
The TKI Era in Chronic Leukemias

 ,
,
Abstract
:1. Introduction
2. Chronic Myeloid Leukemia
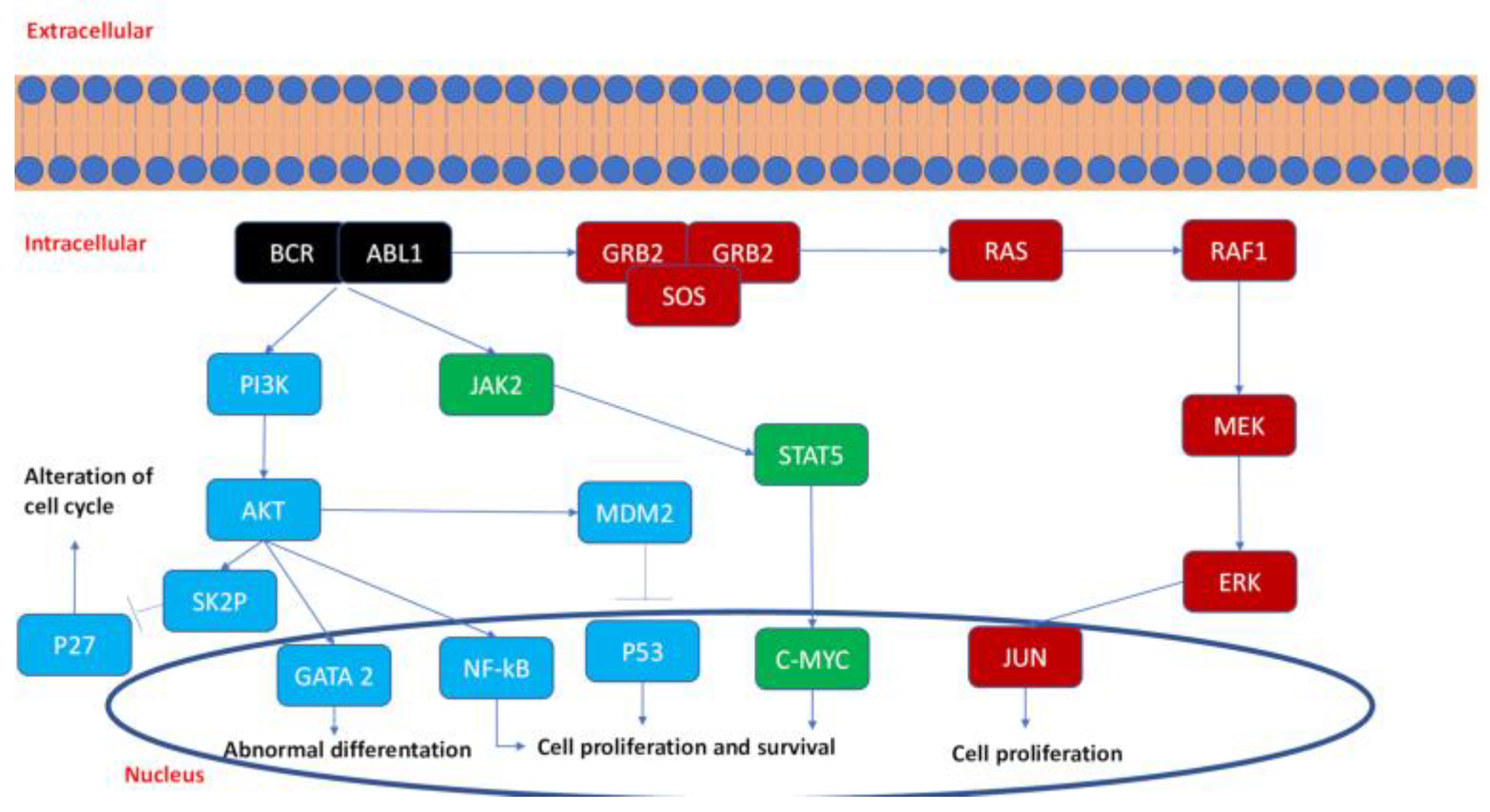
2.1. Kinase Pathways in CML
2.2. TK Inhibition in CML

{kind=link}
{kind=link}
{kind=link}
| N | Clinical Trial | Trial Phase | CML Phase | TKI Dosage | Response | Bcr-AblT315I | |
|---|---|---|---|---|---|---|---|
| Imatinib vs. interferon + low-dose of cytarabine | 1106 | IRIS (NCT00333840) | III | Chronic | 400 mg/die | MCR: 87% CCR: 76% | No |
| Imatinib vs. historic experience | 389 | Retrospective [58] | Accelerated | 600 mg/die | MCR: 49% CCR: 43% | no | |
| Dasatinib vs. imatinib | 519 | Dasision (NCT00481247) | III | Chronic | 100 mg/die | CCR: 83% MMR: 46% | no |
| Dasatinib | 174 | START-A | II | R/R Accelerated | 140 mg/die | MCR: 39% CCR: 32% | no |
| Dasatinib | 109 48 | START-C | II | R/R myeloid blast R/R lymphoid blast | 140 mg/die | MCR: 33% CCR: 23% MCR: 52% CCR: 46% | no |
| Nilotinib vs. imatinib | 846 | ENESTnd (NCT00471497) | III | Chronic | 600mg/die 800 mg/die | CCR: 80% MMR: 44% CCR: 78% MMR: 43% | no |
| Nilotinib | 136 | II [59] | R/R Accelerated | 800 mg/die | MCR: 31% CCR: 19% | no | |
| Bosutinib vs. imatinib | 536 | BFORE (NCT02130557) | III | Chronic | 400 mg/die | CCR: 77% MMR: 47% | no |
| Ponatinib | 267 83 62 | PACE (NCT01207440) | II | Chronic Accelerated Blastic | 45 mg/die | MCR: 56% CCR: 46% MMR: 34% MCR: 39% MCR: 23% | yes |
2.3. TKi Treatment Discontinuation in CML
3. Chronic Lymphocytic Leukemia
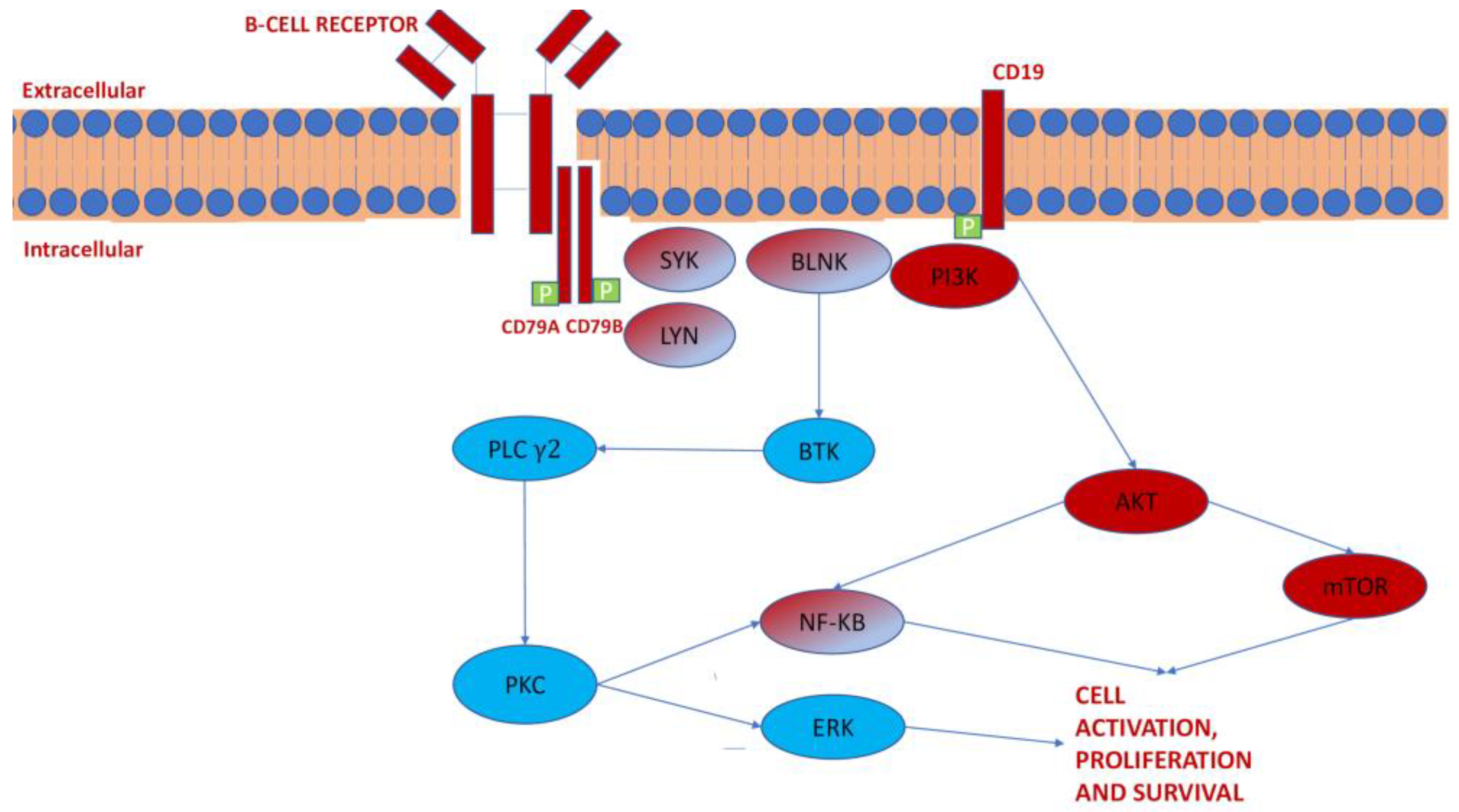
3.1. Kinase Pathways in CLL
3.2. TK Inhibition in CLL
4. Discussion
Funding
Conflicts of Interest
References
- Hubbard, S.R.; Till, J.H. Protein Tyrosine Kinase Structure and Function. Annu. Rev. Biochem. 2000, 69, 373–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine Kinase-Role and Significance in Cancer Review. 2004. Available online: www.medsci.org (accessed on 3 March 2021).
- Heldin, C.-H. Dimerization of cell surface receptors in signal transduction. Cell 1995, 80, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Schlessinger, J.; Ullrich, A. Growth factor signaling by receptor tyrosine kinases. Neuron 1992, 9, 383–391. [Google Scholar] [CrossRef]
- Höglund, M.; Sandin, F.; Simonsson, B. Epidemiology of chronic myeloid leukaemia: An update. Ann. Hematol. 2015, 94, 241–247. [Google Scholar] [CrossRef]
- Hallek, M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Thun, M.J. Cancer Statistics, 2007. CA Cancer J. Clin. 2007, 57, 43–66. [Google Scholar] [CrossRef]
- Kang, Z.-J.; Liu, Y.-F.; Xu, L.-Z.; Long, Z.-J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.-X.; Pan, Y.-J.; Yan, J.-S.; et al. The Philadelphia chromosome in leukemogenesis. Chin. J. Cancer 2016, 35, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Woyach, J.A.; Bojnik, E.; Ruppert, A.S.; Stefanovski, M.R.; Goettl, V.M.; Smucker, K.A.; Smith, L.L.; Dubovsky, J.A.; Towns, W.H.; MacMurray, J.; et al. Bruton’s tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood 2014, 123, 1207–1213. [Google Scholar] [CrossRef]
- Cuní, S.; Pérez-Aciego, P.; Pérez-Chacón, G.; Vargas, J.A.; Sánchez, A.; Martín-Saavedra, F.M.; Ballester, S.; García-Marco, J.; Jordá, J.; Durántez, A. A sustained activation of PI3K/NF-κB pathway is critical for the survival of chronic lymphocytic leukemia B cells. Leukemia 2004, 18, 1391–1400. [Google Scholar] [CrossRef]
- Rowley, J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia identified by Quinacrine Fluorescence and Giemsa Staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Arana-Trejo, R.M.; Ruíz Sánchez, E.; Ignacio-Ibarra, G.; De La Báez Fuente, E.; Garces, O.; Gómez Morales, E.; Castro Granados, M.; Ovilla Martínez, R.; Rubio-Borja, M.E.; Solís Anaya, L.; et al. BCR/ABL p210, p190 and p230 fusion genes in 250 Mexican patients with chronic myeloid leukaemia (CML). Clin. Lab. Haematol. 2002, 24, 145–150. [Google Scholar] [CrossRef]
- Adnan-Awad, S.; Kim, D.; Hohtari, H.; Javarappa, K.K.; Brandstoetter, T.; Mayer, I.; Potdar, S.; Heckman, C.A.; Kytölä, S.; Porkka, K.; et al. Characterization of p190-Bcr-Abl chronic myeloid leukemia reveals specific signaling pathways and therapeutic targets. Leukemia 2021, 35, 1964–1975. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Saglio, G. Molecular Pathways: BCR-ABL. Clin. Cancer Res. 2012, 18, 930–937. [Google Scholar] [CrossRef] [Green Version]
