
mRNA, a Revolution in Biomedicine


1
CICS-UBI—Health Sciences Research Centre, University of Beira Interior, Av. Infante D. Henrique, 6200-506 Covilhã, Portugal
2
Centre de Biophysique Moléculaire (CBM), UPR 4301 CNRS, University of Orléans, 45071 Orléans, France
*
Authors to whom correspondence should be addressed.
Pharmaceutics 2021, 13(12), 2090; https://doi.org/10.3390/pharmaceutics13122090
Submission received: 6 October 2021
/
Revised: 29 November 2021
/
Accepted: 29 November 2021
/
Published: 5 December 2021
(This article belongs to the Collection Women in Pharmaceutics)
Abstract
:The perspective of using messenger RNA (mRNA) as a therapeutic molecule first faced some uncertainties due to concerns about its instability and the feasibility of large-scale production. Today, given technological advances and deeper biomolecular knowledge, these issues have started to be addressed and some strategies are being exploited to overcome the limitations. Thus, the potential of mRNA has become increasingly recognized for the development of new innovative therapeutics, envisioning its application in immunotherapy, regenerative medicine, vaccination, and gene editing. Nonetheless, to fully potentiate mRNA therapeutic application, its efficient production, stabilization and delivery into the target cells are required. In recent years, intensive research has been carried out in this field in order to bring new and effective solutions towards the stabilization and delivery of mRNA. Presently, the therapeutic potential of mRNA is undoubtedly recognized, which was greatly reinforced by the results achieved in the battle against the COVID-19 pandemic, but there are still some issues that need to be improved, which are critically discussed in this review.
1. Introduction
After intensive work unveiling the DNA structure, its function, and potential biomedical applications, scientists changed their target and focused on the study of ribonucleic acid (RNA) [1,2]. Structurally, RNA is composed of a single polynucleotide strand, but some short regions within the RNA molecule can be complementary and form secondary structures (hairpins, loops, among others) [2,3] (Table 1). The secondary structure is determined by the base sequence of the nucleotide chain, so that different RNA molecules can assume different structures. Moreover, as RNA structure determines its function, this diversity is also responsible for RNA engagement in multiple cellular roles [2,4,5].
Cells contain an enormous variety of RNA forms, generally categorized as coding RNAs, which include the messenger RNA (mRNA), and non-coding RNAs (ncRNA) [2,3]. Although only 2% of the RNA transcribed from the human genome will encode proteins, it plays a fundamental role, directly or indirectly, in numerous biological processes [3,6], as it will be discussed later. Recently, with the recognition of the biological value of mRNA, it started to be exploited in a therapeutic perspective [2], gaining now increased relevance as an innovative vaccine for pandemic situations.
2. mRNA Structure and Biogenesis
Several types of RNA play fundamental roles in gene expression, a process responsible for the use of information stored in the DNA sequence to produce effector molecules, such as RNAs and proteins, which are the basis of cell biology [2]. The central dogma of molecular biology states that DNA is transcribed into mRNA, which is later translated into proteins [1]. The mRNA is essentially composed of a complimentary copy of the DNA and serves as a model for the production of proteins, as it contains the information about the amino acids sequence that will compose the target protein [2,7]. In addition, each mRNA molecule presents non-coding or untranslated sequences, which will control the mechanisms of processing and reading of mRNA [2,8].
In bacteria, mRNA is transcribed directly from DNA, and the untranslated region at the 5′ end (5′ UTR) of the mRNA chain, called the Shine-Dalgarno sequence, helps the mRNA bind to the ribosomes [2]. On the other hand, in eukaryotic organisms, the process of gene expression is more complex, involving RNA Polymerase II (Pol II) co for messenger RNA synthesis [9,10]. In this case, a pre-mRNA (the primary transcript) is formed, which is later processed to produce the mature mRNA [4]. In the mature mRNA molecule, each amino acid is encoded by a set of three nucleotides, called a codon [2]. The translation process and the stability of mRNA are determined by specific regions of RNA, which are of high importance for the improved durability of this biomolecule in the cell [8]. Both prokaryotic and eukaryotic mRNAs contain three primary regions [2,11,12], which can be observed in Figure 1. The first, 5′ UTR is a sequence of nucleotides at the 5′ end of the mRNA that does not encode for amino acids [13]. During the processing of pre-mRNA, the addition of a guanine nucleotide with a methyl group (CH3) to the 5′ end (Cap-5′), increases mRNA stability and assists in the transport of mRNA to the cytoplasm and further binding of the mRNA to the ribosome [11,13,14]. This modification facilitates translation, nuclear exportation [8,15], intron removal [2,8] and decreases immunogenicity, benefiting some types of therapies, such as in target proteins replacement. Modifications to the Cap-5′ end, which prevents its removal, may also result in an improvement in mRNA stabilization and resistance to enzymatic degradation [8,11,14]. The second region of the mRNA is the protein coding region, which comprises the codons that determine the amino acid sequence. The protein coding region starts with a first codon which encodes for methionine and finishes with a stop codon [2]. The modification of rare codons in the protein coding sequences with frequent synonymous codons, called optimization of codons, can result in altered levels of expression [8,15] and immunogenicity [16,17]. Both coding and non-coding RNA can be subjected to specific nucleotides’ modification, coined as epi-transcriptomic changes in eukaryotic cells [18]. Those modifications play an important role as they can affect the translation, initiation, stability, localization, or function of RNAs. In 2005, Kariko and Weissmann [19] demonstrated that it is possible to reduce the immunogenicity through chemical modifications of nucleosides of mRNA [8,12,13]. Adding modified nucleosides highly improves mRNA translation and vaccine performance, as described below [8].
The third region, similarly to the 5′ UTR, is a non-coding nucleotide sequence occurring at the 3′ end of the mRNA (3′ UTR) [2,17]. The modification of the 3′ UTR consists in the formation of a polyadenylated “tail” (Poly (A)), in a process called polyadenylation [2,11,12] which is represented in Figure 1. The poly (A) tail promotes inhibition of deadenylation by nucleases, leading to increased stability and a higher translation efficiency [20,21,22]. In addition, this sequence can be advantageous and has been highly explored for the purification of mRNA. Both 5′ and 3′ UTR regions increase the stability of mRNA [2,11], increasing the half-life of mRNA in the cell, which may lead to greater production of proteins [2,13,15]. Moreover, both UTRs of mRNAs are responsible for recruiting RNA-binding proteins, as well as microRNAs (miRNAs), and can profoundly affect translation activity [8,13].
3. Therapeutic Application: mRNA vs. DNA
In the 1990s, intracellular delivery technology and DNA or mRNA production methods were sophisticated enough to support preclinical studies on gene therapy, DNA- or mRNA-based vaccines and cancer immunotherapy [1,8,23]. Both DNA and especially mRNA serve as a model for protein synthesis and, if used in a therapeutic way, allow the cell to produce missing or defective proteins in a given disease or deficiency [15]. Therapies using these nucleic acids are more advantageous than therapies in which an absent or defective protein is directly applied, as they are relatively faster and cheaper to develop, without major size restrictions. In addition, certain proteins, such as membrane proteins, may be difficult or impossible to be effectively produced in vitro. Another advantage of using nucleic acids as biotherapeutics is related to production issues, more specifically the post-translational modifications that are mandatory in proteins and extremely difficult to be guaranteed by the production of proteins in heterologous systems [8,15]. The challenges on post-translation modifications may lead to significant differences between proteins produced endogenously and exogenously (in vitro) and, consequently, cause increased immunogenicity [1,12]. Moreover, the delivery of protein drugs has its limitations, mainly due to their large size and some instability, which makes it more difficult to reach in vivo the concentrations that can induce a therapeutic effect, not to mention the high production costs associated. In this sense, delivering exogenous mRNA into cells seemed to be a very promising way of manipulating protein expression, once it does not integrate cells’ genome, and its natural degradation pathway ensures its temporary activity [1].
Until few years ago, DNA-based therapeutic approaches were proposed as the most appropriate in the treatment of inherited diseases, which were caused by missing or dysfunctional proteins. However, the therapeutic use of DNA requires not only fast delivery of DNA to the cytoplasm but also an efficient entry into the nucleus, since DNA needs to be transcribed into mRNA before the therapeutic protein molecule can be produced [13,15,24]. Furthermore, since most differentiated cells are post-mitotic cells, which do not undergo frequent cell division, nuclear delivery is a major obstacle to DNA-based therapy [15,25]. Conversely, as mRNA enters the cytoplasm, the translation process can start immediately [26], without its transfer into the nucleus being necessary [12,16].
In 1970, mRNA was delivered to cells for the first time through liposomes [1,15]. At that time, mRNA was considered unfeasible for use for gene therapy purposes, due to its instability and immunogenicity [12,13,17]. In addition, at the beginning, mRNA was not recognized as having a relevant role in therapy, as it could not lead to permanent gene therapy [1]. This ideology has only changed in recent years, with the attainment of deeper knowledge about mRNA itself, as well as the identification of stabilizing modifications and the development of improved nucleic acids delivery technologies [13,15,17]. The evolution in this field has lead to an improvement in mRNA translation and half-life, as well as improved transfection efficiency, resulting in a significant reduction in immunogenicity [15,23,24]. Thus, mRNA, as a transient transporter of genetic information capable of inducing proteins expression, started to be investigated to replace DNA or recombinant proteins for therapeutic purposes [8,15,23]. Indeed, it was found that mRNA has a higher flexibility and greater therapeutic utility than almost all other classes of known drugs, mainly because of its transient nature [1,12]. This elevated the interest in mRNA to revolutionize different health areas, such as in vaccination, cell reprogramming, protein replacement therapies, and in the treatment of genetic diseases [8,24,27] in a safer way compared to other gene therapy strategies [1,13]. Since then, mRNA has evolved from a fragile molecule to a therapeutic agent in clinical studies [15]. Overall, this biomolecule, compared to DNA vectors, increases the transfection efficiency of inactive cells [26] and does not present the risk of insertional mutagenesis in the genome [13,25,28], making mRNA a more attractive molecule in protein supplementation therapy [17,24]. Moreover, mRNA enables a faster protein production, allows a higher control over the expression levels, and does not contain additional exogenous sequences, such as antibiotic resistances or viruses-derived promoters, highlighting the safety and effectiveness of mRNA [12,25,29]. The degradation of mRNA can be determined by its structure, thus allowing improved, controllable and temporary pharmacokinetics [1,12,15]. Another key point is the possibility to produce mRNA with different strategies, namely by chemical synthesis or by in vitro transcription, despite the limitations associated with these methods. An alternative that is being exploited is in vivo production, based on recombinant technology. However, for large-scale microbial production, there are still many challenges to overcome in order to achieve a safe process that could be totally accepted by regulatory agencies [12]. The use of mRNA also faces limitations related to its instability under physiological conditions and strong immunogenicity. One of the strategies that can help to solve this problem is the use of a drug delivery system (DDS) capable of protecting mRNA against nucleases and preventing recognition by the immune system, in order to achieve the desired therapeutic results [17,25]. As already mentioned, mRNA has a transient therapeutic effect [13,17]. Although this transient nature of mRNA is beneficial in certain cases, this feature also turns out to be a limitation [30]. In genetic and/or metabolic diseases, in which prolonged treatment is required, repeated administration of this biomolecule is necessary to sustain the therapeutic levels of a protein [8,15,24,30], and this can be stressful for the patient. However, regardless of the treatment regimen (transient or prolonged), the main goal of mRNA therapy is to deliver it successfully to the cytoplasm, for an efficient expression of the target protein, with negligible toxicity and immunogenicity [15]. In recent years, the therapeutic use of mRNAs has shown considerable promise both for prophylaxis (for example, antiviral vaccines) and for the treatment of a wide range of diseases, including myocardial infarction, human immunodeficiency virus (HIV) infection and several types of cancer [1,27], as will be discussed later.
4. Production of mRNA
Since mRNA has been exposed in a positive way to therapeutic actions, the challenges for mRNA production started to be addressed in a more intensive way. The most commonly used methods are the chemical synthesis and enzymatic production by in vitro transcription (IVT) of a linearized plasmid DNA (pDNA) [12,13,31], or by a PCR model [12]. These enzymatic strategies for obtaining mRNA depend on the presence of a bacteriophage promoter, 3′ and 5′ UTR, a protein coding region or open reading frame (ORF) (located between the two regions) [12,28,30], and optionally a Cap-5′ [13] and poly [d (A/T)] sequence [11,12,30]. These sequences (Cap-5′ and poly [d (A/T)]) help to obtain more stable mRNA, more efficient translation and easier transcript purification, due to the poly [d (A/T)] sequence [11,12,13].
The production of mRNA by IVT begins with the addition of all necessary nucleotides (which can be modified or not) [13] and the bacteriophage RNA polymerase (T7, T3 or SP6), which recognizes the promoter in the DNA model [12,25,31]. In eukaryotic cells, mRNAs have a tail on the 3′ UTR of 50 to 250 adenosine residues, and during the synthesis of mRNA, the inclusion of a poly (A) tail is also intended, in order to have a similar structure to endogenous mRNA. It has been reported that gradually increasing the length of the poly (A) tail of the synthetic mRNA to 120 adenosine residues could also increase the protein expression levels [11,12], however, longer chains (>120 adenosine residues) did not elicit the same effect [12]. If the DNA template for the IVT does not contain a poly [d (A/T)] sequence or if it is short, the poly (A) chain can be added post-transcriptionally by the poly (A) polymerase [12,23]. Although this method allows the addition of a poly (A) tail, it is not consistent, as it promotes a variable length of the poly (A) tail and therefore, mRNA molecules that originate will have poly (A) tails with heterogeneous lengths [12]. Synthetic mRNAs with a known and homogeneous poly (A) tail length can be obtained using a pDNA model. Still, it is difficult to know the exact size of this sequence due to the difficulties of sequencing long repetitive chains [12]. Due to these difficulties, pDNA models for mRNA IVT are often cleaved after the poly [(A/T)] sequence by IIS-type restriction enzymes. In this way, cleavage within the recognition sequence is avoided and prevents the existence of nucleotides other than adenines, which could result in mRNAs with a masked poly (A) tail [12].
As mentioned, in addition to IVT from a pDNA, mRNA can also be obtained by a PCR model. This model is produced from the IVT pDNA, using a primer upstream of the bacteriophage promoter and a second primer, which contains a poly (T) sequence, allowing its connection to the poly (A) tail at the 3′ end of the pDNA insert [12]. Regarding the addition of Cap-5′, it can be incorporated in the transcript during IVT, including a dinucleotide m7GpppG as the structural homolog of endogenous Cap-5′ [23]. After IVT, and in view of an adequate therapeutic effect, the synthetic mRNA should be purified, thus seeking the removal of any impurities, among which, the pDNA template, RNA polymerase, unincorporated ribonucleotides [12] and aberrant or double-stranded transcripts (dsRNA), which may have occurred in IVT. These impurities, if not removed from the synthetic mRNA of interest, may lead to unwanted immune responses or induce adverse effects in the organism upon administration [12,23,31]. In addition, if the synthetic mRNA is capped enzymatically, it needs to be purified to remove excess of “Cap” enzymes and molecules [12,25]. Among the most common purification methods used is chromatography [23,31,32]. This has the ability to remove ever-larger by-products, including DNA, which can be degraded by DNases, facilitating purification, as well as abortive transcripts and dsRNA [12]. Thus, a rigorous purification of the mRNA obtained by IVT was recognized as being one of the critical steps for obtaining pure and stable mRNA [23,32].
Globally, these methods allow the preparation of mRNA in a relatively quick and safe manner [24], but their cost is potentially high [33], so it might be beneficial to develop studies for recombinant mRNA production, along with purification strategies in order to obtain a recombinant, pure, safe and cheaper mRNA, which is already being applied to other RNA types [34,35,36].
5. Challenges in mRNA Delivery
To achieve the goal of an effective mRNA-based therapy, meaning the reestablishment of the production of a missing or defective protein, it is necessary that mRNA enter the cells, leading to the target protein expression at therapeutic levels. Usually, DNA and mRNA induce protein expression, whereas small interfering RNAs (siRNAs), microRNAs, oligonucleotides or aptamers provide gene silencing [37].
Due to its hydrophilicity, high molecular weight and negative charge, mRNA has poor cellular uptake in its free form. The knowledge of the mRNA structure allowed the optimization of specific modifications in order to improve the stability of the molecule, increase the efficiency of translation, and reduce immunogenicity. However, it is still necessary to overcome several extra and intracellular barriers for the effective therapeutic use of RNA [38], as presented in Figure 2. Based on the requirements for therapeutic action, the structural components of the mRNA molecule can be modified [13,15,26] (Figure 1). Although mRNA holds great promise in therapy for numerous diseases, the main obstacle is the ability to specifically deliver it to target cells.
One of the limitations of the use of mRNA as therapeutics is its susceptibility to enzymatic degradation, particularly by ubiquitous RNases present in the blood and tissues. Indeed, the existence of these nucleases is a significant obstacle to the systemic application of non-encapsulated mRNA [13,15,17]. In addition to RNases, there are other barriers to be surpassed, such as the cell membrane. Naked mRNA does not have the necessary characteristics for its entry through the phospholipid bilayer, as this biomolecule has a negative charge and a large size and is hardly transported into the cell without its incorporation into a delivery system [15,17,29]. Moreover, in in vivo applications, intravenously administered naked mRNA is rapidly degraded and may even cause an immune response. A study by Islam and colleagues found that the half-life of naked mRNA was about 5 min after intravenous administration, with a significant decrease in serum levels to 10% after 5 min and to 1% after 1 h [39]. Thus, mRNA encapsulation by appropriate delivery systems is an essential requirement for this molecule to overcome those barriers. Adding specific ligands that can recognize receptors on cell membrane will allow the improvement of mRNA delivery into target cells and tissues, with the final aim of achieving an enhanced and specific transfection [17,23,26,30].
Different types of in vivo administration routes have been used for mRNA delivery [15]. As for any type of nucleic acids, mRNA delivery can be achieved using main approaches comprising (i) physical methods that temporarily disrupt the cell membrane; (ii) viral-based approaches that use a virus or viral components capacity for cell transfection, and (iii) non-viral nanosystems [26] represented in Figure 3.
Physical methods are simple and consist of employing a physical force to the membrane barrier to deliver genetic material. The therapeutic application of naked mRNA has mainly been studied ex vivo by physical methods, including electroporation and gene gun [38]. These methods have started to be tested to increase the efficiency of mRNA uptake in vivo, but an increased cell death and limited access to target cells/tissues have been pointed out as disadvantages. For example, RNA complexed with gold particles showed good expression in tissues using gene gun in mouse models, however, no efficiency data are available for larger animals or humans [40,41].
Viral vectors are genetically modified viruses containing genes of a virus partially or completely replaced by antigen-encoding RNA [42,43]. Different viruses, such as picornaviruses [44] and lentivirus [45] have been chosen for mRNA delivery, given their high transfection capacity. In 2014, Hamann and co-workers described foamy virus vectors that allowed an efficient and transient expression of various transgenes by packing and transferring non-viral RNAs (both in vitro and in vivo) [33]. However, the use of recombinant viruses entails inherent risks, such as potential risks of insertion into genome, difficulty in controlling gene expression, limitations in the size of the sequence of interest and the existence of strong immunological side effects. Moreover, viral vectors present high production costs as well as a low packaging capacity [46,47,48]. Due to the risks associated with viral vectors, there has been an increasing number of studies on non-viral delivery systems. These have already demonstrated their enormous potential for the delivery of various nucleic acids, such as pDNA, siRNA and miRNA [26] and explored for the delivery of mRNA [8]. Due to their biocompatible and diversified properties, in addition to their simple formulation with mRNA, with adequate and controlled release kinetics [26], non-viral vectors seem to be the most promising systems. Non-viral systems are mainly made up of synthetic or natural biocompatible components that form complexes with mRNA and vary in composition, physicochemical characteristics, shape, and size [43,49].
