lee, J.-Y.; Lee, K.W.; Kim, K.-T.; Kim, D.-D. Lipid Nanoparticles for Enhancing the Physicochemical Stability and Topical Skin Delivery of Orobol. Pharmaceutics 2020, 12, 845. [Google Scholar] [CrossRef]
- Yotsumoto, K.; Ishii, K.; Kokubo, M.; Yasuoka, S. Improvement of the Skin Penetration of Hydrophobic Drugs by Polymeric Micelles. Int. J. Pharm. 2018, 553, 132–140. [Google Scholar] [CrossRef]
- Cutlar, L.; Zhou, D.; Hu, X.; Duarte, B.; Greiser, U.; Larcher, F.; Wang, W. A Non-Viral Gene Therapy for Treatment of Recessive Dystrophic Epidermolysis Bullosa. Exp. Dermatol. 2016, 25, 818–820. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.; Alshehri, F.; Zhou, D.; Lara-Sáez, I.; Wang, X.; Li, X.; Xu, Q.; Zhang, J.; Wang, W. Efficient and Robust Highly Branched Poly(β-Amino Ester)/Minicircle COL7A1 Polymeric Nanoparticles for Gene Delivery to Recessive Dystrophic Epidermolysis Bullosa Keratinocytes. ACS Appl. Mater. Interfaces 2019, 11, 30661–30672. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Gao, Y.; Aied, A.; Cutlar, L.; Igoucheva, O.; Newland, B.; Alexeeve, V.; Greiser, U.; Uitto, J.; Wang, W. Highly Branched Poly(β-Amino Ester)s for Skin Gene Therapy. J. Control. Release 2016, 244, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Zhou, D.; Alshehri, F.; Lara-Sáez, I.; Lyu, Y.; Creagh-Flynn, J.; Xu, Q.; Zhang, J.; Wang, W. Manipulation of Transgene Expression in Fibroblast Cells by a Multifunctional Linear-Branched Hybrid Poly(β-Amino Ester) Synthesized through an Oligomer Combination Approach. Nano Lett. 2018, 19, 381–391. [Google Scholar] [CrossRef]
- Mostafa, M.; Alaaeldin, E.; Aly, U.F.; Sarhan, H.A. Optimization and Characterization of Thymoquinone-Loaded Liposomes with Enhanced Topical Anti-Inflammatory Activity. AAPS PharmSciTech 2018, 19, 3490–3500. [Google Scholar] [CrossRef] [PubMed]
- Peralta, M.F.; Guzmán, M.L.; Pérez, A.P.; Apezteguia, G.A.; Fórmica, M.L.; Romero, E.L.; Olivera, M.E.; Carrer, D.C. Liposomes Can Both Enhance or Reduce Drugs Penetration through the Skin. Sci. Rep. 2018, 8, 13253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozzuto, G.; Molinari, A. Liposomes as Nanomedical Devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [Green Version]
- Cevc, G.; Blume, G. Lipid Vesicles Penetrate into Intact Skin Owing to the Transdermal Osmotic Gradients and Hydration Force. Biochim. Biophys. Acta 1992, 1104, 226–232. [Google Scholar] [CrossRef]
- Touitou, E.; Godin, B. Ethosomes for Skin Delivery. J. Drug Deliv. Sci. Technol. 2007, 17, 303–308. [Google Scholar] [CrossRef]
- Arsalan, A.; Ahmad, I.; Ali, S.A.; Qadeer, K.; Mahmud, S.; Humayun, F.; Beg, A.E. The Kinetics of Photostabilization of Cyanocobalamin in Liposomal Preparations. Int. J. Chem. Kinet. 2020, 52, 207–217. [Google Scholar] [CrossRef]
- Vitetta, L.; Zhou, J.; Manuel, R.; Dal Forno, S.; Hall, S.; Rutolo, D. Route and Type of Formulation Administered Influences the Absorption and Disposition of Vitamin B12 Levels in Serum. J. Funct. Biomater. 2018, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Castoldi, A.; Herr, C.; Niederstraßer, J.; Labouta, H.I.; Melero, A.; Gordon, S.; Schneider-Daum, N.; Bals, R.; Lehr, C.-M. Calcifediol-Loaded Liposomes for Local Treatment of Pulmonary Bacterial Infections. Eur. J. Pharm. Biopharm. 2017, 118, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Ahad, A.; Al-Saleh, A.A.; Al-Mohizea, A.M.; Al-Jenoobi, F.I.; Raish, M.; Yassin, A.E.B.; Alam, M.A. Formulation and Characterization of Novel Soft Nanovesicles for Enhanced Transdermal Delivery of Eprosartan Mesylate. Saudi Pharm. J. 2017, 25, 1040–1046. [Google Scholar] [CrossRef]