- Ilaria, R.L.; Van Etten, R.A. P210 and P190 Induce the Tyrosine Phosphorylation and DNA Binding Activity of Multiple Specific STAT Family Members. J. Biol. Chem. 1996, 271, 31704–31710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyers, C.L. The Role of MYC in Transformation by BCR-ABL. Leuk. Lymphoma 1993, 11, 45–46. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, L.; Zhang, B.; Li, J.; Dou, X.; Zhao, R.C. TGF-β 1-induced PI3K/Akt/NF- B/MMP9 signalling pathway is activated in Philadelphia chromosome-positive chronic myeloid leukaemia hemangioblasts. J. Biochem. 2011, 149, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, Y.; Fang, S.; Wang, L.; Qi, H.; Zhang, Y.; Zhang, J.; Li, W. BCR/ABL oncogene-induced PI3K signaling pathway leads to chronic myeloid leukemia pathogenesis by impairing immuno-modulatory function of hemangioblasts. Cancer Gene Ther. 2015, 22, 227–237. [Google Scholar] [CrossRef]
- Andreu, E.J.; Lledó, E.; Poch, E.; Ivorra, C.; Albero, M.P.; Martínez-Climent, J.A.; Montiel-Duarte, C.; Rifón, J.; Pérez-Calvo, J.; Arbona, C.; et al. BCR-ABL Induces the Expression of Skp2 through the PI3K Pathway to Promote p27Kip1 Degradation and Proliferation of Chronic Myelogenous Leukemia Cells. Cancer Res. 2005, 65, 3264–3272. [Google Scholar] [CrossRef] [Green Version]
- Kharas, M.G.; Janes, M.R.; Scarfone, V.M.; Lilly, M.B.; Knight, Z.A.; Shokat, K.M.; Fruman, D.A. Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL+ leukemia cells. J. Clin. Investig. 2008, 118, 3038–3050. [Google Scholar] [CrossRef] [Green Version]
- Steelman, L.S.; Pohnert, S.C.; Shelton, J.G.; Franklin, R.A.; Bertrand, F.E.; McCubrey, J.A. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 2004, 18, 189–218. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Hehlmann, R.; Heimpel, H.; Hasford, J.; Kolb, H.J.; Pralle, H.; Hossfeld, D.K.; Queisser, W.; Loffler, H.; Hochhaus, A.; Heinze, B. Randomized comparison of interferon-alpha with busulfan and hydroxyurea in chronic myelogenous leukemia. The German CML Study Group [see comments]. Blood 1994, 84, 4064–4077. [Google Scholar] [CrossRef] [Green Version]
- Chronic granulocytic leukaemia: Comparison of radiotherapy and busulphan therapy. Report of the Medical Research Council’s working party for therapeutic trials in leukaemia. BMJ 1968, 1, 201–208. [CrossRef] [Green Version]
- Verma, D.; Spitzer, G.; Gutterman, J.U.; Zander, A.R.; McCredie, K.B.; Dicke, K.A. Human leukocyte interferon preparation blocks granulopoietic differentiation. Blood 1979, 54, 1423–1427. [Google Scholar] [CrossRef]
- Italian Cooperative Study Group on Chronic Myeloid Leukemia; Tura, S.; Baccarani, M.; Zuffa, E.; Russo, D.; Fanin, R.; Zaccaria, A.; Fiacchini, M. Interferon Alfa-2a as Compared with Conventional Chemotherapy for the Treatment of Chronic Myeloid Leukemia. N. Engl. J. Med. 1994, 330, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Zubay, G.; Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia: Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lympoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 2001, 344, 1038–1042. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, J.A.; Hobbs, G. Tyrosine Kinase Inhibitors in the Treatment of Chronic-Phase CML: Strategies for Frontline Decision-making. Curr. Hematol. Malig. Rep. 2018, 13, 202–211. [Google Scholar] [CrossRef]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr–Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- O’Dwyer, M.E.; Druker, B.J. STI571: An inhibitor of the BCR-ABL tyrosine kinase for the treatment of chronic myelogenous leukaemia. Lancet Oncol. 2000, 1, 207–211. [Google Scholar] [CrossRef]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and Safety of a Specific Inhibitor of the BCR-ABL Tyrosine Kinase in Chronic Myeloid Leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef] [Green Version]
- Branford, S. Molecular monitoring in chronic myeloid leukemia—How low can you go? Hematology 2016, 2016, 156–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, T.P.; Kaeda, J.; Branford, S.; Rudzki, Z.; Hochhaus, A.; Hensley, M.L.; Gathmann, I.; Bolton, A.E.; Van Hoomissen, I.C.; Goldman, J.M.; et al. Frequency of Major Molecular Responses to Imatinib or Interferon Alfa plus Cytarabine in Newly Diagnosed Chronic Myeloid Leukemia. N. Engl. J. Med. 2003, 349, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Litzow, M.R. Imatinib resistance: Obstacles and opportunities. Arch. Pathol. Lab. Med. 2006, 130, 669–679. [Google Scholar] [CrossRef]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.M.; Gattermann, N.; Deininger, M.W.N.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-Year Follow-up of Patients Receiving Imatinib for Chronic Myeloid Leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef]
- Milojkovic, D.; Apperley, J.F. Mechanisms of Resistance to Imatinib and Second-Generation Tyrosine Inhibitors in Chronic Myeloid Leukemia. Clin. Cancer Res. 2009, 15, 7519–7527. [Google Scholar] [CrossRef] [Green Version]
