
Dose Finding and Food Effect Studies of a Novel Abiraterone Acetate Formulation for Oral Suspension in Comparison to a Reference Formulation in Healthy Male Subjects

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Preparation of TOS
2.3. Reconstitution Test for TOS
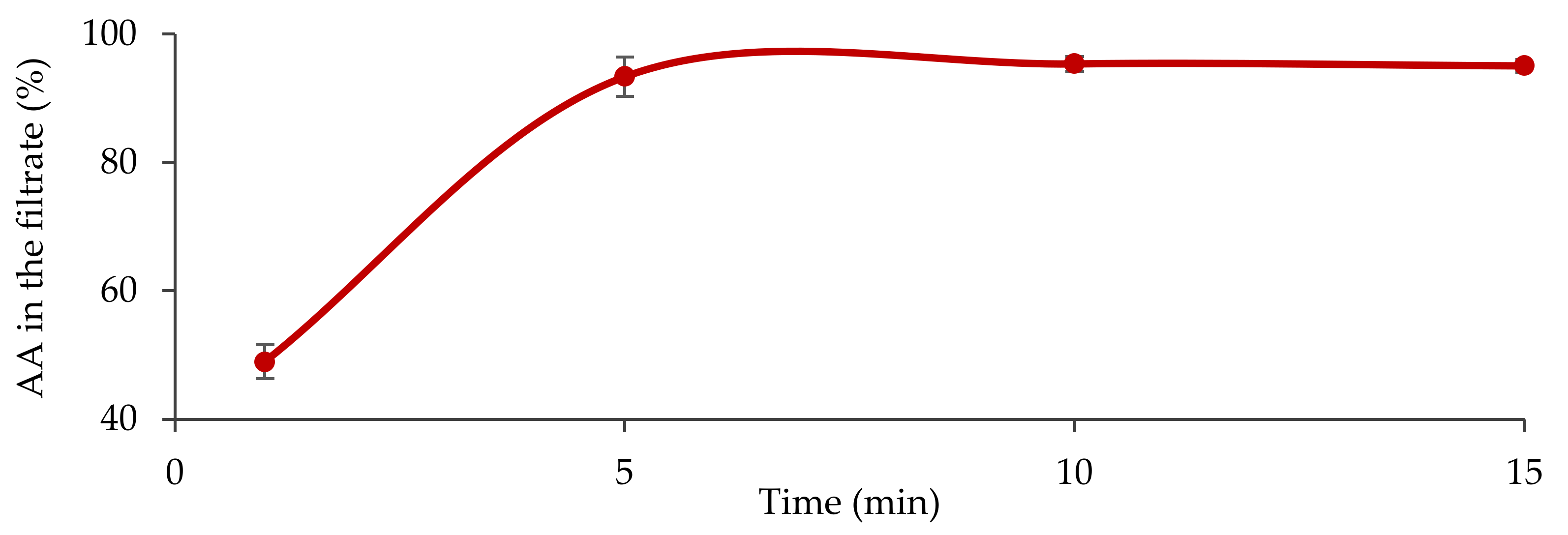
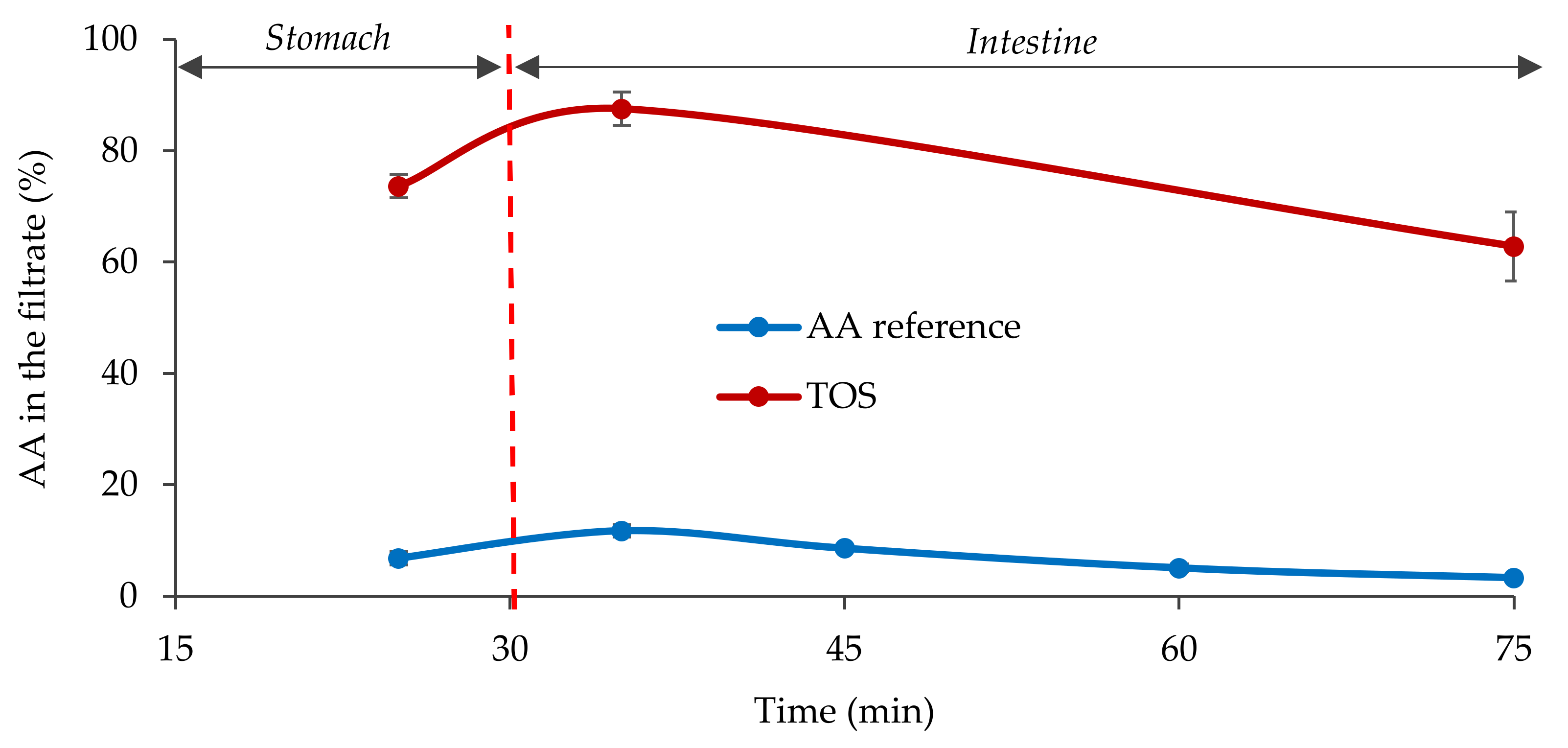
2.4. Fasted State Biorelevant Dissolution Tests
2.5. Quantification of the Active Ingredient in Solution
2.6. Clinical Study Design
2.7. Study Population
2.8. Bioanalytical Method
2.9. Pharmacokinetic Evaluation
2.10. Safety Evaluations
3. Results
3.1. In Vitro Characterization of the Formula
3.2. Subject Disposition and Baseline Characteristics
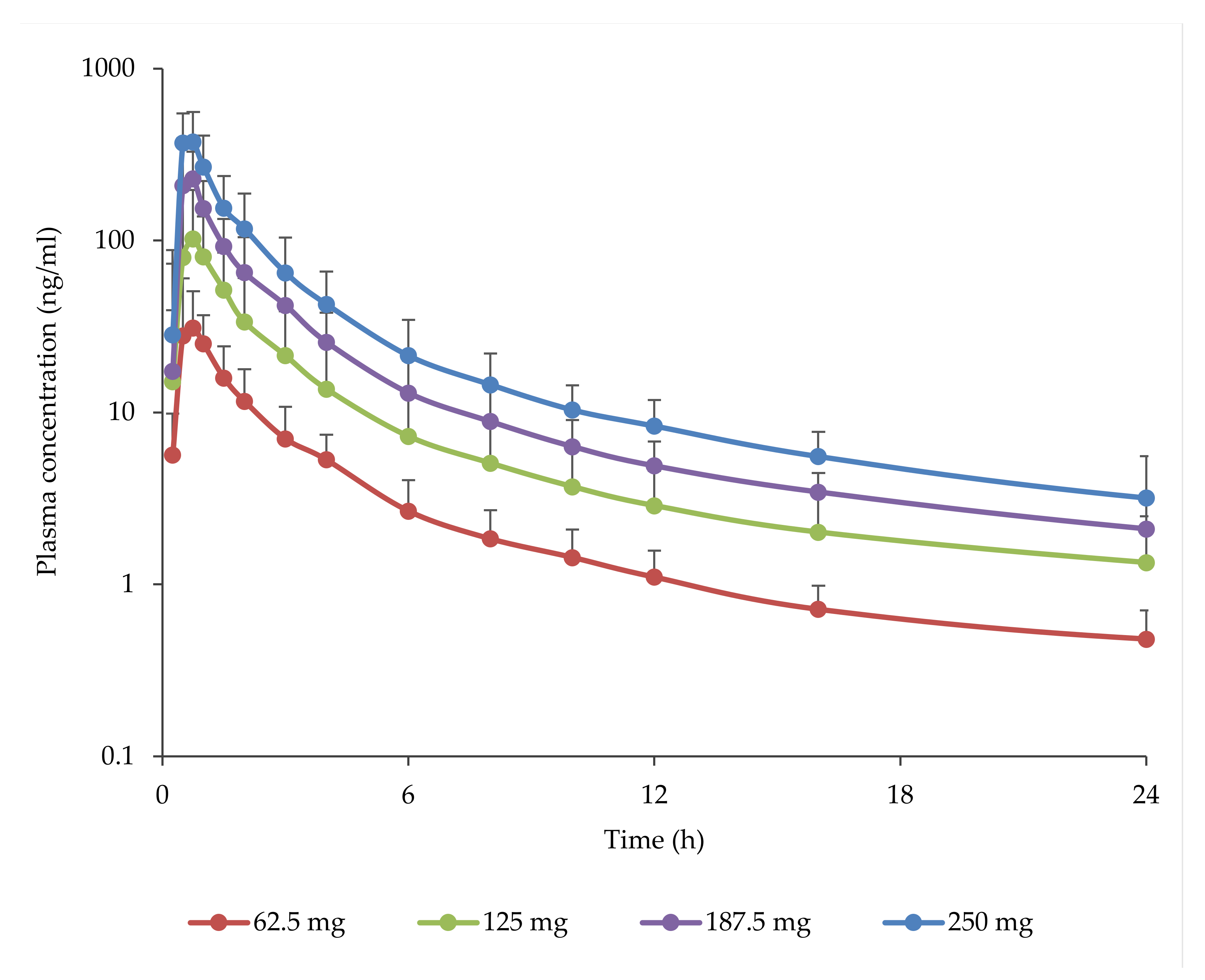
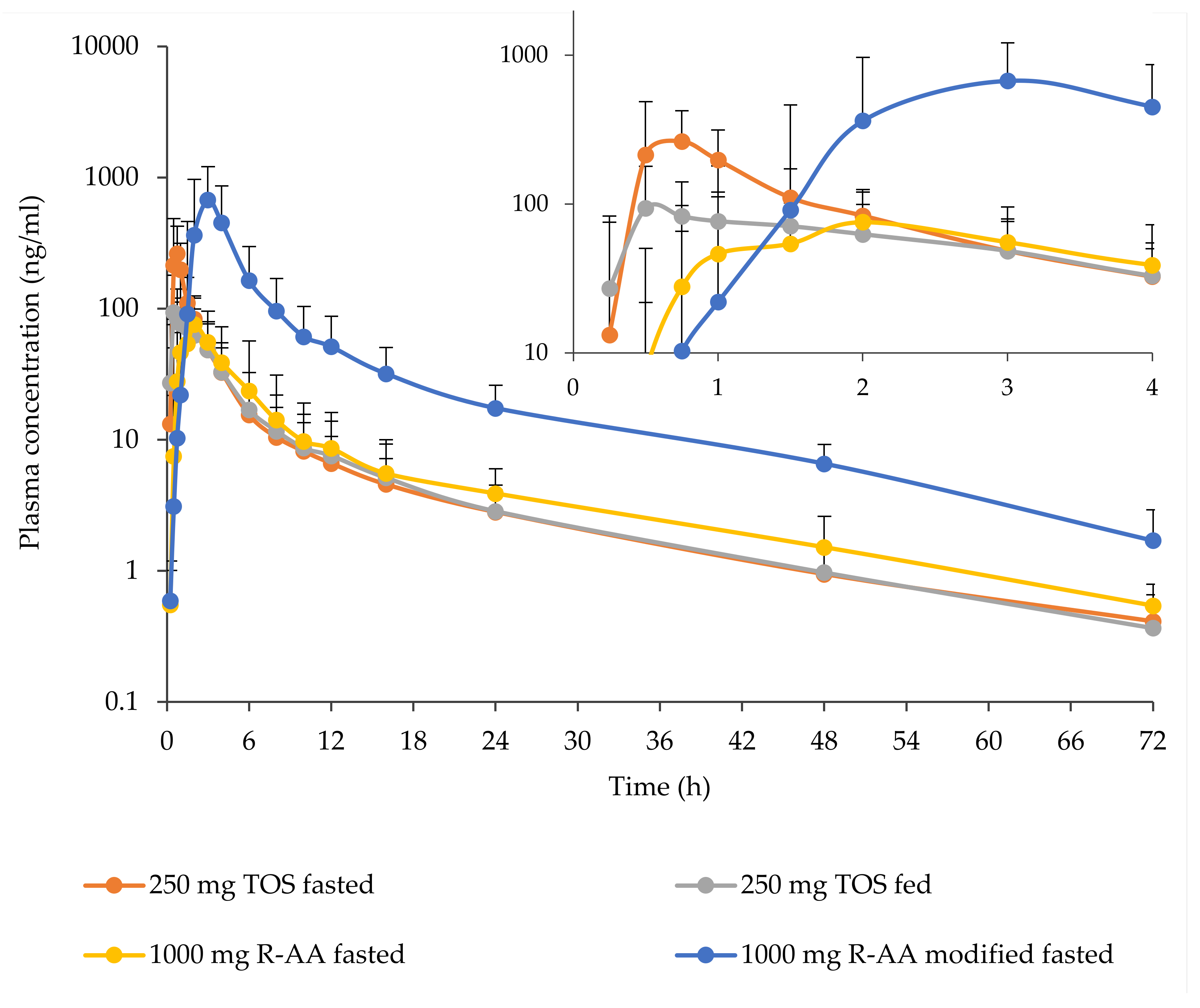
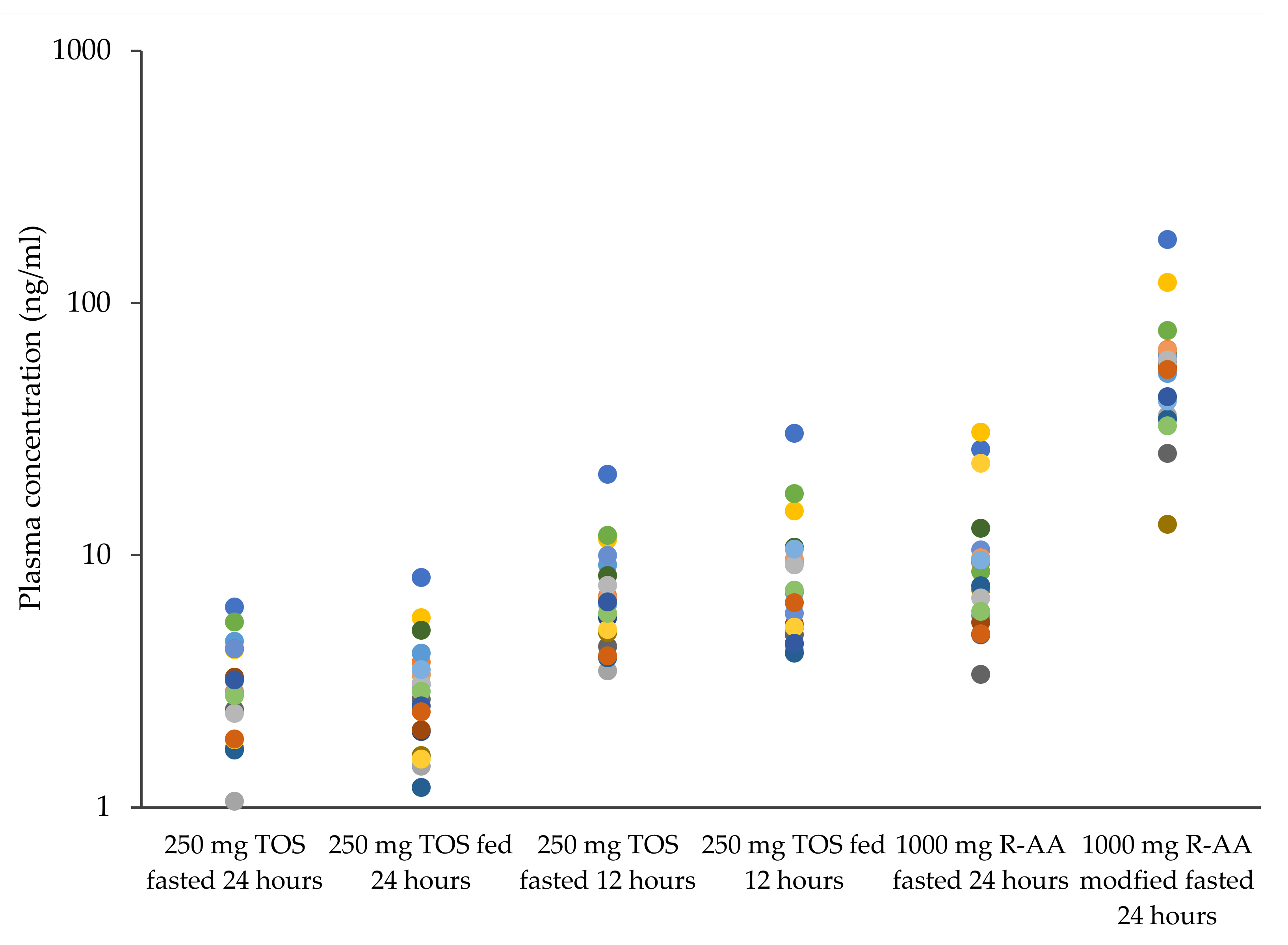
3.3. Pharmacokinetics
3.4. Safety
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AUC | area under the plasma concentration—time curve |
| AUCinf | area under the plasma concentration—time curve from dosing to infinity |
| AUClast | area under the plasma concentration—time curve from dosing to the time of the last measured plasma concentration |
| Cmax | maximal plasma concentration |
| Ct | trough plasma concentration |
| C12 | plasma concentration at 12 h after administration |
| C24 | plasma concentration at 24 h after administration |
| tmax | time to maximal plasma concentration |
| t1/2 | elimination half-life |
| S.D. | standard deviation |
| C.V. | coefficient of variation |
| AE | adverse event |
| mCRPC | metastatic castration resistant prostate cancer |
