
Non-Cationic RGD-Containing Protein Nanocarrier for Tumor-Targeted siRNA Delivery

Abstract
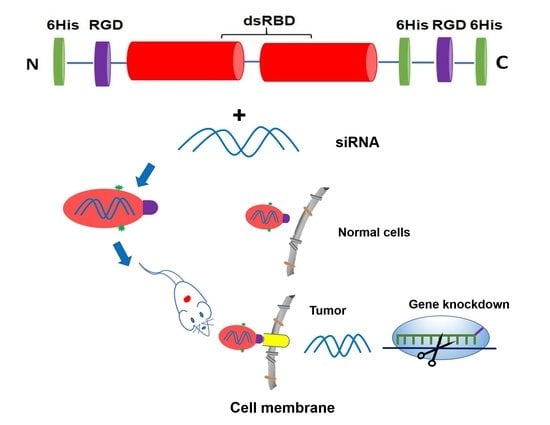
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Mouse
2.3. Construction of Fusion Protein Dual-RGD (Dual-RGD-dsRBD-18his) Expression Plasmid
2.4. Expression and Purification of Dual-RGD Recombinant Protein
2.5. Measurement of Hydrodynamic Diameter and Charge
2.6. SiRNA Binding Assay
2.7. RNase Resistance and Serum Stability
2.8. Western Blot
2.9. Cell-Type Specific Binding Assay
2.10. Cellular Uptake and Endosomal Escape by Confocal Microscopy
2.11. Tumor-Targeting and Biodistribution
2.12. In Vivo Gene Knockdown
2.13. Cell Viability
2.14. In Vivo Toxicity Assay
2.15. Histology Assay
2.16. Statistical Analysis
3. Results
3.1. Construction and Expression of Dual RGD-dsRBD-18his (Dual-RGD) Recombinant Protein
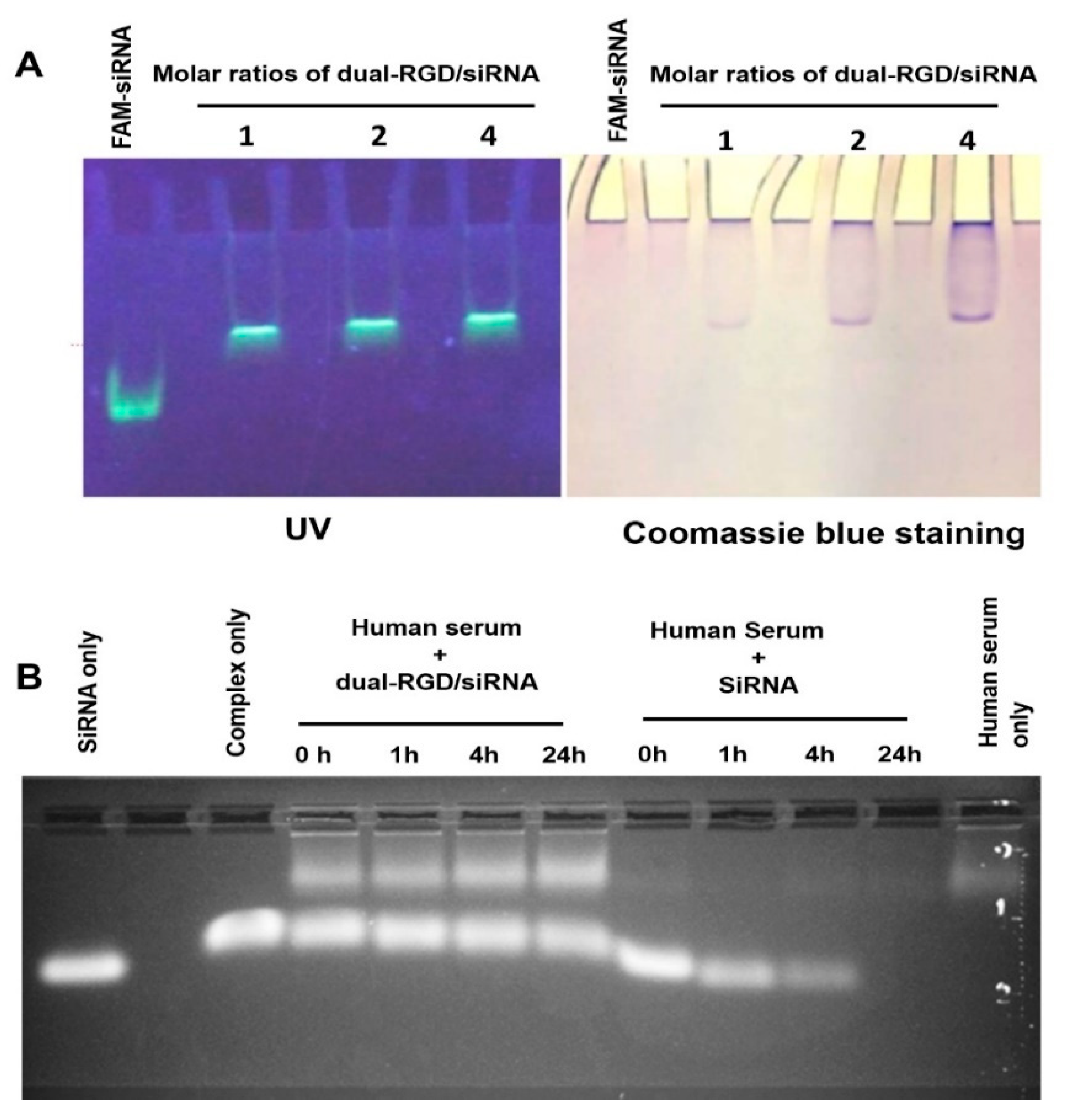
3.2. siRNA Binding Capability
3.3. Protection of siRNA against Nucleolytic Degradation
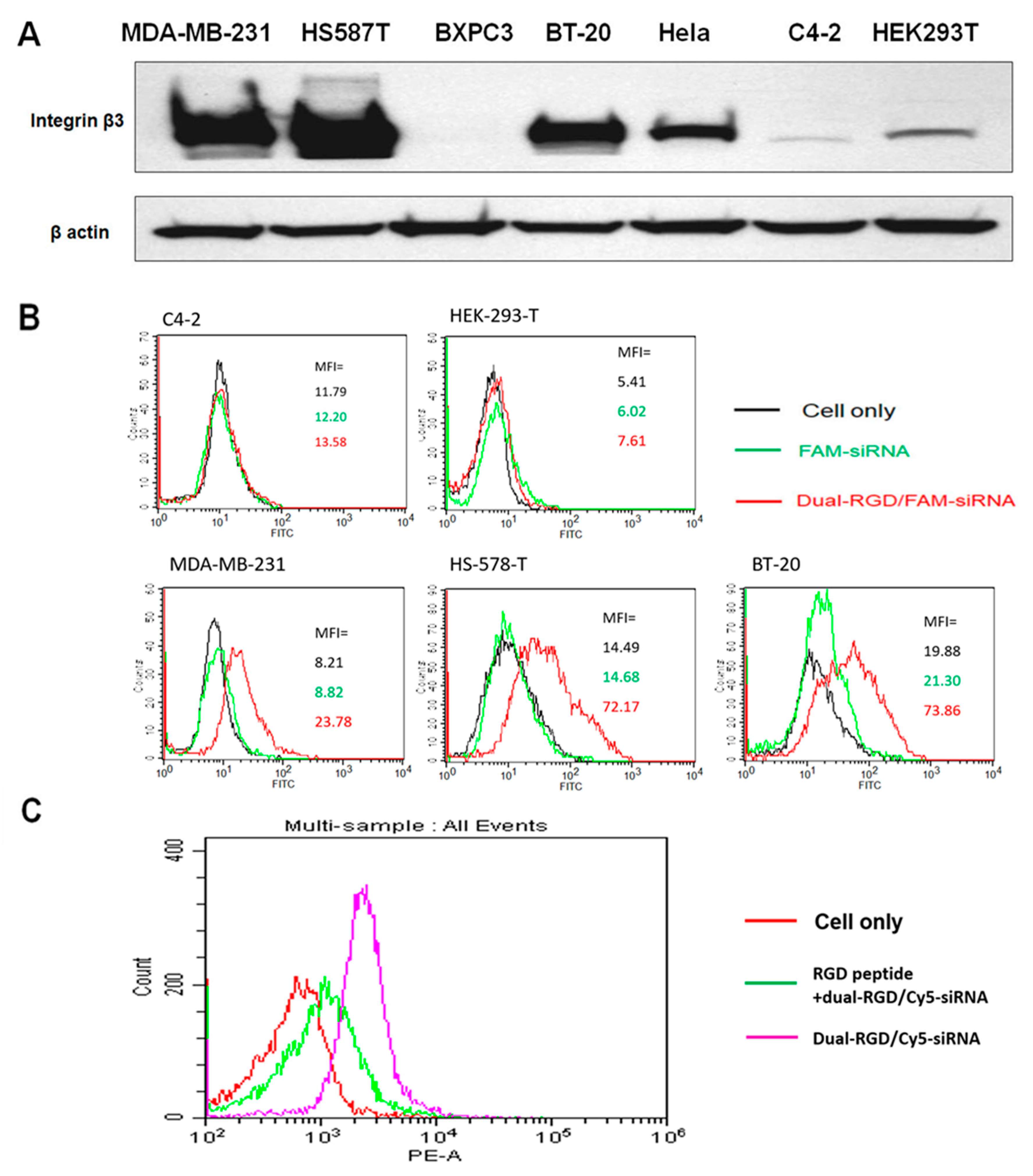
3.4. RGD Binding Specificity
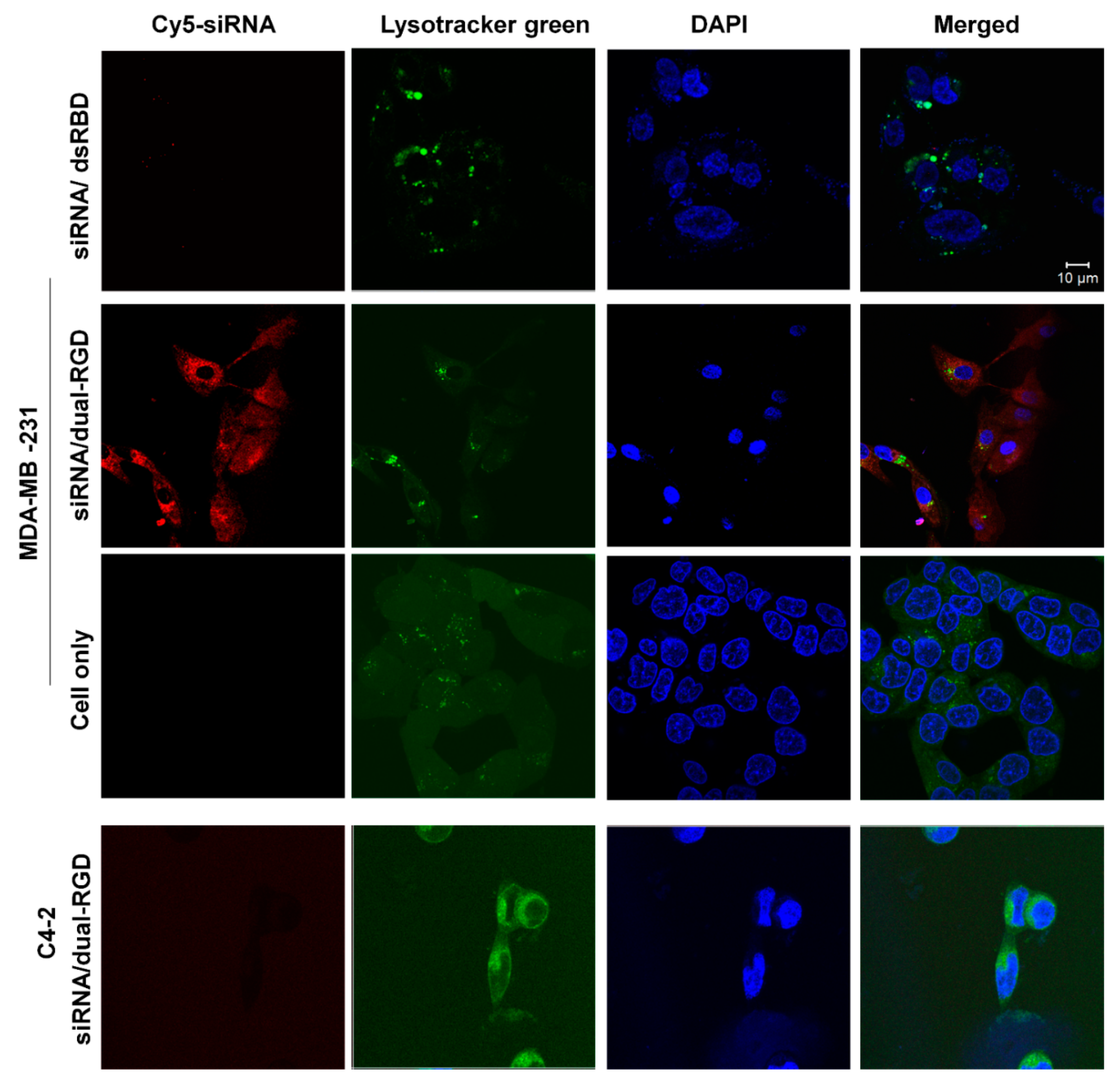
3.5. Cellular Uptake and Subcellular Distribution of Dual-RGD Carrier
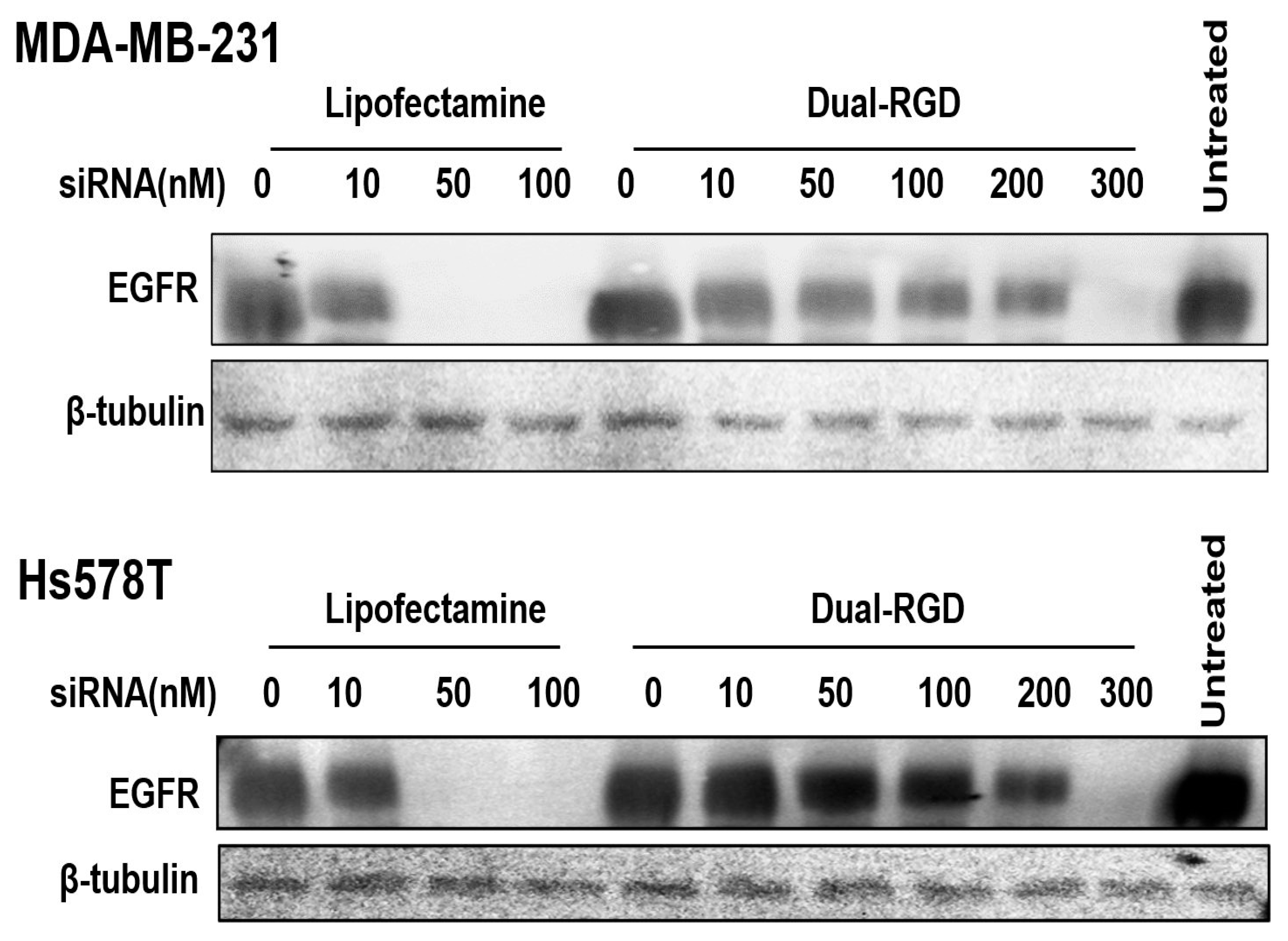
3.6. Target Gene Knockdown In Vitro
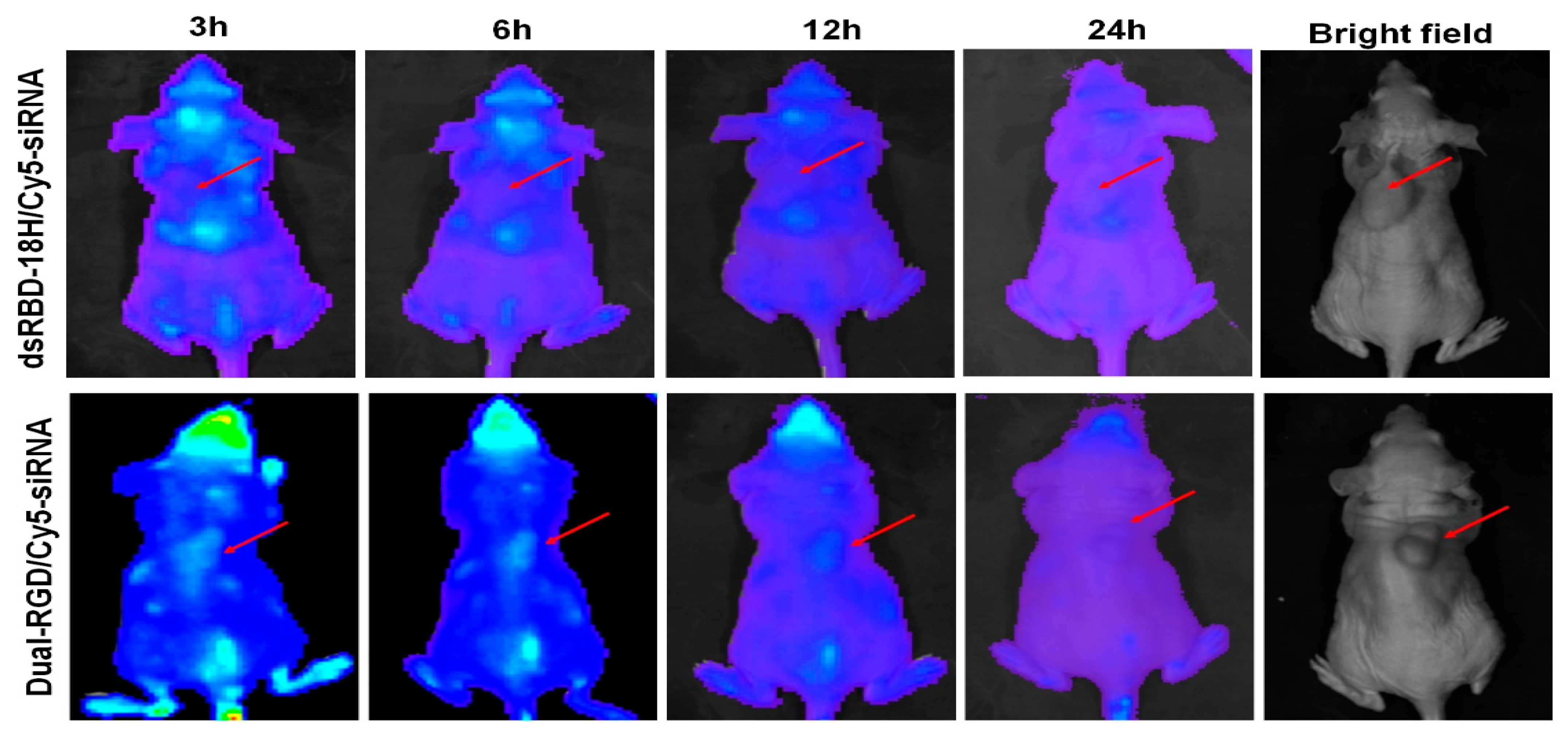
3.7. Tumor-Targeting Capability In Vivo
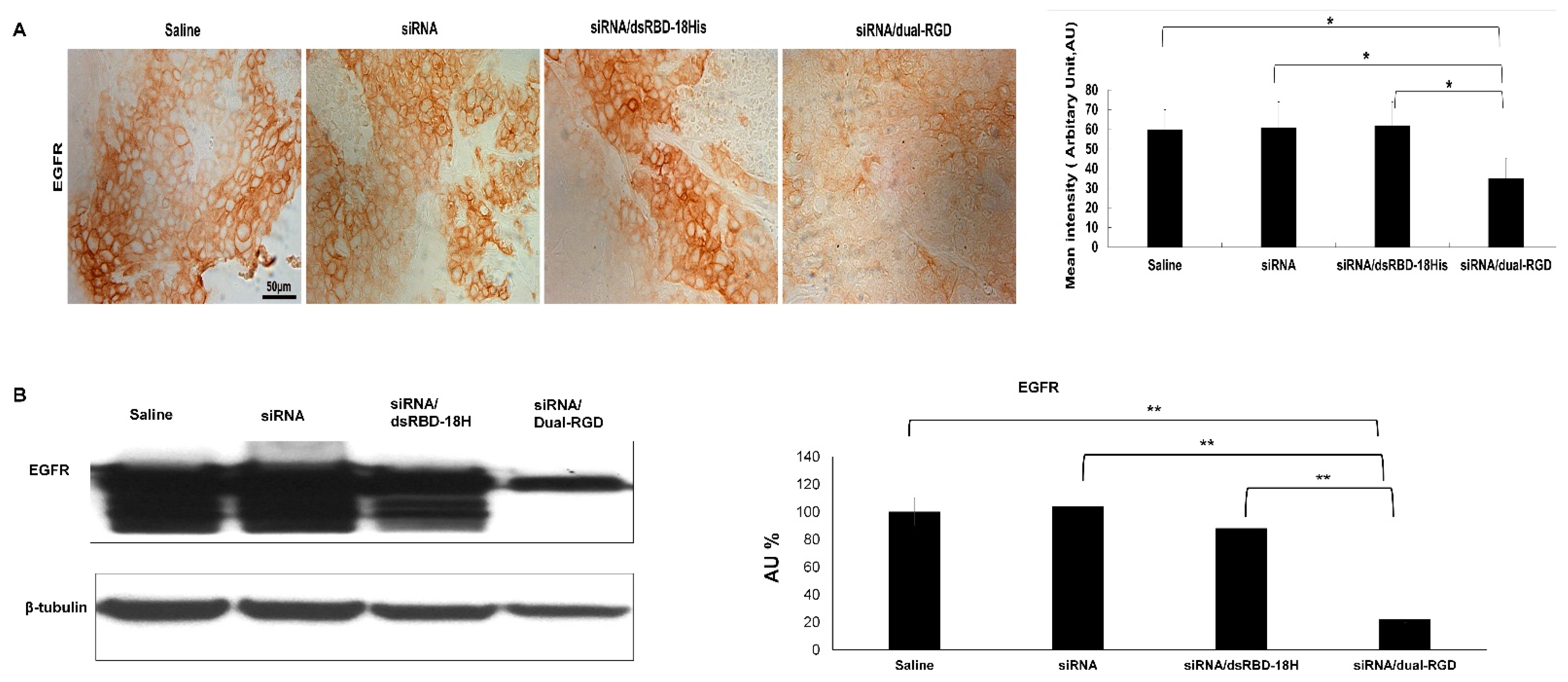
3.8. Target Gene Knockdown In Vivo
3.9. Toxicity Assessment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khvalevsky, E.Z.; Gabai, R.; Rachmut, I.H.; Horwitz, E.; Brunschwig, Z.; Orbach, A.; Shemi, A.; Golan, T.; Domb, A.J.; Yavin, E.; et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20723–20728. [Google Scholar] [CrossRef] [Green Version]
- Byeon, Y.; Lee, J.-W.; Choi, W.S.; Won, J.E.; Kim, G.H.; Kim, M.G.; Wi, T.I.; Lee, J.M.; Kang, T.H.; Jung, I.D.; et al. CD44-targeted PLGA nanoparticles incorporating paclitaxel and FAK siRNA overcome chemoresistance in epithelial ovarian cancer. Cancer Res. 2018, 78, 6247–6256. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.C.; Sun, W.; Khare, P.; Karimi, M.; Wang, X.; Shen, Y.; Ober, R.J.; Ward, E.S. Engineering a HER2-specific antibody–drug conjugate to increase lysosomal delivery and therapeutic efficacy. Nat. Biotechnol. 