- Tokarski, J.S.; Newitt, J.A.; Chang, C.Y.J.; Cheng, J.D.; Wittekind, M.; Kiefer, S.E.; Kish, K.; Lee, F.Y.F.; Borzillerri, R.; Lombardo, L.J.; et al. The Structure of Dasatinib (BMS-354825) Bound to Activated ABL Kinase Domain Elucidates Its Inhibitory Activity against Imatinib-Resistant ABL Mutants. Cancer Res. 2006, 66, 5790–5797. [Google Scholar] [CrossRef] [Green Version]
- Talpaz, M.; Shah, N.P.; Kantarjian, H.; Donato, N.; Nicoll, J.; Paquette, R.; Cortes, J.; O’Brien, S.; Nicaise, C.; Bleickardt, E.; et al. Dasatinib in Imatinib-Resistant Philadelphia Chromosome–Positive Leukemias. N. Engl. J. Med. 2006, 354, 2531–2541. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.; Rousselot, P.; Kim, D.-W.; Ritchie, E.; Hamerschlak, N.; Coutre, S.; Hochhaus, A.; Guilhot, F.; Saglio, G.; Apperley, J.; et al. Dasatinib induces complete hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in blast crisis. Blood 2006, 109, 3207–3213. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H.M.; Saglio, G.; Steegmann, J.L.; Shah, N.P.; Boqué, C.; Chuah, C.; Pavlovsky, C.; Mayer, J.; Cortes, J.; et al. Early response with dasatinib or imatinib in chronic myeloid leukemia: 3-year follow-up from a randomized phase 3 trial (DASISION). Blood 2014, 123, 494–500. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.M.; Shah, N.P.; Cortes, J.E.; Baccarani, M.; Agarwal, M.B.; Undurraga, M.S.; Wang, J.; Ipiña, J.J.K.; Kim, D.-W.; Ogura, M.; et al. Dasatinib or imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: 2-year follow-up from a randomized phase 3 trial (DASISION). Blood 2012, 119, 1123–1129. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.E.; Saglio, G.; Kantarjian, H.M.; Baccarani, M.; Mayer, J.; Boqué, C.; Shah, N.P.; Chuah, C.; Casanova, L.; Bradley-Garelik, B.; et al. Final 5-Year Study Results of DASISION: The Dasatinib Versus Imatinib Study in Treatment-Naïve Chronic Myeloid Leukemia Patients Trial. J. Clin. Oncol. 2016, 34, 2333–2340. [Google Scholar] [CrossRef]
- O’Hare, T.; Eide, C.A.; Deininger, M.W.N. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007, 110, 2242–2249. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Hochhaus, A.; Saglio, G.; De Souza, C.; Flinn, I.W.; Stenke, L.; Goh, Y.-T.; Rosti, G.; Nakamae, H.; Gallagher, N.J.; et al. Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol. 2011, 12, 841–851. [Google Scholar] [CrossRef]
- Hochhaus, A.; Saglio, G.; Hughes, T.; Larson, R.; Kim, D.-W.; Issaragrisil, S.; Le Coutre, P.D.; Etienne, G.; E Dorlhiac-Llacer, P.; E Clark, R.; et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia 2016, 30, 1044–1054. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Hughes, T.P.; Larson, R.A.; Kim, D.-W.; Issaragrisil, S.; le Coutre, P.; Etienne, G.; Boquimpani, C.; Pasquini, R.; Clark, R.E.; et al. Long-term outcomes with frontline nilotinib versus imatinib in newly diagnosed chronic myeloid leukemia in chronic phase: ENESTnd 10-year analysis. Leukemia 2021, 35, 440–453. [Google Scholar] [CrossRef]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Commodore, L.; Huang, W.-S.; Wang, Y.; Thomas, M.; Keats, J.; Xu, Q.; Rivera, V.M.; Shakespeare, W.C.; Clackson, T.; et al. Structural Mechanism of the Pan-BCR-ABL Inhibitor Ponatinib (AP24534): Lessons for Overcoming Kinase Inhibitor Resistance. Chem. Biol. Drug Des. 2010, 77, 1–11. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kim, D.-W.; Pinilla-Ibarz, J.; Le Coutre, P.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. A Phase 2 Trial of Ponatinib in Philadelphia Chromosome–Positive Leukemias. N. Engl. J. Med. 2013, 369, 1783–1796. [Google Scholar] [CrossRef] [Green Version]
- Cross, N.; E White, H.; Müller, M.C.; Saglio, G.; Hochhaus, A. Standardized definitions of molecular response in chronic myeloid leukemia. Leukemia 2012, 26, 2172–2175. [Google Scholar] [CrossRef] [Green Version]
- Lipton, J.H.; Chuah, C.; Guerci-Bresler, A.; Rosti, G.; Simpson, D.; Assouline, S.; Etienne, G.; Nicolini, F.E.; Le Coutre, P.; Clark, R.; et al. Epic: A Phase 3 Trial of Ponatinib Compared with Imatinib in Patients with Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase (CP-CML). Blood 2014, 124, 519. [Google Scholar] [CrossRef]
- Cortes, J.; Kantarjian, H.M.; Kim, D.W.; Khoury, H.J.; Turkina, A.G.; Shen, Z.-X.; Brummendorf, T.H.; Chandy, M.; Arkin, S.; Gambacorti-Passerini, C. Efficacy and Safety of Bosutinib (SKI-606) in Patients with Chronic Phase (CP) Ph+ Chronic Myelogenous Leukemia (CML) with Resistance or Intolerance to Imatinib. Blood 2008, 112, 1098. [Google Scholar] [CrossRef]