| mCSPC | metastatic castration sensitive prostate cancer |
| FDA | Food and Drug Administration |
| PSA | prostate specific antigen |
| PFS | progression free survival |
| SGF | simulated gastric fluid |
| FaSSIF | fasted state simulated intestinal fluid |
| ECG | electrocardiogram |
| QD | once daily |
| GMR | geometric mean ratio |
| LC | liquid chromatography |
| MS | mass spectrometry |
| TOS | tablet for oral suspension |
| R-AA | reference abiraterone acetate |
References
- Available online: https://list.essentialmeds.org/ (accessed on 20 May 2021).
- Stein, M.N.; Goodin, S.; Dipaola, R.S. Abiraterone in prostate cancer: A new angle to an old problem. Clin. Cancer. Res. 2012, 18, 1848–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EMA/CHMP/542871. Assessment Report for Zytiga Procedure No.: EMEA/H/C/002321. Assessment 2011, 44. [Google Scholar]
- Chi, K.N.; Spratlin, J.; Kollmannsberger, C.; North, S.; Pankras, C.; Gonzalez, M.; Bernard, A.; Stieltjes, H.; Peng, L.; Jiao, J.; et al. Food effects on abiraterone pharmacokinetics in healthy subjects and patients with metastatic castration-resistant prostate cancer. J. Clin. Pharmacol. 2015, 55, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- FDA. Yonsa (Abiraterone Acetate) Tablets [Prescribing Information]; Sun Pharma Global: Cranbury, NJ, USA, 2018. [Google Scholar]
- Goldwater, R.; Hussaini, A.; Bosch, B.; Nemeth, P. Comparison of a Novel Formulation of Abiraterone Acetate vs. the Originator Formulation in Healthy Male Subjects: Two Randomized, Open-Label, Crossover Studies. Clin. Pharmacokinet. 