Different types of non-viral systems (polymeric, lipid and hybrid systems) have been developed to protect and improve the delivery of mRNA. [43]. One of the main advantages of polymeric systems is the possibility of their modification to adapt them to the active substance. Biodegradable and mainly cationic polymers are commonly used to develop nanoparticle formulations with nucleic acids, leading to the formation of polyplexes [42,43]. The gold standard of polymeric vehicle, Polyethylenimine (PEI) has been proposed for mRNA delivery but its use has been limited due to its toxicity [50]. New PEI derivatives were designed to improve their biocompatibility and transfection efficiency. For example, a study by Forrest and colleagues described that the toxicity of the PEI polymer is reduced by its conjugation with cyclodextrin [51]. Another example is jetPEI®, a linear PEI marketed as an in vivo mouse transfection reagent designed to improve its biocompatibility and transfection efficiency. This derivative was recently evaluated by Sultana research group, in the administration of mRNA by direct myocardial injection in mice, demonstrating the expression of the protein in the lungs [52]. Moreover, in 2016, Démoulins and co-workers incorporated mRNA encoding influenza virus hemagglutinin and nucleocapsid into PEI nanoparticles in order to improve mRNA vaccines efficacy which was confirmed by both humoral and cellular immune responses induction, in vivo [53]. In addition to PEI, there are other polymers under study. Zhao and colleagues developed a polyethyleneimine-stearic acid (PSA) copolymer to deliver mRNA encoding HIV-1gag into dendritic cells and BALB/c mice. With this polymeric system, and after its subcutaneous injection, Zhao group was able to detect specific antibody levels for anti-HIV-1gag [54]. Another example of a promising polymer is chitosan, a biocompatible cationic glycopolymer that showed ideal conditions for delivering mRNA encoding luciferase [55]. McCullough and colleagues also investigated whether chitosan was able to deliver the self-replicating replicon-RNA encoding the influenza virus hemagglutinin [56]. However, when biodegradable polymers, such as poly Lactic-co-Glycolic Acid (PLGA), are negatively charged, they cannot effectively encapsulate negatively charged mRNAs. Even so, this polymer, when hybridized with cationic lipids, already allows the encapsulation of mRNA [39].
Although widely studied for mRNA complexation, cationic polymers are not as clinically advanced as lipid systems for mRNA-based therapies [42,43]. Various synthetic and naturally derived lipids are used in mRNA delivery. Liposomes have an inherent advantage, as they are able to mimic the composition of the cell membrane, but they are not extensively used as the majority of lipoplex formulations cannot stand the presence of serum and mRNA is more unstable [42,57].
Currently, lipid nanoparticles (LNPs) are considered one of the most developed systems for mRNA delivery and are at the forefront of clinical trials [43]. Typically, LNPs are made up of an ionizable lipid, neutral/auxiliary lipid, cholesterol and PEG lipid. Ionizable lipids are lipids that become cationic at acid pH, allowing them to interact with mRNA through electrostatic interactions, leading to the formation of a complex called lipoplex, while at physiological pH, they have a neutral charge, highly reducing their toxicity [42,43]. Patisiran (ONPATTRO™), the clinically approved siRNA formulation against transthyretin mRNA is made of LNPs containing Dlin-MC3-DMA (MC3 ionizable lipid) [58]. Within LNPs, in addition to ionizable lipids, several studies demonstrate the use of cationic lipids for mRNA encapsulation [59]. N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammonium chloride (DOTMA) was the first synthetic cationic lipid used to complex in vitro transcribed (IVT) mRNA [60]. In addition to this, one of its derivatives, 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) has also been studied for mRNA delivery [61,62]. However, cationic lipids tend to reduce efficacy due to possible nonspecific interactions in vivo, in addition to possible inflammatory responses and toxicity [63] Initially, LNPs were considered promising siRNA delivery systems [64]. Several groups used this same ionizable lipid for LNP dedicated for mRNA delivery [65,66]. Pardi and co-workers report that a single intradermal immunization of LNP encapsulating nucleoside-modified mRNA encoding for the pre-membrane and envelope glycoproteins of a strain of Zika virus elicited potent and durable immune responses in mice and in non-human primates [67]. Moreover, Hekele et al. showed that mRNA encoding the HA antigen of the influenza virus H1N1, encapsulated in LNP made of 1,2-dilinoleyloxy-3-dimethylaminopropane, 1,2-diastearoyl-sn-glycero-3-phosphocholine, cholesterol and PEG-DMG 2000, rapidly induced immune responses in mice [68].
LNPs are now considered as the gold standard of mRNA formulation as they are currently used to prepare COVID-19 vaccines from both Moderna and Pfizer/BioNTech (discussed in Section 6.2).
In order to take advantage of the various delivery systems, the hybrid system, that is, the combination of different materials, such as polymers, lipids, among others, offers greater functionality and flexibility than isolated systems. Lipopolyplexes are an example of such a hybrid system in which the advantages of cationic polymers and lipids for the complexation of nucleic acids are combined [43,69]. The first example of this hybrid system is made of polyethylene glycol (PEG)ylated derivative of histidylated polylysine and L-histidine-(N,N-di-n-hexadecylamine)ethylamide liposomes reported by Mockey and collaborators [70] demonstrated that intravenous injection of MART1 mRNA encapsulated by this lipopolyplex induced a specific vaccination of mice with significant protection against B16F10 melanoma tumor progression. In addition to these most used delivery systems, others have also been explored, such as polypeptides [71,72,73,74], and mineral-coated microparticles [75]. CureVac has developed the RNActive® technology which makes use of protamine as an mRNA delivery system. This is an mRNA vaccine platform based on protamine/mRNA complexes, which is currently under clinical evaluation [72,73,74], and at pre-clinic stage against influenza virus [71]. In 2016, in phase I clinical trials, Kranz and co-workers showed that RNA-lipoplexes, without functionalization of the particles with specific ligands, could precisely target dendritic cells, just by adjusting its net charge. This particular study can be the basis of the development of a class of vaccines for dendritic cells targeting cancer immunotherapy [76]. In 2020, Mai and co-workers developed a cationic liposome/protamine complex (LPC) for nasal administration of mRNA vaccines. In vitro, LPC containing RNA encoding cytokeratin 19 presented significantly greater efficiency in the uptake of vaccine particles and provoked a strong immune response and slowed tumor growth in an aggressive lung cancer model [77].
Regardless of the type of transfection, in vitro (ex vivo) or in vivo, mRNA is translated directly into a functional form, promoting a faster and easier response and therapeutic application [17].
6. mRNA Therapies
Currently, mRNA appears as a very promising and innovative therapeutic approach for diseases associated with functional loss of proteins, through the administration of a synthetic mRNA, which promotes the reestablishment of protein levels and restores its function [17]. Moreover, mRNA can create new cellular functions, for example for passive immunization [13,23], allowing to stimulate the immune system, through the translation of antigenic mRNA for specific cell recognition (e.g., cancer cells) or antibody production [15,24]. The fact that a relatively small amount of encoded antigen, from a synthetic mRNA, can be sufficient to obtain robust signs of efficacy, is one of the main advantages of using this biomolecule in immunotherapy [26]. However, the global success of such mRNA-based treatments depends on a high number of these biomolecules and an effective in vivo delivery to target cells involved in a given disease [13,23,30]. After proving that in vivo mRNA administration is possible and viable, the concept of using mRNA as a therapeutic basis was readily accepted and used [11,13] in a variety of diseases, including diabetes, HIV infection, anemia, hemophilia, myocardial infarction, cancer, asthma, metabolic disorders, fibrosis, skeletal degeneration and neurological disorders, such as Friedreich’s ataxia and Alzheimer’s disease [1,11,17].
Initial mRNA therapies centered its use against cancer, transferring this biomolecule ex vivo and then promoting a localized delivery in vivo (Figure 4). Currently, the most advanced therapies using mRNA are in the clinical testing phase (see Section 6.2). Unlike cancer vaccines and immunotherapies, most protein replacement therapies that use mRNA are still in preclinical development [15].
6.1. Protein Replacement Therapies
Protein replacement therapies performed by mRNAs have enormous potential for treating a wide range of diseases [1,8,17,29] (Table 2).
Application of IVT mRNA for protein replacement therapies relies on the supplementation of proteins that are under-expressed or not functional, as well as on the expression of foreign proteins that can either activate or inhibit certain cellular pathways. Therapies based on mRNA have become more attractive because, contrarily to DNA, mRNA does not enter the nucleus of host cells, and therefore does not present a risk of genome integration or mutagenesis. There are different applications of this type of therapy including genetic and rare diseases [43,78,79]. mRNAs are generally designed to express therapeutic proteins, in such a way as to exhibit no or low immunogenicity, to have prolonged stability and high translation efficiency [8]. Most mRNA-based protein replacement therapies are targeted at certain organs, such as liver, lungs and heart, mainly because currently existing methods are more efficient for the delivery of mRNA to these tissues. The use of this therapy in other organs and cell types requires the development of new delivery strategies, active targeting or different methods of administration [8,29,30].
An example of protein replacement using IVT mRNA is a study performed by Baba and co-workers. In this work, mRNA has shown to be promising in treating neurological disorders by providing proteins and peptides in their native and mature form in neural cells. By using novel mRNA-loaded nanocarriers and carrying out administration through nasal route into mouse models, there was a sustained protein expression for almost two days in nasal tissues. Moreover, upon daily intranasal administration, neurological recovery of olfactory function was enhanced, as well as recovering almost to a nearly normal structure of the olfactory epithelium [80]. In 2017, Ramaswamy and co-workers successfully delivered mRNA through LNP in order to treat a Factor IX (FIX)-deficient mouse as a model of hemophilia B. This study showed that repeated administration of the mRNA-LNPs complex did not cause innate immune responses in hemophilic mice [78]. In 2018, Magadum’s team verified that modified mRNA (modRNA) could induce cardiomyocytes (CMs) proliferation and regeneration by upregulating mutated human follistatin-like protein 1 (hFSTL1). The post-translational modification was hypothesized to be responsible for CM regeneration in vitro with no indications of cardiac hypertrophy. Furthermore, it significantly improved cardiac function, decreased scar size, and also increased capillary density, showing the effectiveness of modRNA in CM proliferation and cardiac regeneration [81]. The same authors, in 2020, also showed that modRNA can induce CM cell cycle by upregulating the glycolytic enzyme pyruvate kinase muscle isoenzyme 2 (Pkm2). This increased expression of the enzyme contributed to re-enforcing the CM cell cycle, which led to cell division and consequently cardiac regeneration [82].
6.2. Immunotherapy
In addition to protein replacement therapy, another branch is immune stimulation against certain diseases. Interleukin 15 (IL-15) cytokine presents a therapeutic anticancer potential, mainly for its immunologic stimulation properties. However, currently used delivery systems with pDNA present low efficiency, and the use of in vitro transcripts could be a better solution. For this, Lei and co-workers, in 2020, verified that through cytokine expression with this mRNA, lymphocyte stimulation was successfully produced and cytotoxicity was triggered in cancer cells. Local or systemic administration of this mRNA induced inhibition of cellular proliferation in several colon cancer models in a safe and efficient way. These results have shown the high therapeutic potential for colorectal cancer immunogenic therapy with this approach [83]. In the same context, Interleukin 2 (IL-2) exerts significant anti-tumor activity. This cytokine is involved in proliferation, differentiation and effector function of T cells and since 1998, it has been approved for the treatment of metastatic melanoma [84]. However, using IL-2 cytokine faces several limitations including the short serum half-life. For this, the use of mRNA expressing IL-2 would prolong the production of the cytokine, thus reducing high and frequent doses. Currently, two nucleoside-modified mRNA LNP encoding for IL-2 are in clinical development for cancer treatment (see review [85]).
Vaccines have been used to provide adequate, specific and short-term immune responses against infectious diseases or cancer. Conventional vaccines consisting of attenuated microorganisms or that contain the majority of virus or bacterial antigens have demonstrated lasting protection against a variety of infectious pathogens, but on rare occasions they can revert to their pathogenic forms [1,8]. More and more epidemic outbreaks are caused by viral infections and in all cases, those are characterized by their unpredictability, high morbidity, exponential spread, and substantial social and economic impact [86]. mRNA vaccines, on the other hand, cannot replicate within the body [1]. Thus, mRNA vaccines have been deeply investigated due to their ability to encode a wide range of antigens, due to the self-adjuvant effects [8,23], as well as for their potential large-scale production in a fast, flexible and low-cost manner [8,23,86]. The development of an mRNA vaccine for specific antigen immunity requires the transfection of antigen-presenting cells, such as dendritic cells [1,8,87], resulting in the induction of humoral and cytotoxic T-cell response [86] which is represented in Figure 5. Because of this, the administration is typically performed by intradermal, intramuscular or subcutaneous injection, as dendritic cells are densely found in skeletal muscle and skin tissue [1,8,29]. In addition to mRNA, DNA was also used for the coding of antigens, but due to the potential for integration into the genome, its use was rather limited [8,23].
In contrast to mRNA, antibody-based cancer therapy faces some challenges related to antibody production problems, low stability in long-term storage, aggregation, and the presence of several impurities intrinsic to the production process. In addition, antibodies, especially bispecific antibodies, have a low serum half-life and continuous administration is required to achieve the therapeutic effect [23]. Thus, the use of mRNA for the generation of therapeutic antibodies in patients represents a promising approach, in order to overcome the limitations of direct use of recombinant antibodies [23].
The development of an mRNA vaccine consists of acquiring genetic information of the infectious agent or the sequence of antigens associated with the tumor. Then, the gene is sequenced, synthesized and cloned into a plasmid. The mRNA is transcribed in vitro and the vaccine is administered to the patient [86]. The mRNA vaccine uses the host cell machinery to translate the corresponding antigen mRNA sequence, thus mimicking the infection or a tumor cell, in order to elicit humoral and cytotoxic immune responses [86,88]. The use of mRNA to induce adaptive immune responses in cancer (examples of vaccines presented in Section 6.2) began in 1995, with the discovery of protective antitumor immunity, which was obtained by intramuscular injection of mRNA from the carcinoembryonic antigen [89]. There are two main types of mRNA immunotherapy against cancer. The first type of immunotherapy works at the cellular level, in the same way as an mRNA vaccine, however, the mRNA encodes tumor-associated antigens. The second type, on the other hand, involves the modification of T cells, with chimeric antigen receptors (CARs), which is called CAR T cell therapy. Billingsley and collaborators demonstrated the C14-4 LNP induced CAR expression at levels equivalent to electroporation, with a substantially reduced cytotoxicity. When compared to electroporated CAR T cells by the lipid system, C14-4 LNP, in a coculture assay with Nalm-6 acute lymphoblastic leukemia cells, Billingsley and collaborators found that both methods induced a strong cancer-killing activity [90]. These results obtained by Billingsley research group show the progress that has been mad and the promising strategies to deliver mRNA to T cells. Usually, in this class of immunotherapy, the patient’s T cells are transfected with synthetic mRNAs, encoding CARs, bind to specific tumor antigens, subsequently eliminating the tumor cells [1].
Since then, mRNA vaccines have been classified into two subtypes: (i) non-amplifying mRNA-based vaccines (also known as mRNA conventional vaccines), that encode the antigen of interest and contain the 5′ and 3′ UTRs [88]; and (ii) self-amplifying mRNA (SAM or saRNA) vaccines [8,13,86,91] that not only encode the antigen, but also the viral replication mechanism, allowing an increase in the amount of intracellular mRNA, consequently leading to a more abundant protein expression [88] (Table 3 and Figure 6). Both types of mRNA vaccines use the translation mechanism of host cells to produce target antigens, in order to induce specific adaptive immune responses [8]. In 2020, He and co-workers developed cationic nanolipoprotein particles (NLPs) to enhance the delivery of large self-amplifying mRNAs (replicons) in vivo. These cationic lipids successfully encapsulated RNA encoding luciferase, protected it from RNase degradation and promoted replicon expression in vivo [92].
The greatest barrier to the usefulness of these vaccines is the need for intracellular delivery [8,13]. However, as already mentioned, through chemical modifications, encapsulation by nanoparticle formulations and through sequence engineering, it is possible to promote an improved targeting, delivery and entrance into the cell, in addition to greater efficiency in translation and enhanced half-life of synthetic mRNA vaccines [8]. Chronologically, mRNA vaccines in dendritic cells for adaptive immunotherapy against cancer and protein replacement therapies were the first therapeutic applications with these biomolecules to enter clinical trials [8,13]. Although therapies based on dendritic cells still represent the majority of clinical trials of mRNA vaccines, vaccination with the use of this biomolecule through non-viral vectors, as well as gene editing, is increasingly being investigated in search of new therapies against diverse diseases [1,8] (Table 4).
The most recent case of immunotherapy associated to mRNA vaccination, in clinical trials, was registered in 2020 and concerns the virus named “Serious Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)”, the former being known as Coronavirus (COVID-19 or 2019-nCoV) [112,113]. SARS-CoV-2 causes an infection in the alveolar epithelial cells of the human respiratory tract [114,115]. SARS-CoV-2 has a large genetic structure. The genome is surrounded by helical nucleocapsid proteins (N) and an outer envelope composed of matrix or membrane glycoproteins (M), envelope proteins (E) and spike glycoproteins (S) (Figure 7), which improve binding to cells, transport and interfere with the immune response of the host [91,116,117]. In addition, the virus has several non-structural proteins (NsPs) that are vital for its life cycle and pathogenic character [91].
The S glycoprotein is part of the outer layer of the virus and is essential for its entry into cells [118]. This protein consists of a receptor binding domain (RBD), that is responsible for specific binding to the angiotensin-converting enzyme 2 (ACE2) receptor, thus allowing the entry of SARS-CoV-2 [114] in the epithelium cells of human lung [119]. In addition, there are studies that indicate that SARS-CoV-2, as well as SARS-CoV, can enter the cell through clathrin-mediated endocytosis [115]. Of all structural proteins, it was found that S glycoprotein induces neutralizing antibodies and it was the main target antigen for the development of the vaccines [117,120].
Given the high transmission of SARS-CoV-2, the World Health Organization (WHO) emphasized the demand for a rapid response to this situation, endeavoring the immediate development of safe and effective prophylactic therapies [112,121]. Due to the great technological advances in sequencing techniques, it was possible to obtain colossal knowledge about SARS-CoV-2 in a very short time, something unprecedented in the history of medicine [113,118].
Vaccines decrease the viral spread and transmission from person to person [88,91], and the development of the SARS-CoV-2 mRNA vaccine was impressively fast [122]. The classical development of vaccines requires an average of about 5 to 10 years, but given the need and technological advances, the development time of the vaccine against SARS-CoV-2 was substantially shorter [88,113].
The approved mRNA vaccines to combat SARS-CoV-2, the vaccines developed by Moderna/NIAID and BioNTech/Fosun Pharma/Pfizer, aim at the expression of the S glycoprotein or RBD subunit [88,91,123]. After transfection of either muscle cells or dendritic cells, the expressed S glycoproteins are presented by the major histocompatibility complex (MHC) class I and II [113]. This process stimulates humoral immunity and leads to the production of neutralizing antibodies against the S glycoprotein by B lymphocytes, preventing viral binding and entry into cells [122] represented in Figure 8. It also induces the generation of specific cytotoxic T cells (CD8+) which can eradicate SARS-CoV-2-infected cells [124,125].
