
Pro-Apoptotic Potential of Pseudevernia furfuracea (L.) Zopf Extract and Isolated Physodic Acid in Acute Lymphoblastic Leukemia Model In Vitro

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection, Identification, Isolation and Characterization
2.2. Cell Culture
2.3. Human Peripheral Blood Samples and T-Lymphocyte Isolation
2.4. Cell Viability Assay
2.5. Flow Cytometric Analyses
2.6. Cell Proliferation Analysis
2.7. Cell Cycle Analysis
2.8. Annexin V/PI Staining for Phosphatidyl Serine Externalization
2.9. Detection of Mitochondrial Membrane Potential (MMP) Changes
2.10. Measurement of Superoxide Anions and Reactive Oxygen Species (ROS)
2.11. Western Blot
2.12. Statistical Analysis
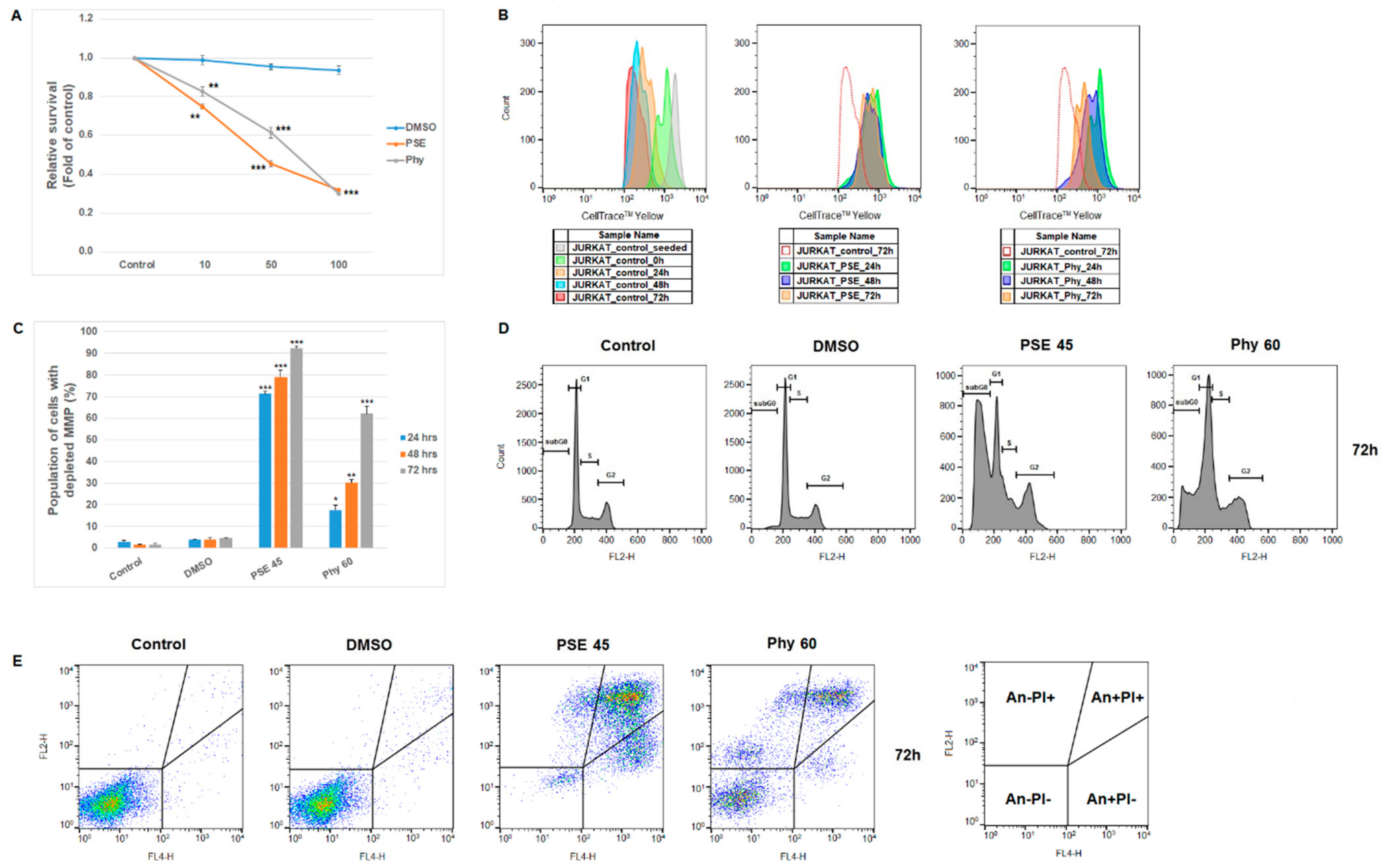
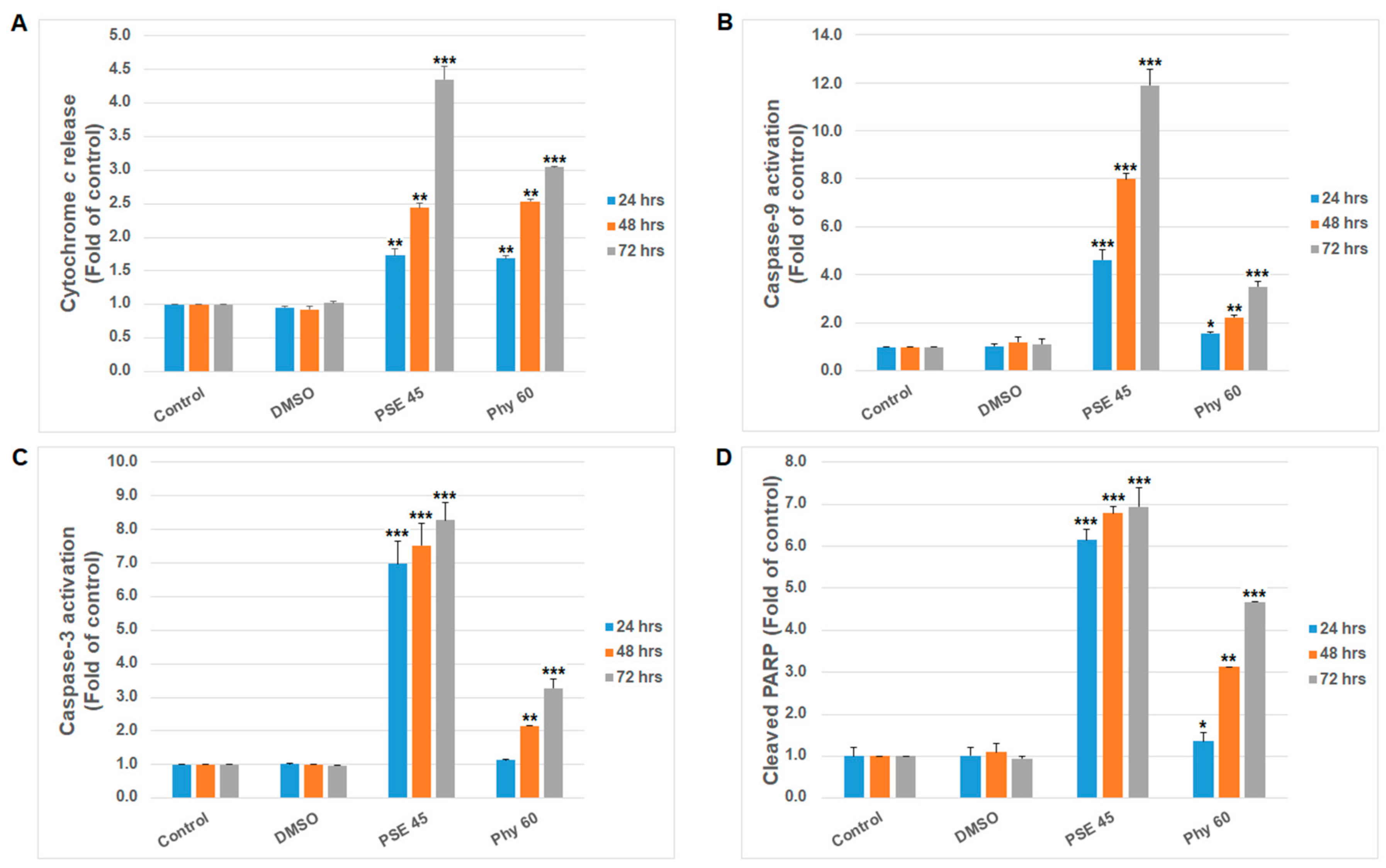
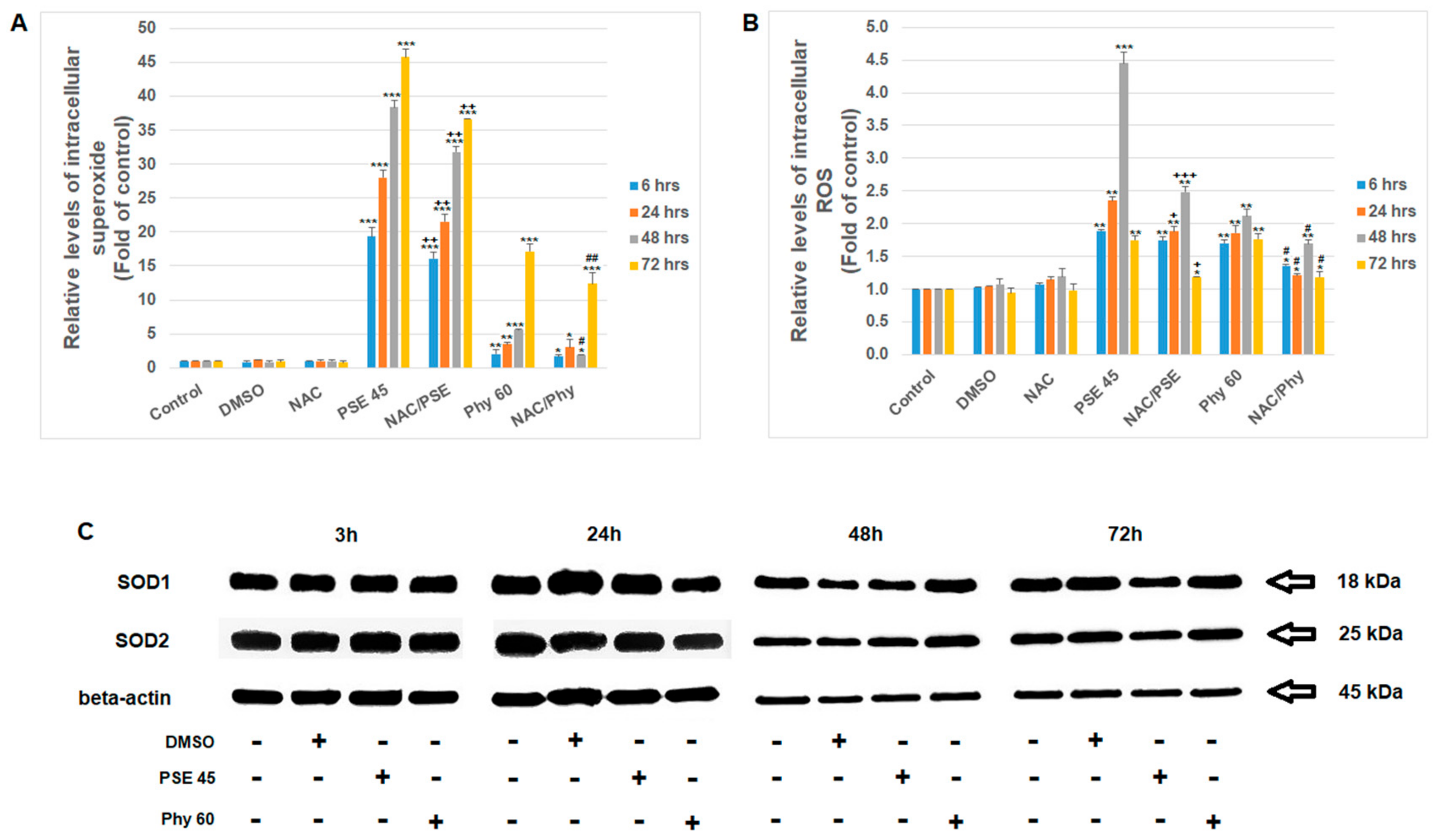
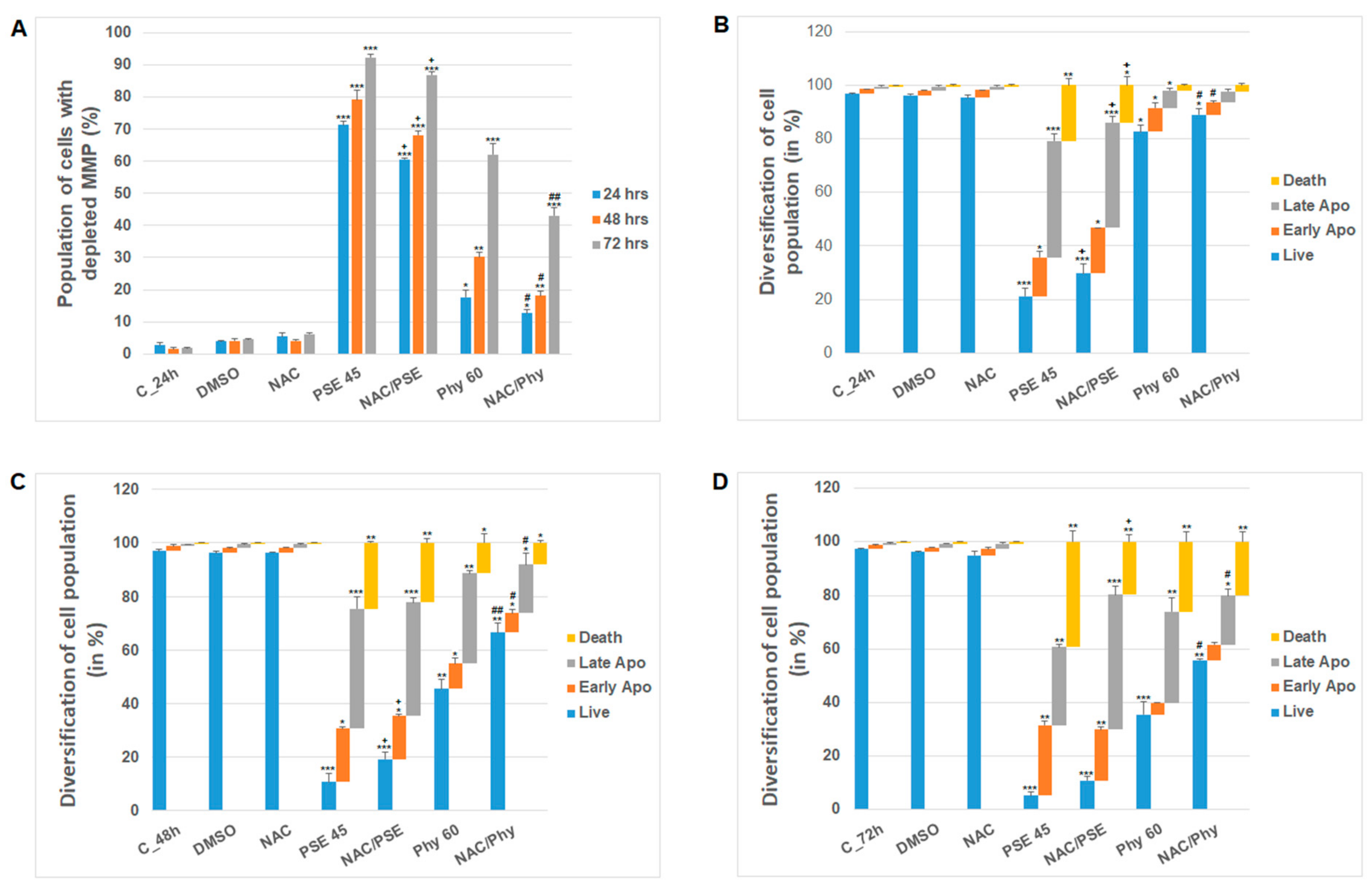
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- PDQ Pediatric Treatment Editorial Board. Childhood Acute Lymphoblastic Leukemia Treatment (PDQ(R)): Health Professional Version. In PDQ Cancer Information Summaries; National Cancer Institute: Bethesda, MD, USA, 2021. [Google Scholar]
- Kadia, T.M.; Gandhi, V. Nelarabine in the treatment of pediatric and adult patients with T-cell acute lymphoblastic leukemia and lymphoma. Expert Rev. Hematol. 2017, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.; Stein, A.; Gokbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foa, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Millan, N.C.; Pastrana, A.; Guitter, M.R.; Zubizarreta, P.A.; Monges, M.S.; Felice, M.S. Acute and sub-acute neurological toxicity in children treated for acute lymphoblastic leukemia. Leuk. Res. 2018, 65, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Van Binsbergen, A.L.; de Haas, V.; van der Velden, V.H.J.; de Groot-Kruseman, H.A.; Fiocco, M.F.; Pieters, R. Efficacy and toxicity of high-risk therapy of the Dutch Childhood Oncology Group in childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2021, e29387. [Google Scholar] [CrossRef] [PubMed]
- Quasthoff, S.; Hartung, H.P. Chemotherapy-induced peripheral neuropathy. J. Neurol. 2002, 249, 9–17. [Google Scholar] [CrossRef]
- Alhosin, M.; Razvi, S.S.I.; Sheikh, R.A.; Khan, J.A.; Zamzami, M.A.; Choudhry, H. Thymoquinone and Difluoromethylornithine (DFMO) Synergistically Induce Apoptosis of Human Acute T Lymphoblastic Leukemia Jurkat Cells Through the Modulation of Epigenetic Pathways. Technol. Cancer Res. Treat. 2020, 19, 1533033820947489. [Google Scholar] [CrossRef] [PubMed]