2017, 56, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Papangelou, A.; Olszanski, A.J.; Stein, C.A.; Bosch, B.; Nemeth, P. The Effect of Food on the Absorption of Abiraterone Acetate from a Fine Particle Dosage Form: A Randomized Crossover Trial in Healthy Volunteers. Oncol. Ther. 2017, 5, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Schultz, H.B.; Meola, T.R.; Thomas, N.; Prestidge, C.A. Oral formulation strategies to improve the bioavailability and mitigate the food effect of abiraterone acetate. Int. J. Pharm. 2020, 577, 119069. [Google Scholar] [CrossRef] [PubMed]
- Carton, E.; Noe, G.; Huillard, O.; Golmard, L.; Giroux, J.; Cessot, A.; Saidu, N.E.B.; Peyromaure, M.; Zerbib, M.; Narjoz, C.; et al. Relation between plasma trough concentration of abiraterone and prostate-specific antigen response in metastatic castration-resistant prostate cancer patients. Eur. J. Cancer 2017, 72, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, T.W.; Graff, J.N.; Zejnullahu, K.; Anantharaman, A.; Zhang, L.; Paz, R.; Premasekharan, G.; Russell, C.; Huang, Y.; Kim, W.; et al. High Dose Abiraterone Acetate in Men with Castration Resistant Prostate Cancer. Clin. Genitourin. Cancer 2017, 15, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Matteucci, M.E.; Brettmann, B.K.; Rogers, T.L.; Elder, E.J.; Williams, R.O.; Johnston, K.P. Design of potent amorphous drug nanoparticles for rapid generation of highly supersaturated media. Mol. Pharm. 2007, 4, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Solymosi, T.; Ötvös, Z.; Angi, R.; Ordasi, B.; Jordán, T.; Semsey, S.; Molnár, L.; Ránky, S.; Filipcsei, G.; Heltovics, G.; et al. Development of an abiraterone acetate formulation with improved oral bioavailability guided by absorption modeling based on in vitro dissolution and permeability measurements. Int. J. Pharm. 