2019, 37, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Calvente, C.J.; Sehgal, A.; Popov, Y.; Kim, Y.O.; Zevallos, V.; Sahin, U.; Diken, M.; Schuppan, D. Specific hepatic delivery of procollagen α1(I) small interfering RNA in lipid-like nanoparticles resolves liver fibrosis. Hepatology 2015, 62, 1285–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, J.; Schaffer, D.V. Antiviral RNAi therapy: Emerging approaches for hitting a moving target. Gene Ther. 2006, 13, 532–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, A.M.; Stolk, J.; Bals, R.; Lickliter, J.D.; Hamilton, J.; Christianson, D.R.; Given, B.D.; Burdon, J.G.; Loomba, R.; Stoller, J.K.; et al. Hepatic-targeted RNA interference provides robust and persistent knockdown of alpha-1 antitrypsin levels in ZZ patients. J. Hepatol. 2018, 69, 378–384. [Google Scholar] [CrossRef]
- Solomon, S.D.; Adams, D.; Kristen, A.; Grogan, M.; Gonzalez-Duarte, A.; Maurer, M.S.; Merlini, G.; Damy, T.; Slama, M.S.; Brannagan, T.H., 3rd; et al. Effects of Patisiran, an RNA Interference Therapeutic, on Cardiac Parameters in Patients with Hereditary Transthyretin-Mediated Amyloidosis. Circulation 2019, 139, 431. [Google Scholar] [CrossRef]
- Lam, J.K.W.; Chow, M.Y.T.; Zhang, Y.; Leung, S.W.S. siRNA versus miRNA as Therapeutics for Gene Silencing. Mol. Ther. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [Green Version]
- Elbashir, S.M.; Lendeckel, W.; Tuschl, T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001, 15, 188–200. [Google Scholar] [CrossRef] [Green Version]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nat. Cell Biol. 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Leung, R.K.; Whittaker, P.A. RNA interference: From gene silencing to gene-specific therapeutics. Pharmacol. Ther. 2005, 107, 222–239. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.-R.; Pottier, M.; Vekris, A.; Opolon, P.; Maksimenko, A.; Malvy, C. Comparison of antisense oligonucleotides and siRNAs in cell culture and in vivo. Biochem. Biophys. Res. Commun. 2002, 296, 1000–1004. [Google Scholar] [CrossRef]