To date, the Pfizer/BioNTech (Comirnaty) vaccine presents 95% of efficiency, while the Moderna (Spikevax) vaccine has 94.5%, Gamaleya product has 92% and the AstraZeneca vaccine, 70% [126,127]. The first two (Pfizer/BioNTech and Moderna) are RNA vaccines that express COVID-19 spike glycoprotein, while the Gamaleya and AstraZeneca vaccines express spike protein from adenovirus vector platforms [128,129].
Both Moderna and Pfizer/BioNTech vaccines are made of a nucleoside-modified (N1 methyl pseudouridine) mRNA formulated in LNPs. These LNPs contain an ionizable lipid, neutral/auxiliary lipid ((phospholipid distearoylphosphatidylcholine) (DSPC)) at physiological pH and cholesterol, which allows to stabilize LNP and increase the efficiency of mRNA delivery, and finally a polyethylene glycol (PEG), which aims to improve colloidal stability, reducing opsonization by plasma proteins. However, these differ in the use of the ionizable lipid, as the Pfizer/BioNTech vaccine used the ionizable lipid ALC-0315 and Moderna vaccine used another ionizable lipid, the SM-102 (Table 5). Although both vaccines use ionizable lipids, both are tertiary amines that are protonated at a low pH, thus allowing for mRNA interaction and protection [130,131] (Table 5). These specific LNPs are therefore essential for a safe and efficient immune response. The mRNA encodes the membrane-anchored, full-length SARS-CoV-19 spike protein and contains mutations for the prefusion conformation, which stabilize the Spike protein.
The LNPs prevent RNA degradation and enable its delivery into host cells after intramuscular injection. Once inside the host cells, mRNA is translated into SARS-CoV-2 spike protein. The expression of this spike antigen induces neutralizing antibodies, as well as cellular immune responses against it, which can confer protection against COVID-19 [132]. The Pfizer/BioNTech vaccine has been recommended to people older than 12 years old, with a dose of 30 μg (0.3 mL) at a cost of $19.50 in the US. Recently, FDA issued emergency use authorization in individuals 5 years of age and older [133]. It consists of a two-dose administration with 21 days between each administration, providing immunogenicity for at least 119 days after the first vaccination and is 95% effective at preventing the SARS-CoV-2 infection. However, the Moderna Vaccine (mRNA-1273) has been recommended to people of or above 18 years of age, with a dose of 50 μg (0.5 mL) at a cost ranging from $32 and $37, in the US [134]. Similarly, it also consists of two shots administered 28 days apart providing immunogenicity for at least 119 days after the first vaccination and is, as referred above, 94.5% effective in the prevention of SARS-CoV-2 infection. It should be noted that age-dependent administration is region-specific, and can vary in different countries [135,136]. Given this, it is safe to say that both vaccines are beneficial in providing immunity against SARS-CoV-2 infection, nevertheless, some allergic responses have been reported. These COVID-19 vaccines can cause mild adverse effects after the first or second dose, including pain, redness, swelling or itching (at the site of vaccine injection), fever, fatigue, headache, muscle pain, nausea, and rarely cause anaphylactic shock. The Pfizer/BioNTech vaccine reports a lower percentage of these adverse effects comparatively with the Moderna vaccine. However, the Moderna vaccine is easier to transport and store (storage between −25 °C and −15 °C) because it is less sensitive when compared to the Pfizer vaccine (stored between −80 °C and −60 °C) [127,137,138].
CVnCoV (CureVac) consists of LNP-encapsulated non-chemically modified mRNA with naturally occurring nucleotides encoding for a full-length S protein that includes two proline mutations (S-2P), which was previously showed to stabilize the conformation of the S proteins for SARS-CoV. The mRNA was codon-optimized in order to provide a higher expression level of S protein and a moderate activation of the immune system [139]. The CureVac vaccine can be distinguished from the previous two candidates by exclusively consisting of non-chemically modified nucleotides and can be applied at comparatively lower doses (12 μg). CureVac company announced preliminary data on 16 June (from a 40,000—person trial), that its two-dose vaccine was only 47% effective at preventing COVID-19, which is half of the efficiency of its previous two rivals. It was expected that this third vaccine candidate would be cheaper and last longer in refrigerated storage than the earlier mRNA vaccines made by Pfizer/BioNTech and Moderna. However, it is suspected that CureVac’s decision not to exchange the biochemical structure of its mRNA, as Pfizer/BioNTech and Moderna did, might be the reason for its poor performance [139,140].
It should be noted that clinical application of mRNA as a therapeutic agent has some limitations due to its instability and the capacity to activate the immune system. Therefore, modifying the in vitro transcribed mRNA structure alongside with the design of suitable nanoparticles is of great importance [43]. This fact comes to be noticed because Pfizer/BioNTech and Moderna vaccines call upon modified RNA, by replacing uridine itself for another nucleotide called pseudouridine (Ψ), which is similar to uridine but contains a natural modification. This modification in exogenous mRNA is thought to decrease inflammatory reactions, while improving translational efficiency and stability. In contrast to Pfizer/BioNTech and Moderna vaccines, CureVac uses normal uridine instead of Ψ, which could be a reason for its poor success once higher doses reflected more severe adverse effects [19,141,142,143]. These improved properties conferred by the incorporation of Ψ make mRNA a promising tool for both gene replacement and vaccination. The innate immune system cells are activated by RNA, since it stimulates Toll-like receptors (TLRs), namely TLR3, TLR7, and TLR8. However, when some modified nucleosides, like, Ψ, 5-methylcytidine (m5C), N6-methyladenosine (m6A), 5-methyluridine (m5U), or 2-thiouridine (s2U) were included into the transcript, most of TLRs were no longer activated, therefore controlling immune activation in vitro and in vivo [19]. These characteristics and the readiness of producing such RNAs by in vitro transcription make Ψ-containing mRNA an important tool for the expression of any protein [144,145,146,147]. Furthermore, codon optimization strategies have been investigated to improve the cost efficiency of recombinant protein production, once most amino acids are encoded by different codons. This is primarily based on the substitution of multiple rare codons by others more frequent, that encode the same amino acid, thus resulting in increased rate and efficiency of protein translation [148]. Another successful modification is the addition of a poly(A) to IVT produced mRNA, which can be directly added during the transcription process (if the DNA template encodes the poly(T) sequence) or can be added post-transcriptionally by enzymatic reactions. Poly(A) tail length influences stability and translation efficiency. Even with a relatively long poly(A) tail that seems to be appropriate, the optimal length can vary depending on the target cell [43,149]. The Kozak sequence plays a major role in the initiation of the translation process and is located at the 5′ UTR. This sequence, defined as “RCCAUGG”, where “R” stands for a purine (A or G), helps drive high levels of translation from the correct start codon, therefore being considered the election sequence for translation initiation in eukaryotes [43,147]. The Pfizer/BioNTech vaccine included a poly(A) chain in their mRNA sequence, as well as an optimized Kozak sequence [147].
Conventional vaccine approaches, such as the use of attenuated and inactivated viruses, successfully provide durable protection against infectious diseases, but they are not able to meet the need for rapid and large-scale development. As already mentioned, although genetic immunization, such as DNA vaccines, has shown to be promising, pDNA delivery raises safety concerns due to the possibility of insertional mutagenesis. Thus, in order to try to obtain a vaccine quickly, safely and effectively, the development of an mRNA vaccine seems to be a reliable approach. This is a safer alternative, as it does not require entry into the nucleus for translation to occur, leading to an improvement in transfection and expression efficiency compared to DNA vaccines. It also presents comparatively lower production costs and capacity for rapid development, because with a simple change of the mRNA sequence, it will lead to the expression of a different protein, which is beneficial given the frequent viral mutations [91,112]. There are currently eight mRNA-based vaccines in clinical development and 22 in pre-clinical studies (Table 6) [150].
6.3. Gene Editing
As previously mentioned, mRNA therapies may also function in gene editing, which can be achieved by encoding nucleases from mRNA for cellular reprogramming [1,13]. Gene editing involves the precision of “cutting” and “pasting” genomic DNA in specific locations, expecting the establishment of a potentially permanent cure for genetic diseases to be a promising therapeutic area for the application of mRNA technology [1,8]. In this therapeutic area, mRNA function is to express programmable nucleases, including zinc finger nucleases (ZFNs), transcription activator effector nucleases (TALENs) [8,13] or CRISPR-Cas9 [1,8]. These genetic engineering tools allow the replacement or modification of gene expression, through the introduction or local deletion of specific modifications in the genome of target cells [8]. This allows the correction of a target gene, by deleting disease-causing mutations or by inserting protective mutations by joining the non-homologous end (NHEJ) [151,152] or even performing a repair or insertion directed to homology (HDR) [151,152,153]. This is schemed in Figure 9. ZFNs and TALENs facilitate the recognition of a sequence by protein–DNA interactions [8,13], however, the complex engineering necessary to create specific domains in proteins directed to DNA recognition and binding greatly restricts its wide application. However, the CRISPR-Cas9 system in Figure 9, is currently the most widely used and characterized gene editing technology [1,8].
In 2020, Jennifer Doudna and Emmanuelle Charpentier won the 2020 Nobel Chemistry Prize for their discovery of a novel and innovative gene-editing technique. CRISPR-Cas9 gene-editing tools allow precise editing of the genome and have countless applications, which scientists aim to use to alter human genes to eliminate diseases and eradicate pathogens. CRISPR-Cas9-mediated gene editing requires only two components: Cas9, a nuclease responsible for DNA cleavage and a short single-stranded RNA guide (sgRNA), which directs DNA cleavage by the nuclease, precisely. Typically, these two components are delivered to cells using a pDNA containing the Cas9 protein and sgRNA genes [1,153]. For more information on the mechanism of action of the CRISPR-Cas9 system, the following literature can be analyzed [151,152,153]. The use of CRISPR-Cas9 technology had only been used to edit the genomes of embryos, zygotes, and cultured cells [154,155], however, this technology has been increasingly used in vivo.
Due to the transient nature of mRNA, the use of this biomolecule can be advantageous in relation to the use of pDNA [1], limiting the presence of nucleases inside cells [13]. In this way, there is a reduction in possible non-specific cleavages which decreases the immune response to the Cas9 protein. In addition to these advantages, it appears that the intracellular presence of the Cas9 protein has been more persistent after mRNA expression compared to the administration of the Cas9/sgRNA ribonucleoprotein complex (Cas9-RNP) [1,8]. As such, co-delivery of mRNA, which encodes Cas9, and sgRNA is an attractive alternative [1]. Cas9 can be administered as mRNA, plasmid DNA or even as a protein. However, for plasmid DNA Cas9 to be functional it must overcome cell and nuclear membrane barriers. Thus, an alternative approach could be the use of Cas9 mRNA. This approach becomes preferable as mRNA only needs to cross the cell membrane to be functional. Liang and collaborators, using the GeneArt commercial system (Thermo) and electroporation, found that in the study of eleven cell lines, the delivery of Cas9 mRNA/gRNA or Cas9 RNPs was superior to the plasmid delivery in all cell lines tested. They also noticed that although the similar cleavage kinetics between Cas9 delivered as plasmid DNA, mRNA and protein to HEK293 cells, in cells transfected with plasmid DNA, the Cas9 protein accumulated over time, while the relatively low expression of Cas9 in mRNA-transfected cells seemed relatively stable for approximately 48 h. However, due to the fast turnover of Cas9 RNP and mRNA compared to the long persistence of Cas9 expressed from plasmids, this could reduce the opportunity for off-target binding and cleavage. When studying this among the six potential off-target sites, it was observed that the use of mRNA and Cas9 RNP had much smaller off-target effects than the use of Cas9 from plasmid DNA [156]. In addition to the use of mRNA in gene editing for the treatment of acquired diseases, Mohsin and colleagues demonstrated by in vitro experiments that Cas9 mRNA/sgRNAs can reduce the sporulation percentage of E. tenella oocysts, as well as their survival rate. These data show that the use of a highly specific sgRNA molecule, when combined with Cas9 mRNA, may be a potentially powerful agent in the development of new therapeutic drugs against parasitic diseases [157]. It should be reinforced that for long-term gene therapy purposes, mRNAs are not sufficiently stable; nevertheless, even transient articulation and ability will make hereditary change perpetual for the activity of Cas9 nuclease. This is the reason why Cas9 mRNA is commonly used, for example in Drosophila, zebrafish, Xenopus and mouse, in both cell culture and model organisms [158,159,160,161]. In addition to the use of non-viral systems, Ling and colleagues found effective and successful delivery using a viral system. These authors used mLP-CRISPR, a lentiviral system that delivers mRNA encoding one of the longest Cas9 proteins (SpCas9) and gRNA simultaneously. By targeting vascular endothelial growth factor A (Vegfa), it was found that with only a single sub injection-retinal of mLP-CRISPR in mouse models, 44% of Vegfa in retinal pigment epithelium was knocked out and the area of choroidal neovascularization was reduced by 63% without inducing off-target edits or anti-Cas9 immune responses [45]. Although CRISPR-Cas9 is most used to control over-expression levels of a particular protein, Qiu and colleagues verified the knockdown of the Angiopoietin-like 3 (Angptl3) gene in a specific and efficient way using the system CRISPR-mRNA Cas9, which led to a significant reduction in serum Angptl3 protein, low-density lipoprotein cholesterol (LDL-C), and triglycerides (TG) levels in wild-type C57BL/6 mice [162], presenting similar results to studies with antisense oligonucleotides [163]. They also verified that the therapeutic effect of genome editing was stable for at least 100 days after the single dose administration [162]. Wang and collaborators demonstrated that CRISPR/Cas9 mRNA-mediated gene editing technology allowed the simultaneous disruption of five genes in mouse embryonic stem cells (ES) with high efficiency, thus verifying that with Cas9 mRNA co-injection and sgRNAs targeting Tet1 and Tet2 in zygotes achieved mutations in both genes with an efficiency of 80% in mice with biallelic mutations [159]. This discovery not only allows us to verify that the CRISPR/Cas9 technology allows mutations in several genes simultaneously, but it will also greatly accelerate the in vivo study of functionally redundant genes and epistatic gene interactions.
Since the discovery of CRISPR-Cas9, the CRISPR revolution has expanded beyond its original use as a genetic engineering tool. New Cas nucleases are being developed to enable faster and more accurate molecular diagnostic platforms for use with next-generation bio-sensing platforms. Abbott and co-workers showed, through bioinformatic analysis, that some different CRISPR-associated RNAs (crRNAs) were able to target over 92% of live influenza A virus strains and over 91% of all coronaviruses. This fact expands CRISPR-Cas13 systems applications beyond diagnostics, such as SHERLOCK, and live-cell RNA imaging [164]. In this context, Cas 13 is an endonuclease that targets and binds sg RNA. Moreover, it demonstrates RNA cis-cleavage activity when activated by a target RNA in different model organisms [165] and is more effective and specific than RNAi in mammalian cells [166,167]. For this purpose, CRISPR-Cas13 has been proposed and used as a tool for SARS-CoV-2 detection [168]. Furthermore, Blanchard and co-workers found that using CRISPR/Cas13a mRNA specific for highly conserved regions of influenza virus and SARS-CoV-2, efficiently degraded influenza RNA in lung tissue when administered after infection, while in hamsters, Cas13a reduced replication of SARS-CoV-2 and reduced the symptoms [169]. Lastly, a promising example for CRISPR-Cas13 application is the study of Rashnonejad and co-workers against Facioscapulohumeral muscular dystrophy. They developed different Cas13b-gRNAs that target various Double Homeobox 4 (DUX4) mRNA parts and verified a decrease over 90% of DUX4 protein in treated cells. Moreover, cell viability was improved, as well as cell death prevention in vitro and in vivo [170]. Overall, the applications of the CRISPR-Cas13 in diagnostics are of interest and will open up new avenues for their in vivo applications, such as RNA knockdown and editing.
7. Conclusions and Future Directions
mRNA holds significant promise in gene therapy, in the control of emerging pandemic infectious diseases, and in diseases where no effective cure or treatment is available, avoiding several problems associated with therapies based on DNA or even based on proteins produced recombinantly. Applications such as immunotherapy and gene editing require protein expression only for limited periods of time, so transient protein expression by mRNA facilitates a wide range of biological processes without the risk of genomic integration. The main factors that prevent the clinical progression of mRNA therapy in chronic diseases are the transient expression and immunogenicity of the mRNA produced by IVT, in addition to the lack of sufficiently effective delivery systems to perform an effective transfection [1]. Although there are still limitations to the use of this biomolecule, over time these barriers have been overcome, through better knowledge of the biomolecule, modifications that can improve mRNA stability and technological evolution on the formulation of new, efficient, safe, and specific administration systems [29]. The development of a targeted non-viral delivery system for mRNA will be the culmination of years of research into therapies using this biomolecule. Finding the best targeting strategy is still the holy grail of nanomedicine. Indeed, vaccination using mRNA vaccines has enormous potential over conventional vaccines. This option offers benefits both in the speed of production and in the cost for developing the mRNA vaccine. Currently, mRNA-based vaccines promise to become the future and revolutionize the world of vaccination for therapeutic and prophylactic applications. The approval of the new mRNA vaccines for COVID-19 provides very good perspectives on the technology. It was an amazing achievement for the mRNA community. That said, the therapeutic use of mRNA has a huge potential to revolutionize medicine as we know it.