2017, 532, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Solymosi, T.; Ötvös, Z.; Angi, R.; Ordasi, B.; Jordán, T.; Molnár, L.; McDermott, J.; Zann, V.; Church, A.; Mair, S.; et al. Novel formulation of abiraterone acetate might allow significant dose reduction and eliminates substantial positive food effect. Cancer Chemother. Pharmacol. 2017, 80, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Kenny, C.; Regan, J.; Balding, L.; Higgins, S.; O’Leary, N.; Kelleher, F.; McDermott, R.; Armstrong, J.; Mihai, A.; Tiernan, E.; et al. Dysphagia prevalence and predictors in cancers outside the head, neck, and upper gastrointestinal tract. J. Pain Symptom. Manag. 2019, 58, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, H.; Takahashi, R.; Watanabe, N.; Oritsu, H.; Shimizu, Y. Prevalence of sarcopenia and its association with dysphagia in cancer patients who require rehabilitation. J. Rehabil. Med. 2017, 49, 682–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, J.; Go, J.T.; Schulze, K.S. Pill properties that cause dysphagia and treatment failure. Curr. Ther. Res. Clin. Exp. 2015, 77, 79–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Center for Drug Evaluation and Research. Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products—General Considerations; Food and Drug Administration, US Department of Health and Human Services: Washington, DC, USA, 2003. [Google Scholar]