- Feriotto, G.; Tagliati, F.; Giriolo, R.; Casciano, F.; Tabolacci, C.; Beninati, S.; Khan, M.T.H.; Mischiati, C. Caffeic Acid Enhances the Anti-Leukemic Effect of Imatinib on Chronic Myeloid Leukemia Cells and Triggers Apoptosis in Cells Sensitive and Resistant to Imatinib. Int. J. Mol. Sci. 2021, 22, 1644. [Google Scholar] [CrossRef]
- Kowalczyk, T.; Sitarek, P.; Toma, M.; Picot, L.; Wielanek, M.; Skala, E.; Sliwinski, T. An Extract of Transgenic Senna obtusifolia L. hairy roots with Overexpression of PgSS1 Gene in Combination with Chemotherapeutic Agent Induces Apoptosis in the Leukemia Cell Line. Biomolecules 2020, 10, 510. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Efferth, T. Repurposing of artemisinin-type drugs for the treatment of acute leukemia. Semin. Cancer Biol. 2021, 68, 291–312. [Google Scholar] [CrossRef]
- Ly, B.T.K.; Ly, D.M.; Linh, P.H.; Son, H.K.; Ha, N.L.; Chi, H.T. Screening of medicinal herbs for cytotoxic activity to leukemia cells. J. Buon 2020, 25, 1989–1996. [Google Scholar]
- Xu, X.; Huang, S.; Xiao, X.; Sun, Q.; Liang, X.; Chen, S.; Zhao, Z.; Huo, Z.; Tu, S.; Li, Y. Challenges and Clinical Strategies of CAR T-Cell Therapy for Acute Lymphoblastic Leukemia: Overview and Developments. Front. Immunol. 2020, 11, 569117. [Google Scholar] [CrossRef]
- Elkhateeb, W.A.; Daba, G.M.; Sheir, D.; Nguyen, T.-D.; Hapuarachchi, K.K.; Thomas, P.W. Mysterious World of Lichens: Highlights on Their History, Applications, and Pharmaceutical Potentials. Nat. Prod. J. 2021, 11, 275–287. [Google Scholar] [CrossRef]
- Verma, N.; Behera, B.C. Future Directions in the Study of Pharmaceutical Potential of Lichens. In Lichen Secondary Metabolites: Bioactive Properties and Pharmaceutical Potential; Ranković, B., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 237–260. [Google Scholar] [CrossRef]
- Goga, M.; Elecko, J.; Marcincinova, M.; Rucova, D.; Backorova, M.; Backor, M. Lichen metabolites: An overview of some secondary metabolites and their biological potential. In Co-Evolution of Secondary Metabolites; Merillon, J.M., Ramawat, K.G., Eds.; Springer: Cham, Switzerland, 2018; pp. 1–36. [Google Scholar] [CrossRef]
