
Lipid Nanoparticles as Delivery Systems for RNA-Based Vaccines


Abstract
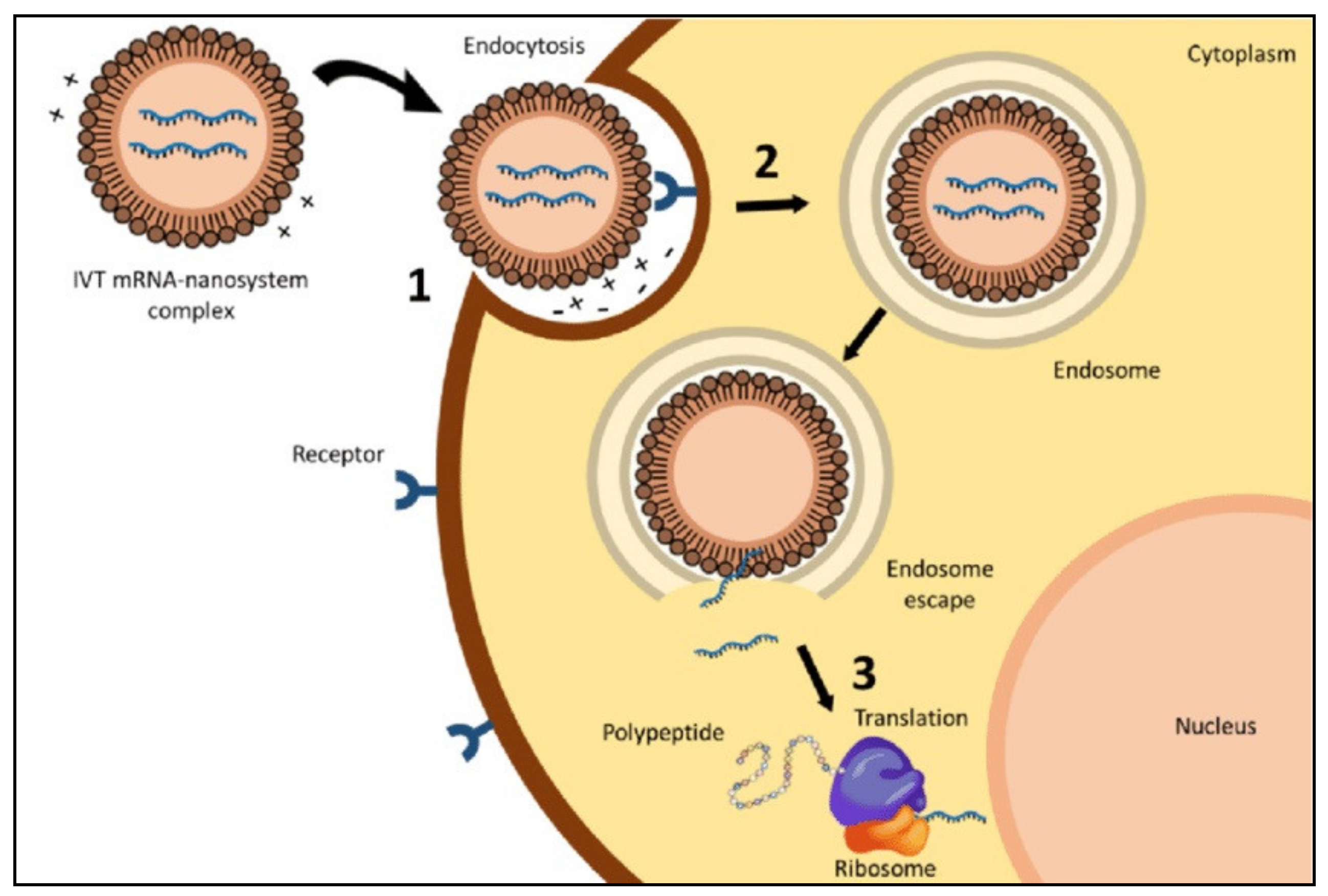
:1. Introduction
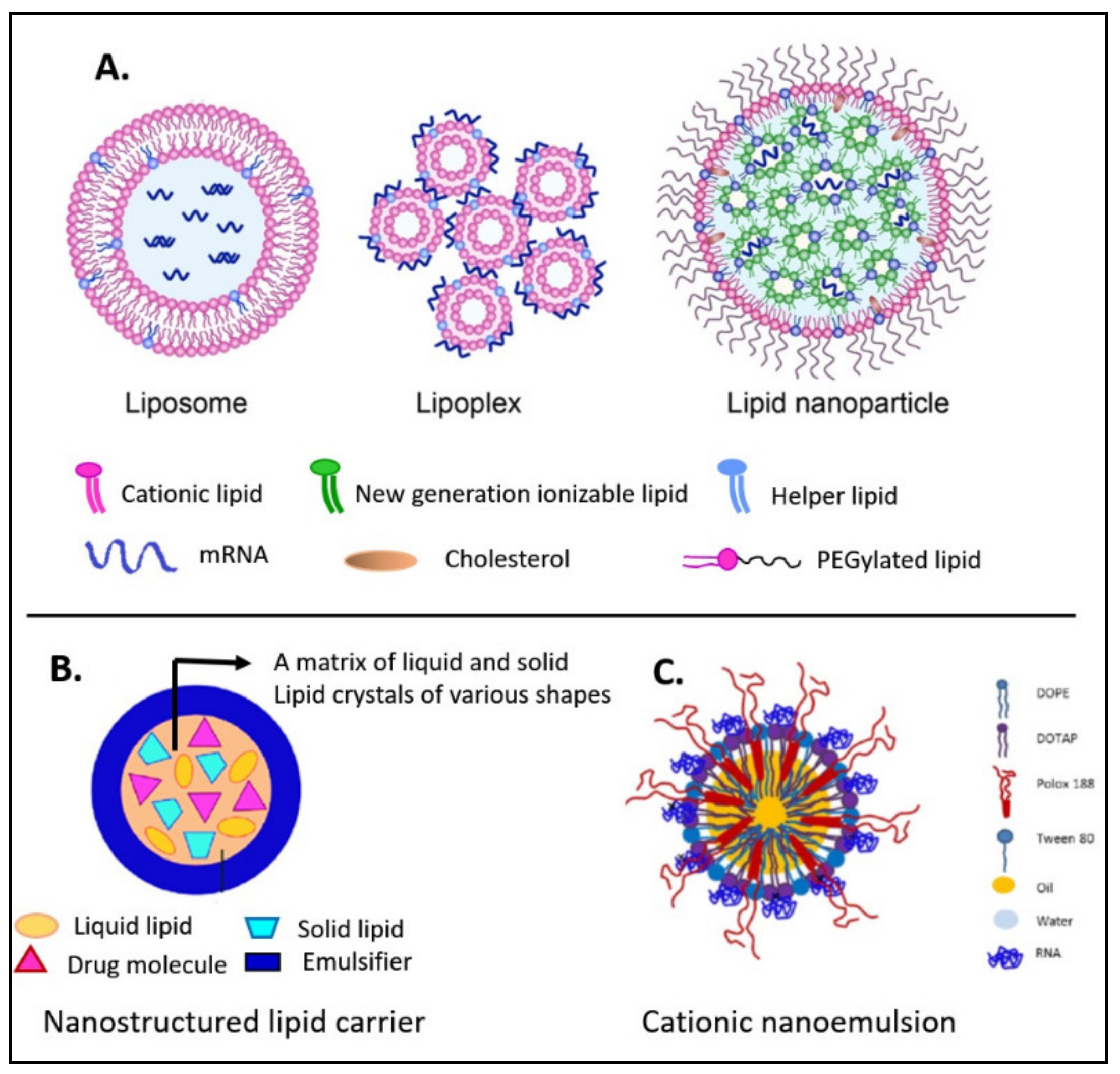
2. Overview of Various Lipid-Based Formulations for the Delivery of Nucleic Acids
2.1. Liposomes
2.2. Lipoplexes
2.3. Cationic Nanoemulsions
2.4. Nanostructured Lipid Carriers
3. Composition of LNPs
3.1. Ionizable Cationic Lipids
3.2. Helper Lipids
3.2.1. PEG–Lipids
3.2.2. Cholesterol
3.2.3. Phosphatidylcholines
4. Methods of Production of LNPs for RNA-Based Vaccines
5. Factors Affecting the In Vivo Delivery and Uptake of RNA/LNP Vaccines
5.1. Route of Administration
5.2. Colloidal Stability of Formulations
5.3. Incorporation of Targeting Moieties
5.4. Incorporation of Adjuvants
6. Pre-Clinical and Clinical Applications of LNPs as Delivery Systems for RNA-Based Vaccines
6.1. RNA/LNP Vaccines against Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection
6.2. RNA/LNP Vaccines against Influenza Virus Infection
6.3. RNA/LNP Vaccines against Rabies Virus Infection
6.4. RNA/LNP Vaccines against Zika Virus Infection
6.5. RNA/LNP Vaccines against Cancer
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito, L.A.; Kommareddy, S.; Maione, D.; Uematsu, Y.; Giovani, C.; Scorza, F.B.; Otten, G.R.; Yu, D.; Mandl, C.W.; Mason, P.W. Self-amplifying mRNA vaccines. In Advances in Genetics; Elsevier: Amsterdam, The Netherlands, 2015; Volume 89, pp. 179–233. [Google Scholar]
- Hekele, A.; Bertholet, S.; Archer, J.; Gibson, D.G.; Palladino, G.; Brito, L.A.; Otten, G.R.; Brazzoli, M.; Buccato, S.; Bonci, A. Rapidly produced SAM® vaccine against H7N9 influenza is immunogenic in mice. Emerging Microb. Infect. 2013, 2, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Pascolo, S. Vaccination with messenger RNA (mRNA). In Toll-Like Receptors (TLRs) and Innate Immunity; Springer: Berlin, Germany, 2008; pp. 221–235. [Google Scholar]
- Pardi, N.; Hogan, M.J.; Naradikian, M.S.; Parkhouse, K.; Cain, D.W.; Jones, L.; Moody, M.A.; Verkerke, H.P.; Myles, A.; Willis, E.; et al. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J. Exp. Med. 2018, 215, 1571–1588. [Google Scholar] [CrossRef] [PubMed]
- Thess, A.; Grund, S.; Mui, B.L.; Hope, M.J.; Baumhof, P.; Fotin-Mleczek, M.; Schlake, T. Sequence-engineered mRNA Without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Mol. Ther. 2015, 23, 1456–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, F.; Lindgren, G.; Lin, A.; Thompson, E.A.; Ols, S.; Röhss, J.; John, S.; Hassett, K.; Yuzhakov, O.; Bahl, K.; et al. Efficient Targeting and Activation of Antigen-Presenting Cells In Vivo after Modified mRNA Vaccine Administration in Rhesus Macaques. Mol. Ther. 2017, 25, 2635–2647. [Google Scholar] [CrossRef] [Green Version]
- Lutz, J.; Lazzaro, S.; Habbeddine, M.; Schmidt, K.E.; Baumhof, P.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Heidenreich, R.; et al. Unmodified mRNA in LNPs constitutes a competitive technology for prophylactic vaccines. NPJ Vaccines 2017, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Magini, D.; Giovani, C.; Mangiavacchi, S.; Maccari, S.; Cecchi, R.; Ulmer, J.B.; De Gregorio, E.; Geall, A.J.; Brazzoli, M.; Bertholet, S. Self-amplifying mRNA vaccines expressing multiple conserved influenza antigens confer protection against homologous and heterosubtypic viral challenge. PLoS One 2016, 11, e0161193. [Google Scholar] [CrossRef]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and Challenges in the Delivery of mRNA-based Vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, S. Human extracellular ribonucleases: Multiplicity, molecular diversity and catalytic properties of the major RNase types. Cell. Mol. Life Sci. CMLS 1998, 54, 785–794. [Google Scholar] [CrossRef]
- Ogris, M.; Brunner, S.; Schüller, S.; Kircheis, R.; Wagner, E. PEGylated DNA/transferrin–PEI complexes: Reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery. Gene Ther. 1999, 6, 595–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouard, D.; Alazard-Dany, N.; Cosset, F.L. Viral vectors: From virology to transgene expression. Br. J. Pharmacol. 2009, 157, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.E. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: From concept to clinic. Mol. Pharm. 2009, 6, 659–668. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hwang, S.J.; Davis, M. New class of polymers for the delivery of macromolecular therapeutics. Bioconjugate Chem. 1999, 10, 1068–1074. [Google Scholar] [CrossRef]
- Lee, H.; Lytton-Jean, A.K.; Chen, Y.; Love, K.T.; Park, A.I.; Karagiannis, E.D.; Sehgal, A.; Querbes, W.; Zurenko, C.S.; Jayaraman, M. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat. Nanotechnol. 2012, 7, 389–393. [Google Scholar] [CrossRef]
- Martin, M.E.; Rice, K.G. Peptide-guided gene delivery. The AAPS J. 2007, 9, E18–E29. [Google Scholar] [CrossRef] [Green Version]
- Pack, D.W.; Hoffman, A.S.; Pun, S.; Stayton, P.S. Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 2005, 4, 581–593. [Google Scholar] [CrossRef]
- Sokolova, V.; Epple, M. Inorganic nanoparticles as carriers of nucleic acids into cells. Angew. Chem. Int. Ed. 2008, 47, 1382–1395. [Google Scholar] [CrossRef]
