
Development of a Model-Informed Dosing Tool to Optimise Initial Antibiotic Dosing—A Translational Example for Intensive Care Units

 , ,
, ,  ,
,
Abstract
:1. Introduction
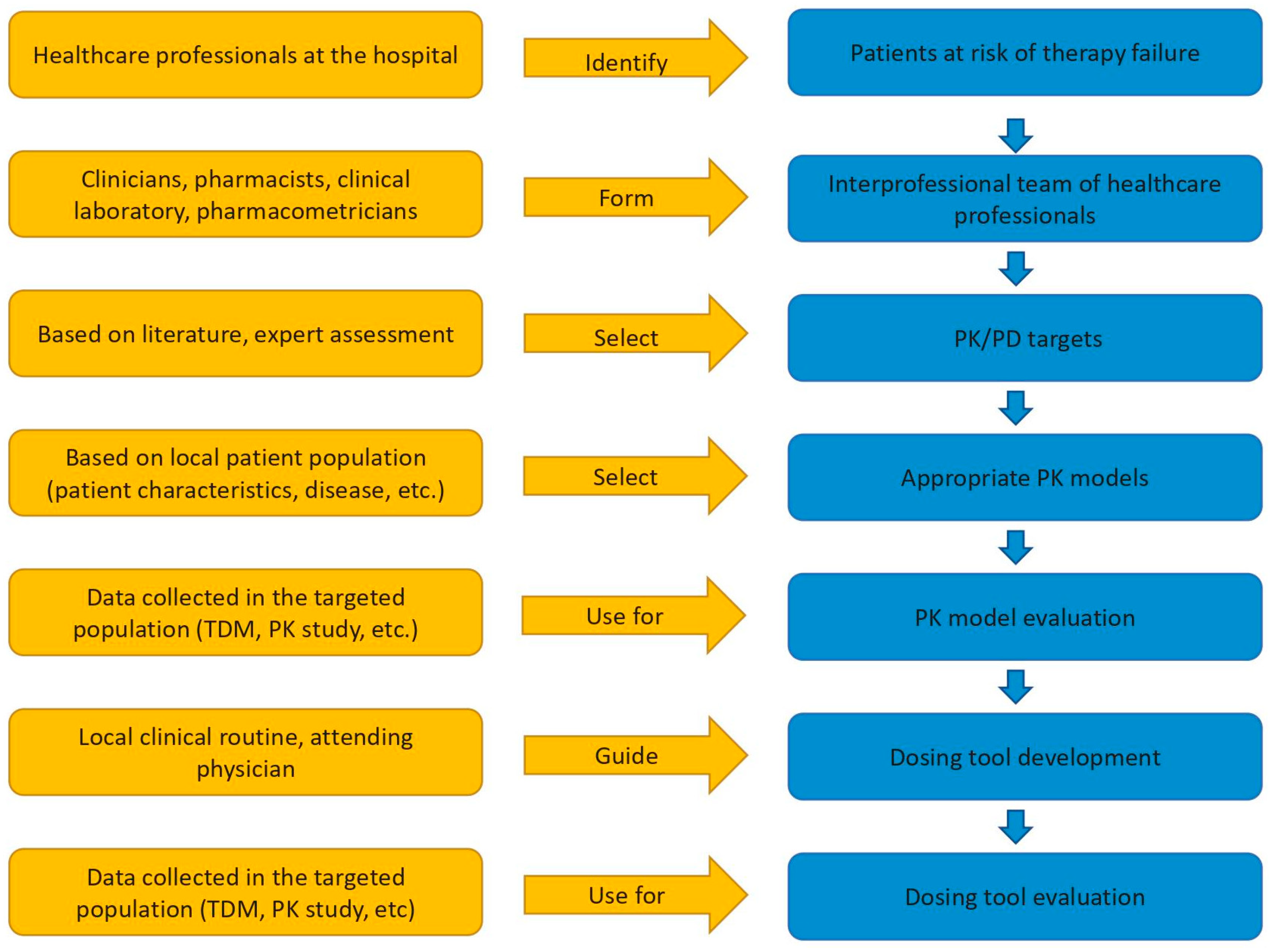
2. Materials and Methods
2.1. Patient Population and Meropenem Concentration Measurements
2.2. Pharmacokinetic Model Selection, Reduction, and Evaluation
2.3. Pharmacokinetic/Pharmacodynamic Targets
2.4. Development of the Dosing Decision Tool
2.4.1. Selection and Evaluation of Dosing Regimens
2.4.2. Integration of Locally Available Pathogen Information
2.5. Retrospective Evaluation of the Dosing Decision Tools Using Real Patient Data
3. Results
3.1. Pharmacokinetic Model Selection, Reduction, and Evaluation
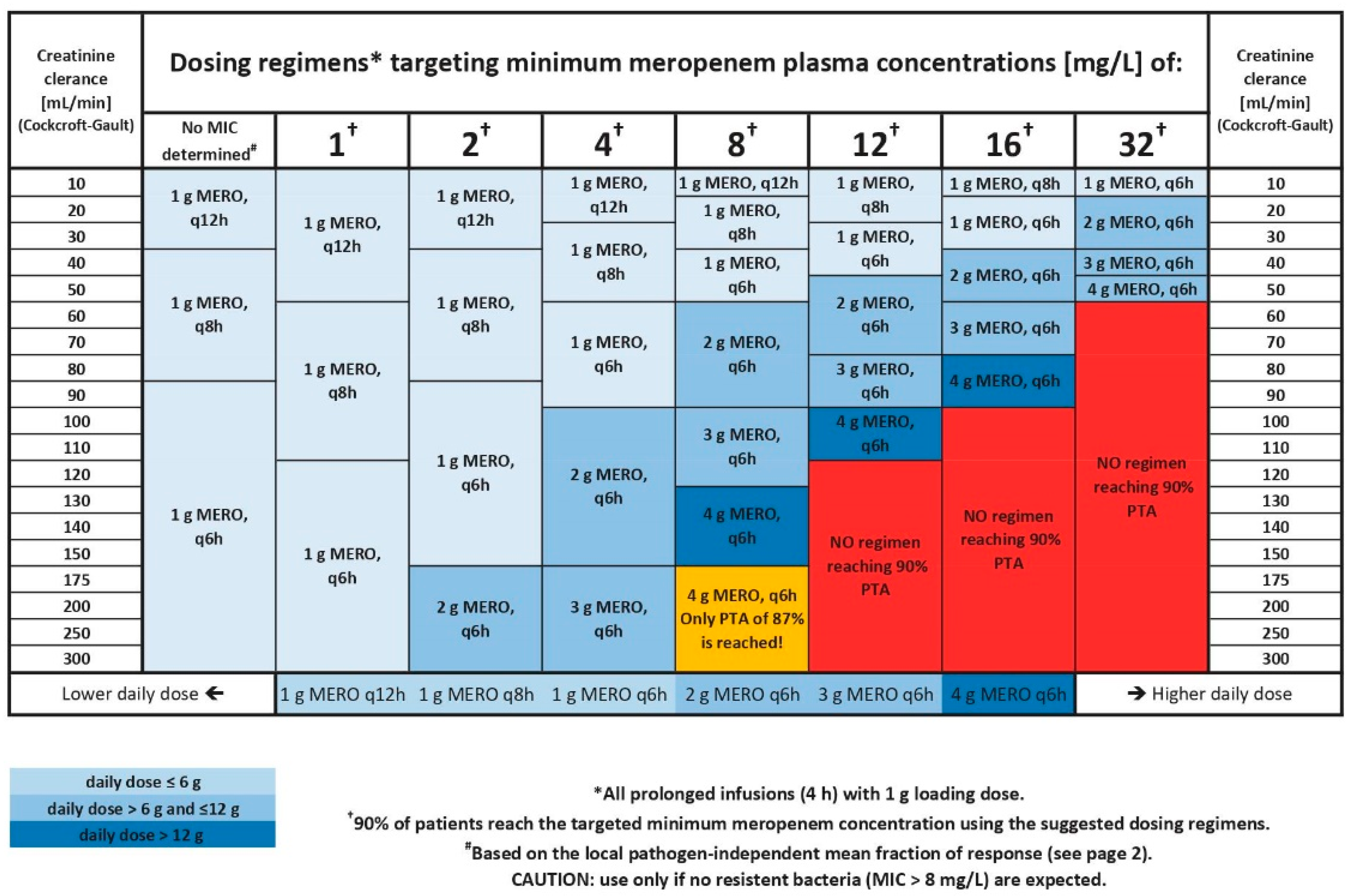
3.2. Development of the Dosing Decision Tool
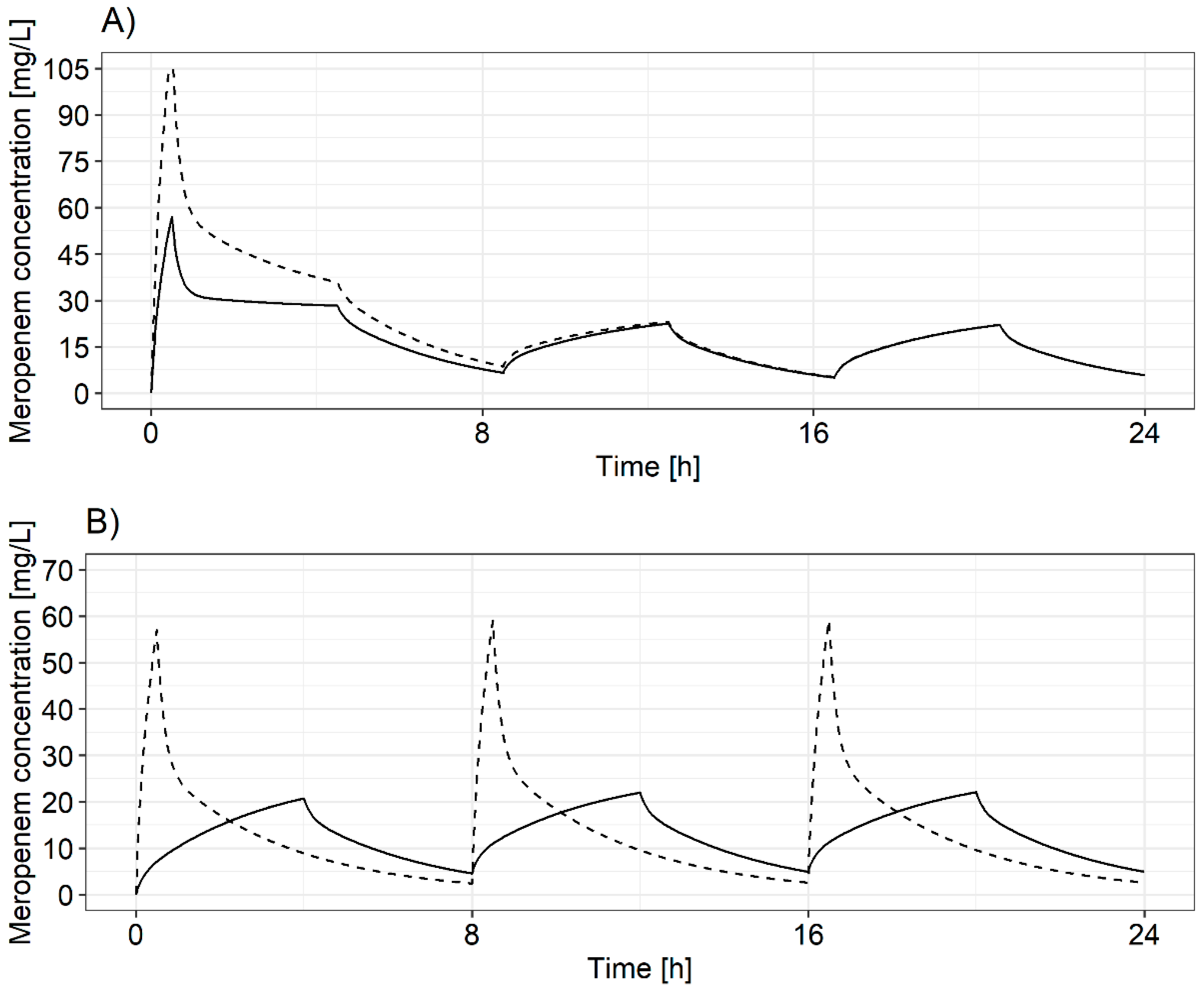