- Touitou, E.; Dayan, N.; Bergelson, L.; Godin, B.; Eliaz, M. Ethosomes—Novel Vesicular Carriers for Enhanced Delivery: Characterization and Skin Penetration Properties. J. Control. Release 2000, 65, 403–418. [Google Scholar] [CrossRef]
- El Maghraby, G.M.; Williams, A.C.; Barry, B.W. Oestradiol Skin Delivery from Ultradeformable Liposomes: Refinement of Surfactant Concentration. Int. J. Pharm. 2000, 196, 63–74. [Google Scholar] [CrossRef]
- Ong, S.G.M.; Ming, L.C.; Lee, K.S.; Yuen, K.H. Influence of the Encapsulation Efficiency and Size of Liposome on the Oral Bioavailability of Griseofulvin-Loaded Liposomes. Pharmaceutics 2016, 8, 25. [Google Scholar] [CrossRef]
- Lin, M.; Qi, X.-R. Purification Method of Drug-Loaded Liposome. In Liposome-Based Drug Delivery Systems; Lu, W.-L., Qi, X.-R., Eds.; Biomaterial Engineering; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar] [CrossRef]
- Panwar, P.; Pandey, B.; Lakhera, P.C.; Singh, K.P. Preparation, Characterization, and In Vitro Release Study of Albendazole-Encapsulated Nanosize Liposomes. Int. J. Nanomed. 2010, 5, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Keller, S.; Heerklotz, H.; Jahnke, N.; Blume, A. Thermodynamics of Lipid Membrane Solubilization by Sodium Dodecyl Sulfate. Biophys. J. 2006, 90, 4509–4521. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Borrell, J.; Pons, M.; Juarez, J.C.; Estelrich, J. The Action of Triton X-100 and Sodium Dodecyl Sulphate on Lipid Layers. Effect on Monolayers and Liposomes. J. Microencapsul. 1990, 7, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, T.; Shoji, Y.; Katayama, K. Anomalous Solubilization Behavior of Dimyristoylphosphatidylcholine Liposomes Induced by Sodium Dodecyl Sulfate Micelles. Anal. Sci. 2012, 28, 345–350. [Google Scholar] [CrossRef] [Green Version]
- Sabeti, B.; Noordin, M.I.; Mohd, S.; Hashim, R.; Dahlan, A.; Javar, H.A. Development and Characterization of Liposomal Doxorubicin Hydrochloride with Palm Oil. Biomed Res. Int. 2014, 2014, 765426. [Google Scholar] [CrossRef]
- Rouser, G.; Fkeischer, S.; Yamamoto, A. Two Dimensional Then Layer Chromatographic Separation of Polar Lipids and Determination of Phospholipids by Phosphorus Analysis of Spots. Lipids 1970, 5, 494–496. [Google Scholar] [CrossRef]
- Jain, S.; Jain, P.; Umamaheshwari, R.B.; Jain, N.K. Transfersomes--a Novel Vesicular Carrier for Enhanced Transdermal Delivery: Development, Characterization, and Performance Evaluation. Drug Dev. Ind. Pharm. 2003, 29, 1013–1026. [Google Scholar] [CrossRef]
- Celia, C.; Trapasso, E.; Cosco, D.; Paolino, D.; Fresta, M. Turbiscan Lab Expert Analysis of the Stability of Ethosomes and Ultradeformable Liposomes Containing a Bilayer Fluidizing Agent. Colloids Surf. B Biointerfaces 2009, 72, 155–160. [Google Scholar] [CrossRef]