- Mintzer, M.A.; Simanek, E.E. Nonviral vectors for gene delivery. Chem. Rev. 2009, 109, 259–302. [Google Scholar] [CrossRef]
- Gomez-Aguado, I.; Rodriguez-Castejon, J.; Vicente-Pascual, M.; Rodriguez-Gascon, A.; Solinis, M.A.; Del Pozo-Rodriguez, A. Nanomedicines to Deliver mRNA: State of the Art and Future Perspectives. Nanomaterials 2020, 10, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Samaridou, E.; Heyes, J.; Lutwyche, P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv. Drug Deliv. Rev. 2020. [Google Scholar] [CrossRef]
- Phua, K.K.; Leong, K.W.; Nair, S.K. Transfection efficiency and transgene expression kinetics of mRNA delivered in naked and nanoparticle format. J. Contr. Rel. 2013, 166, 227–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahl, K.; Senn, J.J.; Yuzhakov, O.; Bulychev, A.; Brito, L.A.; Hassett, K.J.; Laska, M.E.; Smith, M.; Almarsson, O.; Thompson, J.; et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol. Ther. 2017, 25, 1316–1327. [Google Scholar] [CrossRef] [Green Version]
- Lindgren, G.; Ols, S.; Liang, F.; Thompson, E.A.; Lin, A.; Hellgren, F.; Bahl, K.; John, S.; Yuzhakov, O.; Hassett, K.J. Induction of robust B cell responses after influenza mRNA vaccination is accompanied by circulating hemagglutinin-specific ICOS+ PD-1+ CXCR3+ T follicular helper cells. Front. Immunol. 2017, 8, 1539. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Parkhouse, K.; Kirkpatrick, E.; McMahon, M.; Zost, S.J.; Mui, B.L.; Tam, Y.K.; Kariko, K.; Barbosa, C.J.; Madden, T.D.; et al. Nucleoside-modified mRNA immunization elicits influenza virus hemagglutinin stalk-specific antibodies. Nat. Commun. 2018, 9, 3361. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef]
- Richner, J.M.; Jagger, B.W.; Shan, C.; Fontes, C.R.; Dowd, K.A.; Cao, B.; Himansu, S.; Caine, E.A.; Nunes, B.T.D.; Medeiros, D.B.A.; et al. Vaccine Mediated Protection Against Zika Virus-Induced Congenital Disease. Cell 2017, 170, 273–283. [Google Scholar] [CrossRef] [Green Version]
- John, S.; Yuzhakov, O.; Woods, A.; Deterling, J.; Hassett, K.; Shaw, C.A.; Ciaramella, G. Multi-antigenic human cytomegalovirus mRNA vaccines that elicit potent humoral and cell-mediated immunity. Vaccine 2018, 36, 1689–1699. [Google Scholar] [CrossRef]
- Meyer, M.; Huang, E.; Yuzhakov, O.; Ramanathan, P.; Ciaramella, G.; Bukreyev, A. Modified mRNA-Based Vaccines Elicit Robust Immune Responses and Protect Guinea Pigs From Ebola Virus Disease. J. Infect. Dis. 2018, 217, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magné, R.; Gomard, E.; Guillet, J.G.; Lévy, J.P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- Brazzoli, M.; Magini, D.; Bonci, A.; Buccato, S.; Giovani, C.; Kratzer, R.; Zurli, V.; Mangiavacchi, S.; Casini, D.; Brito, L.M. Induction of broad-based immunity and protective efficacy by self-amplifying mRNA vaccines encoding influenza virus hemagglutinin. J. Virolog. 2016, 90, 332–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chahal, J.S.; Khan, O.F.; Cooper, C.L.; McPartlan, J.S.; Tsosie, J.K.; Tilley, L.D.; Sidik, S.M.; Lourido, S.; Langer, R.; Bavari, S. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc. Nat. Acad. Sci. 2016, 113, E4133–E4142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogers, W.M.; Oostermeijer, H.; Mooij, P.; Koopman, G.; Verschoor, E.J.; Davis, D.; Ulmer, J.B.; Brito, L.A.; Cu, Y.; Banerjee, K.; et al. Potent immune responses in rhesus macaques induced by nonviral delivery of a self-amplifying RNA vaccine expressing HIV type 1 envelope with a cationic nanoemulsion. J. Infect. Dis. 2015, 211, 947–955. [Google Scholar] [CrossRef]
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A Cationic Nanoemulsion for the Delivery of Next-generation RNA Vaccines. Mol. Ther. 2014, 22, 2118–2129. [Google Scholar] [CrossRef] [Green Version]
- Maruggi, G.; Chiarot, E.; Giovani, C.; Buccato, S.; Bonacci, S.; Frigimelica, E.; Margarit, I.; Geall, A.; Bensi, G.; Maione, D. Immunogenicity and protective efficacy induced by self-amplifying mRNA vaccines encoding bacterial antigens. Vaccine 2017, 35, 361–368. [Google Scholar] [CrossRef]
- Garcia, A.B.; Siu, E.; Sun, T.; Exler, V.; Brito, L.; Hekele, A.; Otten, G.; Augustijn, K.; Janse, C.J.; Ulmer, J.B. Neutralization of the Plasmodium-encoded MIF ortholog confers protective immunity against malaria infection. Nat. Comm. 2018, 9, 1–13. [Google Scholar]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T. Nonviral delivery of self-amplifying RNA vaccines. Proc. Nat. Acad. Sci. 2012, 109, 14604–14609. [Google Scholar] [CrossRef] [Green Version]