- Solarova, Z.; Liskova, A.; Samec, M.; Kubatka, P.; Busselberg, D.; Solar, P. Anticancer Potential of Lichens’ Secondary Metabolites. Biomolecules 2020, 10, 87. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, M.; Zambare, V.; Malek, L.; Gottardo, C.; Suntres, Z.; Christopher, L. Lichenochemicals: Extraction, purification, characterization, and application as potential anticancer agents. Expert Opin. Drug Discov. 2020, 15, 575–601. [Google Scholar] [CrossRef] [PubMed]
- Backorova, M.; Backor, M.; Mikes, J.; Jendzelovsky, R.; Fedorocko, P. Variable responses of different human cancer cells to the lichen compounds parietin, atranorin, usnic acid and gyrophoric acid. Toxicol. In Vitro 2011, 25, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Burton, J.F.; Cain, B.F. Antileukaemic activity of polyporic acid. Nature 1959, 184 (Suppl. 17), 1326–1327. [Google Scholar] [CrossRef]
- Talapatra, S.K.; Rath, O.; Clayton, E.; Tomasi, S.; Kozielski, F. Depsidones from Lichens as Natural Product Inhibitors of M-Phase Phosphoprotein 1, a Human Kinesin Required for Cytokinesis. J. Nat. Prod. 2016, 79, 1576–1585. [Google Scholar] [CrossRef]
- Ahti, T.; Thell, A. Pseudevernia. In Nordic Lichen Flora Volume 4—Parmeliaceae; Thell, A., Moberg, R., Eds.; Nordic Lichen Society, Uppsala University: Uppsala, Sweden, 2011; Volume 4, pp. 101–102. [Google Scholar]
- Cardile, V.; Graziano, A.C.E.; Avola, R.; Piovano, M.; Russo, A. Potential anticancer activity of lichen secondary metabolite physodic acid. Chem.-Biol. Interact. 2017, 263, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Emsen, B.; Sadi, G.; Bostanci, A.; Aslan, A. In vitro evaluation of cytotoxic, oxidative, genotoxic, and apoptotic activities of physodic acid from Pseudevernia furfuracea in HepG2 and THLE2 cells. Plant Biosyst. 2020, 1–10. [Google Scholar] [CrossRef]
- Paluszczak, J.; Kleszcz, R.; Studzinska-Sroka, E.; Krajka-Kuzniak, V. Lichen-derived caperatic acid and physodic acid inhibit Wnt signaling in colorectal cancer cells. Mol. Cell. Biochem. 2018, 441, 109–124. [Google Scholar] [CrossRef] [Green Version]
- Sahin, E.; Psav, S.D.; Avan, I.; Candan, M.; Sahinturk, V.; Koparal, A.T. Lichen-derived physodic acid exerts cytotoxic and anti-invasive effects in human lung cancer. Rend. Lincei Sci. Fis. Nat. 2021, 32, 511–520. [Google Scholar] [CrossRef]