- Nava, G.; Piñón, E.; Mendoza, L.; Mendoza, N.; Quintanar, D.; Ganem, A. Formulation and in Vitro, Ex Vivo and in Vivo Evaluation of Elastic Liposomes for Transdermal Delivery of Ketorolac Tromethamine. Pharmaceutics 2011, 3, 954–970. [Google Scholar] [CrossRef] [Green Version]
- Akaike, H. Information Theory and an Extension of the Maximum Likelihood Principle. In Selected Papers of Hirotugu Akaike; Parzen, E., Tanabe, K., Kitagawa, G., Eds.; Springer Series in Statistics; Springer: New York, NY, USA, 1998; pp. 199–213. [Google Scholar] [CrossRef]
- Hamilton, D.F.; Ghert, M.; Simpson, A.H.R.W. Interpreting Regression Models in Clinical Outcome Studies. Bone Joint Res. 2015, 4, 152–153. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, T. Rate of Release of Medicaments from Ointment Bases Containing Drugs in Suspension. J. Pharm. Sci. 1961, 50, 874–875. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A Simple Equation for Description of Solute Release I. Fickian and Non-Fickian Release from Non-Swellable Devices in the Form of Slabs, Spheres, Cylinders or Discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A Simple Equation for Description of Solute Release II. Fickian and Anomalous Release from Swellable Devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Kim, H.; Fassihi, R. Application of Binary Polymer System in Drug Release Rate Modulation. 2. Influence of Formulation Variables and Hydrodynamic Conditions on Release Kinetics. J. Pharm. Sci. 1997, 86, 323–328. [Google Scholar] [CrossRef]
- Peppas, N.A.; Sahlin, J.J. A Simple Equation for the Description of Solute Release. III. Coupling of Diffusion and Relaxation. Int. J. Pharm. 1989, 57, 169–172. [Google Scholar] [CrossRef]
- Jain, A.; Jain, S.K. In vitro release kinetics model fitting of liposomes: An insight. Chem. Phys. Lipids 2016, 201, 28–40. [Google Scholar] [CrossRef]
- Carolina Oliveira dos Santos, L.; Spagnol, C.M.; Guillot, A.J.; Melero, A.; Corrêa, M.A. Caffeic Acid Skin Absorption: Delivery of Microparticles to Hair Follicles. Saudi Pharm. J. 2019, 27, 791–797. [Google Scholar] [CrossRef]
- Melero, A.; Hahn, T.; Schaefer, U.F.; Schneider, M. In Vitro Human Skin Segmentation and Drug Concentration-Skin Depth Profiles. Methods Mol. Biol. 2011, 763, 33–50. [Google Scholar] [CrossRef]
- Melero, A.; Ferreira Ourique, A.; Stanisçuaski Guterres, S.; Raffin Pohlmann, A.; Lehr, C.-M.; Ruver Beck, R.C.; Schaefer, U. Nanoencapsulation in Lipid-Core Nanocapsules Controls Mometasone Furoate Skin Permeability Rate and Its Penetration to the Deeper Skin Layers. Skin Pharmacol. Physiol. 2014, 27, 217. [Google Scholar] [CrossRef]
- Shaker, S.; Gardouh, A.R.; Ghorab, M.M. Factors Affecting Liposomes Particle Size Prepared by Ethanol Injection Method. Res. Pharm. Sci. 2017, 12, 346–352. [Google Scholar] [CrossRef]
- Lee, S.-C.; Lee, K.-E.; Kim, J.-J.; Lim, S.-H. The Effect of Cholesterol in the Liposome Bilayer on the Stabilization of Incorporated Retinol. J. Liposome Res. 2005, 15, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Coderch, L.; Fonollosa, J.; De Pera, M.; Estelrich, J.; De La Maza, A.; Parra, J.L. Influence of Cholesterol on Liposome Fluidity by EPR. Relationship with Percutaneous Absorption. J. Control. Release 2000, 68, 85–95. [Google Scholar] [CrossRef]
- Opatha, S.A.T.; Titapiwatanakun, V.; Chutoprapat, R. Transfersomes: A Promising Nanoencapsulation Technique for Transdermal Drug Delivery. Pharmaceutics 2020, 12, 855. [Google Scholar] [CrossRef]