- Chahal, J.S.; Fang, T.; Woodham, A.W.; Khan, O.F.; Ling, J.; Anderson, D.G.; Ploegh, H.L. An RNA nanoparticle vaccine against Zika virus elicits antibody and CD8+ T cell responses in a mouse model. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Khandhar, A.P.; Guderian, J.; Granger, B.; Archer, J.; Archer, M.; Gage, E.; Fuerte-Stone, J.; Larson, E.; Lin, S.; et al. A Nanostructured Lipid Carrier for Delivery of a Replicating Viral RNA Provides Single, Low-Dose Protection against Zika. Mol. The. 2018, 26, 2507–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.J.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Frederick, J.P.; Huang, E.Y.; Burke, K.E.; Mauger, D.M.; Andrianova, E.A.; Farlow, S.J.; Siddiqui, S.; Pimentel, J.; Cheung-Ong, K.; et al. MicroRNAs Enable mRNA Therapeutics to Selectively Program Cancer Cells to Self-Destruct. Nucleic Acid Ther. 2018, 28, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybakova, Y.; Kowalski, P.S.; Huang, Y.; Gonzalez, J.T.; Heartlein, M.W.; DeRosa, F.; Delcassian, D.; Anderson, D.G. mRNA Delivery for Therapeutic Anti-HER2 Antibody Expression In Vivo. Mol. Ther. 2019, 27, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kim, S.Y.; Seo, Y.; Kim, M.H.; Chang, J.; Lee, H. Adjuvant incorporated lipid nanoparticles for enhanced mRNA-mediated cancer immunotherapy. Biomater. Sci. 2020, 8, 1101–1105. [Google Scholar] [CrossRef] [PubMed]
- Midoux, P.; Pichon, C. Lipid-based mRNA vaccine delivery systems. Expert Rev. Vaccines 2015, 14, 221–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajj, K.A.; Whitehead, K.A. Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mat. 2017, 2, 1–17. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Lu, D.; Benjamin, R.; Kim, M.; Conry, R.; Curiel, D. Optimization of methods to achieve mRNA-mediated transfection of tumor cells in vitro and in vivo employing cationic liposome vectors. Cancer Gene Ther. 1994, 1, 245–252. [Google Scholar]
- Zhou, W.-Z.; Hoon, D.; Huang, S.; Fujii, S.; Hashimoto, K.; Morishita, R.; Kaneda, Y. RNA melanoma vaccine: Induction of antitumor immunity by human glycoprotein 100 mRNA immunization. Hum. Gene Ther. 1999, 10, 2719–2724. [Google Scholar] [CrossRef]
- Granot, Y.; Peer, D. Delivering the right message: Challenges and opportunities in lipid nanoparticles-mediated modified mRNA therapeutics-An innate immune system standpoint. Semin. Immunol. 2017, 34, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Felgner, J.H.; Kumar, R.; Sridhar, C.; Wheeler, C.J.; Tsai, Y.J.; Border, R.; Ramsey, P.; Martin, M.; Felgner, P.L. Enhanced gene delivery and mechanism studies with a novel series of cationic lipid formulations. J. Biol. Chem. 1994, 269, 2550–2561. [Google Scholar] [CrossRef]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Nat. Acad. Sci. 1989, 86, 6077–6081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, C.; Rejman, J.; De Haes, W.; Verrier, B.; Van Gulck, E.; Naessens, T.; De Smedt, S.; Bogaert, P.; Grooten, J.; Vanham, G.; et al. Type I IFN Counteracts the Induction of Antigen-Specific Immune Responses by Lipid-Based Delivery of mRNA Vaccines. Mol. Ther. 2013, 21, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Andries, O.; De Filette, M.; Rejman, J.; De Smedt, S.C.; Demeester, J.; Van Poucke, M.; Peelman, L.; Peleman, C.; Lahoutte, T.; Sanders, N.N. Comparison of the gene transfer efficiency of mRNA/GL67 and pDNA/GL67 complexes in respiratory cells. Mol. Pharm. 2012, 9, 2136–2145. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.; Korsholm, K.S.; Rosenkrands, I.; Lindenstrøm, T.; Andersen, P.; Agger, E.M. Cationic liposomes as vaccine adjuvants. Expert Rev. Vaccines 2007, 6, 785–796. [Google Scholar] [CrossRef]
- Lonez, C.; Vandenbranden, M.; Ruysschaert, J.-M. Cationic lipids activate intracellular signaling pathways. Adv. Drug Del. Rev. 2012, 64, 1749–1758. [Google Scholar] [CrossRef]
- Tan, L.; Sun, X. Recent advances in mRNA vaccine delivery. Nano Res. 2018, 11, 5338–5354. [Google Scholar] [CrossRef]
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. Three decades of messenger RNA vaccine development. Nano Today 2019, 28, 100766. [Google Scholar] [CrossRef]
- Semple, S.C.; Klimuk, S.K.; Harasym, T.O.; Dos Santos, N.; Ansell, S.M.; Wong, K.F.; Maurer, N.; Stark, H.; Cullis, P.R.; Hope, M.J. Efficient encapsulation of antisense oligonucleotides in lipid vesicles using ionizable aminolipids: Formation of novel small multilamellar vesicle structures. Biochim. Biophys. Acta (BBA)-Biomembr. 2001, 1510, 152–166. [Google Scholar] [CrossRef] [Green Version]
- Heyes, J.; Palmer, L.; Bremner, K.; MacLachlan, I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J. Controll. Rel. 2005, 107, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.; Stebbing, D.; Crosley, E.J.; Yaworski, E. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Maier, M.A.; Jayaraman, M.; Matsuda, S.; Liu, J.; Barros, S.; Querbes, W.; Tam, Y.K.; Ansell, S.M.; Kumar, V.; Qin, J. Biodegradable lipids enabling rapidly eliminated lipid nanoparticles for systemic delivery of RNAi therapeutics. Mol. Ther. 2013, 21, 1570–1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardi, N.; Secreto, A.J.; Shan, X.; Debonera, F.; Glover, J.; Yi, Y.; Muramatsu, H.; Ni, H.; Mui, B.L.; Tam, Y.K.; et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 2017, 8, 14630. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Lee, R.J. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Deliv. Rev. 2016, 99, 129–137. [Google Scholar] [CrossRef]
- Buyens, K.; De Smedt, S.C.; Braeckmans, K.; Demeester, J.; Peeters, L.; van Grunsven, L.A.; du Jeu, X.d.M.; Sawant, R.; Torchilin, V.; Farkasova, K. Liposome based systems for systemic siRNA delivery: Stability in blood sets the requirements for optimal carrier design. J. Controll. Rel. 2012, 158, 362–370. [Google Scholar] [CrossRef]