- Seklic, D.S.; Obradovic, A.D.; Stankovic, M.S.; Zivanovic, M.N.; Mitrovic, T.L.J.; Stamenkovic, S.M.; Markovic, S.D. Proapoptotic and Antimigratory Effects of Pseudevernia furfuracea and Platismatia glauca on Colon Cancer Cell Lines. Food Technol. Biotech. 2018, 56, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Petrova, K.; Kello, M.; Kuruc, T.; Backorova, M.; Petrovova, E.; Vilkova, M.; Goga, M.; Rucova, D.; Backor, M.; Mojzis, J. Potential Effect of Pseudevernia furfuracea (L.) Zopf Extract and Metabolite Physodic Acid on Tumour Microenvironment Modulation in MCF-10A Cells. Biomolecules 2021, 11, 420. [Google Scholar] [CrossRef] [PubMed]
- Ingelfinger, R.; Henke, M.; Roser, L.; Ulshofer, T.; Calchera, A.; Singh, G.; Parnham, M.J.; Geisslinger, G.; Furst, R.; Schmitt, I.; et al. Unraveling the Pharmacological Potential of Lichen Extracts in the Context of Cancer and Inflammation with a Broad Screening Approach. Front. Pharmacol. 2020, 11, 1322. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, S.; Erkisa, M.; Oran, S.; Ulukaya, E.; Celikler, S.; Ari, F. Lichens exerts an anti-proliferative effect on human breast and lung cancer cells through induction of apoptosis. Drug Chem. Toxicol. 2021, 44, 259–267. [Google Scholar] [CrossRef]
- Goga, M.; Kello, M.; Vilkova, M.; Petrova, K.; Backor, M.; Adlassnig, W.; Lang, I. Oxidative stress mediated by gyrophoric acid from the lichen Umbilicaria hirsuta affected apoptosis and stress/survival pathways in HeLa cells. BMC Complement. Altern. Med. 2019, 19, 221. [Google Scholar] [CrossRef]
- Bessadottir, M.; Eiriksson, F.F.; Becker, S.; Ogmundsdottir, M.H.; Omarsdottir, S.; Thorsteinsdottir, M.; Ogmundsdottir, H.M. Anti-proliferative and pro-apoptotic effects of lichen-derived compound protolichesterinic acid are not mediated by its lipoxygenase-inhibitory activity. Prostaglandis Leukot. Essent. Fat. Acids 2015, 98, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.G.; Zhang, X.; Zhou, B.; Zhang, C.J.; Tu, J.F.; Chen, X.J.; Wang, J.Y.; Gao, H.Q.; Qin, G.M.; Pan, W.S. Usnic Acid Induces Cycle Arrest, Apoptosis, and Autophagy in Gastric Cancer Cells In Vitro and In Vivo. Med. Sci. Monitor 2018, 24, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Yurdacan, B.; Egeli, U.; Eskiler, G.G.; Eryilmaz, I.E.; Cecener, G.; Tunca, B. The role of usnic acid-induced apoptosis and autophagy in hepatocellular carcinoma. Hum. Exp. Toxicol. 2019, 38, 201–215. [Google Scholar] [CrossRef]
- Kosanic, M.; Manojlovic, N.; Jankovic, S.; Stanojkovic, T.; Rankovic, B. Evernia prunastri and Pseudoevernia furfuraceae lichens and their major metabolites as antioxidant, antimicrobial and anticancer agents. Food Chem. Toxicol. 2013, 53, 112–118. [Google Scholar] [CrossRef]