- Second RNAi drug approved. Nat. Biotechnol. 2020, 38, 385. Available online: https://www.nature.com/articles/s41587-020-0494-3 (accessed on 21 October 2021). [CrossRef] [Green Version]
- Priegue, J.M.; Crisan, D.N.; Martinez-Costas, J.; Granja, J.R.; Fernandez-Trillo, F.; Montenegro, J. In Situ Functionalized Polymers for siRNA Delivery. Angew. Chem. Int. Ed. Engl. 2016, 55, 7492. [Google Scholar] [CrossRef] [Green Version]
- Siegwart, D.J.; Whitehead, K.A.; Nuhn, L.; Sahay, G.; Cheng, H.; Jiang, S.; Ma, M.; Lytton-Jean, A.; Vegas, A.; Fenton, P.; et al. Combinatorial synthesis of chemically diverse core-shell nanoparticles for intracellular delivery. Proc. Natl. Acad. Sci. USA 2011, 108, 12996–13001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beloor, J.; Zeller, S.; Choi, C.S.; Lee, S.-K.; Kumar, P. Cationic cell-penetrating peptides as vehicles for siRNA delivery. Ther. Deliv. 2015, 6, 491–507. [Google Scholar] [CrossRef]
- Oe, Y.; Christie, R.J.; Naito, M.; Low, S.A.; Fukushima, S.; Toh, K.; Miura, Y.; Matsumoto, Y.; Nishiyama, N.; Miyata, K.; et al. Actively-targeted polyion complex micelles stabilized by cholesterol and disulfide cross-linking for systemic delivery of siRNA to solid tumors. Biomaterials 2014, 35, 7887–7895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, H.Y.; Liu, S.; Wong, H.L. Nanotoxicity: A key obstacle to clinical translation of siRNA-based nanomedicine. Nanomedicine 2014, 9, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Mi, Y.; Feng, S.S. siRNA-based nanomedicine. Nanomedicine 2013, 8, 859. [Google Scholar] [CrossRef]
- Liu, H.Y.; Gao, X. A universal protein tag for delivery of SiRNA-aptamer chimeras. Sci. Rep. 2013, 3, 3129. [Google Scholar] [CrossRef] [Green Version]
- Ucci, J.W.; Kobayashi, Y.; Choi, G.; Alexandrescu, A.T.; Cole, J.L. Mechanism of Interaction of the Double-Stranded RNA (dsRNA) Binding Domain of Protein Kinase R with Short dsRNA Sequences. Biochemistry 2007, 46, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, P.C.; Cech, T.R. Minor-Groove Recognition of Double-Stranded RNA by the Double-Stranded RNA-Binding Domain from the RNA-Activated Protein Kinase PKR. Biochemistry 1996, 35, 9983–9994. [Google Scholar] [CrossRef]