- Basa-Dénes, O.; Solymosi, T.; Ötvös, Z.; Angi, R.; Ujhelyi, A.; Jordán, T.; Heltovics, G.; Glavinas, H. Investigations of the mechanism behind the rapid absorption of nano-amorphous abiraterone acetate. Eur. J. Pharm. Sci. 2019, 129, 79–86. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Study C01 | Study C02 |
|---|---|---|
| Age (years), mean (range) | 46.4 (25–65) | 41.0 (21–62) |
| Race, n (%) | ||
| White | 12 (100) | 18 (90.0) |
| Black | 0 (0) | 1 (5.0) |
| Asian | 0 (0) | 1(5.0) |
| Other | 0 (0) | 0 (0) |
| Sex, n (%) | ||
| Male | 12 (100) | 20 (100) |
| Height (cm), mean (range) | 176.1 (163–184) | 177.5 (166–187) |
| Weight (kg), mean (range) | 79.30 (64.2–93.7) | 82.70 (67.8–94.0) |
| Body Mass Index (kg/m2), mean (range) | 25.53 (20.2–29.0) | 26.27 (22.6–29.2) |
| Dose Level | 62.5 mg | 125 mg | 187.5 mg | 250 mg |
|---|---|---|---|---|
| No. of Subjects | n = 12 | n = 10 | n = 11 | n = 12 |
| tmax a—h (range) | 0.50 (0.50 to 1.50) | 0.64 (0.50 to 1.50) | 0.70 (0.50 to 1.00) | 0.75 (0.50 to 0.75) |
| Cmax—ng/mL (CV b%) | 43.0 (63.5) | 128 (53.9) | 265 (49.8) | 414 (41.5) |
| C12—ng/mL (CV%) | 1.10 (37.4) | 2.60 (27.8) | 4.91 (39.3) | 8.31 (40.1) |
| C24—ng/mL (CV%) | 0.48 (40.1) | 1.18 (44.6) | 2.10 (47.7) | 3.29 (58.1) |
| tlast a—h (range) | 24.03 (23.07 to 48.02) | 48.00 (24.30 to 72.18) | 70.98 (47.00 to 72.12) | 71.01 (48.00 to 72.23) |
| Clast—ng/mL (CV%) | 0.39 (42.6) | 0.37 (30.7) | 0.41 (41.2) | 0.55 (38.8) |
| AUClast—ng × h/mL (CV%) | 85.1 (43.2) | 242 (37.7) | 506 (40.6) | 844 (42.7) |
| AUCinf—ng × h/mL (CV%) | 91.1 (41.9) | 251 (36.9) | 516 (39.6) | 857 (42.3) |
| t1/2—h (CV%) | 10.38 (40.8) | 14.14 (42.3) | 15.04 (29.7) | 14.05 (33.4) |