- Song, L.; Ahkong, Q.; Rong, Q.; Wang, Z.; Ansell, S.; Hope, M.; Mui, B. Characterization of the inhibitory effect of PEG-lipid conjugates on the intracellular delivery of plasmid and antisense DNA mediated by cationic lipid liposomes. Biochim. Biophys. Acta (BBA) Biomembr. 2002, 1558, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Webb, M.S.; Saxon, D.; Wong, F.M.; Lim, H.J.; Wang, Z.; Bally, M.B.; Choi, L.S.; Cullis, P.R.; Mayer, L.D. Comparison of different hydrophobic anchors conjugated to poly (ethylene glycol): Effects on the pharmacokinetics of liposomal vincristine. Biochim. Biophys. Acta (BBA) Biomembr. 1998, 1372, 272–282. [Google Scholar] [CrossRef] [Green Version]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar] [CrossRef]
- Judge, A.; McClintock, K.; Phelps, J.R.; MacLachlan, I. Hypersensitivity and loss of disease site targeting caused by antibody responses to PEGylated liposomes. Mol. Ther. 2006, 13, 328–337. [Google Scholar] [CrossRef]
- Semple, S.C.; Harasym, T.O.; Clow, K.A.; Ansell, S.M.; Klimuk, S.K.; Hope, M.J. Immunogenicity and rapid blood clearance of liposomes containing polyethylene glycol-lipid conjugates and nucleic acid. J. Pharmacol. Exp. Ther. 2005, 312, 1020–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Ashwanikumar, N.; Robinson, E.; Xia, Y.; Mihai, C.; Griffith, J.P.; Hou, S.; Esposito, A.A.; Ketova, T.; Welsher, K. Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzi, D.; Marchini, C.; Cardarelli, F.; Amenitsch, H.; Garulli, C.; Bifone, A.; Caracciolo, G. Transfection efficiency boost of cholesterol-containing lipoplexes. Biochim. Biophys. Acta (BBA) Biomembr. 2012, 1818, 2335–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenchov, B.G.; MacDonald, R.C.; Siegel, D.P. Cubic phases in phosphatidylcholine-cholesterol mixtures: Cholesterol as membrane “fusogen”. Biophys. J. 2006, 91, 2508–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffs, L.B.; Palmer, L.R.; Ambegia, E.G.; Giesbrecht, C.; Ewanick, S.; MacLachlan, I. A scalable, extrusion-free method for efficient liposomal encapsulation of plasmid DNA. Pharm. Res. 2005, 22, 362–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeki, M.; Saito, T.; Sato, Y.; Yasui, T.; Kaji, N.; Ishida, A.; Tani, H.; Baba, Y.; Harashima, H.; Tokeshi, M. A strategy for synthesis of lipid nanoparticles using microfluidic devices with a mixer structure. RSC Adv. 2015, 5, 46181–46185. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, J.A.; Darjuan, M.M.; Mercer, J.E.; Chen, S.; Van Der Meel, R.; Thewalt, J.L.; Tam, Y.Y.C.; Cullis, P.R. On the formation and morphology of lipid nanoparticles containing ionizable cationic lipids and siRNA. ACS Nano 2018, 12, 4787–4795. [Google Scholar] [CrossRef] [Green Version]
- Leung, A.K.; Hafez, I.M.; Baoukina, S.; Belliveau, N.M.; Zhigaltsev, I.V.; Afshinmanesh, E.; Tieleman, D.P.; Hansen, C.L.; Hope, M.J.; Cullis, P.R. Lipid nanoparticles containing siRNA synthesized by microfluidic mixing exhibit an electron-dense nanostructured core. J. Phys. Chem. C 2012, 116, 18440–18450. [Google Scholar] [CrossRef]
- Broos, K.; Van der Jeught, K.; Puttemans, J.; Goyvaerts, C.; Heirman, C.; Dewitte, H.; Verbeke, R.; Lentacker, I.; Thielemans, K.; Breckpot, K. Particle-mediated intravenous delivery of antigen mRNA results in strong antigen-specific T-cell responses despite the induction of type I interferon. Mol. Ther.-Nucleic Acids 2016, 5, e326. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Controll. Rel. 2015, 217, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Imm. 2008, 38, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Oussoren, C.; Storm, G. Liposomes to target the lymphatics by subcutaneous administration. Adv. Drug Deliv. Rev. 2001, 50, 143–156. [Google Scholar] [CrossRef]
- Reddy, S.T.; Rehor, A.; Schmoekel, H.G.; Hubbell, J.A.; Swartz, M.A. In vivo targeting of dendritic cells in lymph nodes with poly (propylene sulfide) nanoparticles. J. Controll. Rel. 2006, 112, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Yang, Q.; Bagby, T.R.; Forrest, M.L. Lymphatic drug delivery using engineered liposomes and solid lipid nanoparticles. Adv. Drug Deliv. Rev. 2011, 63, 901–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swartz, M.A.; Hubbell, J.A.; Reddy, S.T. Lymphatic drainage function and its immunological implications: From dendritic cell homing to vaccine design. In Proceedings of Seminars in Immunology; Academic Press: Cambridge, MA, USA, 2008; pp. 147–156. [Google Scholar]