- Fernández-García, R.; Lalatsa, A.; Statts, L.; Bolás-Fernández, F.; Ballesteros, M.P.; Serrano, D.R. Transferosomes as Nanocarriers for Drugs across the Skin: Quality by Design from Lab to Industrial Scale. Int. J. Pharm. 2020, 573, 118817. [Google Scholar] [CrossRef]
- Ascenso, A.; Raposo, S.; Batista, C.; Cardoso, P.; Mendes, T.; Praça, F.G.; Bentley, M.V.L.B.; Simões, S. Development, Characterization, and Skin Delivery Studies of Related Ultradeformable Vesicles: Transfersomes, Ethosomes, and Transethosomes. Int. J. Nanomed. 2015, 10, 5837–5851. [Google Scholar] [CrossRef] [Green Version]
- Wu, I.Y.; Bala, S.; Škalko-Basnet, N.; di Cagno, M.P. Interpreting Non-Linear Drug Diffusion Data: Utilizing Korsmeyer-Peppas Model to Study Drug Release from Liposomes. Eur. J. Pharm. Sci. 2019, 138, 105026. [Google Scholar] [CrossRef]
- Carreras, J.J.; Tapia-Ramirez, W.E.; Sala, A.; Guillot, A.J.; Garrigues, T.M.; Melero, A. Ultraflexible Lipid Vesicles Allow Topical Absorption of Cyclosporin A. Drug Deliv. Transl. Res. 2020, 10, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Han, D.; Cai, C.; Tang, X. An Overview of Liposome Lyophilization and Its Future Potential. J. Control. Release 2010, 142, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Magarkar, A.; Dhawan, V.; Kallinteri, P.; Viitala, T.; Elmowafy, M.; Róg, T.; Bunker, A. Cholesterol Level Affects Surface Charge of Lipid Membranes in Saline Solution. Sci. Rep. 2014, 4, 5005. [Google Scholar] [CrossRef] [Green Version]
- Abdulbaqi, I.M.; Darwis, Y.; Khan, N.A.K.; Assi, R.A.; Khan, A.A. Ethosomal Nanocarriers: The Impact of Constituents and Formulation Techniques on Ethosomal Properties, in Vivo Studies, and Clinical Trials. Int. J. Nanomed. 2016, 11, 2279–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iskandaryah, I.; Masrijal, C.; Harmita, H. Effects of sonication on size distribution and entrapment of lynestrenol transferosome. Int. J. Appl. Pharm. 2020, 245–247. [Google Scholar] [CrossRef]
- Xu, X.; Khan, M.A.; Burgess, D.J. Predicting Hydrophilic Drug Encapsulation inside Unilamellar Liposomes. Int. J. Pharm. 2012, 423, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Kirby, C.; Gregoriadis, G. Dehydration-Rehydration Vesicles: A Simple Method for High Yield Drug Entrapment in Liposomes. Nat. Biotechnology 1984, 2, 979–984. [Google Scholar] [CrossRef]
- Melero, A.; Garrigues, T.M.; Almudever, P.; Villodre, A.M.; Lehr, C.M.; Schäfer, U. Nortriptyline Hydrochloride Skin Absorption: Development of a Transdermal Patch. Eur. J. Pharm. Biopharm. 2008, 69, 588–596. [Google Scholar] [CrossRef]
- Wu, C.-S.; Guo, J.-H.; Lin, M.-J. Stability Evaluation of PH-Adjusted Goat Milk for Developing Ricotta Cheese with a Mixture of Cow Cheese Whey and Goat Milk. Foods 2020, 9, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristiano, M.C.; Froiio, F.; Spaccapelo, R.; Mancuso, A.; Nisticò, S.P.; Udongo, B.P.; Fresta, M.; Paolino, D. Sulforaphane-Loaded Ultradeformable Vesicles as A Potential Natural Nanomedicine for the Treatment of Skin Cancer Diseases. Pharmaceutics 2019, 12, 6. [Google Scholar] [CrossRef] [Green Version]