- Cortes, J.E.; Gambacorti-Passerini, C.; Deininger, M.W.; Mauro, M.J.; Chuah, C.; Kim, D.-W.; Dyagil, I.; Glushko, N.; Milojkovic, D.; Le Coutre, P.; et al. Bosutinib Versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia: Results From the Randomized BFORE Trial. J. Clin. Oncol. 2018, 36, 231–237. [Google Scholar] [CrossRef]
- Schoepfer, J.; Jahnke, W.; Berellini, G.; Buonamici, S.; Cotesta, S.; Cowan-Jacob, S.W.; Dodd, S.; Drueckes, P.; Fabbro, D.; Gabriel, T.; et al. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J. Med. Chem. 2018, 61, 8120–8135. [Google Scholar] [CrossRef] [Green Version]
- Hughes, F.T.P.; Goh, M.Y.-T.; Ottmann, O.G.; Minami, H.; Rea, D.; Lang, F.; Mauro, M.J.; DeAngelo, D.J.; Talpaz, M.; Hochhaus, A.; et al. Expanded Phase 1 Study of ABL001, a Potent, Allosteric Inhibitor of BCR-ABL, Reveals Significant and Durable Responses in Patients with CML-Chronic Phase with Failure of Prior TKI Therapy. Blood 2016, 128, 625. [Google Scholar] [CrossRef]
- Saglio, G.; Hughes, T.P.; Geissler, J.; Kapoor, S.; Longin, A.-S.; Mukherjee, A.; Cortes, J.E. Randomized, Open-Label, Multicenter, Phase 2 Study of Asciminib (ABL001) As an Add-on to Imatinib Versus Continued Imatinib Versus Switch to Nilotinib in Patients with Chronic Myeloid Leukemia in Chronic Phase Who Have Not Achieved a Deep Molecular Response with Frontline Imatinib. Blood 2019, 134, 5910. [Google Scholar] [CrossRef]
- Hochhaus, A.; Boquimpani, C.; Rea, M.D.; Minami, M.Y.; Lomaia, E.; Voloshin, S.; Turkina, A.G.; Kim, D.-W.; Apperley, F.J.; Cortes, J.E.; et al. Efficacy and Safety Results from ASCEMBL, a Multicenter, Open-Label, Phase 3 Study of Asciminib, a First-in-Class STAMP Inhibitor, vs. Bosutinib (BOS) in Patients (Pts) with Chronic Myeloid Leukemia in Chronic Phase (CML-CP) Previously Treated with ≥2 Tyrosine Kinase Inhibitors (TKIs). Blood 2020, 136, LBA-4. [Google Scholar] [CrossRef]
- Kantarjian, H.; Talpaz, M.; O’Brien, S.; Giles, F.; Faderl, S.; Verstovsek, S.; Garcia-Manero, G.; Shan, J.; Rios, M.B.; Champlin, R.; et al. Survival benefit with imatinib mesylate therapy in patients with accelerated-phase chronic myelogenous leukemia—Comparison with historic experience. Cancer 2005, 103, 2099–2108. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.J.; Abruzzese, E.; Rosti, G.; Kim, N.-W.; Bhatia, R.; Bosly, A.; Goldberg, S.; Kam, G.L.; Jagasia, M.; Mendrek, W.; et al. Nilotinib is active in chronic and accelerated phase chronic myeloid leukemia following failure of imatinib and dasatinib therapy. Leukemia 2010, 24, 1299–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, D.M.; Branford, S.; Seymour, J.F.; Schwarer, A.P.; Arthur, C.; Yeung, D.T.; Dang, P.; Goyne, J.M.; Slader, C.; Filshie, R.J.; et al. Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: Results from the TWISTER study. Blood 2013, 122, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Mahon, F.-X.; Réa, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Shah, N.P.; García-Gutiérrez, V.; Jiménez-Velasco, A.; Larson, S.; Saussele, S.; Rea, D.; Mahon, F.-X.; Levy, M.Y.; Gómez-Casares, M.T.; Pane, F.; et al. Dasatinib discontinuation in patients with chronic-phase chronic myeloid leukemia and stable deep molecular response: The DASFREE study. Leuk. Lymphoma 2020, 61, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Saussele, S.; Richter, J.; Guilhot, J.; Gruber, F.X.; Hjorth-Hansen, H.; Almeida, A.; Janssen, J.J.W.M.; Mayer, J.; Koskenvesa, P.; Panayiotidis, P.; et al. Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO-SKI): A prespecified interim analysis of a prospective, multicentre, non-randomised, trial. Lancet Oncol. 2018, 19, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.P. NCCN Guidelines Updates: Discontinuing TKI Therapy in the Treatment of Chronic Myeloid Leukemia. J. Natl. Compr. Cancer Netw. 2019, 17, 611–613. [Google Scholar]
- Herishanu, Y.; Pérez-Galán, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-κB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011, 117, 563–574. [Google Scholar] [CrossRef]
- Dal Porto, J.M.; Gauld, S.B.; Merrell, K.T.; Mills, D.; Pugh-Bernard, A.E.; Cambier, J. B cell antigen receptor signaling 101. Mol. Immunol. 2004, 41, 599–613. [Google Scholar] [CrossRef]
- Vihinen, M.; Mattsson, P.T.; Smith, C.I. Bruton tyrosine kinase BTK in X-linked agammaglobulinemia XLA. Front. Biosci. 2000, 5, 917–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crofford, L.J.; Nyhoff, L.E.; Sheehan, J.H.; Kendall, P.L. The role of Bruton’s tyrosine kinase in autoimmunity and implications for therapy. Expert Rev. Clin. Immunol. 2016, 12, 763–773. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Ma, J.; Guo, A.; Lu, P.; Leonard, J.P.; Coleman, M.P.; Liu, M.; Buggy, J.J.; Furman, R.R.; Wang, Y.L. BTK inhibition targets in vivo CLL proliferation through its effects on B-cell receptor signaling activity. Leukemia 2013, 28, 649–657. [Google Scholar] [CrossRef]