Author Contributions
Conceptualization, B.B., C.P. and F.S.; validation, C.P. and F.S.; investigation, B.B., R.C., N.L., C.P. and F.S.; writing—original draft preparation, B.B., R.C. and N.L.; writing—review & editing, B.B., R.C., N.L., C.P. and F.S.; supervision, C.P. and F.S.; project administration, C.P. and F.S.; Funding acquisition, C.P. and F.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Portuguese Foundation for Science and Technology (FCT), through the project UIDB/00709/2020, and by the project PTDC/BII-BBF/29496/2017 (PUREmiRSILs) funded by FEDER, through COMPETE2020—Programa Operacional Competitividade e Internacionalização (POCI), and by national funds (OE), through FCT/MCTES and FEDER EX010229 (C. Pichon). N. Laroui is funded by the European commission via FEDER EX010229.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
Carapito R. acknowledges FCT for the Ph.D fellowship (2020.07980.BD). Baptista B. acknowledges a fellowship within the project PUREmiRSILs and the fellowship BID/ICI-FC/CICS/Santander Universidades-UBI/2021, within the Santander Universidades-UBI/2021 Protocol.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hajj, K.A.; Whitehead, K.A. Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2017, 2. [Google Scholar] [CrossRef]
- Pierce, B. Genetics: A Conceptual Approach, 6th ed.; W.H. Freeman and Company: New York, NY, USA, 2017. [Google Scholar]
- Nguyen, T.C.; Zaleta-Rivera, K.; Huang, X.; Dai, X.; Zhong, S. RNA, Action through Interactions. Trends Genet. 2018, 34, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Saldi, T.; Riemondy, K.; Erickson, B.; Bentley, D.L. Alternative RNA structures formed during transcription depend on elongation rate and modify RNA processing. Mol. Cell 2021, 81, 1789–1801.e5. [Google Scholar] [CrossRef]
- Lehman, N. RNA in evolution. Wiley Interdiscip. Rev. RNA 2010, 1, 202–213. [Google Scholar] [CrossRef]
- Chujo, T.; Yamazaki, T.; Hirose, T. Architectural RNAs (arcRNAs): A class of long noncoding RNAs that function as the scaffold of nuclear bodies. Biochim. Biophys. Acta (BBA)—Bioenerg. 2016, 1859, 139–146. [Google Scholar] [CrossRef]
- Bicknell, A.A.; Ricci, E.P. When mRNA translation meets decay. Biochem. Soc. Trans. 2017, 45, 339–351. [Google Scholar] [CrossRef]
- Kowalski, P.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [Green Version]
- Kornberg, R.D. The molecular basis of eukaryotic transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 12955–12961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detke, S.; Stein, J.; Stein, G. Synthesis of histone messenger RNAs by RNA polymerase II in nuclei from S phase HeLa S3cells. Nucleic Acids Res. 1978, 5, 1515–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlake, T.; Thess, A.; Thran, M.; Jordan, I. mRNA as novel technology for passive immunotherapy. Cell. Mol. Life Sci. 2018, 76, 301–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Mc Cafferty, S.; Combes, F.; Huysmans, H.; De Temmerman, J.; Gitsels, A.; Vanrompay, D.; Catani, J.P.; Sanders, N.N. mRNA therapeutics deliver a hopeful message. Nano Today 2018, 23, 16–39. [Google Scholar] [CrossRef]
- Granot-Matok, Y.; Kon, E.; Dammes, N.; Mechtinger, G.; Peer, D. Therapeutic mRNA delivery to leukocytes. J. Control. Release 2019, 305, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Mao, Y.; Ji, Q.; Dersh, D.; Yewdell, J.W.; Qian, S.-B. Decoding mRNA translatability and stability from the 5′ UTR. Nat. Struct. Mol. Biol. 2020, 27, 1–8. [Google Scholar] [CrossRef]
- Trepotec, Z.; Lichtenegger, E.; Plank, C.; Aneja, M.K.; Rudolph, C. Delivery of mRNA Therapeutics for the Treatment of Hepatic Diseases. Mol. Ther. 2019, 27, 794–802. [Google Scholar] [CrossRef] [Green Version]
- Magadum, A.; Kaur, K.; Zangi, L. mRNA-Based Protein Replacement Therapy for the Heart. Mol. Ther. 2019, 27, 785–793. [Google Scholar] [CrossRef] [Green Version]
- Sahu, I.; Haque, A.K.M.A.; Weidensee, B.; Weinmann, P.; Kormann, M.S.D. Recent Developments in mRNA-Based Protein Supplementation Therapy to Target Lung Diseases. Mol. Ther. 2019, 27, 803–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grozhik, A.V.; Jaffrey, S.R. Distinguishing RNA modifications from noise in epitranscriptome maps. Nat. Chem. Biol. 2018, 14, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Eichhorn, S.W.; Subtelny, A.O.; Kronja, I.; Kwasnieski, J.C.; Orr-Weaver, T.L.; Bartel, D.P. mRNA poly(A)-tail changes specified by deadenylation broadly reshape translation in Drosophila oocytes and early embryos. eLife 2016, 5, e16955. [Google Scholar] [CrossRef] [PubMed]
- Wiederhold, K.; Passmore, L.A. Cytoplasmic deadenylation: Regulation of mRNA fate. Biochem. Soc. Trans. 2010, 38, 1531–1536. [Google Scholar] [CrossRef] [Green Version]
- Dreyfus, M.; Régnier, P. The poly (A) tail of mRNAs: Bodyguard in eukaryotes, scavenger in bacteria. Cell 2002, 111, 611–613. [Google Scholar] [CrossRef] [Green Version]
- Schlake, T.; Thran, M.; Fiedler, K.; Heidenreich, R.; Petsch, B.; Fotin-Mleczek, M. mRNA: A Novel Avenue to Antibody Therapy? Mol. Ther. 2019, 27, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Weng, Y.; Li, C.; Yang, T.; Hu, B.; Zhang, M.; Guo, S.; Xiao, H.; Liang, X.-J.; Huang, Y. The challenge and prospect of mRNA therapeutics landscape. Biotechnol. Adv. 2020, 40, 107534. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Perche, F.; Ikegami, M.; Uchida, S.; Kataoka, K.; Itaka, K. Messenger RNA-based therapeutics for brain diseases: An animal study for augmenting clearance of beta-amyloid by intracerebral administration of neprilysin mRNA loaded in polyplex nanomicelles. J. Control. Release 2016, 235, 268–275. [Google Scholar] [CrossRef]
- Guan, S.; Rosenecker, J. Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems. Gene Ther. 2017, 24, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Park, S.-J.; Yim, Y.; Kim, J.; Choi, C.; Won, C.; Min, D.-H. Recent Advances in RNA Therapeutics and RNA Delivery Systems Based on Nanoparticles. Adv. Ther. 2018, 1. [Google Scholar] [CrossRef]
- Cao, J.; An, D.; Galduroz, M.; Zhuo, J.; Liang, S.; Eybye, M.; Frassetto, A.; Kuroda, E.; Funahashi, A.; Santana, J.; et al. mRNA Therapy Improves Metabolic and Behavioral Abnormalities in a Murine Model of Citrin Deficiency. Mol. Ther. 2019, 27, 1242–1251. [Google Scholar] [CrossRef] [Green Version]
- Midoux, P.; Pichon, C. Lipid-based mRNA vaccine delivery systems. Expert Rev. Vaccines 2014, 14, 221–234. [Google Scholar] [CrossRef] [Green Version]
- An, D.; Frassetto, A.; Jacquinet, E.; Eybye, M.; Milano, J.; DeAntonis, C.; Nguyen, V.; Laureano, R.; Milton, J.; Sabnis, S.; et al. Long-term efficacy and safety of mRNA therapy in two murine models of methylmalonic acidemia. EBioMedicine 2019, 45, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.-J. Developing mRNA-vaccine technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.A. A Comparison of Plasmid DNA and mRNA as Vaccine Technologies. Vaccines 2019, 7, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, A.-M.; Batra, N.; Tu, M.-J.; Sweeney, C. Novel approaches for efficient in vivo fermentation production of noncoding RNAs. Appl. Microbiol. Biotechnol. 2020, 104, 1927–1937. [Google Scholar] [CrossRef] [PubMed]
- Pereira, P.; Pedro, A.Q.; Queiroz, J.A.; Figueiras, A.R.; Sousa, F. New insights for therapeutic recombinant human miRNAs heterologous production: Rhodovolum sulfidophilum vs. Escherichia coli. Bioengineered 2017, 8, 670–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, P.; Pedro, A.Q.; Tomás, J.; Maia, C.J.; Queiroz, J.A.; Figueiras, A.; Sousa, F. Advances in time course extracellular production of human pre-miR-29b from Rhodovulum sulfidophilum. Appl. Microbiol. Biotechnol. 2016, 100, 3723–3734. [Google Scholar] [CrossRef] [PubMed]
- Baptista, B.; Riscado, M.; Queiroz, J.; Pichon, C.; Sousa, F. Non-coding RNAs: Emerging from the discovery to therapeutic applications. Biochem. Pharmacol. 2021, 189, 114469. [Google Scholar] [CrossRef]
- Roy, I.; Stachowiak, M.K.; Bergey, E.J. Nonviral gene transfection nanoparticles: Function and applications in the brain. Nanomed. Nanotechnol. Biol. Med. 2008, 4, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.A.; Xu, Y.; Tao, W.; Ubellacker, J.M.; Lim, M.; Aum, D.; Lee, G.Y.; Zhou, K.; Zope, H.; Yu, M.; et al. Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat. Biomed. Eng. 2018, 2, 850–864. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Ramamoorth, M.; Narvekar, A. Non viral vectors in gene therapy- an overview. J. Clin. Diagn. Res. 2015, 9, GE01–GE06. [Google Scholar] [CrossRef]
- Uddin, M.N.; Roni, M.A. Challenges of Storage and Stability of mRNA-Based COVID-19 Vaccines. Vaccines 2021, 9, 1033. [Google Scholar] [CrossRef]
- Gómez-Aguado, I.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Rodríguez-Gascón, A.; Aspiazu, M.; Ángeles, S.; Del Pozo-Rodríguez, A. Nanomedicines to Deliver mRNA: State of the Art and Future Perspectives. Nanomaterials 2020, 10, 364. [Google Scholar] [CrossRef] [Green Version]
- Rozovics, J.M.; Chase, A.J.; Cathcart, A.L.; Chou, W.; Gershon, P.D.; Palusa, S.; Wilusz, J.; Semler, B.L. Picornavirus Modification of a Host mRNA Decay Protein. mBio 2012, 3, e00431-12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, S.; Yang, S.; Hu, X.; Yin, D.; Dai, Y.; Qian, X.; Wang, D.; Pan, X.; Hong, J.; Sun, X.; et al. Lentiviral delivery of co-packaged Cas9 mRNA and a Vegfa-targeting guide RNA prevents wet age-related macular degeneration in mice. Nat. Biomed. Eng. 2021, 5, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and Challenges in the Delivery of mRNA-Based Vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liang, H.; Liu, J.; Wang, Z. Poly (amidoamine) (PAMAM) dendrimer mediated delivery of drug and pDNA/siRNA for cancer therapy. Int. J. Pharm. 2018, 546, 215–225. [Google Scholar] [CrossRef]
- Kaur, I.P.; Sharma, G.; Singh, M.; Sandhu, S.K.; Deol, P.; Yadav, M.; Yakhmi, J.V. Nanobiomaterials as gene-delivery vehicles. Nanobiomater. Drug Deliv. 2016, 9, 447–486. [Google Scholar] [CrossRef]
- Hassett, K.J.; Higgins, J.; Woods, A.; Levy, B.; Xia, Y.; Hsiao, C.J.; Acosta, E.; Almarsson, O.; Moore, M.J.; Brito, L.A. Impact of lipid nanoparticle size on mRNA vaccine immunogenicity. J. Control. Release 2021, 335, 237–246. [Google Scholar] [CrossRef]
- Godbey, W.T.; Wu, K.K.; Mikos, A.G. Size matters: Molecular weight affects the efficiency of poly(ethylenimine) as a gene delivery vehicle. J. Biomed. Mater. Res. 1999, 45, 268–275. [Google Scholar] [CrossRef]
- Forrest, M.L.; Gabrielson, N.; Pack, D.W. Cyclodextrin-polyethylenimine conjugates for targeted in vitro gene delivery. Biotechnol. Bioeng. 2004, 89, 416–423. [Google Scholar] [CrossRef]
- Sultana, N.; Magadum, A.; Hadas, Y.; Kondrat, J.; Singh, N.; Youssef, E.; Calderon, D.; Chepurko, E.; Dubois, N.; Hajjar, R.J.; et al. Optimizing Cardiac Delivery of Modified mRNA. Mol. Ther. 2017, 25, 1306–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggli, N.; Demoulins, T.; Pichon, C.; Suter, R.; Guzmán, C.A.; Ebensen, T.; Midoux, P.; McCullough, K.; Milona, P.; Schulze, K.; et al. Polyethylenimine-based polyplex delivery of self-replicating RNA vaccines. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 711–722. [Google Scholar] [CrossRef]
- Zhao, M.; Li, M.; Zhang, Z.; Gong, T.; Sun, X. Induction of HIV-1 gag specific immune responses by cationic micelles mediated delivery of gag mRNA. Drug Deliv. 2015, 23, 2596–2607. [Google Scholar] [CrossRef]
- Lallana, E.; de la Rosa, J.M.R.; Tirella, A.; Pelliccia, M.; Gennari, A.; Stratford, I.J.; Puri, S.; Ashford, M.; Tirelli, N. Chitosan/Hyaluronic Acid Nanoparticles: Rational Design Revisited for RNA Delivery. Mol. Pharm. 2017, 14, 2422–2436. [Google Scholar] [CrossRef]
- McCullough, K.C.; Bassi, I.; Milona, P.; Suter, R.; Thomann-Harwood, L.; Englezou, P.; Démoulins, T.; Ruggli, N. Self-replicating Replicon-RNA Delivery to Dendritic Cells by Chitosan-nanoparticles for Translation In Vitro and In Vivo. Mol. Ther.—Nucleic Acids 2014, 3, e173. [Google Scholar] [CrossRef]
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The Limitless Future of RNA Therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 628137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Goel, V.; Robbie, G.J. Pharmacokinetics of Patisiran, the First Approved RNA Interference Therapy in Patients With Hereditary Transthyretin-Mediated Amyloidosis. J. Clin. Pharmacol. 2019, 60, 573–585. [Google Scholar] [CrossRef] [Green Version]
- Gerhardt, A.; Voigt, E.; Archer, M.; Reed, S.; Larson, E.; Van Hoeven, N.; Kramer, R.; Fox, C.; Casper, C. A Thermostable, Flexible RNA Vaccine Delivery Platform for Pandemic Response. bioRxiv 2021. [Google Scholar] [CrossRef]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef] [Green Version]
- Sayour, E.J.; De Leon, G.; Pham, C.; Grippin, A.; Kemeny, H.; Chua, J.; Huang, J.; Sampson, J.; Sanchez-Perez, L.; Flores, C.; et al. Systemic activation of antigen-presenting cells via RNA-loaded nanoparticles. OncoImmunology 2016, 6, e1256527. [Google Scholar] [CrossRef] [Green Version]
- Verbeke, R.; Lentacker, I.; Wayteck, L.; Breckpot, K.; Van Bockstal, M.; Descamps, B.; Vanhove, C.; De Smedt, S.C.; Dewitte, H. Co-delivery of nucleoside-modified mRNA and TLR agonists for cancer immunotherapy: Restoring the immunogenicity of immunosilent mRNA. J. Control Release 2017, 266, 287–300. [Google Scholar] [CrossRef]
- Basha, G.; Novobrantseva, T.I.; Rosin, N.; Tam, Y.Y.C.; Hafez, I.M.; Wong, M.K.; Sugo, T.; Ruda, V.M.; Qin, J.; Klebanov, B.; et al. Influence of Cationic Lipid Composition on Gene Silencing Properties of Lipid Nanoparticle Formulations of siRNA in Antigen-Presenting Cells. Mol. Ther. 2011, 19, 2186–2200. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Sedic, M.; Senn, J.J.; Lynn, A.; Laska, M.; Smith, M.; Platz, S.J.; Bolen, J.; Hoge, S.; Bulychev, A.; Jacquinet, E.; et al. Safety Evaluation of Lipid Nanoparticle–Formulated Modified mRNA in the Sprague-Dawley Rat and Cynomolgus Monkey. Vet. Pathol. 2017, 55, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Arteta, M.Y.; Kjellman, T.; Bartesaghi, S.; Wallin, S.; Wu, X.; Kvist, A.J.; Dabkowska, A.; Székely, N.; Radulescu, A.; Bergenholtz, J.; et al. Successful reprogramming of cellular protein production through mRNA delivered by functionalized lipid nanoparticles. Proc. Natl. Acad. Sci. USA 2018, 115, E3351–E3360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardi, N.; Hogan, M.; Pelc, R.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Hekele, A.; Bertholet, S.; Archer, J.; Gibson, D.G.; Palladino, G.; Brito, L.A.; Otten, G.R.; Brazzoli, M.; Buccato, S.; Bonci, A.; et al. Rapidly produced SAM® vaccine against H7N9 influenza is immunogenic in mice. Emerg. Microbes Infect. 2013, 2, 1–7. [Google Scholar] [CrossRef]
- Guevara, M.L.; Persano, F.; Persano, S. Advances in lipid nanoparticles for mRNA-based cancer immunotherapy. Front. Chem. 2020, 8, 963. [Google Scholar] [CrossRef]
- Mockey, M.; Bourseau, E.; Chandrashekhar, V.; Chaudhuri, A.; Lafosse, S.; Le Cam, E.; Quesniaux, V.F.J.; Ryffel, B.; Pichon, C.; Midoux, P. mRNA-based cancer vaccine: Prevention of B16 melanoma progression and metastasis by systemic injection of MART1 mRNA histidylated lipopolyplexes. Cancer Gene Ther. 2007, 14, 802–814. [Google Scholar] [CrossRef] [Green Version]
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.-J.; Stitz, L.; et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 2012, 30, 1210–1216. [Google Scholar] [CrossRef]
- Kübler, H.; Scheel, B.; Gnad-Vogt, U.; Miller, K.; Schultze-Seemann, W.; Dorp, F.V.; Parmiani, G.; Hampel, C.; Wedel, S.; Trojan, L.; et al. Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: A first-in-man phase I/IIa study. J. Immunother. Cancer 2015, 3, 26. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, M.; Schröder, A.; Scheel, B.; Hong, H.S.; Muth, A.; von Boehmer, L.; Zippelius, A.; Mayer, F.; Reck, M.; Atanackovic, D.; et al. A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol. Immunother. 2019, 68, 799–812. [Google Scholar] [CrossRef]
- Armbruster, N.; Jasny, E.; Petsch, B. Advances in RNA Vaccines for Preventive Indications: A Case Study of a Vaccine Against Rabies. Vaccines 2019, 7, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, A.S.; Yu, X.; Umhoefer, J.M.; Chamberlain, C.S.; Wildenauer, L.A.; Diarra, G.M.; Hacker, T.A.; Murphy, W.L. Single-dose mRNA therapy via biomaterial-mediated sequestration of overexpressed proteins. Sci. Adv. 2020, 6, eaba2422. [Google Scholar] [CrossRef] [PubMed]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Mai, Y.; Guo, J.; Zhao, Y.; Ma, S.; Hou, Y.; Yang, J. Intranasal delivery of cationic liposome-protamine complex mRNA vaccine elicits effective anti-tumor immunity. Cell. Immunol. 2020, 354, 104143. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Tonnu, N.; Tachikawa, K.; Limphong, P.; Vega, J.B.; Karmali, P.P.; Chivukula, P.; Verma, I.M. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E1941–E1950. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Baba, M.; Itaka, K.; Kondo, K.; Yamasoba, T.; Kataoka, K. Treatment of neurological disorders by introducing mRNA in vivo using polyplex nanomicelles. J. Control. Release 2015, 201, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Magadum, A.; Singh, N.; Kurian, A.A.; Sharkar, M.T.K.; Chepurko, E.; Zangi, L. Ablation of a Single N-Glycosylation Site in Human FSTL 1 Induces Cardiomyocyte Proliferation and Cardiac Regeneration. Mol. Ther.-Nucleic Acids 2018, 13, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Magadum, A.; Singh, N.; Kurian, A.A.; Munir, I.; Mehmood, T.; Brown, K.; Sharkar, M.T.K.; Chepurko, E.; Sassi, Y.; Oh, J.G.; et al. Pkm2 Regulates Cardiomyocyte Cell Cycle and Promotes Cardiac Regeneration. Circulation 2020, 141, 1249–1265. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Zhang, X.; Men, K.; Gao, Y.; Yang, X.; Wu, S.; Duan, X.; Wei, Y.; Tong, R. Efficient Colorectal Cancer Gene Therapy with IL-15 mRNA Nanoformulation. Mol. Pharm. 2020, 17. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in cancer immunotherapy. OncoImmunology 2016, 5, e1163462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Türeci, Ö.; Vormehr, M.; Kranz, L.M. mRNA therapeutics in cancer immunotherapy. Mol. Cancer 2021, 20, 69. [Google Scholar] [CrossRef]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delehedde, C.; Even, L.; Midoux, P.; Pichon, C.; Perche, F. Intracellular Routing and Recognition of Lipid-Based mRNA Nanoparticles. Pharmaceutics 2021, 13, 945. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.-I.; Wu, C.-J.; Tu, Y.-F.; Chi, C.-Y. Fighting COVID-19: A quick review of diagnoses, therapies, and vaccines. Biomed. J. 2020, 43, 341–354. [Google Scholar] [CrossRef]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar]