| Dose Level | - | 62.5 mg | 125 mg | 187.5 mg | 250 mg |
|---|---|---|---|---|---|
| No. of Subjects | - | n = 12 | n = 10 | n = 11 | n = 12 |
| Cmax (ng/mL): | geometric mean | 43 | 128 | 265 | 414 |
| β (90% CI) | 1.64 (1.43, 1.84) | ||||
| 2β (90% CI) | 3.11 (2.70, 3.59) | ||||
| AUClast (ng × h/mL): | geometric mean | 85.1 | 242 | 506 | 844 |
| β (90% CI) | 1.65 (1.52, 1.77) | ||||
| 2β (90% CI) | 3.14 (2.88, 3.42) | ||||
| AUCinf (ng × h/mL): | geometric mean | 91.1 | 251 | 516 | 857 |
| β (90% CI) | 1.61 (1.49, 1.73) | ||||
| 2β (90% CI) | 3.05 (2.81, 3.32) | ||||
| Dose Level | 250 mg TOS | 250 mg TOS | 1000 mg R-AA | 1000 mg R-AA |
|---|---|---|---|---|
| Status | fasted | Fed | fasted | modified fasted |
| No. of Subjects | n = 19 b | n = 20 | n = 20 | n = 20 |
| tmax a – h (range) | 0.50 (0.50 to 1.00) | 0.50 (0.50 to 2.00) | 1.50 (0.75 to 6.00) | 3.00 (2.00 to 4.00) |
| Cmax – ng/mL (CV%) | 306 (60.4) | 127 (48.0) | 105 (58.6) | 821 (72.3) |
| C12 – ng/mL (CV%) | 6.80 (44.6) [n = 18] | 7.55 (57.8) | 8.60 (64.3) | 51.3 (58.8) |
| C24 – ng/mL (CV%) | 2.80 (46.9) | 2.84 (51.1) | 3.89 (49.6) | 17.4 (52.4) |
| tlast – h (range) | 72.00 (48.00 to 72.08) | 72.02 (48.00 to 72.18) | 72.00 (48.00 to 72.15) | 72.03 (72.00 to 72.47) |
| Clast – ng/mL (CV%) | 0.425 (47.8) | 0.372 (41.6) | 0.542 (42.9) | 1.70 (62.6) |
| AUClast – ng × h/mL (CV%) | 625 (45.7) | 485 (44.6) | 532 (57.7) | 3460 (55.3) |
| AUCinf – ng × h/mL (CV%) | 636 (45.1) | 495 (44.0) | 547 (56.3) | 3510 (55.0) |
| t1/2—h (CV%) | 15.145 (21.8) | 14.624 (22.5) | 16.081 (16.5) | 13.829 (17.5) |