- Wiestner, A. Emerging role of kinase-targeted strategies in chronic lymphocytic leukemia. Hematology 2012, 2012, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Johnson, A.J.; Byrd, J.C. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood 2012, 120, 1175–1184. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Huynh, L.; Apgar, J.; Tang, L.; Rassenti, L.; Weiss, A.; Kipps, T.J. ZAP-70 enhances IgM signaling independent of its kinase activity in chronic lymphocytic leukemia. Blood 2008, 111, 2685–2692. [Google Scholar] [CrossRef]
- Zhang, Y.-R.; Lyu, P.; Xiong, W.-J.; Liu, X.-X.; Liu, H.-M.; Cui, R.; Wang, Q.; Chen, W.-M.; Qiu, L.-G.; Yi, S.-H. TOSO interacts with SYK and enhances BCR pathway activation in chronic lymphocytic leukemia. Chin. Med. J. 2020, 133, 2090–2097. [Google Scholar] [CrossRef]
- Jaglowski, S.M.; Byrd, J.C. Rituximab in Chronic Lymphocytic Leukemia. Semin. Hematol. 2010, 47, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Cartron, G.; De Guibert, S.; Dilhuydy, M.-S.; Morschhauser, F.; Leblond, V.; Dupuis, J.; Mahe, B.; Bouabdallah, R.; Lei, G.; Wenger, M.; et al. Obinutuzumab (GA101) in relapsed/refractory chronic lymphocytic leukemia: Final data from the phase 1/2 GAUGUIN study. Blood 2014, 124, 2196–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Clarke, M.; Wheatley, K.; Peto, R. Chemotherapeutic Options in Chronic Lymphocytic Leukemia: A Meta-analysis of the Randomized Trials. J. Natl. Cancer Inst. 1999, 91, 861–868. [Google Scholar] [CrossRef]
- Niederle, N.; Megdenberg, D.; Balleisen, L.; Heit, W.; Knauf, W.; Weiß, J.; Freier, W.; Hinke, A.; Ibach, S.; Eimermacher, H. Bendamustine compared to fludarabine as second-line treatment in chronic lymphocytic leukemia. Ann. Hematol. 2013, 92, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; O’Brien, S.; Wierda, W.; Kantarjian, H.; Wen, S.; Do, K.-A.; Thomas, D.A.; Cortes, J.; Lerner, S.; Keating, M.J. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood 2008, 112, 975–980. [Google Scholar] [CrossRef] [Green Version]
- Herman, S.E.M.; Gordon, A.L.; Hertlein, E.; Ramanunni, A.; Zhang, X.; Jaglowski, S.; Flynn, J.; Jones, J.; Blum, K.A.; Buggy, J.J.; et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood 2011, 117, 6287–6296. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.E.M.; Gordon, A.L.; Wagner, A.J.; Heerema, N.A.; Zhao, W.; Flynn, J.M.; Jones, J.; Andritsos, L.; Puri, K.D.; Lannutti, B.J.; et al. Phosphatidylinositol 3-kinase-δ inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010, 116, 2078–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Tian, D.; Ren, X.; Ding, S.; Jia, M.; Xin, M.; Thareja, S. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: A mini-review. Eur. J. Med. Chem. 2018, 151, 315–326. [Google Scholar] [CrossRef] [PubMed]
- De Claro, R.A.; McGinn, K.M.; Verdun, N.; Lee, S.-L.; Chiu, H.-J.; Saber, H.; Brower, M.E.; Chang, C.G.; Pfuma, E.; Habtemariam, B.; et al. FDA Approval: Ibrutinib for Patients with Previously Treated Mantle Cell Lymphoma and Previously Treated Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2015, 21, 3586–3590. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, S.; Jones, J.A.; Coutre, S.E.; Mato, A.R.; Hillmen, P.; Tam, C.; Österborg, A.; Siddiqi, T.; Thirman, M.J.; Furman, R.R.; et al. Ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia with 17p deletion (RESONATE-17): A phase 2, open-label, multicentre study. Lancet Oncol. 2016, 17, 768–778. [Google Scholar] [CrossRef]
- Zenz, T.; Mohr, J.; Edelmann, J.; Sarno, A.; Hoth, P.; Heuberger, M.; Helfrich, H.; Mertens, D.; Döhner, H.; Stilgenbauer, S. Treatment resistance in chronic lymphocytic leukemia–the role of the p53 pathway. Leuk. Lymphoma 2009, 50, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2013, 369, 32–42. [Google Scholar] [CrossRef]
- Thompson, P.A.; O’Brien, S.M.; Wierda, W.G.; Ferrajoli, A.; Stingo, F.C.; Smith, S.C.; Burger, J.A.; Estrov, Z.; Jain, N.; Kantarjian, H.M.; et al. Complex karyotype is a stronger predictor than del(17p) for an inferior outcome in relapsed or refractory chronic lymphocytic leukemia patients treated with ibrutinib-based regimens. Cancer 2015, 121, 3612–3621. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A.; Tedeschi, A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Bairey, O.; Hillmen, P.; Bartlett, N.L.; Li, J.; et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2015, 373, 2425–2437. [Google Scholar] [CrossRef]