3.2.1. Selection and Evaluation of Dosing Regimens
3.2.2. Integration of Locally Available Pathogen Information
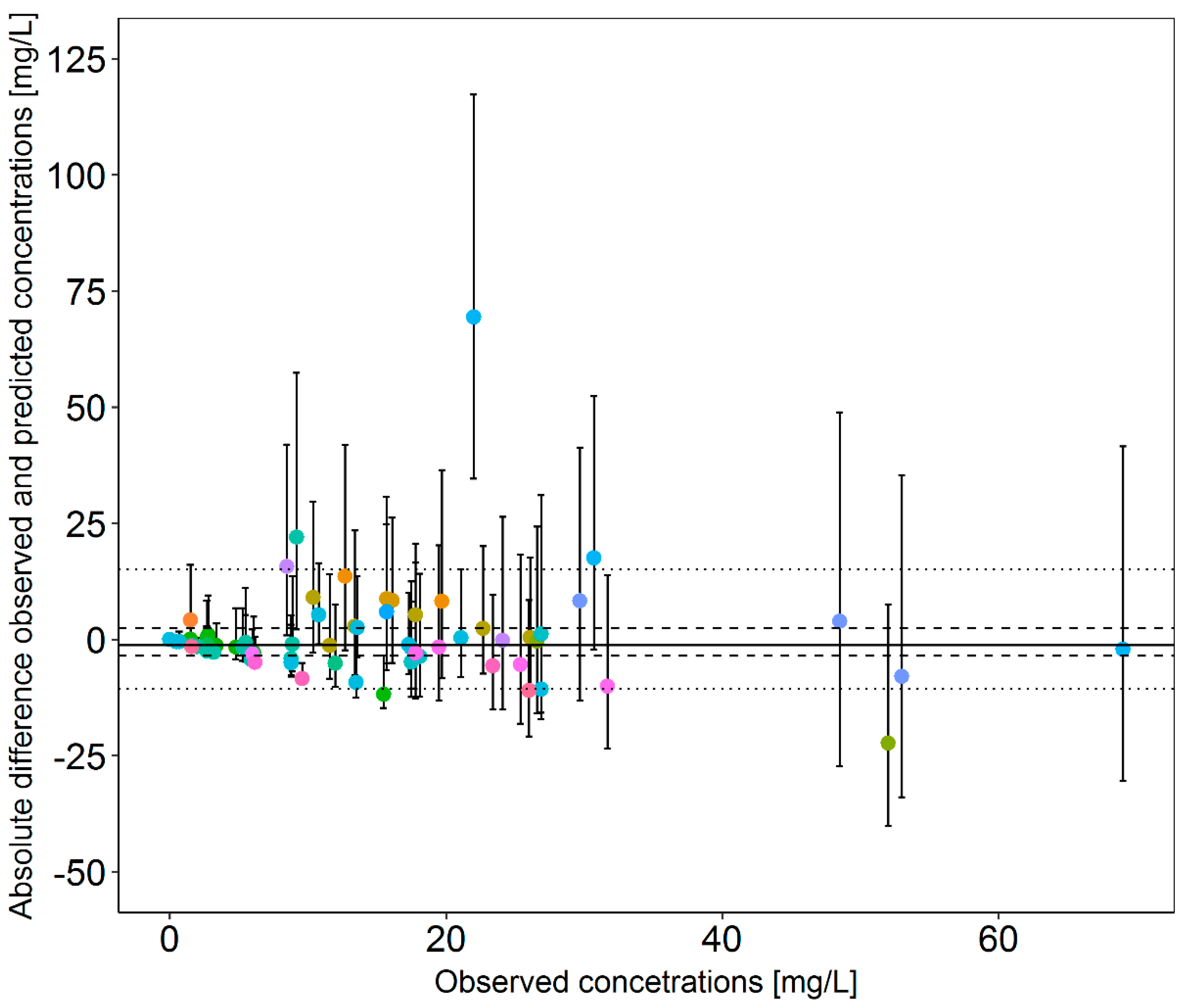
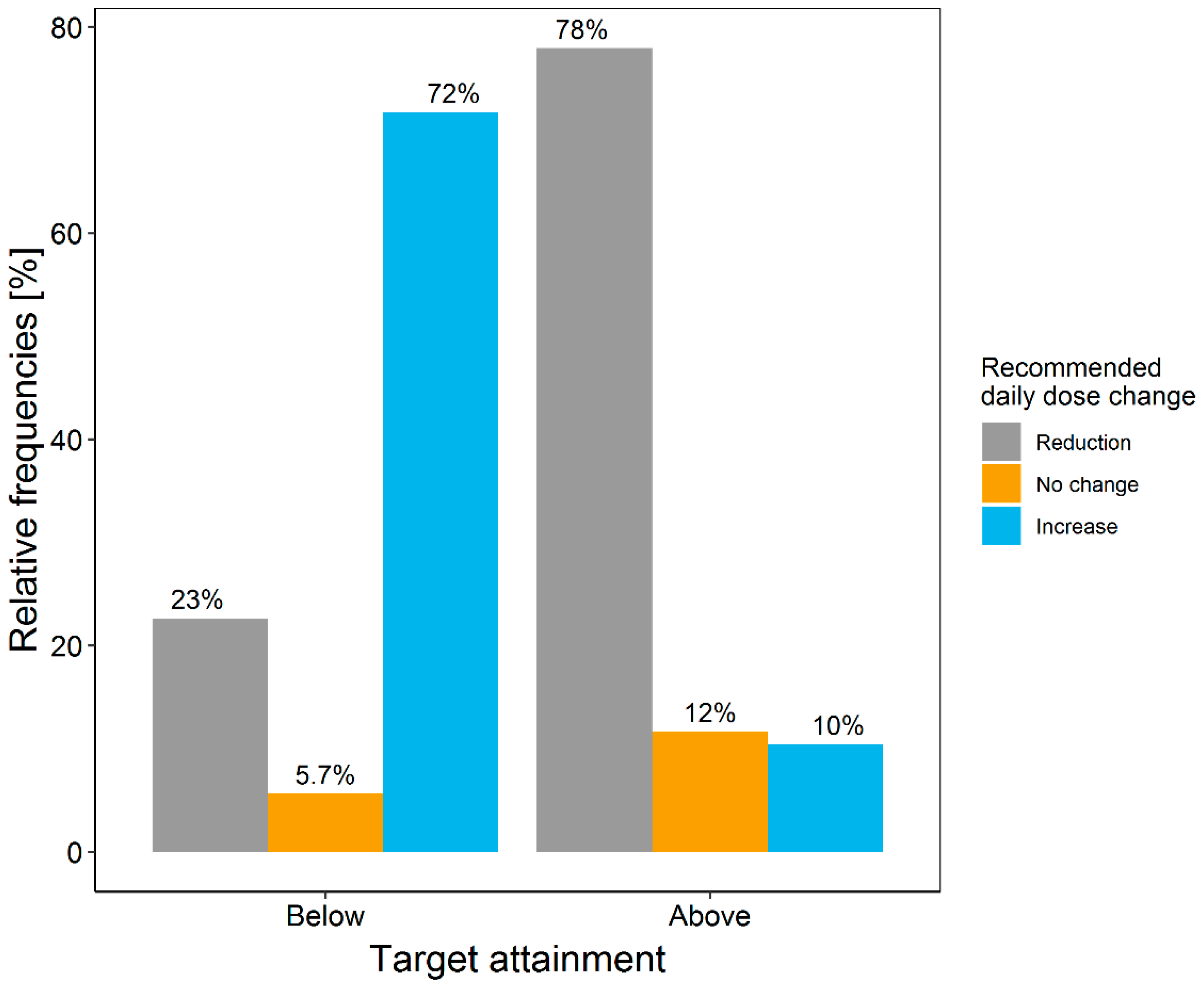
3.3. Retrospective Evaluation of the Dosing Decision Tools Using Real Patient Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levy Hara, G.; Kanj, S.S.; Pagani, L.; Abbo, L.; Endimiani, A.; Wertheim, H.F.L.; Amábile-Cuevas, C.; Tattevin, P.; Mehtar, S.; Lopes Cardoso, F.; et al. Ten Key Points for the Appropriate use of Antibiotics in Hospitalised Patients: A Consensus from the Antimicrobial Stewardship and Resistance Working Groups of the International Society of Chemotherapy. Int. J. Antimicrob. Agents 2016, 48, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A.; Paul, S.K.; Akova, M.; Bassetti, M.; De Waele, J.J.; Dimopoulos, G.; Kaukonen, K.M.; Koulenti, D.; Martin, C.; Montravers, P.; et