- Billingsley, M.M.; Singh, N.; Ravikumar, P.; Zhang, R.; June, C.H.; Mitchell, M.J. Ionizable Lipid Nanoparticle-Mediated mRNA Delivery for Human CAR T Cell Engineering. Nano Lett. 2020, 20, 1578–1589. [Google Scholar] [CrossRef]
- Belete, T.M. A review on Promising vaccine development progress for COVID-19 disease. Vacunas 2020, 21, 121–128. [Google Scholar] [CrossRef]
- He, W.; Evans, A.C.; Rasley, A.; Bourguet, F.; Peters, S.; Kamrud, K.I.; Wang, N.; Hubby, B.; Felderman, M.; Gouvis, H.; et al. Cationic HDL mimetics enhance in vivo delivery of self-replicating mRNA. Nanomed. Nanotechnol. Biol. Med. 2020, 24, 102154. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, L.; Almutairi, M.M.; Hotez, P.J.; Pollet, J. Enlisting the mRNA Vaccine Platform to Combat Parasitic Infections. Vaccines 2019, 7, 122. [Google Scholar] [CrossRef] [Green Version]
- Alton, E.W.F.W.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R.; et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015, 3, 684–691. [Google Scholar] [CrossRef] [Green Version]
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Backert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: An open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 2017, 390, 1511–1520. [Google Scholar] [CrossRef]
- Fotin-Mleczek, M.; Duchardt, K.M.; Lorenz, C.; Pfeiffer, R.; Ojkić-Zrna, S.; Probst, J.; Kallen, K.-J. Messenger RNA-based Vaccines with Dual Activity Induce Balanced TLR-7 Dependent Adaptive Immune Responses and Provide Antitumor Activity. J. Immunother. 2011, 34, 1–15. [Google Scholar] [CrossRef]
- Feldman, R.A.; Fuhr, R.; Smolenov, I.; Ribeiro, A.; Panther, L.; Watson, M.; Senn, J.J.; Smith, M.; Almarsson, Ӧ.; Pujar, H.S.; et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 2019, 37, 3326–3334. [Google Scholar] [CrossRef]
- Bos, R.; Marquardt, K.L.; Cheung, J.; Sherman, L.A. Functional differences between low- and high-affinity CD8+ T cells in the tumor environment. OncoImmunology 2012, 1, 1239–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, L.; Hinterberger, M.; Wirnsberger, G.; Kyewski, B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat. Rev. Immunol. 2009, 9, 833–844. [Google Scholar] [CrossRef]
- Leal, L.; Guardo, A.C.; Morón-López, S.; Salgado, M.; Mothe, B.; Heirman, C.; Pannus, P.; Vanham, G.; van den Ham, H.J.; Gruters, R.; et al. Phase I clinical trial of an intranodally administered mRNA-based therapeutic vaccine against HIV-1 infection. AIDS 2018, 32, 2533–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jong, W.; Aerts, J.; Allard, S.; Brander, C.; Buyze, J.; Florence, E.; van Gorp, E.; Vanham, G.; Leal, L.; Mothe, B. iHIVARNA phase IIa, a randomized, placebo-controlled, double-blinded trial to evaluate the safety and immunogenicity of iHIVARNA-01 in chronically HIV-infected patients under stable combined antiretroviral therapy. Trials 2019, 20, 1–10. [Google Scholar]
- Gandhi, R.T.; Kwon, D.S.; Macklin, E.A.; Shopis, J.R.; McLean, A.P.; McBrine, N.; Flynn, T.; Peter, L.; Sbrolla, A.; Kaufmann, D.E. Immunization of HIV-1-infected persons with autologous dendritic cells transfected with mRNA encoding HIV-1 Gag and Nef: Results of a randomized, placebo-controlled clinical trial. J. Acquir. Immune Defic. Syndr. 2016, 71, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyte, J.A.; Mu, L.; Aamdal, S.; Kvalheim, G.; Dueland, S.; Hauser, M.; Gullestad, H.P.; Ryder, T.; Lislerud, K.; Hammerstad, H.; et al. Phase I/II trial of melanoma therapy with dendritic cells transfected with autologous tumor-mRNA. Cancer Gene Ther. 2006, 13, 905–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wecker, M.; Gilbert, P.; Russell, N.; Hural, J.; Allen, M.; Pensiero, M.; Chulay, J.; Chiu, Y.-L.; Karim, S.S.A.; Burke, D.S. Phase I Safety and Immunogenicity Evaluations of an Alphavirus Replicon HIV-1 Subtype C gag Vaccine in Healthy HIV-1-Uninfected Adults. Clin. Vaccine Immunol. 2012, 19, 1651–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Ding, W.; Zhu, D.; Yu, L.; Jiang, X.; Wang, X.; Zhang, C.; Wang, L.; Ji, T.; Liu, D.; et al. TALEN-mediated targeting of HPV oncogenes ameliorates HPV-related cervical malignancy. J. Clin. Investig. 2014, 125, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Yu, L.; Zhu, D.; Ding, W.; Wang, X.; Zhang, C.; Wang, L.; Jiang, X.; Shen, H.; He, D.; et al. Disruption of HPV16-E7 by CRISPR/Cas System Induces Apoptosis and Growth Inhibition in HPV16 Positive Human Cervical Cancer Cells. BioMed Res. Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef]
- Su, S.; Hu, B.; Shao, J.; Shen, B.; Du, J.; Du, Y.; Zhou, J.; Yu, L.; Zhang, L.; Chen, F.; et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci. Rep. 2016, 6, 20070. [Google Scholar] [CrossRef] [Green Version]
- Quan, L.; Chen, X.; Liu, A.; Zhang, Y.; Guo, X.; Yan, S.; Liu, Y. PD-1 Blockade Can Restore Functions of T-Cells in Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma In Vitro. PLoS ONE 2015, 10, e0136476. [Google Scholar] [CrossRef]
- DiGiusto, D.L.; Cannon, P.M.; Holmes, M.C.; Li, L.; Rao, A.; Wang, J.; Lee, G.; Gregory, P.; Kim, K.A.; Hayward, S.B.; et al. Preclinical development and qualification of ZFN-mediated CCR5 disruption in human hematopoietic stem/progenitor cells. Mol. Ther.—Methods Clin. Dev. 2016, 3, 16067. [Google Scholar] [CrossRef] [Green Version]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.-M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef]
- Ahammad, I.; Lira, S.S. Designing a novel mRNA vaccine against SARS-CoV-2: An immunoinformatics approach. Int. J. Biol. Macromol. 2020, 162, 820–837. [Google Scholar] [CrossRef]
- Khuroo, M.S.; Khuroo, M.; Khuroo, M.S.; Sofi, A.A.; Khuroo, N.S. COVID-19 Vaccines: A Race Against Time in the Middle of Death and Devastation! J. Clin. Exp. Hepatol. 2020, 10, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Velavan, T.P.; Meyer, C.G. The COVID-19 epidemic. Trop. Med. Int. Health 2020, 25, 278–280. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Wang, X. COVID-19: A new challenge for human beings. Cell. Mol. Immunol. 2020, 17, 555–557. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [Green Version]
- Koirala, A.; Joo, Y.J.; Khatami, A.; Chiu, C.; Britton, P.N. Vaccines for COVID-19: The current state of play. Paediatr. Respir. Rev. 2020, 35, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Caddy, S. Developing a vaccine for COVID-19. BMJ 2020, 369, m1790. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Shen, H.-M. Targeting the Endocytic Pathway and Autophagy Process as a Novel Therapeutic Strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724–1731. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Lurie, N.; Saville, M.; Hatchett, R.; Halton, J. Developing COVID-19 Vaccines at Pandemic Speed. N. Engl. J. Med. 2020, 382, 1969–1973. [Google Scholar] [CrossRef]
- Thanh Le, T.; Andreadakis, Z.; Kumar, A.; Gómez Román, R.; Tollefsen, S.; Saville, M.; Mayhew, S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Corey, B.L.; Mascola, J.R.; Fauci, A.S.; Collins, F.S. A strategic approach to COVID-19 vaccine R&D. Science 2020, 368, 948–950. [Google Scholar] [CrossRef]
- Kowalzik, F.; Schreiner, D.; Jensen, C.; Teschner, D.; Gehring, S.; Zepp, F. mRNA-Based Vaccines. Vaccines 2021, 9, 390. [Google Scholar] [CrossRef] [PubMed]
- Oberhardt, V.; Luxenburger, H.; Kemming, J.; Schulien, I.; Ciminski, K.; Giese, S.; Csernalabics, B.; Lang-Meli, J.; Janowska, I.; Staniek, J.; et al. Rapid and stable mobilization of CD8+ T cells by SARS-CoV-2 mRNA vaccine. Nature 2021, 597, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Kim, J.H.; Marks, F.; Clemens, J.D. Looking beyond COVID-19 vaccine phase 3 trials. Nat. Med. 2021, 27, 205–211. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Comirnaty Epar Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/comirnaty-epar-product-information_en.pdf (accessed on 18 November 2021).
- Spikevax previously COVID-19 vaccine Moderna Epar Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/spikevax-previously-covid-19-vaccine-moderna-epar-product-information_en.pdf (accessed on 18 November 2021).
- Lamb, Y.N. BNT162b2 mRNA COVID-19 Vaccine: First Approval. Drugs 2021, 81, 495–501. [Google Scholar] [CrossRef]
- Fact Sheet for Healthcare Providers Administering Vaccine (Vaccination Providers)—Emergency Use Authorization (EUA) of the Pfizer-Biontech COVID-19 Vaccine to Prevent Coronavirus Disease 2019 (COVID-19). Available online: https://www.fda.gov/media/153713/download (accessed on 26 November 2021).
- Fact Sheet for Healthcare Providers Administering Vaccine. Moderna. U.S. Food Drug Adm. 2020, 8, 55. Available online: https://www.fda.gov/media/144637/download (accessed on 8 May 2021).
- Comirnaty (COVID-19 mRNA Vaccine [Nucleoside Modified])—An Overview of Comirnaty and Why It Is Authorised in the EU. Available online: https://www.ema.europa.eu/en/documents/overview/comirnaty-epar-medicine-overview_en.pdf (accessed on 26 November 2021).
- Spikevax (COVID-19 mRNA Vaccine [Nucleoside Modified])—An Overview of Spikevax and Why It Is Authorised in the EU. Available online: https://www.ema.europa.eu/en/documents/overview/spikevax-previously-covid-19-vaccine-moderna-epar-medicine-overview_en.pdf (accessed on 26 November 2021).
- Widge, A.T.; Rouphael, N.G.; Jackson, L.A.; Anderson, E.J.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J.; et al. Durability of Responses after SARS-CoV-2 mRNA-1273 Vaccination. N. Engl. J. Med. 2020, 384, 80–82. [Google Scholar] [CrossRef]
- Meo, S.A.; Bukhari, I.A.; Akram, J.; Meo, A.S.; Klonoff, D.C. COVID-19 vaccines: Comparison of biological, pharmacological characteristics and adverse effects of Pfizer/BioNTech and Moderna Vaccines. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1663–1669. [Google Scholar] [PubMed]
- Kremsner, P.G.; Mann, P.; Kroidl, A.; Leroux-Roels, I.; Schindler, C.; Gabor, J.J.; Schunk, M.; Leroux-Roels, G.; Bosch, J.J.; Fendel, R.; et al. Safety and immunogenicity of an mRNA-lipid nanoparticle vaccine candidate against SARS-CoV-2. Wien. Klin. Wochenschr. 2021, 133, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.; Roth, N.; Schwendt, K.; Fotin-Mleczek, M.; Mueller, S.O.; Petsch, B. mRNA-based SARS-CoV-2 vaccine candidate CVnCoV induces high levels of virus-neutralising antibodies and mediates protection in rodents. npj Vaccines 2021, 6, 57. [Google Scholar] [CrossRef]
- Abu-Raddad, L.J.; Chemaitelly, H.; Butt, A.A. Effectiveness of the BNT162b2 COVID-19 Vaccine against the B.1.1.7 and B.1.351 Variants. N. Engl. J. Med. 2021, 385, 187–189. [Google Scholar] [CrossRef]
- Sheikh, A.; McMenamin, J.; Taylor, B.; Robertson, C.; Public Health Scotland and the EAVE II Collaborators. SARS-CoV-2 Delta VOC in Scotland: Demographics, risk of hospital admission, and vaccine effectiveness. Lancet 2021, 397, 2461–2462. [Google Scholar] [CrossRef]
- Oliver, S.E.; Gargano, J.W.; Marin, M.; Wallace, M.; Curran, K.G.; Chamberland, M.; McClung, N.; Campos-Outcalt, D.; Morgan, R.L.; Mbaeyi, S.; et al. The Advisory Committee on Immunization Practices’ Interim Recommendation for Use of Moderna COVID-19 Vaccine - United States, December 2020. MMWR Morb. Mortal. Wkly. Rep. 2021, 69, 1653–1656. [Google Scholar] [CrossRef]
- Vaidyanathan, S.; Azizian, K.T.; Haque, A.A.; Henderson, J.M.; Hendel, A.; Shore, S.; Antony, J.S.; Hogrefe, R.I.; Kormann, M.S.; Porteus, M.H.; et al. Uridine Depletion and Chemical Modification Increase Cas9 mRNA Activity and Reduce Immunogenicity without HPLC Purification. Mol. Ther.—Nucleic Acids 2018, 12, 530–542. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector with Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Eyler, D.E.; Franco, M.K.; Batool, Z.; Wu, M.Z.; Dubuke, M.L.; Dobosz-Bartoszek, M.; Jones, J.; Polikanov, Y.S.; Roy, B.; Koutmou, K.S. Pseudouridinylation of mRNA coding sequences alters translation. Proc. Natl. Acad. Sci. USA 2019, 116, 23068–23074. [Google Scholar] [CrossRef] [PubMed]
- Nance, K.D.; Meier, J.L. Modifications in an Emergency: The Role of N1-Methylpseudouridine in COVID-19 Vaccines. ACS Cent. Sci. 2021, 7, 748–756. [Google Scholar] [CrossRef]
- Alexaki, A.; Hettiarachchi, G.K.; Athey, J.C.; Katneni, U.; Simhadri, V.; Hamasaki-Katagiri, N.; Nanavaty, P.; Lin, B.; Takeda, K.; Freedberg, D.; et al. Effects of codon optimization on coagulation factor IX translation and structure: Implications for protein and gene therapies. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, O.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef]
- OMS. COVID-19 Vaccine Tracker and Landscape. Available online: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (accessed on 1 October 2021).
- Ghosh, D.; Venkataramani, P.; Nandi, S.; Bhattacharjee, S. CRISPR–Cas9 a boon or bane: The bumpy road ahead to cancer therapeutics. Cancer Cell Int. 2019, 19, 1–10. [Google Scholar] [CrossRef]
- Janssen, J.M.; Chen, X.; Liu, J.; Gonçalves, M.A. The Chromatin Structure of CRISPR-Cas9 Target DNA Controls the Balance between Mutagenic and Homology-Directed Gene-Editing Events. Mol. Ther.-Nucleic Acids 2019, 16, 141–154. [Google Scholar] [CrossRef] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.B.; Zhang, S.; Kos, P.; Xiong, H.; Zhou, K.; Perelman, S.S.; Zhu, H.; Siegwart, D.J. Non-Viral CRISPR/Cas Gene Editing In Vitro and In Vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9 mRNA and sgRNA. Angew. Chem. Int. Ed. Engl. 2017, 56, 1059–1063. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Potter, J.; Kumar, S.; Zou, Y.; Quintanilla, R.; Sridharan, M.; Carte, J.; Chen, W.; Roark, N.; Ranganathan, S.; et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 2015, 208, 44–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohsin, M.; Li, Y.; Zhang, X.; Wang, Y.; Huang, Z.; Yin, G.; Zhang, Z. Development of CRISPR-CAS9 based RNA drugs against Eimeria tenella infection. Genomics 2021, 113, 4126–4135. [Google Scholar] [CrossRef] [PubMed]
- Gratz, S.J.; Cummings, A.M.; Nguyen, J.N.; Hamm, D.C.; Donohue, L.K.; Harrison, M.M.; Wildonger, J.; O’Connor-Giles, K.M. Genome Engineering of Drosophila with the CRISPR RNA-Guided Cas9 Nuclease. Genetics 2013, 194, 1029–1035. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [Green Version]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.-R.J.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Shen, B.; Cui, Y.; Chen, Y.; Wang, J.; Wang, L.; Kang, Y.; Zhao, X.; Si, W.; Li, W.; et al. Generation of Gene-Modified Cynomolgus Monkey via Cas9/RNA-Mediated Gene Targeting in One-Cell Embryos. Cell 2014, 156, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, M.; Glass, Z.; Chen, J.; Haas, M.; Jin, X.; Zhao, X.; Rui, X.; Ye, Z.; Li, Y.; Zhang, F.; et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Graham, M.J.; Lee, R.G.; Brandt, T.A.; Tai, L.-J.; Fu, W.; Peralta, R.; Yu, R.; Hurh, E.; Paz, E.; McEvoy, B.W.; et al. Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. N. Engl. J. Med. 2017, 377, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Abbott, T.R.; Dhamdhere, G.; Liu, Y.; Lin, X.; Goudy, L.; Zeng, L.; Chemparathy, A.; Chmura, S.; Heaton, N.S.; Debs, R.; et al. Development of CRISPR as an Antiviral Strategy to Combat SARS-CoV-2 and Influenza. Cell 2020, 181, 865.e12–876.e12. [Google Scholar] [CrossRef]
- Guo, X.; Rahman, J.A.; Wessels, H.-H.; Méndez-Mancilla, A.; Haro, D.; Chen, X.; Sanjana, N.E. Transcriptome-wide Cas13 guide RNA design for model organisms and viral RNA pathogens. Cell Genom. 2021, 1, 100001. [Google Scholar] [CrossRef]
- Kushawah, G.; Hernandez-Huertas, L.; Abugattas-Nuñez Del Prado, J.; Martinez-Morales, J.R.; DeVore, M.L.; Hassan, H.; Moreno-Sanchez, I.; Tomas-Gallardo, L.; Diaz-Moscoso, A.; Monges, D.E.; et al. CRISPR-Cas13d Induces Efficient mRNA Knockdown in Animal Embryos. Dev. Cell 2020, 54, 805.e7–817.e7. [Google Scholar] [CrossRef]
- Xu, D.; Cai, Y.; Tang, L.; Han, X.; Gao, F.; Cao, H.; Qi, F.; Kapranov, P. A CRISPR/Cas13-based approach demonstrates biological relevance of vlinc class of long non-coding RNAs in anticancer drug response. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Patchsung, M.; Jantarug, K.; Pattama, A.; Aphicho, K.; Suraritdechachai, S.; Meesawat, P.; Sappakhaw, K.; Leelahakorn, N.; Ruenkam, T.; Wongsatit, T.; et al. Clinical validation of a Cas13-based assay for the detection of SARS-CoV-2 RNA. Nat. Biomed. Eng. 2020, 4, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, E.L.; Vanover, D.; Bawage, S.S.; Tiwari, P.M.; Rotolo, L.; Beyersdorf, J.; Peck, H.E.; Bruno, N.C.; Hincapie, R.; Michel, F.; et al. Treatment of influenza and SARS-CoV-2 infections via mRNA-encoded Cas13a in rodents. Nat. Biotechnol. 2021, 39, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Rashnonejad, A.; Chermahini, G.A.; Wallace, L.; Harper, S.O. 8DUX4 mRNA silencing with CRISPR-Cas13 gene therapy as a prospective treatment for Facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2019, 29, S40. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of eukaryotic mRNA, integrating the fundamental regions for its processing, such as 5′ UTR, protein coding region, and 3′ UTR. Schematization of polyadenylation process and introduction of Cap at the 5′ end, as well as its biological and structural functions. P—Phosphate group; N—Nucleotide; CH3—Methyl group; A—Adenine; G—Guanine; UTR—Untranslated region.