- Leventis, P.A.; Grinstein, S. The Distribution and Function of Phosphatidylserine in Cellular Membranes. Annu. Rev. Biophys. 2010, 39, 407–427. [Google Scholar] [CrossRef]
- Dorstyn, L.; Akey, C.W.; Kumar, S. New insights into apoptosome structure and function. Cell Death Differ. 2018, 25, 1194–1208. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.J.; Yu, X.C.; Asara, J.M.; Heuser, J.E.; Ludtke, S.J.; Akey, C.W. The Holo-Apoptosome: Activation of Procaspase-9 and Interactions with Caspase-3. Structure 2011, 19, 1084–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soldani, C.; Scovassi, A.I. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: An update. Apoptosis 2002, 7, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Popovici, V.; Bucur, L.; Vochita, G.; Gherghel, D.; Mihai, C.T.; Rambu, D.; Calcan, S.I.; Costache, T.; Cucolea, I.E.; Matei, E.; et al. In Vitro Anticancer Activity and Oxidative Stress Biomarkers Status Determined by Usnea barbata (L.) FH Wigg. Dry Extracts. Antioxidants 2021, 10, 1141. [Google Scholar] [CrossRef]
- Tang, J.Y.; Wu, K.H.; Wang, Y.Y.; Farooqi, A.A.; Huang, H.W.; Yuan, S.S.F.; Jian, R.I.; Tsao, L.Y.; Chen, P.A.; Chang, F.R.; et al. Methanol Extract of Usnea barbata Induces Cell Killing, Apoptosis, and DNA Damage against Oral Cancer Cells through Oxidative Stress. Antioxidants 2020, 9, 694. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.L.; Akey, I.V.; Akey, C.W.; Kranz, R.G. Oxidized or Reduced Cytochrome c and Axial Ligand Variants All Form the Apoptosome in Vitro. Biochemistry 2017, 56, 2766–2769. [Google Scholar] [CrossRef]
- Huang, P.; Feng, L.; Oldham, E.A.; Keating, M.J.; Plunkett, W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature 2000, 407, 390–395. [Google Scholar] [CrossRef]
- Sahin, N.; Emsen, B.; Aslan, A.; Sadi, G. Antioxidant potential of Pseudevernia furfuracea (L.) Zopf and its secondary metabolites on hepatocellular carcinoma cells: Regulation of antioxidant enzymes. Anatol. J. Bot. 2021, 5, 127–133. [Google Scholar] [CrossRef]
- Ari, F.; Aztopal, N.; Oran, S.; Bozdemir, S.; Celikler, S.; Ozturk, S.; Ulukaya, E. Parmelia sulcata Taylor and Usnea filipendula Stirt induce apoptosis-like cell death and DNA damage in cancer cells. Cell Prolif. 2014, 47, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.J.; Yang, L.X. Gamma-H2AX—A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar] [PubMed]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Lanz, M.C.; Dibitetto, D.; Smolka, M.B. DNA damage kinase signaling: Checkpoint and repair at 30 years. EMBO J. 2019, 38, e101801. [Google Scholar] [CrossRef]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef]