- Moreno, C.; Greil, R.; Demirkan, F.; Tedeschi, A.; Anz, B.; Larratt, L.; Šimkovič, M.; Samoilova, O.; Novak, J.; Ben-Yehuda, D.; et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2018, 20, 43–56. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Wang, X.V.; Kay, N.E.; Hanson, C.A.; O’Brien, S.; Barrientos, J.; Jelinek, D.F.; Braggio, E.; Leis, J.F.; Zhang, C.C.; et al. Ibrutinib–Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 432–443. [Google Scholar] [CrossRef]
- Woyach, J.A.; Ruppert, A.S.; Heerema, N.A.; Zhao, W.; Booth, A.M.; Ding, W.; Bartlett, N.L.; Brander, D.M.; Barr, P.M.; Rogers, K.A.; et al. Ibrutinib Regimens versus Chemoimmunotherapy in Older Patients with Untreated CLL. N. Engl. J. Med. 2018, 379, 2517–2528. [Google Scholar] [CrossRef]
- Munir, T.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Barr, P.M.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Final analysis from RESONATE: Up to six years of follow-up on ibrutinib in patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma. Am. J. Hematol. 2019, 94, 1353–1363. [Google Scholar] [CrossRef] [Green Version]
- Chanan-Khan, A.; Cramer, P.; Demirkan, F.; Fraser, G.; Silva, R.S.; Grosicki, S.; Pristupa, A.; Janssens, A.; Mayer, J.; Bartlett, N.L.; et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): A randomised, double-blind, phase 3 study. Lancet Oncol. 2015, 17, 200–211. [Google Scholar] [CrossRef]
- Fraser, G.; Cramer, P.; Demirkan, F.; Silva, R.S.; Grosicki, S.; Pristupa, A.; Janssens, A.; Mayer, J.; Bartlett, N.; Dilhuydy, M.-S.; et al. Updated results from the phase 3 HELIOS study of ibrutinib, bendamustine, and rituximab in relapsed chronic lymphocytic leukemia/small lymphocytic lymphoma. Leukemia 2018, 33, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, B.T.; Ellmeier, W.; Watson, S.P. Tec regulates platelet activation by GPVI in the absence of Btk. Blood 2003, 102, 3592–3599. [Google Scholar] [CrossRef] [Green Version]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Burger, J.A.; Blum, K.A.; Coleman, M.; Wierda, W.G.; Jones, J.A.; Zhao, W.; Heerema, N.A.; et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015, 125, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Paydas, S. Management of adverse effects/toxicity of ibrutinib. Crit. Rev. Oncol. 2019, 136, 56–63. [Google Scholar] [CrossRef]
- Mato, A.R.; Nabhan, C.; Thompson, M.C.; Lamanna, N.; Brander, D.M.; Hill, B.; Howlett, C.; Skarbnik, A.; Cheson, B.D.; Zent, C.; et al. Toxicities and outcomes of 616 ibrutinib-treated patients in the United States: A real-world analysis. Haematologica 2018, 103, 874–879. [Google Scholar] [CrossRef]
- Sharman, J.P.; Banerji, V.; Fogliatto, L.M.; Herishanu, Y.; Munir, T.; Walewska, R.; Follows, G.; Karlsson, K.; Ghia, P.; Corbett, G.; et al. ELEVATE TN: Phase 3 Study of Acalabrutinib Combined with Obinutuzumab (O) or Alone Vs O Plus Chlorambucil (Clb) in Patients (Pts) with Treatment-Naive Chronic Lymphocytic Leukemia (CLL). Blood 2019, 134, 31. [Google Scholar] [CrossRef]
- Ghia, P.; Pluta, A.; Wach, M.; Lysak, D.; Kozak, T.; Šimkovič, M.; Kaplan, P.; Kraychok, I.; Illes, A.; de la Serna, J.; et al. ASCEND: Phase III, Randomized Trial of Acalabrutinib Versus Idelalisib Plus Rituximab or Bendamustine Plus Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2020, 38, 2849–2861. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Hillmen, P.; Ghia, P.; Kater, A.P.; Chanan-Khan, A.; Furman, R.R.; O’Brien, S.; Yenerel, M.N.; Illés, A.; Kay, N.; et al. Acalabrutinib Versus Ibrutinib in Previously Treated Chronic Lymphocytic Leukemia: Results of the First Randomized Phase III Trial. J. Clin. Oncol. 2021, 39, 3441–3452. [Google Scholar] [CrossRef] [PubMed]
- Hillmen, P.; Brown, J.R.; Eichhorst, B.F.; Lamanna, N.; O’Brien, S.M.; Qiu, L.; Salmi, T.; Hilger, J.; Wu, K.; Cohen, A.; et al. ALPINE: Zanubrutinib versus ibrutinib in relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma. Future Oncol. 2020, 16, 517–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danilov, A.V.; Herbaux, C.; Walter, H.S.; Hillmen, P.; Rule, S.A.; Kio, E.A.; Karlin, L.; Dyer, M.J.; Mitra, S.S.; Yi, P.C.; et al. Phase Ib Study of Tirabrutinib in Combination with Idelalisib or Entospletinib in Previously Treated Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2020, 26, 2810–2818. [Google Scholar] [CrossRef] [Green Version]
- Kaul, M.; End, P.; Cabanski, M.; Schuhler, C.; Jakab, A.; Kistowska, M.; Kinhikar, A.; Maiolica, A.; Sinn, A.; Fuhr, R.; et al. Remibrutinib (LOU064): A selective potent oral BTK inhibitor with promising clinical safety and pharmacodynamics in a randomized phase I trial. Clin. Transl. Sci. 2021, 14, 1756–1768. [Google Scholar] [CrossRef]