Figure 1.
Schematic representation of eukaryotic mRNA, integrating the fundamental regions for its processing, such as 5′ UTR, protein coding region, and 3′ UTR. Schematization of polyadenylation process and introduction of Cap at the 5′ end, as well as its biological and structural functions. P—Phosphate group; N—Nucleotide; CH3—Methyl group; A—Adenine; G—Guanine; UTR—Untranslated region.

Figure 2.
Schematic representation of Extra- and Intracellular Barriers for nucleic acids delivery. (I)—Endo and exonuclease degradation; (II)—Interaction and binding with erythrocytes; (III)—Binding and aggregation via serum protein complexation; (IV)—Immune activation to delivered nucleic acids.
Figure 2.
Schematic representation of Extra- and Intracellular Barriers for nucleic acids delivery. (I)—Endo and exonuclease degradation; (II)—Interaction and binding with erythrocytes; (III)—Binding and aggregation via serum protein complexation; (IV)—Immune activation to delivered nucleic acids.

Figure 3.
Schematic representation of different delivery strategies for mRNA therapies. There are represented 3 methods for the delivery of mRNA: physical methods, such as electroporation and gene gun; viral delivery systems, that uses recombinant virus; and non-viral delivery systems, such as lipid nanoparticles, polyplex, lipoplex and lipopolyplex.
Figure 3.
Schematic representation of different delivery strategies for mRNA therapies. There are represented 3 methods for the delivery of mRNA: physical methods, such as electroporation and gene gun; viral delivery systems, that uses recombinant virus; and non-viral delivery systems, such as lipid nanoparticles, polyplex, lipoplex and lipopolyplex.

Figure 4.
1 (Green)—Extraction of the patient’s cells by biopsy; 2—In vitro cell growth of cells from biopsy; 3—In vitro cell transfection by encapsulated and/or naked synthetic mRNA; 4—Cell growth of transfected cells and undergoing mRNA translation; 5—Autograft with mRNA transfected cells for therapeutic protein production. 1′ (Blue)—Systemic (intravenous or intramuscular) or localized (target organ or tissue) delivery of nanoparticle and mRNA formulations into the patient.
Figure 4.
1 (Green)—Extraction of the patient’s cells by biopsy; 2—In vitro cell growth of cells from biopsy; 3—In vitro cell transfection by encapsulated and/or naked synthetic mRNA; 4—Cell growth of transfected cells and undergoing mRNA translation; 5—Autograft with mRNA transfected cells for therapeutic protein production. 1′ (Blue)—Systemic (intravenous or intramuscular) or localized (target organ or tissue) delivery of nanoparticle and mRNA formulations into the patient.

Figure 5.
Antigen processing and presentation by dendritic cells, for adaptive immune system activation, following subcutaneous injection of a mRNA vaccine. A synthetic mRNA is internalized by antigen presenting dendritic cells, where the mRNA is translated. Then, the antigen is exposed by class I or II major histocompatibility complex (MHC) molecules and is later recognized by CD8+ or CD4+ T cells, activating chemical and humoral responses.
Figure 5.
Antigen processing and presentation by dendritic cells, for adaptive immune system activation, following subcutaneous injection of a mRNA vaccine. A synthetic mRNA is internalized by antigen presenting dendritic cells, where the mRNA is translated. Then, the antigen is exposed by class I or II major histocompatibility complex (MHC) molecules and is later recognized by CD8+ or CD4+ T cells, activating chemical and humoral responses.

Figure 6.