- Han, X.; Cheng, K.; Xu, Y.; Wang, Y.; Min, H.; Zhang, Y.; Zhao, X.; Zhao, R.; Anderson, G.J.; Ren, L.; et al. Modularly Designed Peptide Nanoprodrug Augments Antitumor Immunity of PD-L1 Checkpoint Blockade by Targeting Indoleamine 2,3-Dioxygenase. J. Am. Chem. Soc. 2020, 142, 2490–2496. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, S.H.; Choe, J.; Park, T.G. Intracellular small interfering RNA delivery using genetically engineered double-stranded RNA binding protein domain. J. Gene Med. 2009, 11, 804–812. [Google Scholar] [CrossRef]
- Zhu, L.; Guo, N.; Li, Q.; Ma, Y.; Jacboson, O.; Lee, S.; Choi, H.S.; Mansfield, J.R.; Niu, G.; Chen, X. Dynamic PET and Optical Imaging and Compartment Modeling using a Dual-labeled Cyclic RGD Peptide Probe. Theranostics 2012, 2, 746–756. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.B.; Chen, K.; Chen, X. (68) Ga-labeled multimeric RGD peptides for microPET imaging of integrin alpha(v)beta (3) expression. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1100. [Google Scholar] [CrossRef] [PubMed]
- Kang, F.; Wang, Z.; Li, G.; Wang, S.; Liu, D.; Zhang, M.; Zhao, M.; Yang, W.; Wang, J. Inter-heterogeneity and intra-heterogeneity of alphavbeta3 in non-small cell lung cancer and small cell lung cancer patients as revealed by (68) Ga-RGD2 PET imaging. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1520. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Du, P.; Jiang, S.-H.; Jin, G.-H.; Huang, Q.; Hua, Z.-C. Enhancement of antitumor properties of TRAIL by targeted delivery to the tumor neovasculature. Mol. Cancer Ther. 2008, 7, 851–861. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Kang, C.; Liu, F.; Zhou, Y.; Luo, L.; Qiao, H. RGD Peptide-Based Target Drug Delivery of Doxorubicin Nanomedicine. Drug Dev. Res. 2017, 78, 283–291. [Google Scholar] [CrossRef]
- Jin, Z.H.; Furukawa, T.; Degardin, M.; Sugyo, A.; Tsuji, A.B.; Yamasaki, T.; Kawamura, K.; Fujibayashi, Y.; Zhang, M.R.; Boturyn, D.; et al. alphaVbeta3 Integrin-Targeted Radionuclide Therapy with 64Cu-cyclam-RAFT-c(-RGDfK-)4. Mol. Cancer Ther. 2016, 15, 2076. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chen, K.; Cai, W.; Li, Z.; He, L.; Kashefi, A.; Chen, X. Integrin-targeted imaging and therapy with RGD4C-TNF fusion protein. Mol. Cancer Ther. 2008, 7, 1044–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nat. Cell Biol. 1984, 309, 30–33. [Google Scholar] [CrossRef]