- Sharma, S.; Galanina, N.; Guo, A.; Lee, J.; Kadri, S.; Van Slambrouck, C.; Long, B.; Wang, W.; Ming, M.; Furtado, L.V.; et al. Identification of a structurally novel BTK mutation that drives ibrutinib resistance in CLL. Oncotarget 2016, 7, 68833–68841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estupiñán, H.Y.; Wang, Q.; Berglöf, A.; Schaafsma, G.C.P.; Shi, Y.; Zhou, L.; Mohammad, D.K.; Yu, L.; Vihinen, M.; Zain, R.; et al. BTK gatekeeper residue variation combined with cysteine 481 substitution causes super-resistance to irreversible inhibitors acalabrutinib, ibrutinib and zanubrutinib. Leukemia 2021, 35, 1317–1329. [Google Scholar] [CrossRef]
- Hamasy, A.; Wang, Q.; Blomberg, K.E.M.; Mohammad, D.K.; Yu, L.; Vihinen, M.; Berglöf, A.; Smith, C.I.E. Substitution scanning identifies a novel, catalytically active ibrutinib-resistant BTK cysteine 481 to threonine (C481T) variant. Leukemia 2017, 31, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woyach, J.A.; Furman, R.R.; Liu, T.-M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.-H.; Steggerda, S.M.; Versele, M.; et al. Resistance Mechanisms for the Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.I.E.; Burger, J.A. Resistance Mutations to BTK Inhibitors Originate From the NF-κB but Not From the PI3K-RAS-MAPK Arm of the B Cell Receptor Signaling Pathway. Front. Immunol. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Reiff, S.D.; Mantel, R.; Smith, L.L.; Greene, J.T.; Muhowski, E.M.; Fabian, C.A.; Goettl, V.M.; Tran, M.; Harrington, B.K.; Rogers, K.A.; et al. The BTK Inhibitor ARQ 531 Targets Ibrutinib-Resistant CLL and Richter Transformation. Cancer Discov. 2018, 8, 1300–1315. [Google Scholar] [CrossRef]
- Chan, P.; Yu, J.; Chinn, L.; Prohn, M.; Huisman, J.; Matzuka, B.; Hanley, W.; Tuckwell, K.; Quartino, A. Population Pharmacokinetics, Efficacy Exposure-response Analysis, and Model-based Meta-analysis of Fenebrutinib in Subjects with Rheumatoid Arthritis. Pharm. Res. 2020, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Fabian, C.A.; Reiff, S.D.; Guinn, D.; Neuman, L.; Fox, J.A.; Wilson, W.; Byrd, J.C.; Woyach, J.A.; Johnson, A.J. Abstract 1207: SNS-062 Demonstrates Efficacy in Chronic Lymphocytic Leukemia In Vitro and Inhibits C481S Mutated Bruton Tyrosine Kinase; American Association for Cancer Research: Philadelphia, PA, USA, 2017. [Google Scholar]
- Woyach, J.; Stephens, D.D.M.; Flinn, I.W.; Bhat, S.A.; Savage, R.E.; Chai, F.; Eathiraj, S.; Granlund, L.; Szuszkiewicz, L.A.; Schwartz, B.; et al. Final Results of Phase 1, Dose Escalation Study Evaluating ARQ 531 in Patients with Relapsed or Refractory B-Cell Lymphoid Malignancies. Blood 2019, 134, 4298. [Google Scholar] [CrossRef]
- Byrd, J.C.; Smith, S.; Wagner-Johnston, N.; Sharman, J.; Chen, A.I.; Advani, R.; Augustson, B.; Marlton, P.; Commerford, S.R.; Okrah, K.; et al. First-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget 2018, 9, 13023–13035. [Google Scholar] [CrossRef] [Green Version]
- Mato, A.R.; Shah, N.N.; Jurczak, W.; Cheah, C.Y.; Pagel, J.M.; Woyach, J.A.; Fakhri, B.; Eyre, T.A.; Lamanna, N.; Patel, M.R.; et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): A phase 1/2 study. Lancet 2021, 397, 892–901. [Google Scholar] [CrossRef]
- Barrientos, J.C.; Furman, R.R.; Leonard, J.; Flinn, I.; Rai, K.R.; De Vos, S.; Schreeder, M.T.; Wagner-Johnston, N.D.; Sharman, J.P.; Boyd, T.E.; et al. Update on a phase I study of the selective PI3Kδ inhibitor idelalisib (GS-1101) in combination with rituximab and/or bendamustine in patients with relapsed or refractory CLL. J. Clin. Oncol. 2013, 31, 7017. [Google Scholar] [CrossRef]
- O’Brien, S.M.; Lamanna, N.; Kipps, T.J.; Flinn, I.; Zelenetz, A.D.; Burger, J.A.; Keating, M.; Mitra, S.; Holes, L.; Yu, A.S.; et al. A phase 2 study of idelalisib plus rituximab in treatment-naïve older patients with chronic lymphocytic leukemia. Blood 2015, 126, 2686–2694. [Google Scholar] [CrossRef] [Green Version]
- Lampson, B.L.; Kasar, S.N.; Matos, T.R.; Morgan, E.A.; Rassenti, L.; Davids, M.S.; Fisher, D.C.; Freedman, A.S.; Jacobson, C.A.; Armand, P.; et al. Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-mediated hepatotoxicity. Blood 2016, 128, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.A.; Robak, T.; Brown, J.R.; Awan, F.T.; Badoux, X.; Coutre, S.; Loscertales, J.; Taylor, K.; Vandenberghe, E.; Wach, M.; et al. Efficacy and safety of idelalisib in combination with ofatumumab for previously treated chronic lymphocytic leukaemia: An open-label, randomised phase 3 trial. Lancet Haematol. 2017, 4, e114–e126. [Google Scholar] [CrossRef]
- Zelenetz, A.D.; Barrientos, J.C.; Brown, J.R.; Coiffier, B.; Delgado, J.; Egyed, M.; Ghia, P.; Illés, P.Á.; Jurczak, W.; Marlton, P.; et al. Idelalisib or placebo in combination with bendamustine and rituximab in patients with relapsed or refractory chronic lymphocytic leukaemia: Interim results from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2017, 18, 297–311. [Google Scholar] [CrossRef] [Green Version]