(A)—Schematic structure of conventional non-amplifying mRNA vaccine. (B)—Schematic structure of self-amplifying mRNA vaccine (replicon). UTR—Untranslated region; nsP—Non-structural proteins; A—Adenine; G—Guanine; P—Phosphate group.
Figure 6.
(A)—Schematic structure of conventional non-amplifying mRNA vaccine. (B)—Schematic structure of self-amplifying mRNA vaccine (replicon). UTR—Untranslated region; nsP—Non-structural proteins; A—Adenine; G—Guanine; P—Phosphate group.

Figure 7.
Scheme of the structure of SARS-CoV-2, with different viral proteins indicated.

Figure 8.
Actuation mechanism of the main mRNA vaccines against SARS-CoV-2. This process begins with the injection, in the patient’s deltoid muscle, of the mRNA usually encapsulated in lipid nanoparticles (LNPs). LNPs loaded with the mRNA encoding the SARS-CoV-2 spike glycoprotein (S), can reach the apical lymph nodes where they transfect dendritic cells. After entry and release of the cell endosome, the mRNA sequence is expressed and post-translational modifications occur. Subsequently, the S glycoprotein is transported and presented in the cell membrane of immune cells (antigen presenting cells). The S glycoproteins incites a specific cytotoxic and humoral immune response, leading to the production of antibodies against SARS-CoV-2, with the aim of achieving immunization against COVID-19.
Figure 8.
Actuation mechanism of the main mRNA vaccines against SARS-CoV-2. This process begins with the injection, in the patient’s deltoid muscle, of the mRNA usually encapsulated in lipid nanoparticles (LNPs). LNPs loaded with the mRNA encoding the SARS-CoV-2 spike glycoprotein (S), can reach the apical lymph nodes where they transfect dendritic cells. After entry and release of the cell endosome, the mRNA sequence is expressed and post-translational modifications occur. Subsequently, the S glycoprotein is transported and presented in the cell membrane of immune cells (antigen presenting cells). The S glycoproteins incites a specific cytotoxic and humoral immune response, leading to the production of antibodies against SARS-CoV-2, with the aim of achieving immunization against COVID-19.

Figure 9.
Schematic of CRISPR-Cas9-mediated genome editing. A CRISPR-Cas9 endonuclease is directed to a DNA sequence by means of a single guide RNA sequence (sgRNA), resulting in double strand cleavage. Subsequently they are repaired by non-homologous final union (NHEJ) or homology-directed repair (HDR). NHEJ repair provides errors, often leads to insertion or deletion mutations, which can lead to genome instability. Alternatively, in the presence of an exogenous donor DNA model, it can be repaired through error-free HDR, projecting precise DNA changes.
Figure 9.
Schematic of CRISPR-Cas9-mediated genome editing. A CRISPR-Cas9 endonuclease is directed to a DNA sequence by means of a single guide RNA sequence (sgRNA), resulting in double strand cleavage. Subsequently they are repaired by non-homologous final union (NHEJ) or homology-directed repair (HDR). NHEJ repair provides errors, often leads to insertion or deletion mutations, which can lead to genome instability. Alternatively, in the presence of an exogenous donor DNA model, it can be repaired through error-free HDR, projecting precise DNA changes.


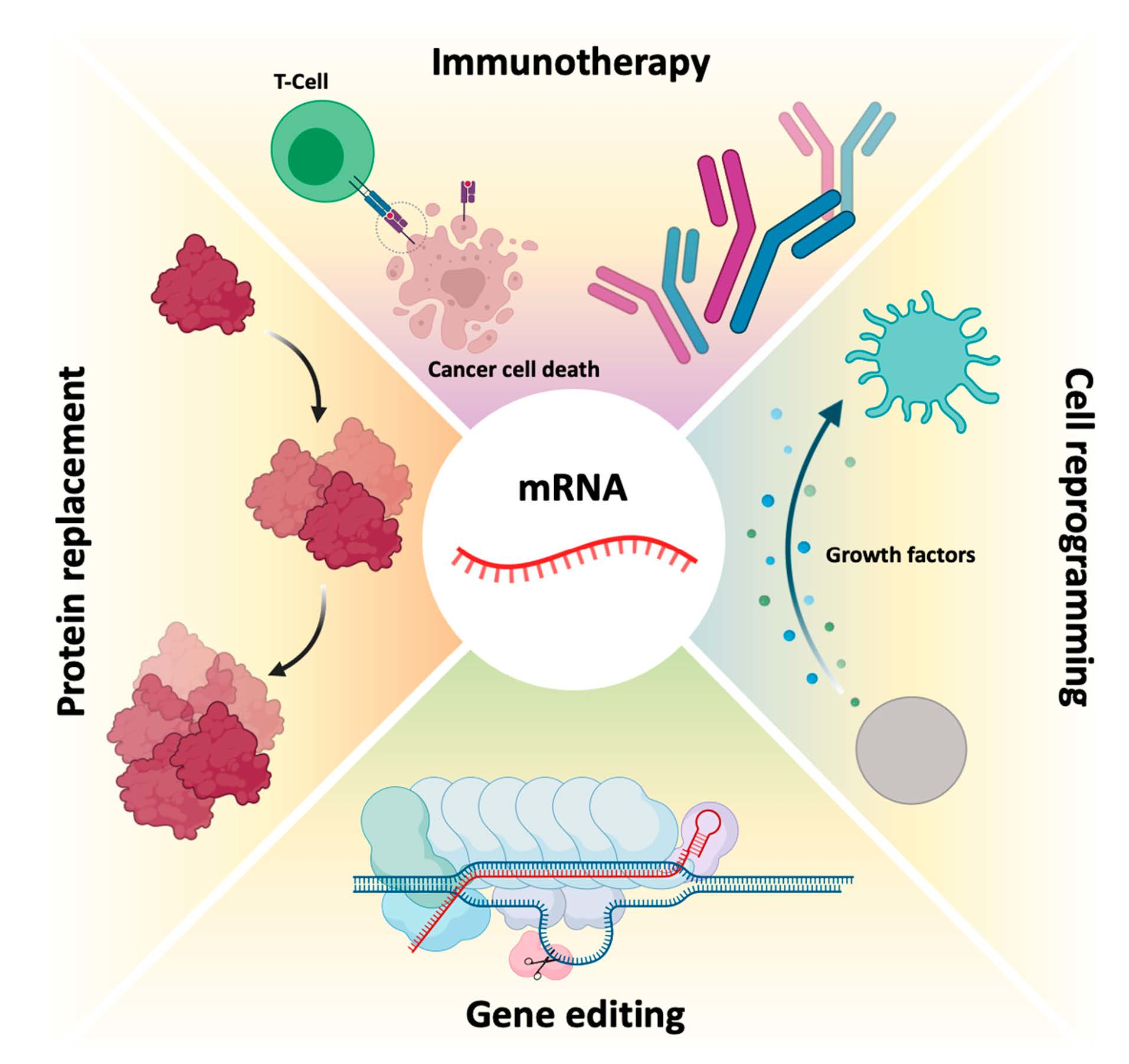{kind=link}
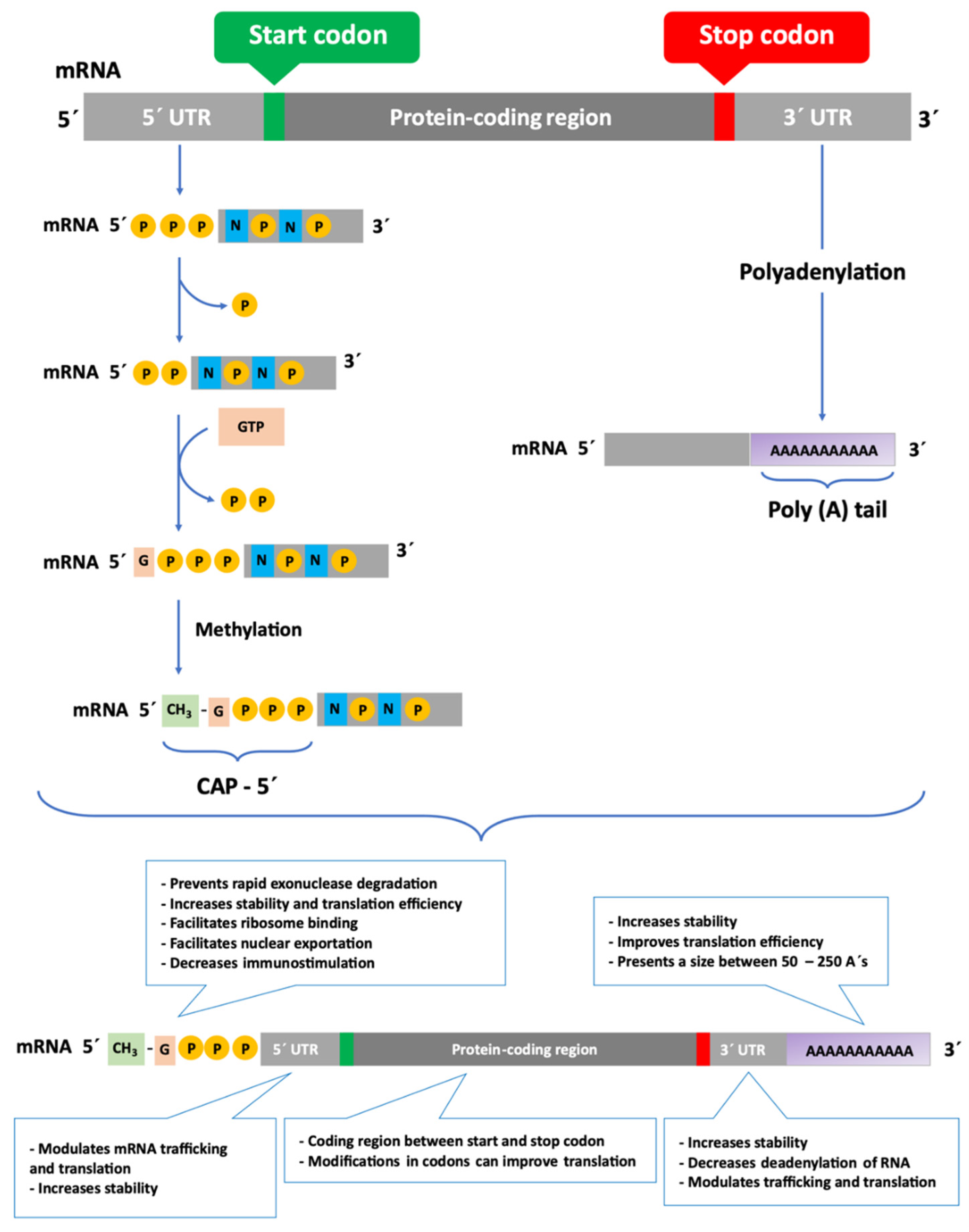{kind=link}
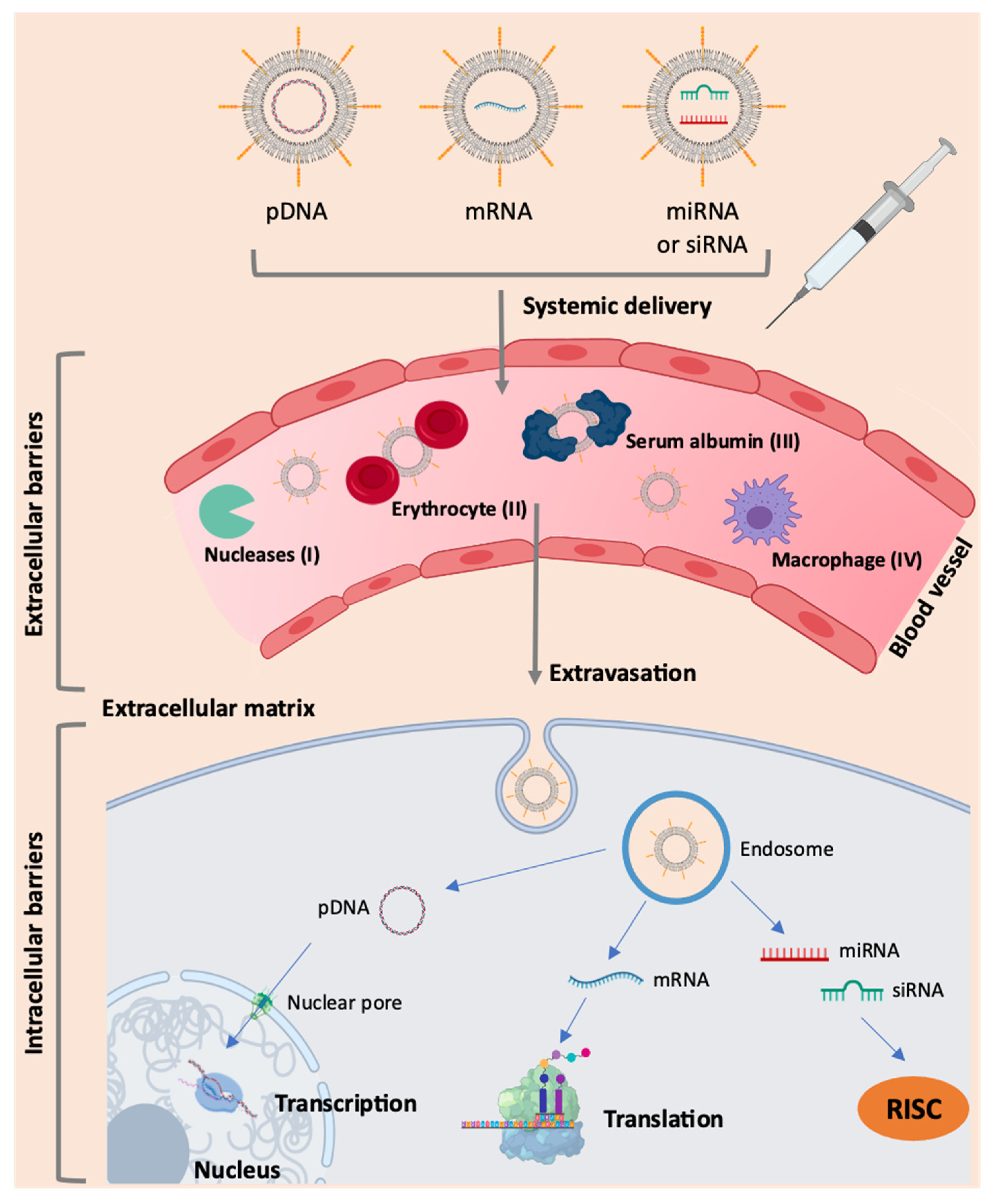{kind=link}
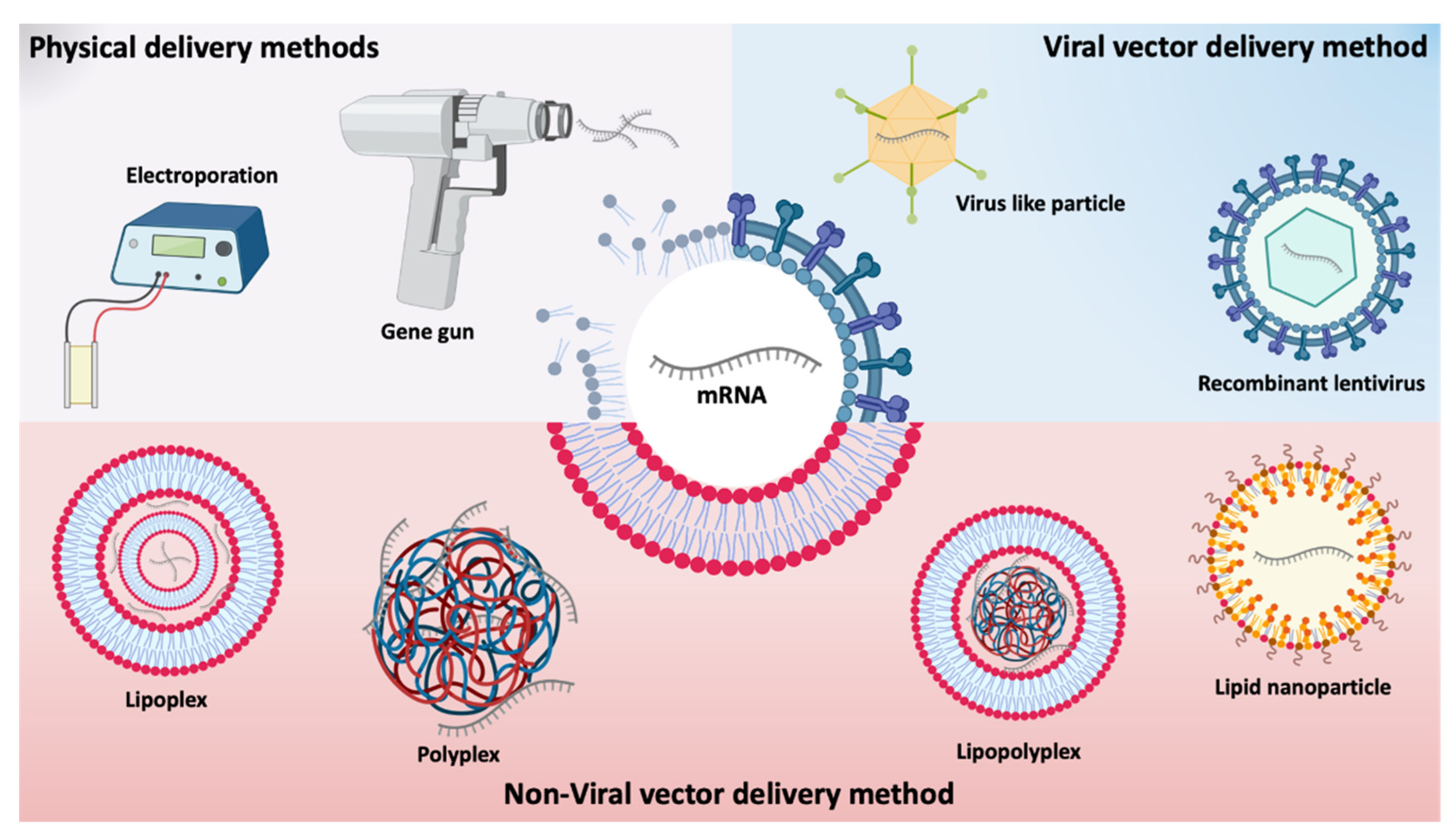{kind=link}
{kind=link}
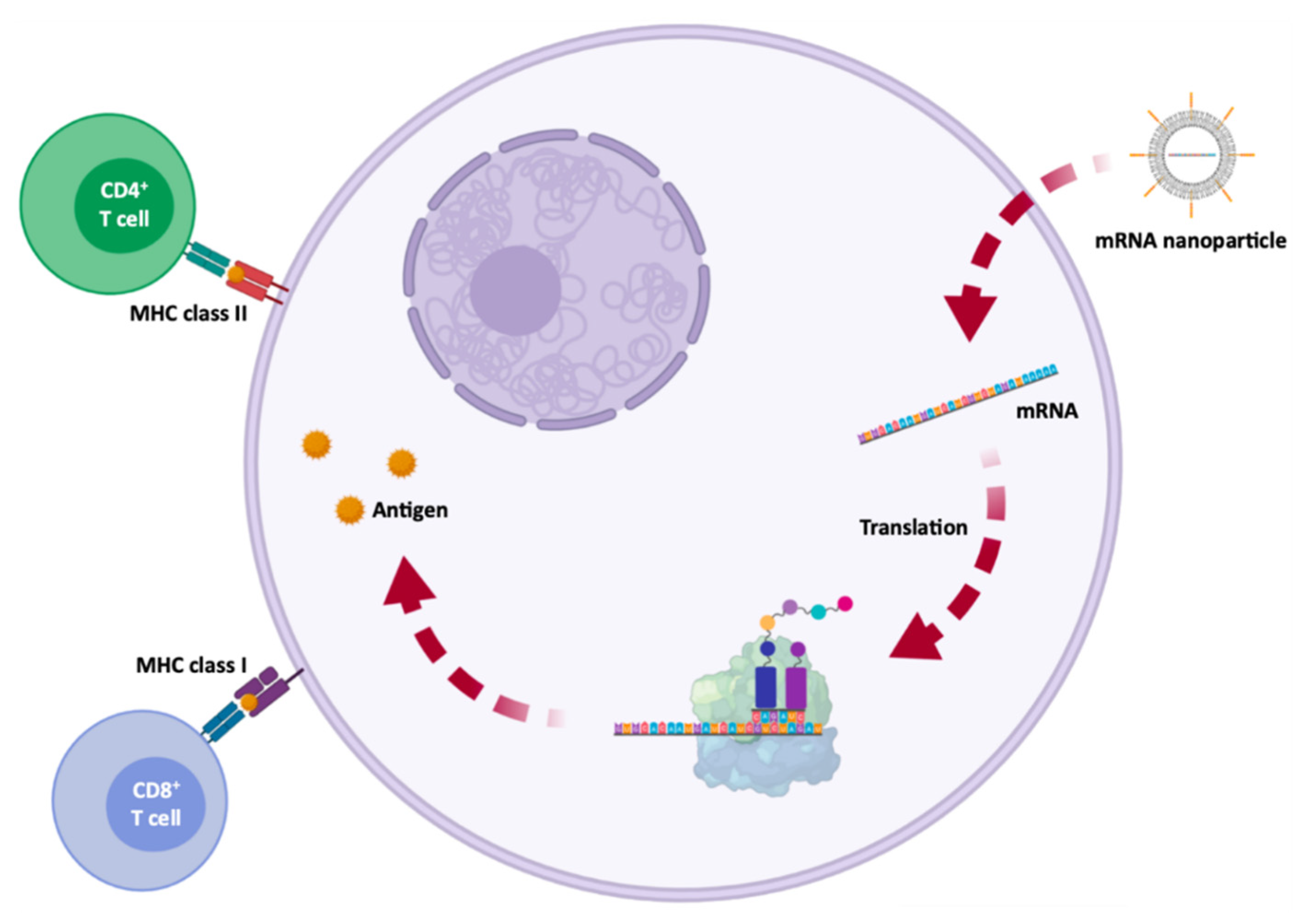{kind=link}
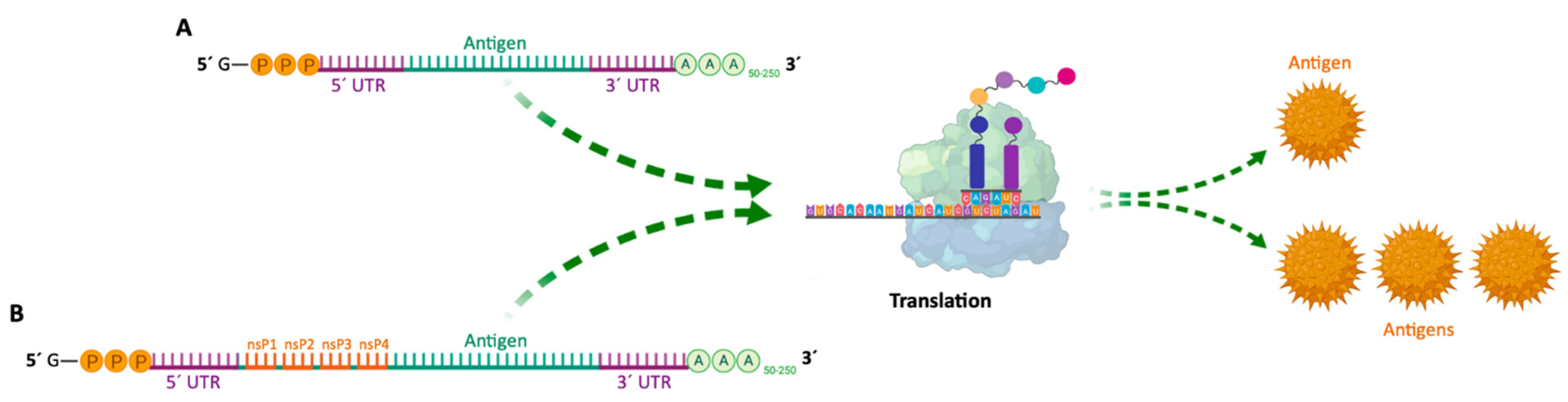{kind=link}
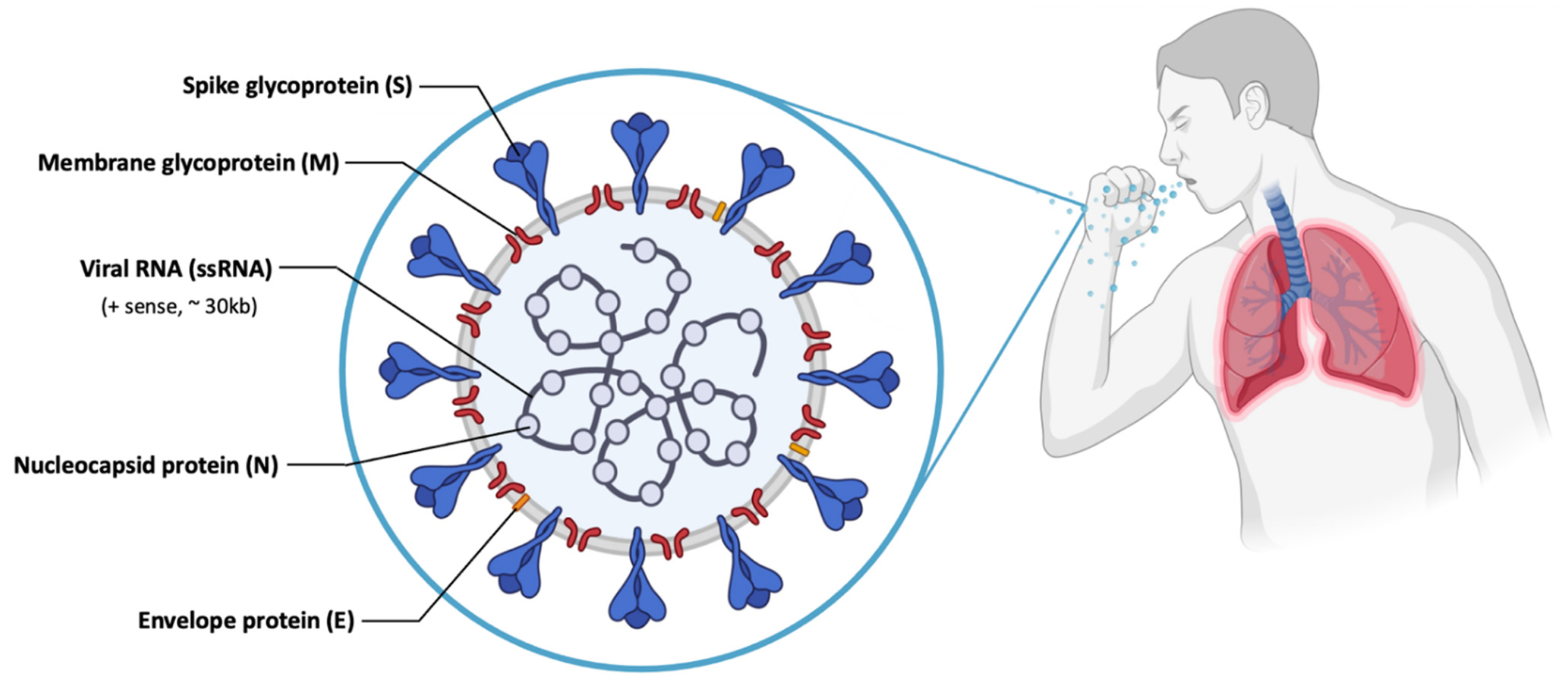{kind=link}
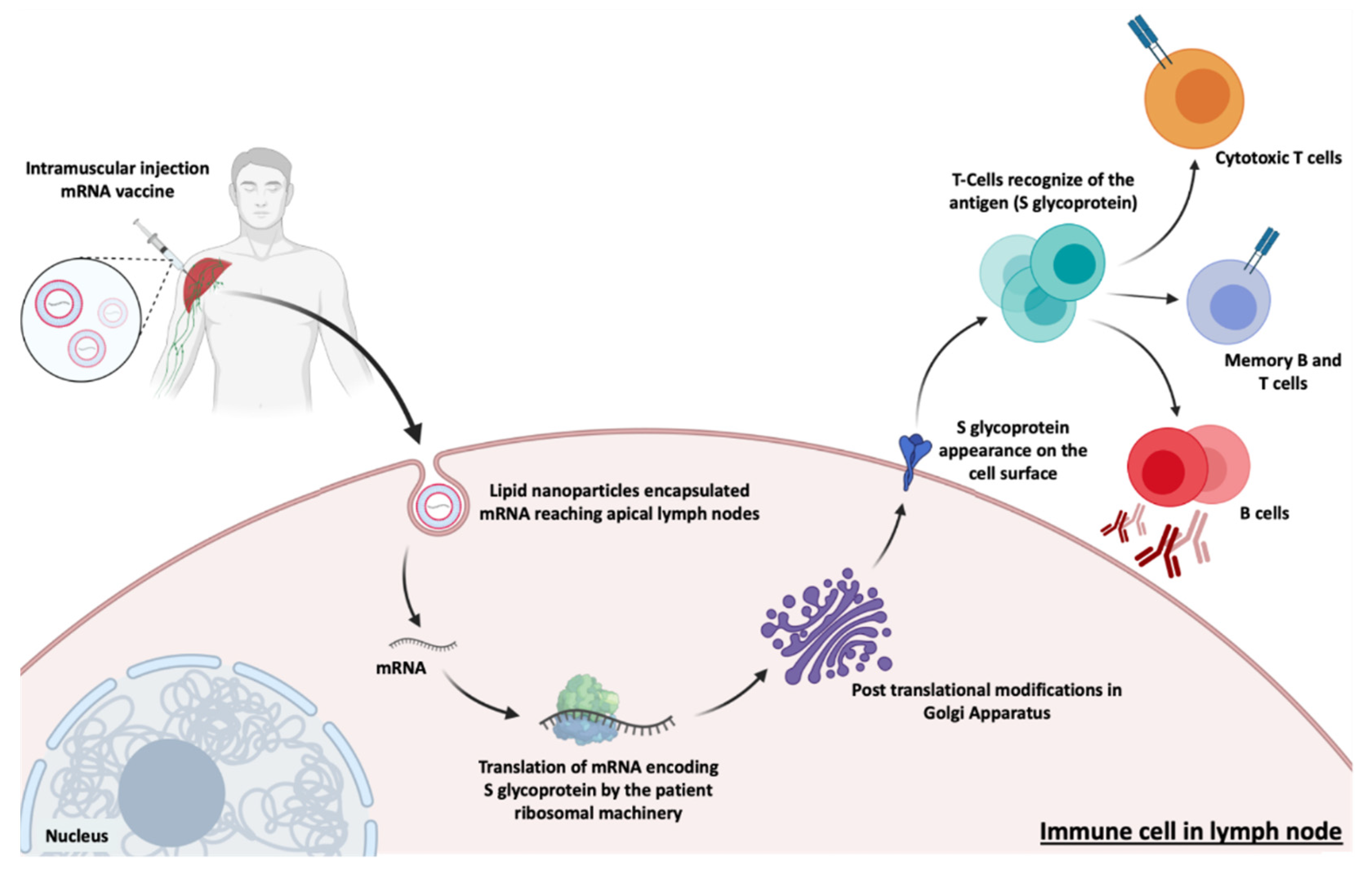{kind=link}
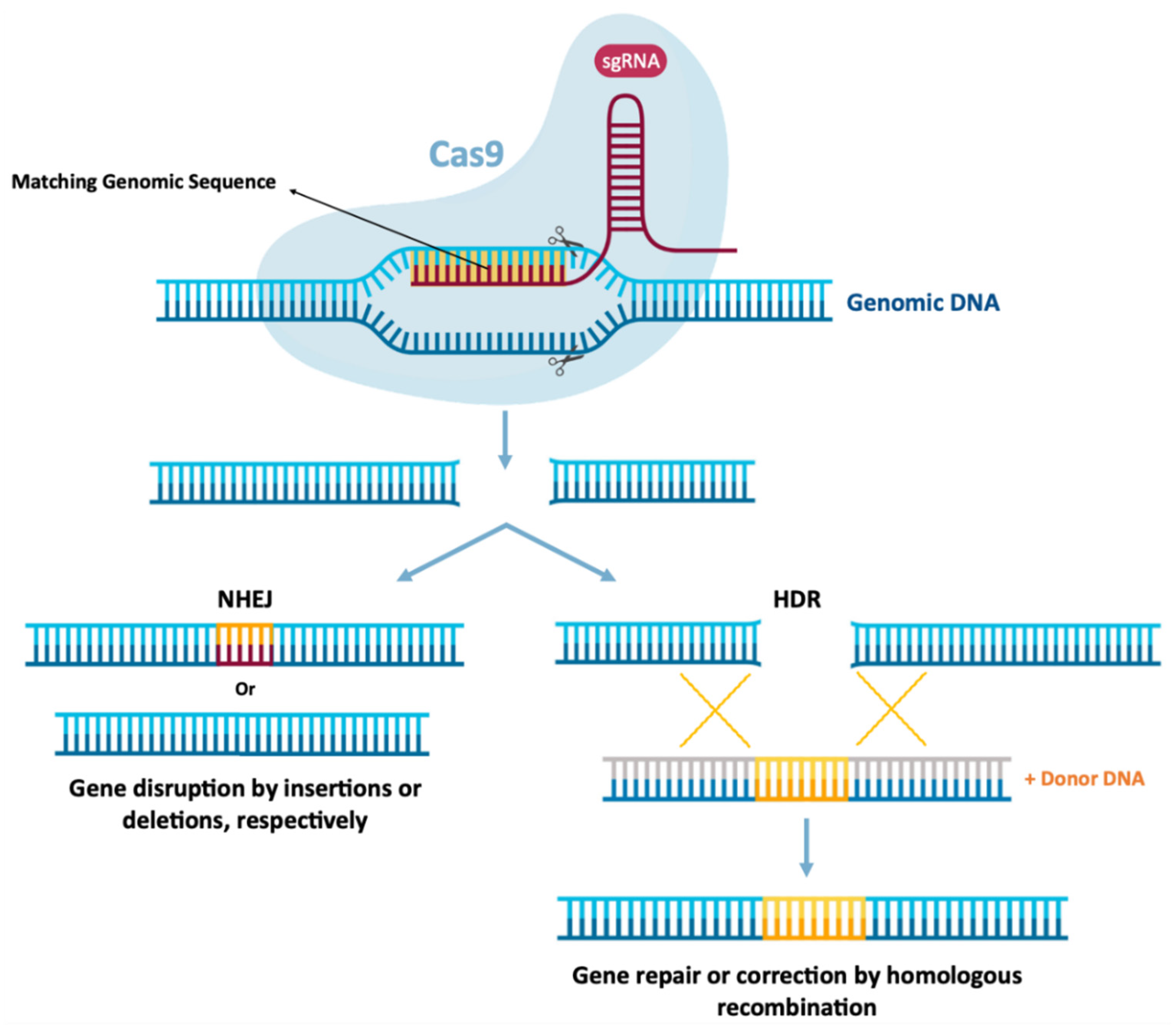{kind=link}
Table 1.