- Elsana, H.; Olusanya, T.O.B.; Carr-Wilkinson, J.; Darby, S.; Faheem, A.; Elkordy, A.A. Evaluation of Novel Cationic Gene Based Liposomes with Cyclodextrin Prepared by Thin Film Hydration and Microfluidic Systems. Sci. Rep. 2019, 9, 15120. [Google Scholar] [CrossRef] [Green Version]
- Liang, T.; Guan, R.; Quan, Z.; Tao, Q.; Liu, Z.; Hu, Q. Cyanidin-3-o-Glucoside Liposome: Preparation via a Green Method and Antioxidant Activity in GES-1 Cells. Food Res. Int. 2019, 125, 108648. [Google Scholar] [CrossRef] [PubMed]
- Muppidi, K.; Pumerantz, A.S.; Wang, J.; Betageri, G. Development and Stability Studies of Novel Liposomal Vancomycin Formulations. Int. Sch. Res. Not. 2012, 2012, 636743. [Google Scholar] [CrossRef] [Green Version]
- Franzé, S.; Selmin, F.; Samaritani, E.; Minghetti, P.; Cilurzo, F. Lyophilization of Liposomal Formulations: Still Necessary, Still Challenging. Pharmaceutics 2018, 10, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Liu, X.; Fahr, A. Skin Penetration and Deposition of Carboxyfluorescein and Temoporfin from Different Lipid Vesicular Systems: In Vitro Study with Finite and Infinite Dosage Application. Int. J. Pharm. 2011, 408, 223–234. [Google Scholar] [CrossRef]
- Cunha, S.; Costa, C.P.; Loureiro, J.A.; Alves, J.; Peixoto, A.F.; Forbes, B.; Sousa Lobo, J.M.; Silva, A.C. Double Optimization of Rivastigmine-Loaded Nanostructured Lipid Carriers (NLC) for Nose-to-Brain Delivery Using the Quality by Design (QbD) Approach: Formulation Variables and Instrumental Parameters. Pharmaceutics 2020, 12, 599. [Google Scholar] [CrossRef]
- Spagnol, C.M.; Zaera, A.M.; Isaac, V.L.B.; Corrêa, M.A.; Salgado, H.R.N. Release and Permeation Profiles of Spray-Dried Chitosan Microparticles Containing Caffeic Acid. Saudi Pharm. J. 2018, 26, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Gulati, K.; Kant, K.; Findlay, D.; Losic, D. Periodically Tailored Titania Nanotubes for Enhanced Drug Loading and Releasing Performances. J. Mater. Chem. B 2015, 3, 2553–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nounou, M.M.; El-Khordagui, L.K.; Khalafallah, N.A.; Khalil, S.A. In Vitro Release of Hydrophilic and Hydrophobic Drugs from Liposomal Dispersions and Gels. Acta Pharm. 2006, 56, 311–324. [Google Scholar]
- Leite, N.B.; Martins, D.B.; Fazani, V.E.; Vieira, M.R.; Dos Santos Cabrera, M.P. Cholesterol Modulates Curcumin Partitioning and Membrane Effects. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2320–2328. [Google Scholar] [CrossRef]
- Lu, T.; ten Hagen, T.L.M. A Novel Kinetic Model to Describe the Ultra-Fast Triggered Release of Thermosensitive Liposomal Drug Delivery Systems. J. Control. Release 2020, 324, 669–678. [Google Scholar] [CrossRef]
- Kaur, G.; Grewal, J.; Jyoti, K.; Jain, U.K.; Chandra, R.; Madan, J. Chapter 15—Oral Controlled and Sustained Drug Delivery Systems: Concepts, Advances, Preclinical, and Clinical Status. In Drug Targeting and Stimuli Sensitive Drug Delivery Systems; Grumezescu, A.M., Ed.; William Andrew Publishing: Norwich, NY, USA, 2018; pp. 567–626. [Google Scholar] [CrossRef]
- Brazel, C.S.; Huang, X. The Cost of Optimal Drug Delivery: Reducing and Preventing the Burst Effect in Matrix Systems. In Carrier-Based Drug Delivery; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2004; Volume 879. [Google Scholar] [CrossRef]