- Pierschbacher, M.; Hayman, E.G.; Ruoslahti, E. Synthetic peptide with cell attachment activity of fibronectin. Proc. Natl. Acad. Sci. USA 1983, 80, 1224–1227. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Liu, C.W.; Puglisi, J.D. Specific Recognition of HIV TAR RNA by the dsRNA Binding Domains (dsRBD1–dsRBD2) of PKR. J. Mol. Biol. 2006, 358, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Yu, X.; Liu, H.; Wu, D.; She, J.-X. Co-targeting EGFR and survivin with a bivalent aptamer-dual siRNA chimera effectively suppresses prostate cancer. Sci. Rep. 2016, 6, 30346. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Ghamande, S.; Liu, H.; Xue, L.; Zhao, S.; Tan, W.; Zhao, L.; Tang, S.C.; Wu, D.; Korkaya, H.; et al. Targeting EGFR/HER2/HER3 with a Three-in-One Aptamer-siRNA Chimera Confers Superior Activity against HER2(+) Breast Cancer. Mol. Ther. Nucleic Acids 2018, 10, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, S.; Bu, X.-Y.; Khankaldyyan, V.; Gonzales-Gomez, I.; McComb, J.G.; Laug, W.E. Effect of the angiogenesis inhibitor cilengitide (emd 121974) on glioblastoma growth in nude mice. Neurosurgery 2006, 59, 1304–1312. [Google Scholar] [CrossRef]
- Burke, P.A.; DeNardo, S.J.; Miers, L.A.; Lamborn, K.R.; Matzku, S.; DeNardo, G.L. Cilengitide targeting of alpha(v)beta(3) integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts. Cancer Res. 2002, 62, 4263. [Google Scholar]
- Eguchi, A.; Meade, B.R.; Chang, Y.C.; Fredrickson, C.T.; Willert, K.; Puri, N.; Dowdy, S.F. Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein. Nat. Biotechnol. 2009, 27, 567. [Google Scholar] [CrossRef] [PubMed]
- Clemens, M.J.; Elia, A. The Double-Stranded RNA-Dependent Protein Kinase PKR: Structure and Function. J. Interf. Cytokine Res. 1997, 17, 503–524. [Google Scholar] [CrossRef]