- Zhuang, Y.; Ma, Y.; Wang, C.; Hai, L.; Yan, C.; Zhang, Y.; Liu, F.; Cai, L. PEGylated cationic liposomes robustly augment vaccine-induced immune responses: Role of lymphatic trafficking and biodistribution. J. Controll. Rel. 2012, 159, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Xu, Z.; Miao, L.; Huang, L. mRNA Vaccine with Antigen-Specific Checkpoint Blockade Induces an Enhanced Immune Response against Established Melanoma. Mol. Ther. 2018, 26, 420–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monopoli, M.P.; Åberg, C.; Salvati, A.; Dawson, K.A. Biomolecular coronas provide the biological identity of nanosized materials. Nat. Nanotechnol. 2012, 7, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Braeckmans, K.; Buyens, K.; Bouquet, W.; Vervaet, C.; Joye, P.; Vos, F.D.; Plawinski, L.; Doeuvre, L.c.; Angles-Cano, E.; Sanders, N.N.; et al. Sizing Nanomatter in Biological Fluids by Fluorescence Single Particle Tracking. Nano Lett. 2010, 10, 4435–4442. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; De Smedt, S.C.; Remaut, K. Fluorescence Correlation Spectroscopy to find the critical balance between extracellular association and intracellular dissociation of mRNA complexes. Acta Biomater. 2018, 75, 358–370. [Google Scholar] [CrossRef]
- Li, S.; Tseng, W.; Stolz, D.B.; Wu, S.; Watkins, S.; Huang, L. Dynamic changes in the characteristics of cationic lipidic vectors after exposure to mouse serum: Implications for intravenous lipofection. Gene Ther. 1999, 6, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Schöttler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailänder, V.; Wurm, F.R. Protein adsorption is required for stealth effect of poly (ethylene glycol)-and poly (phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016, 11, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Wasungu, L.; Nomden, A.; Stuart, M.C.; Polushkin, E.; Engberts, J.B.; Hoekstra, D. Interference of poly (ethylene glycol)–lipid analogues with cationic-lipid-mediated delivery of oligonucleotides; role of lipid exchangeability and non-lamellar transitions. Biochem. J. 2002, 366, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Neagu, M.; Piperigkou, Z.; Karamanou, K.; Engin, A.B.; Docea, A.O.; Constantin, C.; Negrei, C.; Nikitovic, D.; Tsatsakis, A. Protein bio-corona: Critical issue in immune nanotoxicology. Arch. Toxicol. 2017, 91, 1031–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghimi, S.M.; Simberg, D. Complement activation turnover on surfaces of nanoparticles. Nano Today 2017, 15, 8–10. [Google Scholar] [CrossRef] [Green Version]
- Grenier, P.; de Oliveira Viana, I.M.; Lima, E.M.; Bertrand, N. Anti-polyethylene glycol antibodies alter the protein corona deposited on nanoparticles and the physiological pathways regulating their fate in vivo. J. Controll. Rel. 2018, 287, 121–131. [Google Scholar] [CrossRef]
- Phua, K.K. Towards targeted delivery systems: Ligand conjugation strategies for mRNA nanoparticle tumor vaccines. J. Imm. Res. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Baudino, L.; Sardini, A.; Ruseva, M.M.; Fossati-Jimack, L.; Cook, H.T.; Scott, D.; Simpson, E.; Botto, M. C3 opsonization regulates endocytic handling of apoptotic cells resulting in enhanced T-cell responses to cargo-derived antigens. Proc. Nat. Acad. Sci. USA 2014, 111, 1503–1508. [Google Scholar] [CrossRef] [Green Version]
- Zelenay, S.; Keller, A.M.; Whitney, P.G.; Schraml, B.U.; Deddouche, S.; Rogers, N.C.; Schulz, O.; Sancho, D.; Reis e Sousa, C. The dendritic cell receptor DNGR-1 controls endocytic handling of necrotic cell antigens to favor cross-priming of CTLs in virus-infected mice. J. Clin. Invest. 2012, 122, 1615–1627. [Google Scholar] [CrossRef] [Green Version]
- Perche, F.; Benvegnu, T.; Berchel, M.; Lebegue, L.; Pichon, C.; Jaffres, P.A.; Midoux, P. Enhancement of dendritic cells transfection in vivo and of vaccination against B16F10 melanoma with mannosylated histidylated lipopolyplexes loaded with tumor antigen messenger RNA. Nanomedicine 2011, 7, 445–453. [Google Scholar] [CrossRef]
- Parhiz, H.; Shuvaev, V.V.; Pardi, N.; Khoshnejad, M.; Kiseleva, R.Y.; Brenner, J.S.; Uhler, T.; Tuyishime, S.; Mui, B.L.; Tam, Y.K.; et al. PECAM-1 directed re-targeting of exogenous mRNA providing two orders of magnitude enhancement of vascular delivery and expression in lungs independent of apolipoprotein E-mediated uptake. J. Controll. Rel. 2018, 291, 106–115. [Google Scholar] [CrossRef]
- Kedmi, R.; Veiga, N.; Ramishetti, S.; Goldsmith, M.; Rosenblum, D.; Dammes, N.; Hazan-Halevy, I.; Nahary, L.; Leviatan-Ben-Arye, S.; Harlev, M.; et al. A modular platform for targeted RNAi therapeutics. Nat. Nanotechnol. 2018, 13, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Veiga, N.; Goldsmith, M.; Granot, Y.; Rosenblum, D.; Dammes, N.; Kedmi, R.; Ramishetti, S.; Peer, D. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Al Zaki, A.; Hui, J.Z.; Muzykantov, V.R.; Tsourkas, A. Multifunctional nanoparticles: Cost versus benefit of adding targeting and imaging capabilities. Science 2012, 338, 903–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvati, A.; Pitek, A.S.; Monopoli, M.P.; Prapainop, K.; Bombelli, F.B.; Hristov, D.R.; Kelly, P.M.; Åberg, C.; Mahon, E.; Dawson, K.A. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat. Nanotechnol. 2013, 8, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Tenzer, S.; Storck, W.; Hobernik, D.; Raker, V.K.; Fischer, K.; Decker, S.; Dzionek, A.; Krauthauser, S.; Diken, M.; et al. Protein corona-mediated targeting of nanocarriers to B cells allows redirection of allergic immune responses. J. Allergy Clin. Immunol. 2018, 142, 1558–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavano, R.; Gabrielli, L.; Lubian, E.; Fedeli, C.; Visentin, S.; Polverino De Laureto, P.; Arrigoni, G.; Geffner-Smith, A.; Chen, F.; Simberg, D.; et al. C1q-Mediated Complement Activation and C3 Opsonization Trigger Recognition of Stealth Poly(2-methyl-2-oxazoline)-Coated Silica Nanoparticles by Human Phagocytes. ACS Nano 2018, 12, 5834–5847. [Google Scholar] [CrossRef]