| (A) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Test | 1000 mg R-AA (Fasted) | ||||||||
| Parameter | Test | Prandial State | n | Adj Geo Mean a | n | Adj Geo Mean a | Ratio b (%) | 90% CI c | p-Value d |
| Cmax (ng/mL) | 250 mg TOS | Fasted | 19 | 314 | 20 | 105 | 299.83 | (238.25, 377.33) | <0.001 |
| AUClast (ng × h/mL) | 250 mg TOS | Fasted | 19 | 646 | 20 | 532 | 121.46 | (105.18, 140.25) | 0.028 |
| AUCinf (ng × h/mL) | 250 mg TOS | Fasted | 19 | 657 | 20 | 547 | 120.19 | (104.23, 138.59) | 0.035 |
| Cmax (ng/mL) | 250 mg TOS | Fed | 20 | 127 | 20 | 105 | 121.11 | (96.61, 151.82) | 0.16 |
| AUClast (ng × h/mL) | 250 mg TOS | Fed | 20 | 485 | 20 | 532 | 91.24 | (79.21, 105.10) | 0.28 |
| AUCinf (ng × h/mL) | 250 mg TOS | Fed | 20 | 495 | 20 | 547 | 90.46 | (78.65, 104.05) | 0.24 |
| (B) | |||||||||
| Test | 1000 mg R-AA (Modified Fasted) | ||||||||
| Parameter | Test | Prandial State | n | Adj Geo Mean a | n | Adj Geo Mean a | Ratio b (%) | 90% CI c | p-Value d |
| Cmax (ng/mL) | 250 mg TOS | Fasted | 19 | 314 | 20 | 821 | 38.31 | (30.44, 48.21) | <0.001 |
| AUClast (ng × h/mL) | 250 mg TOS | Fasted | 19 | 646 | 20 | 3460 | 18.64 | (16.14, 21.53) | <0.001 |
| AUCinf (ng × h/mL) | 250 mg TOS | Fasted | 19 | 657 | 20 | 3510 | 18.74 | (16.25, 21.60) | <0.001 |
| Cmax (ng/mL) | 250 mg TOS | Fed | 20 | 127 | 20 | 821 | 15.47 | (12.34, 19.40) | <0.001 |
| AUClast (ng × h/mL) | 250 mg TOS | Fed | 20 | 485 | 20 | 3460 | 14.00 | (12.16, 16.13) | <0.001 |
| AUCinf (ng × h/mL) | 250 mg TOS | Fed | 20 | 495 | 20 | 3510 | 14.10 | (12.26, 16.22) | <0.001 |