- Chong, K.; Feng, L.; Schappert, K.; Meurs, E.; Donahue, T.; Friesen, J.; Hovanessian, A.; Williams, B. Human p68 kinase exhibits growth suppression in yeast and homology to the translational regulator GCN2. EMBO J. 1992, 11, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Johnston, D.S.; Brown, N.H.; Gall, J.G.; Jantsch, M. A conserved double-stranded RNA-binding domain. Proc. Natl. Acad. Sci. USA 1992, 89, 10979–10983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, S.R.; Mathews, M.B. Two RNA-binding motifs in the double-stranded RNA-activated protein kinase, DAI. Genes Dev. 1992, 6, 2478–2490. [Google Scholar] [CrossRef]
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, N.B.; Kerr, I.M.; Williams, B.; Hovanessian, A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef]
- Lee, K.; Kunkeaw, N.; Jeon, S.H.; Lee, I.; Johnson, B.H.; Kang, G.-Y.; Bang, J.Y.; Park, H.S.; Leelayuwat, C.; Lee, Y.S. Precursor miR-886, a novel noncoding RNA repressed in cancer, associates with PKR and modulates its activity. RNA 2011, 17, 1076–1089. [Google Scholar] [CrossRef] [Green Version]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2018, 11, 480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, T.; Hunt, T.; Jackson, R.J.; Robertson, H.D. The characteristics of inhibition of protein synthesis by double-stranded ribonucleic acid in reticulocyte lysates. J. Biol. Chem. 1975, 250, 409–417. [Google Scholar] [CrossRef]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity Generated by the Tumor Microenvironment Drives Local Invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Creusat, G.; Rinaldi, A.S.; Weiss, E.; Elbaghdadi, R.; Remy, J.S.; Mulherkar, R.; Zuber, G. Proton sponge trick for pH-sensitive disassembly of polyethylenimine-based siRNA delivery systems. Bioconjug. Chem. 2010, 21, 994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, W.; Li, J.; Corey, E.; Gao, X. Publisher Correction: A ribonucleoprotein octamer for targeted siRNA delivery. Nat. Biomed. Eng. 2018, 2, 622. [Google Scholar] [CrossRef] [PubMed]