- Jacobi, U.; Kaiser, M.; Toll, R.; Mangelsdorf, S.; Audring, H.; Otberg, N.; Sterry, W.; Lademann, J. Porcine Ear Skin: An in Vitro Model for Human Skin. Skin Res. Technol. 2007, 13, 19–24. [Google Scholar] [CrossRef]
- Surber, C.; Schwarb, F.P.; Smith, E.W. Tape-Stripping Technique. J. Toxicol. Cutan. Ocul. Toxicol. 2001, 20, 461–474. [Google Scholar] [CrossRef]
- Abd, E.; Roberts, M.S.; Grice, J.E. A Comparison of the Penetration and Permeation of Caffeine into and through Human Epidermis after Application in Various Vesicle Formulations. Skin Pharmacol. Physiol. 2016, 29, 24–30. [Google Scholar] [CrossRef]
- Guillot, A.J.; Cordeiro, A.S.; Donnelly, R.F.; Montesinos, M.C.; Garrigues, T.M.; Melero, A. Microneedle-Based Delivery: An Overview of Current Applications and Trends. Pharmaceutics 2020, 12, 569. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components (% w/v) | Formulations | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| L1 | L2 | L3 | T1c | T2c | T1d | T2d | E1 | E2 | |
| Phospholipon 90G ® | 4.5 | 4.5 | 4.5 | 4.5 | 4.5 | 4.5 | 4.5 | 3 | 3 |
| Cholesterol | 0.135 | 0.135 | 0.135 | - | - | - | - | - | - |
| Tween 80 | - | - | - | 0.675 | 0.675 | 0.675 | 0.675 | - | - |
| B12 1 | 1 1 | 0.2 2 | 0.2 1 | 1 1 | 0.2 2 | 1 1 | 0.2 2 | 0.2 2 | 0.2 3 |
| Reconstitution solvent (mL) | 10 mL PBS | 10 mL PBS | 10 mL PBS | 10 mL PBS | 10 mL PBS | 10 mL PBS | 10 mL PBS | 20 mL Water | 20 mL Water |
| Purification method 4 | C | C | C | C | C | D | D | C | C |
| Formulations | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| L1 | L2 | L3 | T1c | T2c | T1d | T2d | E1 | E2 | |
| Size (nm) | 283 ± 6 | 278 ± 13 | 275 ± 9 | 175 ± 5 | 169 ± 10 | 177 ± 4 | 171 ± 3 | 150 ± 5 | 141 ± 11 |
| PDI | 0.269 ± 0.07 | 0.205 ± 0.002 | 0.215 ± 0.009 | 0.239 ± 0.02 | 0.261 ± 0.03 | 0.223 ± 0.01 | 0.244 ± 0.03 | 0.200 ± 0.003 | 0.193 ± 0.01 |
| Zeta potential (mV) | −11.2 ± 0.12 | −10.1 ± 0.3 | −9.55 ± 0.84 | −4.77 ± 0.13 | −5.01 ± 0.21 | −5.51 ± 0.17 | −5.17 ± 0.37 | −4.63 ± 0.75 | −5.35 ± 1.58 |
| Drug loading (mg per mL) | 1.9 ± 0.3 | 0.75 ± 0.07 | 0.17 ± 0.09 | 0.52 ± 0.02 | 0.14 ± 0.04 | 2.2 ± 0.2 | 0.60 ± 0.02 | 0.22 ± 0.03 | 0.23 ± 0.04 |
| PC (%) | 80 ± 4 | 84 ± 7 | 77 ± 2 | 33 ± 3 | 30 ± 0.9 | 86 ± 8 | 82 ± 5 | 66 ± 2 | 60 ± 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guillot, A.J.; Jornet-Mollá, E.; Landsberg, N.; Milián-Guimerá, C.; Montesinos, M.C.; Garrigues, T.M.; Melero, A. Cyanocobalamin Ultraflexible Lipid Vesicles: Characterization and In Vitro Evaluation of Drug-Skin Depth Profiles. Pharmaceutics 2021, 13, 418. https://doi.org/10.3390/pharmaceutics13030418
Guillot AJ, Jornet-Mollá E, Landsberg N, Milián-Guimerá C, Montesinos MC, Garrigues TM, Melero A. Cyanocobalamin Ultraflexible Lipid Vesicles: Characterization and In Vitro Evaluation of Drug-Skin Depth Profiles. Pharmaceutics. 2021; 13(3):418. https://doi.org/10.3390/pharmaceutics13030418
Chicago/Turabian StyleGuillot, Antonio José, Enrique Jornet-Mollá, Natalia Landsberg, Carmen Milián-Guimerá, M. Carmen Montesinos, Teresa M. Garrigues, and Ana Melero. 2021. "Cyanocobalamin Ultraflexible Lipid Vesicles: Characterization and In Vitro Evaluation of Drug-Skin Depth Profiles" Pharmaceutics 13, no. 3: 418. https://doi.org/10.3390/pharmaceutics13030418