| Test | Reference (250 mg TOS) (fasted) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameter | Test | Prandial State | n | Adj Geo Mean a | n | Adj Geo Mean a | Ratio (%) b | 90% CI c | p-Value d |
| Cmax (ng/mL) | 250 mg TOS | Fed | 20 | 127 | 19 | 314 | 40.39 | (32.10, 50.83) | <0.001 |
| AUClast (ng × h/mL) | 250 mg TOS | Fed | 20 | 485 | 19 | 646 | 75.12 | (65.05, 86.75) | 0.002 |
| AUCinf (ng × h/mL) | 250 mg TOS | Fed | 20 | 495 | 19 | 657 | 75.26 | (65.27, 86.79) | 0.002 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jordán, T.; Basa-Dénes, O.; Angi, R.; Orosz, J.; Ötvös, Z.; Ujhelyi, A.; Filipcsei, G.; Molnár, L.; Solymosi, T.; Glavinas, H.; et al. Dose Finding and Food Effect Studies of a Novel Abiraterone Acetate Formulation for Oral Suspension in Comparison to a Reference Formulation in Healthy Male Subjects. Pharmaceutics 2021, 13, 2171. https://doi.org/10.3390/pharmaceutics13122171
Jordán T, Basa-Dénes O, Angi R, Orosz J, Ötvös Z, Ujhelyi A, Filipcsei G, Molnár L, Solymosi T, Glavinas H, et al. Dose Finding and Food Effect Studies of a Novel Abiraterone Acetate Formulation for Oral Suspension in Comparison to a Reference Formulation in Healthy Male Subjects. Pharmaceutics. 2021; 13(12):2171. https://doi.org/10.3390/pharmaceutics13122171
Chicago/Turabian StyleJordán, Tamás, Orsolya Basa-Dénes, Réka Angi, János Orosz, Zsolt Ötvös, Andrea Ujhelyi, Genovéva Filipcsei, László Molnár, Tamás Solymosi, Hristos Glavinas, and et al. 2021. "Dose Finding and Food Effect Studies of a Novel Abiraterone Acetate Formulation for Oral Suspension in Comparison to a Reference Formulation in Healthy Male Subjects" Pharmaceutics 13, no. 12: 2171. https://doi.org/10.3390/pharmaceutics13122171