- Prokopiou, E.M.; Ryder, S.; Walsh, J.J. Tumour vasculature targeting agents in hybrid/conjugate drugs. Angiogenesis 2013, 16, 503–524. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Cooper, C.R.; Chay, C.H.; Pienta, K.J. The role of alpha(v)beta(3) in prostate cancer progression. Neoplasia 2002, 4, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Bachelier, R.; Treilleux, I.; Pujuguet, P.; Peyruchaud, O.; Baron, R.; Clement-Lacroix, P.; Clezardin, P. Tumor alphavbeta3 integrin is a therapeutic target for breast cancer bone metastases. Cancer Res. 2007, 67, 5821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Li, Y.; Shen, Y.; Wang, A.; Wang, S.; Xie, T. The Functions and Applications of RGD in Tumor Therapy and Tissue Engineering. Int. J. Mol. Sci. 2013, 14, 13447–13462. [Google Scholar] [CrossRef] [Green Version]
- Klubo-Gwiezdzinska, J.; Chen, X. Targeting Integrins with Radiolabeled RGD Analogues for Radiotheranostics of Metastatic Radioactive Iodine Nonresponsive Thyroid Cancer: New Avenues in Personalized Medicine. Thyroid 2020, 30, 476–478. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.; Kolb, H.C.; Zhang, J.J.; Bélanger, M.-J.; Stubbs, J.B.; Stabin, M.G.; Hostetler, E.D.; Alpaugh, R.K.; von Mehren, M.; Walsh, J.C.; et al. Biodistribution and Radiation Dosimetry of the Integrin Marker 18F-RGD-K5 Determined from Whole-Body PET/CT in Monkeys and Humans. J. Nucl. Med. 2012, 53, 787–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Niu, G.; Wu, H.; Chen, X. Clinical Application of Radiolabeled RGD Peptides for PET Imaging of Integrin alphavbeta3. Theranostics 2016, 6, 78. [Google Scholar] [CrossRef] [Green Version]
- Pasqualini, R.; Koivunen, E.; Kain, R.; Lahdenranta, J.; Sakamoto, M.; Stryhn, A.; Ashmun, R.A.; Shapiro, L.H.; Arap, W.; Ruoslahti, E. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000, 60, 722–727. [Google Scholar] [PubMed]
- Hood, J.D.; Bednarski, M.; Frausto, R.; Guccione, S.; Reisfeld, R.A.; Xiang, R.; Cheresh, D.A. Tumor Regression by Targeted Gene Delivery to the Neovasculature. Science 2002, 296, 2404–2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, T.D.; Kalogeropoulos, S.; Nguyen, T.; Liu, S.; Bartis, J.; Ellars, C.; Edwards, S.; Onthank, D.; Silva, P.; Yalamanchili, P.; et al. Design, synthesis, and evaluation of radiolabeled integrin alpha v beta 3 receptor antagonists for tumor imaging and radiotherapy. Cancer Biother. Radiopharm. 2003, 18, 627. [Google Scholar] [CrossRef] [PubMed]