Structural comparison of RNA and DNA molecules.
| Characteristics | DNA  | RNA  |
|---|---|---|
| Type of sugar | Deoxyribose | Ribose |
| Bases | A, T, C, G | A, U, C, G |
| Double or single stranded | Double | Single |
| Secondary structure | Double helix | Many types |
| Stability | Stable | Easily degraded |
Table 2.
Therapeutic approaches based on mRNA and their functions.
| Therapeutic Approach | Objective/Function |
|---|---|
| Protein Replacement | Restore function, increase expression or replace protein in rare monogenic diseases |
| Cell reprogramming | Modulate cellular behavior by expressing transcription and/or growth factors |
| Immunotherapies | Elicit specific immune responses against target cells, for example through therapeutic antibodies |
Table 3.
Advantages and disadvantages of non-amplifying mRNA Vaccines and SAM Vaccines.
| mRNA Vaccines | Structure | Advantages | Disadvantages | References |
|---|---|---|---|---|
| Non-amplifying mRNA Vaccines | Basic structure of the mRNA, with a coding region for the desired antigens. | - Relatively small mRNA size (~2–3 kb). - Absence of additional proteins, minimizing unwanted immune interactions. - Relatively easy to produce and amplify. - Simplified sequence engineering. - Direct antigen expression. | - Potential toxicity from modified nucleotides. - Short duration of expression. - Need for high RNA doses. - Low antigen quantity. | [8,86] |
| SAM Vaccines | Encode a manipulated RNA virus genome (replicon). It generally contains two different protein coding regions, one encoding nonstructural proteins involved in mRNA capping and replication, and the other in antigen expression. | - High yield of target antigen. - Enhanced and prolonged antigen expression. - Lower effective RNA doses (more safe). - Intrinsic adjuvant effect. - Potential apoptosis of vaccine-carrying cells due to vaccine self-amplification (enhanced cross-presentation). - Option for single-vector delivery of multiple or complex antigens. | - RNA replicons are not able to tolerate many of the synthetic nucleotide modifications and sequence alterations. - Inclusion of unrelated proteins, which may increase unwanted immunogenicity. - Large replicon size (~10 kb), decreasing cell internalization efficiency. - Interaction between nsPs and host factors yet to be addressed. - Longer RNA length (more difficult production). - Potential elevated inflammation. | [8,13,29,86,93] |
Table 4.
Clinical trials for RNA-Based Protein Therapy (Protein replacement, cell reprogramming, immunotherapy) and gene editing.
Table 4.
Clinical trials for RNA-Based Protein Therapy (Protein replacement, cell reprogramming, immunotherapy) and gene editing.
| Name | Therapetic Modality | Protein Target | Administration Method | Delivery Vehicle | Disease | Sponsor Institution | ClinicalTrials.gov Identifer | Phase | Therapeutic Approach | References |
|---|---|---|---|---|---|---|---|---|---|---|
| MRT5005 | mRNA | CFTR | Inhalation | LNPs | Cystic fibrosis | Translate Bio | NCT03375047 | I/II | Protein Replacement | [94] |
| AZD8601 | mRNA | VEGF-A | Intracardiac injection | Naked mRNA | Heart failure | AstraZeneca | NCT03370887 | II | Cell reprogramming | [15] |
| CV7201 | mRNA | Rabies virus glycoprotein | I.D or I.M | RNActive, protamine | Rabies | CureVac | NCT02241135 | I | Immunotherapy | [95] |
| CV7202 | mRNA | Rabies virus glycoprotein | I.M | LNPs | Rabies | NCT03713086 | I | Immunotherapy | [13] | |
| CV9201 | mRNA | TAAs | I.D | RNActive, protamine | NSCLC | NCT00923312 | I/II | Immunotherapy | [96] | |
| CV9202 | mRNA | TAAs | I.D | RNActive, protamine | NSCLC | NCT03164772 | I/II | Immunotherapy | [8] | |
| CV9104 | mRNA | TAAs | I.D | RNActive, protamine | Prostate carcinoma | NCT02140138 | II | Immunotherapy | ||
| HARE-40 | mRNA | HPV antigen CD40 | I.D | Naked RNA | HPV-driven squamous cell carcinoma | BioNTech | NCT03418480 | I/II | Immunotherapy | |
| Lipo-MERIT | mRNA | TAAs: NYESO-1, MAGE-A3, tyrosinase, and TPTE | I.V | Lipo-MERIT, DOTMA(DOTAP)/ DOPE lipoplex | Advanced melanoma | NCT02410733 | I | Immunotherapy | ||
| IVAC | mRNA | 3 TAAs selected from a warehouse and p53 RNA; Neo-Ag based on NGS screening | I.V | Lipo-MERIT, DOTMA(DOTAP)/DOPE lipoplex | TNBC | BioNTech | NCT02316457 | I | Immunotherapy | [8] |
| RBL001/RBL002 | mRNA | TAAs | Ultrasound guided I.N | Naked mRNA | Melanoma | NCT01684241 | I | Immunotherapy | ||
| IVAC MUTANOME | mRNA | Neo-Ag | Ultrasound guided I.N | Naked mRNA | Melanoma | NCT02035956 | I | Immunotherapy | ||
| RO7198457 | mRNA | Neo-Ag | I.V | Naked mRNA | Melanoma; NSCLC; Bladder cancer | NCT03289962 | I | Immunotherapy | ||
| mRNA-1325 | mRNA | Zika virus antigen | I.D | LNPs | Zika virus | Moderna | NCT03014089 | I | Immunotherapy | |
| mRNA-1653 | mRNA | hMPV and hPIV type 3 vaccine | I.D | LNPs | hMPV and hPIV infection | NCT03392389 | I | Immunotherapy | ||
| VAL-506440 | mRNA | H10N8 antigen | I.D | LNPs | Influenza | Moderna | NCT03076385 | I | Immunotherapy | [97] |
| VAL-339851 | mRNA | H7 influenza antigen | I.D | LNPs | Influenza | NCT03345043 | I | Immunotherapy | ||
| mRNA-1647/1443 | mRNA | CMV glycoprotein H pentamer complex | I.D | LNPs | CMV infection | NCT03382405 | I | Immunotherapy | [8] | |
| mRNA-2416 | mRNA | Human OX40L | I.D | LNPs | Solid tumor malignancies or lymphoma | NCT03323398 | I | Immunotherapy | ||
| mRNA-4157 | mRNA | Neo-Ag | Intratumoral | LNPs | Solid tumor | NCT03313778 | I | Immunotherapy | ||
| mRNA-4650 | mRNA | Neo-Ag | I.M | Naked mRNA | Melanoma; Colon cancer; GI cancer; Genitourinary cancer; HCC | NCT03480152 | I/II | Immunotherapy | [98,99] | |
| mRNA-1388 | mRNA | VAL-181388 | I.M | LNPs | CHIKV | NCT03325075 | I | Immunotherapy | [12] | |
| mRNA-2752 | mRNA | OX40L, IL-23, and IL-36γ | Intratumoral | LNPs | Solid tumor or lymphoma | Moderna/AstraZeneca | NCT03739931 | I | Immunotherapy | [13] |
| iHIVARNA-01 | mRNA | Trimix (CD40L, CD70 and caTLR4 RNA—mRNA-transfected) | I.N | Naked mRNA | HIV infection | Hospital Clínic de Barcelona | NCT02413645 | I | Immunotherapy | [100] |
| mRNA | I.N | Naked mRNA | HIV infection | Erasmus Medical Center | NCT02888756 | II | Immunotherapy | [101] | ||
| - | mRNA | CT7, MAGE-A3, and WT1 mRNA-electroporated LCs | I.D | DC-loaded mRNA | Malignant melanoma | Memorial Sloan Kettering Cancer Center | NCT01995708 | I | Immunotherapy | [8] |
| - | mRNA | HIV-1 Gag- and Nef-transfected DCs | I.D | DC-loaded mRNA | HIV infection | Massachusetts General Hospital | NCT00833781 | I/II | Immunotherapy | [102] |
| - | mRNA | Neo-Ag | S.C | Naked mRNA | Solid tumor malignancies or lymphoma | Changhai Hospital Stemirna Therapeutics | NCT03468244 | N.A | Immunotherapy | [8] |
| - | mRNA | TAA for melanoma (Melan-A, MAGE-A1, MAGE-A3, survivin, GP100, and tyrosinase) | I.D | Naked mRNA | Melanoma | University Hospital Tuebingen | NCT00204516 | I/II | Immunotherapy | [12] |
| - | mRNA | TAA-transfected DC | I.D or I.N | DC-loaded mRNA | Malignant melanoma | Oslo University Hospital | NCT01278940 | I/II | Immunotherapy | [103] |
| - | mRNA | I.D | DC-loaded mRNA | Prostate cancer | NCT01278914 | I/II | Immunotherapy | [8] | ||
| AVX601 | Replicon | Alphavirus replicon vaccine expressing CMV genes | I.M or S.C | - | CMV | AlphaVax | NCT00439803 | I | Immunotherapy | [8] |
| AVX502 | Replicon | Alphavirus replicon vaccine expressing an influenza HA protein | I.M or S.C | - | Influenza | NCT00440362; NCT00706732 | I/II | Immunotherapy | ||
| AVX101 | Replicon | Alphavirus replicon, HIV-1 subtype C Gag vaccine | I.M or S.C | - | HIV infections | NCT00097838; NCT00063778 | I | Immunotherapy | [104] | |
| AVX701 | Replicon | Alphavirus replicon encoding the protein | I.M or S.C | - | Colon cancer; CRC; Breast cancer; Lung cancer; Pancreatic cancer | NCT01890213; NCT00529984 | I/II | Immunotherapy | [8] | |
| NY-ESO-1 | CRISPR-Cas9 | PD-1 and TCR | Ex vivo | Autologous T cells | Multiple myeloma; Synovial sarcoma; Melanoma | University of Pennsylvania | NCT03399448 | I | Gene Editing | [8] |
| CRISPR/TALEN-HPV E6/E7 | CRISPR/Cas9, TALEN | E6 and E7 | N.A | Plasmid DNA in gel | Cervical intraepithelial neoplasia | First Affiliated Hospital, Sun Yat-Sen University | NCT03057912 | I | Gene Editing | [105,106] |
| CTX001 | CRISPR-Cas9 | BCL11A | Ex vivo | Modified CD34+ hHSPCs | ß-thalassemia | Vertex Pharmaceuticals Incorporated | NCT03655678 | I/II | Gene Editing | [8] |
| - | CRISPR-Cas9 | PD-1 and TCR | Ex vivo | CAR-T cells | Mesothelin positive multiple solid tumors | Chinese PLA General Hospital | NCT03545815 | I | Gene Editing | |
| - | CRISPR-Cas9 | CD19 and CD20 | Ex vivo | Dual specificity CAR-T cells | ß cell leukemia and lymphoma | NCT03398967 | I/II | Gene Editing | ||
| UCART019 | CRISPR-Cas9 | CD19 | Ex vivo | CAR-T cells | ß cell leukemia and lymphoma | NCT03166878 | I/II | Gene Editing | [107] | |
| - | CRISPR-Cas9 | PD-1 | Ex vivo | Cytotoxic T lymphocytes | EBV-associated malignancies | Yang Yang | NCT03044743 | I/II | Gene Editing | [108,109] |
| SB-728mR-HSPC | ZFN mRNA | CCR5 | Ex vivo (mRNA) | CD34+ hHSPCs | HIV | City of Hope Medical Center | NCT02500849 | I | Gene Editing | [110] |
| SB-728mR-T | ZFN mRNA | CCR5 | Ex vivo (mRNA) | T cells | HIV | Sangamo Therapeutics | NCT02225665 | I/II | Gene Editing | [111] |
CFTR—Cystic fibrosis transmembrane conductance regulator; LNPs—Lipid nanoparticles; VEGF-A—Vascular endothelial growth factor A; I.D.—Intradermal; I.M.—Intramuscular; TAAs—Tumor-associated antigens; NSCLC—Non-small-cell lung carcinoma; HPV—Human Papillomavirus; I.N.—Intranodal; MAGE-A—Melanoma-associated antigen-A; TPTE—Putative tyrosine-protein phosphatase; I.V—Intravenous; Neo-Ag—Neo-antigen; NGS—Next-Generation Sequencing; TNBC—Triple-negative breast cancer; hMPV—Human metapneumovirus; hPIVs—Human parainfluenza viruses; CMV—Cytomegalovirus; GI—Gastrointestinal; HCC—Hepatocellular cancer; CHIKV—Chikungunya virus; IL—Interleukin; HIV—Human immunodeficiency virus; WT1—Wilms’ tumor 1; LCs—Langerhans cells; DC—Dendritic cell; S.C.—Subcutaneous; N.A—Not applicable; GP100—Glycoprotein 100; HA—Hemagglutinin; CRC—Colorectal cancer; PD-1—Programmed cell death protein 1; TLR—Toll-like receptor; BCL11A—B-cell lymphoma/leukemia 11A; hHSPCs—Human hematopoietic stem and progenitor cells; CAR—Chimeric antigen receptor; EBV—Epstein-Barr virus; ZFN—Zinc-finger nucleases; CCR5—C-C Motif Chemokine Receptor 5.
| Lipid Name | Role | Abbreviation | Molar Lipid Ratios (%) (Ionizable Cationic Lipid:Neutral Lipid:Cholesterol:PEG-ylated Lipid) |
|---|---|---|---|
| BNT162b2 vaccine | |||
| 4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate | ionizable cationic lipid | ALC-0315 | 46.3:9.4:42.7:1.6 |
| 1,2-Distearoyl-sn-glycero-3-phosphocholine | helper lipid | DSPC | |
| cholesterol | helper lipid | Chol | |
| 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide | PEG-lipid | ALC-0159 | |
| mRNA-1273 vaccine | |||
| heptadecan-9-yl 8-((2-hydroxyethyl)[6-oxo-6-(undecyloxy)hexyl]amino)octanoate | ionizable cationic lipid | SM-102 | 50:10:38.5:1.5 |
| 1,2-distearoyl-sn-glycero-3-phosphocholine | helper lipid | DSPC | |
| cholesterol | helper lipid | Chol | |
| 1,2-Dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 | PEG-lipid | PEG2000-DMG | |
Table 6.
mRNA vaccines and new candidates for COVID-19 [150].
Table 6.
mRNA vaccines and new candidates for COVID-19 [150].
| Name | Therapetic Modality | Protein Target | Administration Method | Delivery Vehicle | Developer | ClinicalTrials.gov Identifer, EU Clinical Trials Register or Chinese Clinical Trial Register | Phase |
|---|---|---|---|---|---|---|---|
| mRNA-1273 | mRNA | Spike Glycoprotein | Intramuscular | LNP | Moderna/NIAID | EUCTR2021-002327-38-NL | IV |
| BNT162b2 | mRNA | RBD/Spike Glycoprotein | Intramuscular | LNP | Pfizer/BioNTech + Fosun Pharma | NCT04760132 | IV |
| CVnCoV Vaccine | mRNA | Spike Glycoprotein | Intramuscular | LNP | CureVac AG | NCT04674189 | III |
| ARCT-021 | mRNA | Spike Glycoprotein | Intramuscular | LNP | Arcturus Therapeutics | NCT04668339 | II |
| LNP-nCoVsaRNA | mRNA | Spike Glycoprotein | Intramuscular | LNP | Imperial College London | ISRCTN17072692 | I |
| SARS-CoV-2 mRNA vaccine (ARCoV) | mRNA | RBD | Intramuscular | LNP | AMS/Walvax Biotechnology and Suzhou Abogen Biosciences | NCT04847102 | III |
| ChulaCov19 mRNA vaccine | mRNA | Spike Glycoprotein | Intramuscular | LNP | Chulalongkorn University | NCT04566276 | I |
| PTX-COVID19-B, mRNA vaccine | mRNA | Spike Glycoprotein | Intramuscular | LNP | Providence therapeutics | NCT04765436 | I |
| saRNA formulated in a NLC | mRNA | - | - | NLC | Infectious Disease Research Institute/Amyris, Inc. | - | Pre-Clinical |
| LNP-encapsulated mRNA encoding S | mRNA | Spike Glycoprotein | - | LNP | Max-Planck-Institute of Colloids and Interfaces | - | Pre-Clinical |
| Self-amplifying RNA | mRNA | - | - | - | Gennova | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | Selcuk University | - | Pre-Clinical |
| LNP-mRNA | mRNA | - | - | LNP | Translate Bio/Sanofi Pasteur | - | Pre-Clinical |
| LNP-mRNA | mRNA | - | - | LNP | CanSino Biologics/Precision NanoSystems | - | Pre-Clinical |
| LNP-encapsulated mRNA cocktail encoding VLP | mRNA | - | - | LNP | Fudan University/Shanghai JiaoTong University/RNACure Biopharma | - | Pre-Clinical |
| LNP-encapsulated mRNA encoding RBD | mRNA | RBD | - | LNP | Fudan University/Shanghai JiaoTong University/RNACure Biopharma | - | Pre-Clinical |
| Replicating Defective SARS-CoV-2 derived RNAs | mRNA | - | - | - | Centro Nacional Biotecnología (CNB-CSIC), Spain | - | Pre-Clinical |
| LNP-encapsulated mRNA | mRNA | - | - | LNP | University of Tokyo/Daiichi-Sankyo | - | Pre-Clinical |
| Liposome-encapsulated mRNA | mRNA | - | - | LNP | BIOCAD | - | Pre-Clinical |
| Several mRNA candidates | mRNA | - | - | - | RNAimmune, Inc. | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | FBRI SRC VB VECTOR, Rospotrebnadzor, Koltsovo | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | China CDC/Tongji University/Stermina | - | Pre-Clinical |
| mRNA in an intranasal delivery system | mRNA | - | Intranasal | - | eTheRNA | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | Greenlight Biosciences | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | IDIBAPS-Hospital Clinic, Spain | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | Providence Therapeutics | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | Cell Tech Pharmed | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | ReNAP Co. | - | Pre-Clinical |
| D614G variant LNP-encapsulated mRNA | mRNA | - | - | LNP | Globe Biotech Ltd. | - | Pre-Clinical |
| Encapsulated mRNA | mRNA | - | - | - | CEA | - | Pre-Clinical |
LNPs—Lipid nanoparticles; NIAID—National Institute of Allergy and Infectious Diseases; RBD—Receptor-binding domain; AMS—Academy of Military Science.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Baptista, B.; Carapito, R.; Laroui, N.; Pichon, C.; Sousa, F. mRNA, a Revolution in Biomedicine. Pharmaceutics 2021, 13, 2090. https://doi.org/10.3390/pharmaceutics13122090
AMA Style
Baptista B, Carapito R, Laroui N, Pichon C, Sousa F. mRNA, a Revolution in Biomedicine. Pharmaceutics. 2021; 13(12):2090. https://doi.org/10.3390/pharmaceutics13122090
Chicago/Turabian StyleBaptista, Bruno, Rita Carapito, Nabila Laroui, Chantal Pichon, and Fani Sousa. 2021. "mRNA, a Revolution in Biomedicine" Pharmaceutics 13, no. 12: 2090. https://doi.org/10.3390/pharmaceutics13122090
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.