al. DALI: Defining Antibiotic Levels in Intensive Care Unit Patients: Are Current ß-lactam Antibiotic Doses Sufficient for Critically Ill Patients? Clin. Infect. Dis. 2014, 58, 1072–1083. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A.; Abdul-Aziz, M.H.; Lipman, J.; Mouton, J.W.; Vinks, A.A.; Felton, T.W.; Hope, W.W.; Farkas, A.; Neely, M.N.; Schentag, J.J.; et al. Individualised Antibiotic Dosing for Patients Who are Critically Ill: Challenges and Potential Solutions. Lancet Infect. Dis. 2014, 14, 498–509. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, C. Antibiotic dosing in critical illness. J. Antimicrob. Chemother. 2011, 66, 25–31. [Google Scholar] [CrossRef]
- Vossen, M.G.; Ehmann, L.; Pferschy, S.; Maier-Salamon, A.; Haidinger, M.; Weiser, C.; Wenisch, J.M.; Saria, K.; Kajahn, C.; Jilch, S.; et al. Elimination of Doripenem during Dialysis and Pharmacokinetic Evaluation of Posthemodialysis Dosing for Patients Undergoing Intermittent Renal Replacement Therapy. Antimicrob. Agents Chemother. 2018, 62, e02430-17. [Google Scholar] [CrossRef] [Green Version]
- Kees, M.G.; Minichmayr, I.K.; Moritz, S.; Beck, S.; Wicha, S.G.; Kees, F.; Kloft, C.; Steinke, T. Population Pharmacokinetics of Meropenem during Continuous Infusion in Surgical ICU patients. J. Clin. Pharmacol. 2016, 56, 307–315. [Google Scholar] [CrossRef]
- Minichmayr, I.K.; Schaeftlein, A.; Kuti, J.L.; Zeitlinger, M.; Kloft, C. Clinical Determinants of Target Non-Attainment of Linezolid in Plasma and Interstitial Space Fluid: A Pooled Population Pharmacokinetic Analysis with Focus on Critically Ill Patients. Clin. Pharmacokinet. 2017, 56, 617–633. [Google Scholar] [CrossRef]