- Janssen, M.L.; Oyen, W.J.; Dijkgraaf, I.; Massuger, L.F.; Frielink, C.; Edwards, D.S.; Rajopadhye, M.; Boonstra, H.; Corstens, F.H.; Boerman, O.C. Tumor targeting with radiolabeled alpha(v)beta(3) integrin binding peptides in a nude mouse model. Cancer Res. 2002, 62, 6146. [Google Scholar] [PubMed]
- Zhang, Y.-F.; Wang, J.-C.; Bian, D.-Y.; Zhang, X.; Zhang, Q. Targeted delivery of RGD-modified liposomes encapsulating both combretastatin A-4 and doxorubicin for tumor therapy: In vitro and in vivo studies. Eur. J. Pharm. Biopharm. 2010, 74, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, X.; Zhang, L.; Qian, L.; Liu, C.; Zheng, J.; Jiang, Y. Development of biodegradable polymeric implants of RGD-modified PEG-PAMAM-DOX conjugates for long-term intratumoral release. Drug Deliv. 2015, 22, 389–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertilaccio, M.T.; Grioni, M.; Sutherland, B.W.; Degl’Innocenti, E.; Freschi, M.; Jachetti, E.; Greenberg, N.M.; Corti, A.; Bellone, M. Vasculature-targeted tumor necrosis factor-alpha increases the therapeutic index of doxorubicin against prostate cancer. Prostate 2008, 68, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Ponzoni, M. Tumor Vascular Targeting with Tumor Necrosis Factor alpha and Chemotherapeutic Drugs. Ann. N. Y. Acad. Sci. 2004, 1028, 104–112. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Xue, L.; Zhao, J.; Zhao, S.; Wu, D.; Liu, H.Y. Non-Cationic RGD-Containing Protein Nanocarrier for Tumor-Targeted siRNA Delivery. Pharmaceutics 2021, 13, 2182. https://doi.org/10.3390/pharmaceutics13122182
Yu X, Xue L, Zhao J, Zhao S, Wu D, Liu HY. Non-Cationic RGD-Containing Protein Nanocarrier for Tumor-Targeted siRNA Delivery. Pharmaceutics. 2021; 13(12):2182. https://doi.org/10.3390/pharmaceutics13122182
Chicago/Turabian StyleYu, Xiaolin, Lu Xue, Jing Zhao, Shuhua Zhao, Daqing Wu, and Hong Yan Liu. 2021. "Non-Cationic RGD-Containing Protein Nanocarrier for Tumor-Targeted siRNA Delivery" Pharmaceutics 13, no. 12: 2182. https://doi.org/10.3390/pharmaceutics13122182