- Harper, J.W.; Elledge, S.J.; Keyomarsi, K.; Dynlacht, B.; Tsai, L.H.; Zhang, P.; Dobrowolski, S.; Bai, C.; Connell-Crowley, L.; Swindell, E.; et al. Inhibition of cyclin-dependent kinases by p21. Mol. Biol. Cell 1995, 6, 387–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Ji, P.; Jiang, H.; Rekhtman, K.; Bloom, J.; Ichetovkin, M.; Pagano, M.; Zhu, L. An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol. Cell 2004, 16, 47–58. [Google Scholar] [CrossRef]
- Singh, N.; Nambiar, D.; Kale, R.K.; Singh, R.P. Usnic acid inhibits growth and induces cell cycle arrest and apoptosis in human lung carcinoma A549 cells. Nutr. Cancer 2013, 65 (Suppl. 1), 36–43. [Google Scholar] [CrossRef]
- Suh, S.S.; Kim, T.K.; Kim, J.E.; Hong, J.M.; Nguyen, T.T.T.; Han, S.J.; Youn, U.J.; Yim, J.H.; Kim, I.C. Anticancer Activity of Ramalin, a Secondary Metabolite from the Antarctic Lichen Ramalina terebrata, against Colorectal Cancer Cells. Molecules 2017, 22, 1361. [Google Scholar] [CrossRef]
- Ghate, N.B.; Chaudhuri, D.; Sarkar, R.; Sajem, A.L.; Panja, S.; Rout, J.; Mandal, N. An antioxidant extract of tropical lichen, Parmotrema reticulatum, induces cell cycle arrest and apoptosis in breast carcinoma cell line MCF-7. PLoS ONE 2013, 8, e82293. [Google Scholar] [CrossRef]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef]
- Kuo, Y.C.; Huang, K.Y.; Yang, C.H.; Yang, Y.S.; Lee, W.Y.; Chiang, C.W. Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J. Biol. Chem. 2008, 283, 1882–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymonowicz, K.; Oeck, S.; Malewicz, N.M.; Jendrossek, V. New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response. Cancers 2018, 10, 78. [Google Scholar] [CrossRef] [Green Version]
- Backorova, M.; Jendzelovsky, R.; Kello, M.; Backor, M.; Mikes, J.; Fedorocko, P. Lichen secondary metabolites are responsible for induction of apoptosis in HT-29 and A2780 human cancer cell lines. Toxicol. In Vitro 2012, 26, 462–468. [Google Scholar] [CrossRef]
- Bessadottir, M.; Skuladottir, E.A.; Gowan, S.; Eccles, S.; Omarsdottir, S.; Oegmundsdottir, H.M. Effects of anti-proliferative lichen metabolite, protolichesterinic acid on fatty acid synthase, cell signalling and drug response in breast cancer cells. Phytomedicine 2014, 21, 1717–1724. [Google Scholar] [CrossRef]