- Felton, T.W.; Hope, W.W.; Roberts, J.A. How Severe Is Antibiotic Pharmacokinetic Variability in Critically Ill Patients and What Can Be Done about it? Diagn. Microbiol. Infect. Dis. 2014, 79, 441–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, J.A.; Ulldemolins, M.; Roberts, M.S.; McWhinney, B.; Ungerer, J.; Paterson, D.L.; Lipman, J. Therapeutic Drug Monitoring of β-Lactams in Critically Ill Patients: Proof of Concept. Int. J. Antimicrob. Agents 2010, 36, 332–339. [Google Scholar] [CrossRef]
- Tabah, A.; de Waele, J.; Lipman, J.; Zahar, J.R.; Cotta, M.O.; Barton, G.; Timsit, J.F.; Roberts, J.A. The ADMIN-ICU Survey: A Survey on Antimicrobial Dosing and Monitoring in ICUs. J. Antimicrob. Chemother. 2015, 70, 2671–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paviour, S.; Hennig, S.; Staatz, C.E. Usage and Monitoring of Intravenous Tobramycin in Cystic Fibrosis in Australia and the UK. J. Pharm. Pract. Res. 2016, 46, 15–21. [Google Scholar] [CrossRef]
- Darwich, A.S.; Ogungbenro, K.; Vinks, A.A.; Powell, J.R.; Reny, J.; Marsousi, N.; Daali, Y.; Fairman, D. Why Has Model-Informed Precision Dosing Not Yet Become Common Clinical Reality? Lessons from the Past and a Roadmap for the Future. Clin. Pharmacol. Ther. 2017, 101, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Liebchen, U.; Paal, M.; Scharf, C.; Schroeder, I.; Grabein, B.; Zander, J.; Siebers, C.; Zoller, M. The ONTAI Study—A Survey on Antimicrobial Dosing and the Practice of Therapeutic Drug Monitoring in German Intensive Care Units. J. Crit. Care 2020, 60, 260–266. [Google Scholar] [CrossRef]
- EuCAST. EuCAST Breakpoints. The European Committee on Antimicrobial Susceptibility Testing. 2018. Available online: https://eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_8.0_Breakpoint_Tables.pdf (accessed on 3 December 2021).