- Reddy, S.T.; van der Vlies, A.J.; Simeoni, E.; Angeli, V.; Randolph, G.J.; O’Neil, C.P.; Lee, L.K.; Swartz, M.A.; Hubbell, J.A. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007, 25, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef]
- Verbeke, R.; Lentacker, I.; Wayteck, L.; Breckpot, K.; Van Bockstal, M.; Descamps, B.; Vanhove, C.; De Smedt, S.C.; Dewitte, H. Co-delivery of nucleoside-modified mRNA and TLR agonists for cancer immunotherapy: Restoring the immunogenicity of immunosilent mRNA. J. Controll. Rel. 2017, 266, 287–300. [Google Scholar] [CrossRef]
- Pektor, S.; Hilscher, L.; Walzer, K.C.; Miederer, I.; Bausbacher, N.; Loquai, C.; Schreckenberger, M.; Sahin, U.; Diken, M.; Miederer, M. In vivo imaging of the immune response upon systemic RNA cancer vaccination by FDG-PET. EJNMMI Res. 2018, 8, 80. [Google Scholar] [CrossRef]
- Leroux-Roels, G. Unmet needs in modern vaccinology: Adjuvants to improve the immune response. Vaccine 2010, 28, C25–C36. [Google Scholar] [CrossRef] [PubMed]
- Kedmi, R.; Ben-Arie, N.; Peer, D. The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomaterials 2010, 31, 6867–6875. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, S.B.; McBurney, W.T.; Young, K.; Hanley, T.; Boyd, B.J.; Rades, T.; Hook, S. Cubosomes containing the adjuvants imiquimod and monophosphoryl lipid A stimulate robust cellular and humoral immune responses. J. Controll. Rel. 2013, 165, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Bal, S.M.; Hortensius, S.; Ding, Z.; Jiskoot, W.; Bouwstra, J.A. Co-encapsulation of antigen and Toll-like receptor ligand in cationic liposomes affects the quality of the immune response in mice after intradermal vaccination. Vaccine 2011, 29, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erikçi, E.; Gursel, M.; Gürsel, İ. Differential immune activation following encapsulation of immunostimulatory CpG oligodeoxynucleotide in nanoliposomes. Biomaterials 2011, 32, 1715–1723. [Google Scholar] [CrossRef]
- Wu, T.Y.-H.; Singh, M.; Miller, A.T.; De Gregorio, E.; Doro, F.; D’Oro, U.; Skibinski, D.A.; Mbow, M.L.; Bufali, S.; Herman, A.E. Rational design of small molecules as vaccine adjuvants. Sci. Translational Med. 2014, 6, ra160–ra263. [Google Scholar] [CrossRef]
- Jensen, S.; Thomsen, A.R. Sensing of RNA viruses: A review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012, 86, 2900–2910. [Google Scholar] [CrossRef] [Green Version]
- Jackson, N.A.; Kester, K.E.; Casimiro, D.; Gurunathan, S.; DeRosa, F. The promise of mRNA vaccines: A biotech and industrial perspective. Npj Vaccines 2020, 5, 1–6. [Google Scholar] [CrossRef]
- Frederiksen, L.S.F.; Zhang, Y.; Foged, C.; Thakur, A. The Long Road Toward COVID-19 Herd Immunity: Vaccine Platform Technologies and Mass Immunization Strategies. Front. Immun. 2020, 11, 1–26. [Google Scholar] [CrossRef]
- Richner, J.M.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.M.; Julander, J.G.; Tang, W.W.; Shresta, S.; Pierson, T.C. Modified mRNA vaccines protect against Zika virus infection. Cell 2017, 168, 1114–1125. [Google Scholar] [CrossRef] [Green Version]
- Scorza, F.B.; Pardi, N. New kids on the block: RNA-based influenza virus vaccines. Vaccines 2018, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, R.A.; Fuhr, R.; Smolenov, I.; Ribeiro, A.M.; Panther, L.; Watson, M.; Senn, J.J.; Smith, M.; Almarsson, Ö.; Pujar, H.S. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 2019, 37, 3326–3334. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. New Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, C.; Leroux–Roels, I.; Huang, K.B.; Bica, M.A.; Loeliger, E.; Schoenborn-Kellenberger, O.; Walz, L.; Leroux-Roels, G.; von Sonnenburg, F.; Oostvogels, L. Proof-of-concept of a low-dose unmodified mRNA-based rabies vaccine formulated with lipid nanoparticles in human volunteers: A phase 1 trial. Vaccine 2021. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Mc Cafferty, S.; Combes, F.; Huysmans, H.; De Temmerman, J.; Gitsels, A.; Vanrompay, D.; Portela Catani, J.; Sanders, N.N. mRNA therapeutics deliver a hopeful message. Nano Today 2018, 23, 16–39. [Google Scholar] [CrossRef]
- Laczko, D.; Hogan, M.J.; Toulmin, S.A.; Hicks, P.; Lederer, K.; Gaudette, B.T.; Castano, D.; Amanat, F.; Muramatsu, H.; Oguin, T.H., 3rd; et al. A Single Immunization with Nucleoside-Modified mRNA Vaccines Elicits Strong Cellular and Humoral Immune Responses against SARS-CoV-2 in Mice. Immunity 2020, 53, 724–732. [Google Scholar] [CrossRef]
- Lu, J.; Lu, G.; Tan, S.; Xia, J.; Xiong, H.; Yu, X.; Qi, Q.; Li, L.; Yu, H.; Xia, N.; et al. A COVID-19 mRNA vaccine encoding SARS-CoV-2 virus-like particles induces a strong antiviral-like immune response in mice. Cell Res. 2020, 30, 936–939. [Google Scholar] [CrossRef]
- McKay, P.F.; Hu, K.; Blakney, A.K.; Samnuan, K.; Brown, J.C.; Penn, R.; Zhou, J.; Bouton, C.R.; Rogers, P.; Polra, K.; et al. Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nat. Commun. 2020, 11, 3523. [Google Scholar] [CrossRef]