- Dincsoy, A.B.; Duman, D.C. Changes in apoptosis-related gene expression profiles in cancer cell lines exposed to usnic acid lichen secondary metabolite. Turk. J. Biol. 2017, 41, 484–493. [Google Scholar] [CrossRef] [Green Version]
- Emsen, B.; Togar, B.; Turkez, H.; Aslan, A. Effects of two lichen acids isolated from Pseudevernia furfuracea (L.) Zopf in cultured human lymphocytes. Z. Nat. C 2018, 73, 303–312. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies | |
|---|---|
| Cleaved Caspase-3 (Asp175) (5A1A) Rabbit mAb PE conjugate | Cell Signaling Technology® |
| Cleaved Caspase-9 (Asp315) (D8I9E) Rabbit mAb PE conjugate | |
| Cleaved PARP (Asp214) (D64E10) XP Rabbit mAb PE conjugate | |
| Phospho-Histone H2A.X (Ser139) (20E3) Rabbit mAb Alexa Fluor 647 conjugate | |
| Phospho-p53 (Ser15) (16G8) Mouse mAb PE conjugate | |
| p21 Waf1/Cip1 (12D1) Rabbit mAb | |
| Anti-phospho-ATM mAb PE conjugate | Merck Millipore |
| Anti-phospho-SMC1 mAb Alexa Fluor® 488 conjugate | |
| Cytochrome c (6H2) Mouse mAb FITC conjugate | Thermo Scientific |
| Anti-Oxoguanine 8 (2Q2311) Mouse mAb | Abcam |
| Secondary Conjugated Antibodies | |
| Goat anti-Mouse IgG Alexa Fluor® 488 conjugate | Thermo Scientific |
| Goat anti-Rabbit IgG Alexa Fluor® 488 conjugate | |
| Primary Antibodies | Mr (kDa) | Source/Origin | Company |
|---|---|---|---|
| JNK1 | 46 | Mouse | Thermo Scientific |
| Phospho SAPK/JNK (Thr183/Tyr185) | 46 | Mouse | Cell Signaling Technology® |
| Phospho Rb (Ser807/811) | 110 | Rabbit | |
| Phospho p38 MAPK (Thr180/Tyr182) | 43 | Rabbit | |
| p38 MAPK | 43 | Rabbit | |
| Phospho Akt (Thr308) | 60 | Rabbit | |
| Akt (pan) | 60 | Rabbit | |
| Phospho PI3K p85 (Tyr458)/p55 (Tyr199) | 85, 60 | Rabbit | |
| PI3K p85 | 85 | Rabbit | |
| p27 Kip1 | 27 | Rabbit | |
| SOD1/CuZnSOD | 18 | Rabbit | |
| β-actin | 45 | Mouse | |
| SOD2/MnSOD | 25 | Mouse | Abcam |
| Secondary Antibodies | Mr (kDa) | Source/Origin | Company |
| Anti-Mouse IgG HRP | - | Goat | Cell Signaling Technology® |
| Anti-Rabbit IgG HRP | - | Goat |
| Jurkat | Live (An−/PI−) | Early Apo (An+/PI−) | Late Apo (An+/PI+) | Death (An−/PI+) |
|---|---|---|---|---|
| CTRL 24 h | 96.8 ± 0.3 | 1.8 ± 0.1 | 1.0 ± 0.3 | 0.5 ± 0.1 |
| DMSO | 96.2 ± 0.6 | 1.8 ± 0.2 | 1.5 ± 0.3 | 0.5 ± 0.2 |
| PSE 45 | 21.2 ± 2.9 *** | 14.5 ± 2.1 * | 43.5 ± 2.6 *** | 20.9 ± 2.4 ** |
| Phy 60 | 82.7 ± 2.4 * | 8.8 ± 1.9 * | 6.5 ± 0.8 * | 2.0 ± 0.2 |