- Ewig, S.; Höffken, G.; Kern, W.; Rohde, G.; Flick, H.; Krause, R.; Ott, S.; Bauer, T.; Dalhoff, K.; Gatermann, S.; et al. Behandlung von erwachsenen Patienten mit ambulant erworbener Pneumonie und Prävention—Update 2016. Pneumologie 2016, 70, 151–200. [Google Scholar] [CrossRef]
- Linden, P. Safety Profile of Meropenem: An Updated Review of Over 6000 Patients Treated with Meropenem. Drug Saf. 2007, 30, 657–668. [Google Scholar] [CrossRef]
- Chimata, M.; Nagase, M.; Suzuki, Y.; Shimomura, M.; Kakuta, S. Pharmacokinetics of Meropenem in Patients with Various Degrees of Renal Function, Including Patients with End-Stage Renal Disease. Antimicrob. Agents Chemother. 1993, 37, 229–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolau, D.P. Pharmacokinetic and Pharmacodynamic Properties of Meropenem. Clin. Infect. Dis. 2008, 47, S32–S40. [Google Scholar] [CrossRef] [Green Version]
- Brendel, K.; Comets, E.; Laffont, C.; Laveille, C.; Mentré, F. Metrics for External Model Evaluation with an Application to the Population Pharmacokinetics of Gliclazide. Pharm. Res. 2006, 23, 2036–2049. [Google Scholar] [CrossRef] [Green Version]
- Mentré, F.; Escolano, S. Prediction Discrepancies for the Evaluation of Nonlinear Mixed-Effects Models. J. Pharmacokinet. Pharmacodyn. 2006, 33, 345–367. [Google Scholar] [CrossRef]
- Lindbom, L.; Pihlgren, P.; Jonsson, N. PsN-Toolkit—A Collection of Computer Intensive Statistical Methods for Non-Linear Mixed Effect Modeling Using NONMEM. Comput. Methods Progr. Biomed. 2005, 79, 241–257. [Google Scholar] [CrossRef]
- Comets, E.; Brendel, K.; Mentré, F. Computing Normalised Prediction Distribution Errors to Evaluate Nonlinear Mixed-Effect Models: The NPDE Add-On Package for R. Comput. Methods Progr. Biomed. 2007, 90, 154–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Du, X.; Kuti, J.L.; Nicolau, D.P. Clinical Pharmacodynamics of Meropenem in Patients with Lower Respiratory Tract Infections. Antimicrob. Agents Chemother. 2007, 51, 1725–1730. [Google Scholar] [CrossRef] [Green Version]
- Ariano, R.E.; Zelenitsky, S.A.; Nyhlén, A.; Sitar, D.S. An Evaluation of an Optimal Sampling Strategy for Meropenem in Febrile Neutropenics. J. Clin. Pharmacol. 2005, 45, 832–835. [Google Scholar] [CrossRef]
- Drlica, K. The Mutant Selection Window and Antimicrobial Resistance. J. Antimicrob. Chemother. 2003, 52, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, W.A. The Pharmacology of Meropenem, a New Carbapenem Antibiotic. Clin. Infect. Dis. 1997, 24, S266–S275. [Google Scholar] [CrossRef] [Green Version]
- Guilhaumou, R.; Benaboud, S.; Bennis, Y.; Dahyot-Fizelier, C.; Dailly, E.; Gandia, P.; Goutelle, S.; Lefeuvre, S.; Mongardon, N.; Roger, C.; et al. Optimization of the Treatment with Beta-Lactam Antibiotics in Critically Ill Patients—Guidelines from the French Society of Pharmacology and Therapeutics (Société Française de Pharmacologie et Thérapeutique—SFPT) and the French Society of Anaesthesia. Crit. Care 2019, 23, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Cockcroft, D.W.; Gault, M.H. Prediction of Creatinine Clearance from Serum Creatinine. Nephron 1976, 16, 31–41. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency Committee for Medicinal Products for Human Use (CHMP). Guideline on the Use of Pharmacokinetics and Pharmacodynamics in the Development of Antimicrobial Medicinal Products. 2016, pp. 1–17. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-use-pharmacokinetics-pharmacodynamics-development-antimicrobial-medicinal-products_en.pdf (accessed on 10 May 2021).