- Vangapandu, H.V.; Jain, N.; Gandhi, V. Duvelisib: A phosphoinositide-3 kinase δ/γ inhibitor for chronic lymphocytic leukemia. Expert Opin. Investig. Drugs 2017, 26, 625–632. [Google Scholar] [CrossRef] [Green Version]
- Flinn, I.W.; O’Brien, S.; Kahl, B.; Patel, M.; Oki, Y.; Foss, F.F.; Porcu, P.; Jones, J.; Burger, J.A.; Jain, N.; et al. Duvelisib, a novel oral dual inhibitor of PI3K-δ,γ, is clinically active in advanced hematologic malignancies. Blood 2018, 131, 877–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flinn, I.W.; Hillmen, P.; Montillo, M.; Nagy, Z.; Illés, A.; Etienne, G.; Delgado, J.; Kuss, B.J.; Tam, C.S.; Gasztonyi, Z.; et al. The phase 3 DUO trial: Duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood 2018, 132, 2446–2455. [Google Scholar] [CrossRef] [Green Version]
- Forero-Torres, A.; Ramchandren, R.; Yacoub, A.; Wertheim, M.S.; Edenfield, W.J.; Caimi, P.; Gutierrez, M.; Akard, L.; Escobar, C.; Call, J.; et al. Parsaclisib, a potent and highly selective PI3Kδ inhibitor, in patients with relapsed or refractory B-cell malignancies. Blood 2019, 133, 1742–1752. [Google Scholar] [CrossRef] [Green Version]
- Mato, A.R.; Ghosh, N.; Schuster, S.J.; Lamanna, N.; Pagel, J.M.; Flinn, I.W.; Barrientos, J.C.; Rai, K.R.; Reeves, J.A.; Cheson, B.D.; et al. Phase 2 study of the safety and efficacy of umbralisib in patients with CLL who are intolerant to BTK or PI3Kδ inhibitor therapy. Blood 2021, 137, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Murali, I.; Kasar, S.; McWilliams, E.M.; Itchaki, G.; Tyekucheva, S.; Livitz, D.; Leshchiner, I.; Dong, S.; Fernandes, S.M.; Getz, G.; et al. Activating MAPK Pathway Mutations Mediate Primary Resistance to PI3K Inhibitors in Chronic Lymphocytic Leukemia (CLL). Blood 2018, 132, 587. [Google Scholar] [CrossRef]
- Iyengar, S.; Clear, A.; Bödör, C.; Maharaj, L.; Lee, A.; Calaminici, M.; Matthews, J.; Iqbal, S.; Auer, R.; Gribben, J.; et al. P110α-mediated constitutive PI3K signaling limits the efficacy of p110δ-selective inhibition in mantle cell lymphoma, particularly with multiple relapse. Blood 2013, 121, 2274–2284. [Google Scholar] [CrossRef] [Green Version]
- Woyach, J.A.; Johnson, A.J. Targeted therapies in CLL: Mechanisms of resistance and strategies for management. Blood 2015, 126, 471–477. [Google Scholar] [CrossRef] [Green Version]
- Fürstenau, M.; De Silva, N.; Eichhorst, B.; Hallek, M. Minimal Residual Disease Assessment in CLL: Ready for Use in Clinical Routine? HemaSphere 2019, 3, e287. [Google Scholar] [CrossRef] [PubMed]
- Izzo, B.; Gottardi, E.M.; Errichiello, S.; Daraio, F.; Baratè, C.; Galimberti, S. Monitoring Chronic Myeloid Leukemia: How Molecular Tools May Drive Therapeutic Approaches. Front. Oncol. 2019, 9, 833. [Google Scholar] [CrossRef] [Green Version]
- Maddocks, K.J.; Ruppert, A.S.; Lozanski, G.; Heerema, N.A.; Zhao, W.; Abruzzo, L.V.; Lozanski, A.; Davis, M.; Gordon, A.L.; Smith, L.L.; et al. Etiology of Ibrutinib Therapy Discontinuation and Outcomes in Patients with Chronic Lymphocytic Leukemia. JAMA Oncol. 2015, 1, 80–87. [Google Scholar] [CrossRef]



| TKI | Strong Resistance | Mild–Moderate Resistance |
|---|---|---|
| Imatinib | Y253–E255–T315 | M244–L248–G250–Q252–F317–M351–M355–F359–H396 |
| Dasatinib | T315 | V299–F317 |
| Nilotinib | T315 | L248–Y253–E255–F359 |
| Bosutinib | T315–V299 | L248–G250–E255–F317 |
| Ponatinib | T315–E255 | |
| Asciminib | A337–W464–P465–V468–I502 |
| Parameter | Odds Ratio (95%CI) | p Value |
|---|---|---|
| Age at stop of TKI (years) | 1.9 (0.95–1.26) | 0.21 |
| Interferon pretreatment | 2.50 (1.43–4.36) | 0.0013 |
| Duration of interferon pretreatment (years) | 1.38 (1.12–1.69) | 0.0022 |
| Duration of TKI treatment (years) | 1.16 (1.08–1.25) | <0.0001 |
| DMR duration while receiving TKI (years) | 1.16 (1.08–1.25) | 0.00011 |
| Time of TKI treatment before DMR (years) | 1.02 (0.93–1.13) | 0.66 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Novellis, D.; Cacace, F.; Caprioli, V.; Wierda, W.G.; Mahadeo, K.M.; Tambaro, F.P. The TKI Era in Chronic Leukemias. Pharmaceutics 2021, 13, 2201. https://doi.org/10.3390/pharmaceutics13122201
De Novellis D, Cacace F, Caprioli V, Wierda WG, Mahadeo KM, Tambaro FP. The TKI Era in Chronic Leukemias. Pharmaceutics. 2021; 13(12):2201. https://doi.org/10.3390/pharmaceutics13122201
Chicago/Turabian StyleDe Novellis, Danilo, Fabiana Cacace, Valeria Caprioli, William G. Wierda, Kris M. Mahadeo, and Francesco Paolo Tambaro. 2021. "The TKI Era in Chronic Leukemias" Pharmaceutics 13, no. 12: 2201. https://doi.org/10.3390/pharmaceutics13122201