| CTRL 48 h | 97.0 ± 0.6 | 1.8 ± 0.6 | 0.6 ± 0.1 | 0.5 ± 0.1 |
| DMSO | 96.2 ± 0.6 | 1.8 ± 0.4 | 1.5 ± 0.2 | 0.5 ± 0.1 |
| PSE 45 | 10.7 ± 3.2 *** | 19.9 ± 0.9 * | 44.8 ± 4.5 *** | 24.7 ± 0.4 ** |
| Phy 60 | 45.6 ± 3.6 ** | 9.4 ± 2.0 * | 33.7 ± 1.0 ** | 11.4 ± 3.5 * |
| CTRL 72 h | 97.3 ± 0.4 | 1.4 ± 0.2 | 0.8 ± 0.2 | 0.4 ± 0.1 |
| DMSO | 96.3 ± 0.2 | 1.5 ± 0.1 | 1.5 ± 0.1 | 0.8 ± 0.2 |
| PSE 45 | 5.3 ± 1.4 *** | 26.3 ± 1.7 ** | 29.2 ± 0.9 ** | 39.3 ± 4.0 ** |
| Phy 60 | 35.6 ± 4.6 *** | 4.2 ± 0.1 | 34.2 ± 1.1 ** | 26.0 ± 2.6 ** |
| Jurkat | Live (An−/PI−) | Early Apo (An+/PI−) | Late Apo (An+/PI+) | Death (An−/PI+) |
|---|---|---|---|---|
| CTRL 24 h | 96.8 ± 0.3 | 1.8 ± 0.1 | 1.0 ± 0.3 | 0.5 ± 0.1 |
| DMSO | 96.2 ± 0.6 | 1.8 ± 0.2 | 1.5 ± 0.3 | 0.5 ± 0.2 |
| PSE 45 | 21.2 ± 2.9 *** | 14.5 ± 2.1 * | 43.5 ± 2.6 *** | 20.9 ± 2.4 ** |
| Phy 60 | 82.7 ± 2.4 * | 8.8 ± 1.9 * | 6.5 ± 0.8 * | 2.0 ± 0.2 |
| CTRL 48 h | 97.0 ± 0.6 | 1.8 ± 0.6 | 0.6 ± 0.1 | 0.5 ± 0.1 |
| DMSO | 96.2 ± 0.6 | 1.8 ± 0.4 | 1.5 ± 0.2 | 0.5 ± 0.1 |
| PSE 45 | 10.7 ± 3.2 *** | 19.9 ± 0.9 * | 44.8 ± 4.5 *** | 24.7 ± 0.4 ** |
| Phy 60 | 45.6 ± 3.6 ** | 9.4 ± 2.0 * | 33.7 ± 1.0 ** | 11.4 ± 3.5 * |
| CTRL 72 h | 97.3 ± 0.4 | 1.4 ± 0.2 | 0.8 ± 0.2 | 0.4 ± 0.1 |
| DMSO | 96.3 ± 0.2 | 1.5 ± 0.1 | 1.5 ± 0.1 | 0.8 ± 0.2 |
| PSE 45 | 5.3 ± 1.4 *** | 26.3 ± 1.7 ** | 29.2 ± 0.9 ** | 39.3 ± 4.0 ** |
| Phy 60 | 35.6 ± 4.6 *** | 4.2 ± 0.1 | 34.2 ± 1.1 ** | 26.0 ± 2.6 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kello, M.; Kuruc, T.; Petrova, K.; Goga, M.; Michalova, Z.; Coma, M.; Rucova, D.; Mojzis, J. Pro-Apoptotic Potential of Pseudevernia furfuracea (L.) Zopf Extract and Isolated Physodic Acid in Acute Lymphoblastic Leukemia Model In Vitro. Pharmaceutics 2021, 13, 2173. https://doi.org/10.3390/pharmaceutics13122173
Kello M, Kuruc T, Petrova K, Goga M, Michalova Z, Coma M, Rucova D, Mojzis J. Pro-Apoptotic Potential of Pseudevernia furfuracea (L.) Zopf Extract and Isolated Physodic Acid in Acute Lymphoblastic Leukemia Model In Vitro. Pharmaceutics. 2021; 13(12):2173. https://doi.org/10.3390/pharmaceutics13122173
Chicago/Turabian StyleKello, Martin, Tomas Kuruc, Klaudia Petrova, Michal Goga, Zuzana Michalova, Matus Coma, Dajana Rucova, and Jan Mojzis. 2021. "Pro-Apoptotic Potential of Pseudevernia furfuracea (L.) Zopf Extract and Isolated Physodic Acid in Acute Lymphoblastic Leukemia Model In Vitro" Pharmaceutics 13, no. 12: 2173. https://doi.org/10.3390/pharmaceutics13122173