- Ehmann, L.; Zoller, M.; Minichmayr, I.K.; Scharf, C.; Huisinga, W.; Zander, J.; Kloft, C. Development of a Dosing Algorithm for Meropenem in Critically Ill Patients Based on a Population Pharmacokinetic/Pharmacodynamic Analysis. Int. J. Antimicrob. Agents 2019, 54, 309–317. [Google Scholar] [CrossRef]
- Harbarth, S.; Garbino, J.; Pugin, J.; Romand, J.A.; Lew, D.; Pittet, D. Inappropriate Initial Antimicrobial Therapy and Its Effect on Survival in a Clinical Trial of Immunomodulating Therapy for Severe Sepsis. Am. J. Med. 2003, 115, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Ehmann, L.; Zoller, M.; Minichmayr, I.K.; Scharf, C.; Maier, B.; Schmitt, M.V.; Hartung, N.; Huisinga, W.; Vogeser, M.; Frey, L.; et al. Role of Renal Function in Risk Assessment of Target Non-Attainment after Standard Dosing of Meropenem in Critically Ill Patients: A Prospective Observational Study. Crit. Care 2017, 21, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minichmayr, I.K.; Roberts, J.A.; Frey, O.R.; Roehr, A.C.; Kloft, C.; Brinkmann, A. Development of a Dosing Nomogram for Continuous-Infusion Meropenem in Critically Ill Patients Based on a Validated Population Pharmacokinetic Model. J. Antimicrob. Chemother. 2018, 73, 1330–1339. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dosing Regimen | Dose Per Infusion [mg] | Infusion Duration [h] | Dosing Interval [h] | Total Daily Dose [mg] |
|---|---|---|---|---|
| 1 | 1000 | 4 | 6 | 4000 |
| 2 | 1000 | 4 | 8 | 3000 |
| 3 | 1000 | 4 | 12 | 2000 |
| 4 | 2000 | 4 | 6 | 8000 |
| 5 | 2000 | 4 | 8 | 6000 |
| 6 | 2000 | 4 | 12 | 4000 |
| 7 | 3000 | 4 | 6 | 12,000 |
| 8 | 3000 | 4 | 8 | 9000 |
| 9 | 3000 | 4 | 12 | 6000 |
| 10 | 4000 | 4 | 6 | 16,000 |
| 11 | 4000 | 4 | 8 | 12,000 |
| 12 | 4000 | 4 | 12 | 8000 |
| 13 | 4000 | 24 | 24 | 4000 |
| 14 | 6000 | 24 | 24 | 6000 |
| 15 | 8000 | 24 | 24 | 8000 |
| Patient Characteristic | Charité Universitätsmedizin-Berlin | Ehmann et al. |
|---|---|---|
| Categorical | n (%) | n (%) |
| No. of patients | 81 | 42 |
| No. of meropenem samples | 306 | 1376 |
| Male | 55 (67.9) | 27 (56.3) |
| No. of extracorporeal membrane oxygenation | 8 (9.88) | 6 (12.5) |
| Continuous (unit) | Median (5th–95th percentile) | Median (5th–95th percentile) |
| Age (years) | 64.0 (40.0–81.0) | 55.5 (32.0–69.9) |
| Weight (kg) | 75.0 (48.0–116) | 70.5 (47.4–121) |
| Creatinine clearance # (mL/min) | 74.4 (24.7–253) | 80.8 (24.8–191) |
| Serum albumin concentration (g/dL) | 2.68 (2.00–3.60) | 2.80 (2.20–3.56) |
| Total Daily Dose [mg] | Probability of Target Attainment, % | ||
|---|---|---|---|
| PI, 6 h Interval | PI, 8 h Interval | CI, 24 h Interval | |
| 4000 | 75.0 | - | 16.2 |
| 6000 | - | 58.0 | 67.7 |
| 8000 | 97.0 | - | 67.6 |
| 9000 | - | 76.8 | - |
| 12,000 | 99.4 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weinelt, F.A.; Stegemann, M.S.; Theloe, A.; Pfäfflin, F.; Achterberg, S.; Schmitt, L.; Huisinga, W.; Michelet, R.; Hennig, S.; Kloft, C. Development of a Model-Informed Dosing Tool to Optimise Initial Antibiotic Dosing—A Translational Example for Intensive Care Units. Pharmaceutics 2021, 13, 2128. https://doi.org/10.3390/pharmaceutics13122128
Weinelt FA, Stegemann MS, Theloe A, Pfäfflin F, Achterberg S, Schmitt L, Huisinga W, Michelet R, Hennig S, Kloft C. Development of a Model-Informed Dosing Tool to Optimise Initial Antibiotic Dosing—A Translational Example for Intensive Care Units. Pharmaceutics. 2021; 13(12):2128. https://doi.org/10.3390/pharmaceutics13122128
Chicago/Turabian StyleWeinelt, Ferdinand Anton, Miriam Songa Stegemann, Anja Theloe, Frieder Pfäfflin, Stephan Achterberg, Lisa Schmitt, Wilhelm Huisinga, Robin Michelet, Stefanie Hennig, and Charlotte Kloft. 2021. "Development of a Model-Informed Dosing Tool to Optimise Initial Antibiotic Dosing—A Translational Example for Intensive Care Units" Pharmaceutics 13, no. 12: 2128. https://doi.org/10.3390/pharmaceutics13122128