- Tai, W.; Zhang, X.; Drelich, A.; Shi, J.; Hsu, J.C.; Luchsinger, L.; Hillyer, C.D.; Tseng, C.K.; Jiang, S.; Du, L. A novel receptor-binding domain (RBD)-based mRNA vaccine against SARS-CoV-2. Cell Res 2020, 30, 932–935. [Google Scholar] [CrossRef]
- Zhang, N.N.; Li, X.F.; Deng, Y.Q.; Zhao, H.; Huang, Y.J.; Yang, G.; Huang, W.J.; Gao, P.; Zhou, C.; Zhang, R.R.; et al. A Thermostable mRNA Vaccine against COVID-19. Cell 2020, 182, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.S.; Flynn, B.; Foulds, K.E.; Francica, J.R.; Boyoglu-Barnum, S.; Werner, A.P.; Flach, B.; O’Connell, S.; Bock, K.W.; Minai, M.; et al. Evaluation of the mRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. New Engl. J. Med. 2020, 383, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- “Moderna Inc.”. Moderna’s COVID-19 Vaccine Candidate Meets its Primary Efficacy Endpoint in the First Interim Analysis of the Phase 3 COVE Study. Available online: https://investors.modernatx.com/news-releases/news-release-details/modernas-covid-19-vaccine-candidate-meets-its-primary-efficacy (accessed on 18 November 2020).
- Chauhan, G.; Madou, M.J.; Kalra, S.; Chopra, V.; Ghosh, D.; Martinez-Chapa, S.O. Nanotechnology for COVID-19: Therapeutics and Vaccine Research. ACS Nano 2020, 14, 7760–7782. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.D.; Shukla, S.; Chung, Y.H.; Beiss, V.; Chan, S.K.; Ortega-Rivera, O.A.; Wirth, D.M.; Chen, A.; Sack, M.; Pokorski, J.K.; et al. COVID-19 vaccine development and a potential nanomaterial path forward. Nat. Nanotechnol. 2020, 15, 646–655. [Google Scholar] [CrossRef] [PubMed]
- “Pfizer Inc.”. Pfizer and Biontech announce vaccine candidate against COVID-19 achieved success in first interim analysis from phase 3 study. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-biontech-announce-vaccine-candidate-against (accessed on 18 November 2020).
- Krammer, F.; Palese, P. Influenza virus hemagglutinin stalk-based antibodies and vaccines. Curr. Opin. Virol. 2013, 3, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Omri, A.; Krishnan, L.; McCluskie, M.J. Potential applications of nanoparticles in cancer immunotherapy. Hum. Vaccines Immunotherapeutics 2017, 13, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Miao, L.; Sui, J.; Hao, Y.; Huang, G. Nanoparticle cancer vaccines: Design considerations and recent advances. Asian J. Pharm. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Melief, C.J.; van Hall, T.; Arens, R.; Ossendorp, F.; van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Miller, M.; Sahin, U.; Derhovanessian, E.; Kloke, B.; Simon, P.; Bukur, V.; Albrecht, C.; Paruzynski, A.; Löwer, M.; Kuhn, A. 6O IVAC MUTANOME: A first-in-human phase I clinical trial targeting individual mutant neoantigens for the treatment of melanoma. Ann. Oncol. 2017, 28, xi1–xi2. [Google Scholar] [CrossRef] [Green Version]
- McNamara, M.A.; Nair, S.K.; Holl, E.K. RNA-Based Vaccines in Cancer Immunotherapy. J. Immunol. Res. 2015, 2015, 794528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melhem, N.M.; Liu, X.D.; Boczkowski, D.; Gilboa, E.; Barratt-Boyes, S.M. Robust CD4+ and CD8+ T cell responses to SIV using mRNA-transfected DC expressing autologous viral Ag. Eur. J. Immun. 2007, 37, 2164–2173. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Kariko, K.; Tureci, O. mRNA-based therapeutics—Developing a new class of drugs. Nat Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Van Nuffel, A.M.; Benteyn, D.; Wilgenhof, S.; Pierret, L.; Corthals, J.; Heirman, C.; Van Der Bruggen, P.; Coulie, P.G.; Neyns, B.; Thielemans, K. Dendritic cells loaded with mRNA encoding full-length tumor antigens prime CD4+ and CD8+ T cells in melanoma patients. Mol. Ther. 2012, 20, 1063–1074. [Google Scholar] [CrossRef] [Green Version]
- Lai, I.; Swaminathan, S.; Baylot, V.; Mosley, A.; Dhanasekaran, R.; Gabay, M.; Felsher, D.W. Lipid nanoparticles that deliver IL-12 messenger RNA suppress tumorigenesis in MYC oncogene-driven hepatocellular carcinoma. J. ImmunoTher. Cancer 2018, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Delivery Formulation | mRNA | Lipids Used | Encoded Antigen | In Vivo Animal Model | Delivery Route | Obtained Immunity Response | References |
|---|---|---|---|---|---|---|---|
| Infectious diseases | |||||||
| LNP | Conventional, sequence-optimized | Ionizable amino lipid, phospholipid, cholesterol and a PEGylated lipid, | HA (influenza virus) | Mice, NHPs | IM | Humoral, cellular, innate | [9] |
| LNP | Conventional, nucleoside-modified | Ionizable lipid: DSPC: cholesterol: PEG-lipid | HA (influenza virus) | Mice, ferret, NHPs | ID, IM | Humoral, cellular, innate, protection | [6,8,27,28,29] |
| LNP | Conventional, nucleoside-modified | Ionizable lipid: DSPC: cholesterol: PEG-lipid | prM-E (Zika virus) | Mice, NHPs | ID, IM | Humoral, protection | [30,31] |
| LNP | Conventional, nucleoside-modified | Ionizable lipid: DSPC: cholesterol: PEG lipid | PC, gB, pp65 (HCMV) | Mice, NHPs | IM | Humoral, cellular | [32] |
| LNP | Conventional, nucleoside-modified | Ionizable lipid: DSPC: cholesterol: PEG-lipid | gp (Ebola virus) | Guinea pigs | IM | Humoral, protection | [33] |
| LNP | Conventional, nucleoside-modified | Ionizable cationic lipid (Acuitas Therapeutics)/phosphatidylcholine/cholesterol/ polyethylene glycol/lipid | Env (HIV) | Mice, NHPs | ID | Humoral, cellular | [6] |
| Liposomes | Conventional, unmodified | Cholesterol/dipalmitoyl phosphatidylcholine/phosphatidylserine | Nucleoprotein (influenza virus) | Mice | SC | Cellular | [34] |
| CNE, LNP, MDNP | Self-amplifying | Squalene, DOTAP, sorbitan trioleate and polysorbate 80 | HA (influenza virus) | Mice, ferrets | IM | Humoral, cellular, protection | [4,35,36] |
| CNE | Self-amplifying | Squalene, Span 85, DOTAP | gp140 (HIV) | Mice, rabbit, NHPs | IM | Humoral, cellular | [37,38] |
| CNE | Self-amplifying | Squalene, Span 85, DOTAP | gB, pp65-IE1 (HCMV) | NHPs | IM | Humoral, cellular | [38] |
| CNE | Self-amplifying | Squalene, DOTAP, sorbitan trioleate and polysorbate 80 | SLOdm, BP-2a (streptococci) | Mice | IM | Humoral, protection | [39] |
| CNE | Self-amplifying | Squalene, DOTAP, sorbitan trioleate and polysorbate 80 | PMIF (malaria) | Mice | IM | Humoral, cellular, protection | [40] |
| CNE, LNP | Self-amplifying | DSPC, cholesterol, DMG PEG 2000, DLinDMA | F (RSV) | Mice, cotton rats | IM | Humoral, cellular, protection | [38,41] |
| LNP | Self-amplifying | - | gp (rabies), gH/gL (HCMV) | Mice | IM | Humoral | [3] |
| LNP | Self-amplifying | DLinDMA: DSPC: DMG PEG 2000: cholesterol | NP, M1 (influenza virus) | Mice | IM | Humoral, cellular, protection | [10] |
| MDNP, NLC | Self-amplifying | Modified dendrimer and DMG PEG 2000 | prM-E (Zika virus) | Mice, guinea pigs | IM | Humoral | [42,43] |
| MDNP | Self-amplifying | Modified dendrimer and 1 DMG PEG 2000 | gp (Ebola virus) | Mice | IM | Humoral, cellular, protection | [36] |
| MDNP | Self-amplifying | Modified dendrimer and DMG PEG 2000 | Six antigens (Toxoplasma gondii) | Mice | IM | Protection | [36] |
| Cancer immunotherapy | |||||||
| LNP | Conventional, nucleoside-modified or Conventional, unmodified | Ovalbumin (OVA) or tumor-associated antigens TRP2 and gp100 | Mice | SC | [44] | ||
| LNP | Conventional, nucleoside-modified | Ionizable lipid: DSPC: cholesterol: PEG-lipid | Apoptic proteins, Caspase or PUMA | Mice | IV | [45] | |
| LNP | In vitro transcribed | Anti-HER2 Antibody | Mice | IV | [46] | ||
| LNP | In vitro transcribed | Ionizable lipid, cholesterol, DOPE, C16-polyethylene glycol2000 (PEG-lipid) | Ovalbumin (OVA) | Mice | IM | Cellular, innate immune response | [47] |
| Sponsoring Manufacturer | mRNA Vaccine | Delivery System | Target | Trial Number | Stage | Status | Reference |
|---|---|---|---|---|---|---|---|
| Infectious diseases | |||||||
| Moderna Therapeutics/National Institute of Allergy and Infectious Diseases (NIAID) | mRNA-1273 (perfusion stabilized S protein mRNA vaccine) | LNP | COVID-19 | NCT04470427 | Phase III | Active, not recruiting | [126] |
| BioNTech / Pfizer | BNT162 (3 LNP–mRNA vaccines) | LNP | COVID-19 | NCT04537949 | Phase III | Recruiting | [126] |
| CureVac | CV7202 (sequence-optimized) | LNP | Rabies | NCT03713086 | Phase I | Active, not recruiting, PCD: January 2022 | [127] |
| Moderna Therapeutics | mRNA-1440 (nucleoside-modified) | LNP | Influenza H10N8 | NCT03076385 | Phase I | Completed PCD: October 2018 | [27,125] |
| Moderna Therapeutics | mRNA-1851 (nucleoside-modified) | LNP | Influenza H7N9 | NCT03345043 | Phase I | Active, not recruiting, PCD: February 2020 | [27,125] |
| Moderna Therapeutics | mRNA-1653 (nucleoside-modified) | LNP | HMPV/HPIV3 | NCT03392389 | Phase I | Completed, PCD: July 2019 | [2] |
| Moderna Therapeutics | mRNA-1325 (nucleoside-modified) | LNP | Zika | NCT03014089 | Phase I | Completed, PCD: July 2019 | [123] |
| Moderna Therapeutics | mRNA-1893 | Zika | NCT04064905 | Phase I | Active, not recruiting, PCD: February 2021 | [123] | |
| Moderna Therapeutics | mRNA-1647 and mRNA-1443 (nucleoside-modified) | LNP | HCMV | NCT03382405 | Phase I | Active, not recruiting, PCD: July 2020 | [2] |
| Moderna Therapeutics | mRNA-1388 (nucleoside-modified) | LNP | Chikungunya | NCT03325075 | Phase I | Completed, PCD: November 2019 | [2] |
| Cancer immunotherapy | |||||||
| BioNTech RNA Pharmaceuticals GmbH | mRNA lipoplex (Lipo–MERIT) | Liposomes | TAAs (advanced melanoma) | NCT02410733 | Phase I | Active, not recruiting | [128] |
| BioNTech AG | mRNA lipoplex (TNBC–MERIT) | Liposomes | TAAs (triple-negative breast cancer) | NCT02316457 | Phase I | Active, not recruiting | [129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldosari, B.N.; Alfagih, I.M.; Almurshedi, A.S. Lipid Nanoparticles as Delivery Systems for RNA-Based Vaccines. Pharmaceutics 2021, 13, 206. https://doi.org/10.3390/pharmaceutics13020206
Aldosari BN, Alfagih IM, Almurshedi AS. Lipid Nanoparticles as Delivery Systems for RNA-Based Vaccines. Pharmaceutics. 2021; 13(2):206. https://doi.org/10.3390/pharmaceutics13020206
Chicago/Turabian StyleAldosari, Basmah N., Iman M. Alfagih, and Alanood S. Almurshedi. 2021. "Lipid Nanoparticles as Delivery Systems for RNA-Based Vaccines" Pharmaceutics 13, no. 2: 206. https://doi.org/10.3390/pharmaceutics13020206