
Modified Gold Nanoparticles to Overcome the Chemoresistance to Gemcitabine in Mutant p53 Cancer Cells




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Procedure
2.2.1. Synthesis of AuNPs
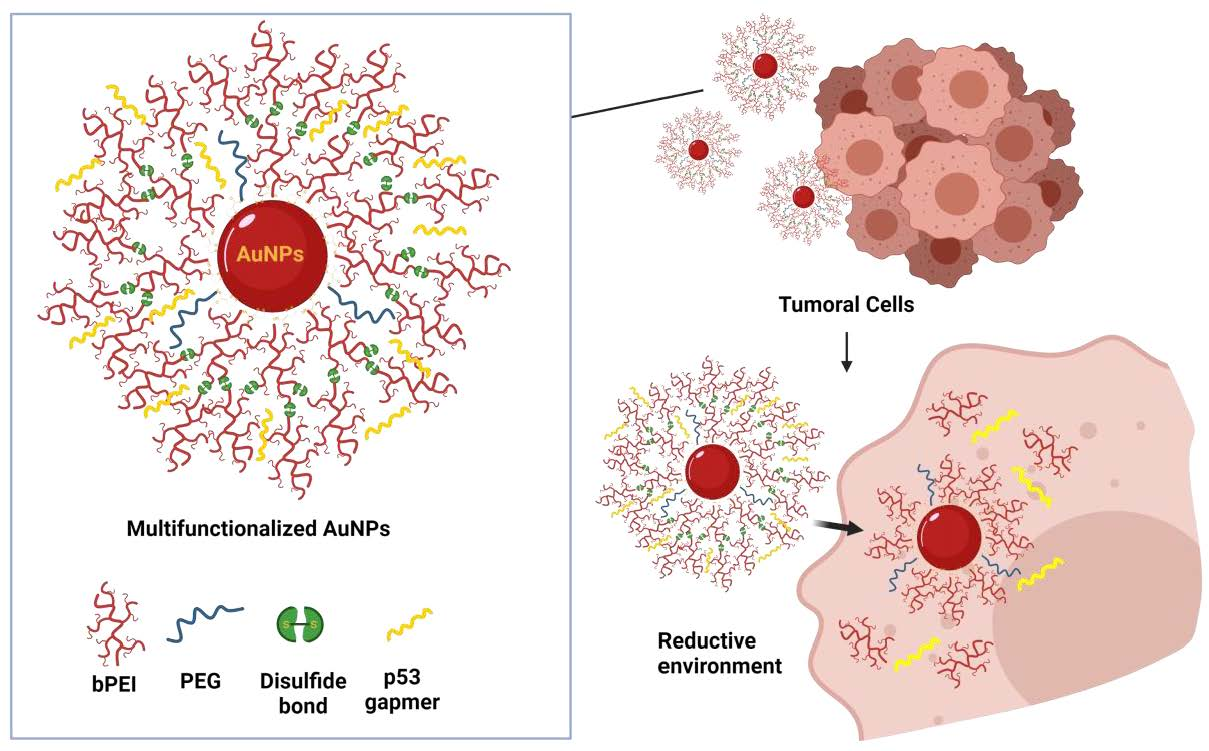
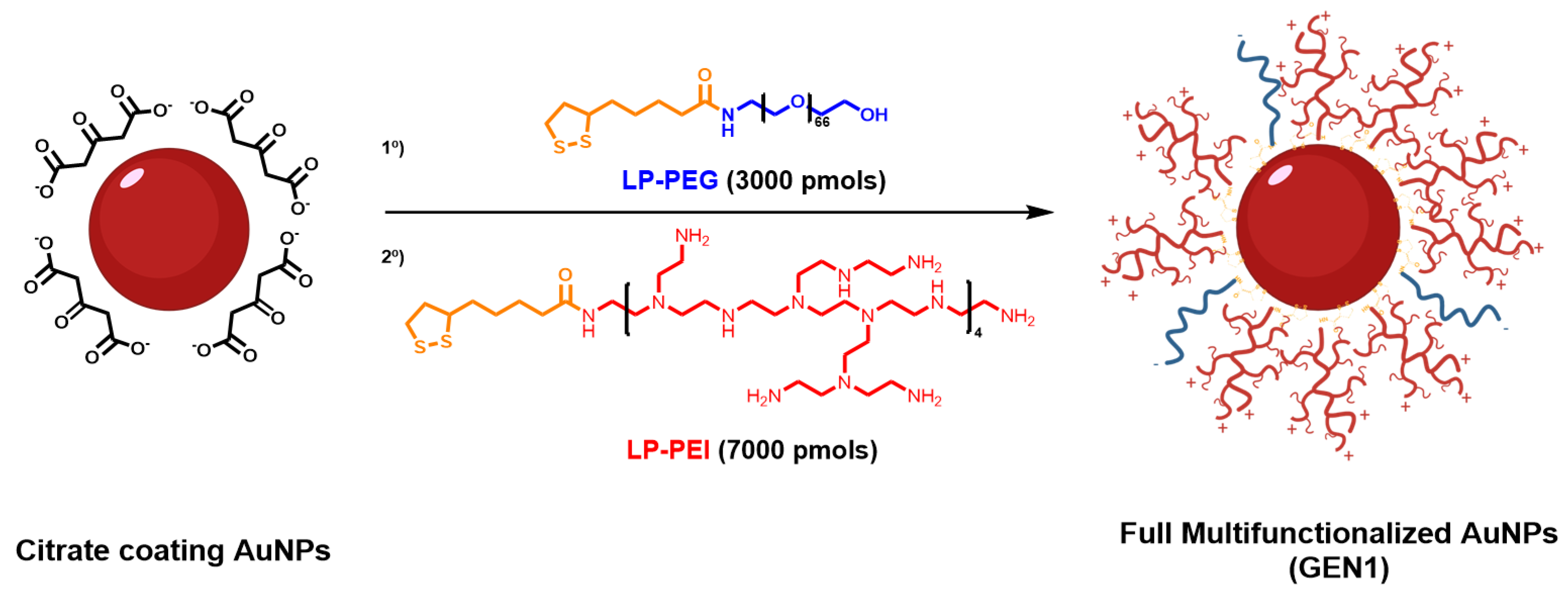
2.2.2. AuNPs Multifunctionalization
2.2.3. AuNPs GEN1
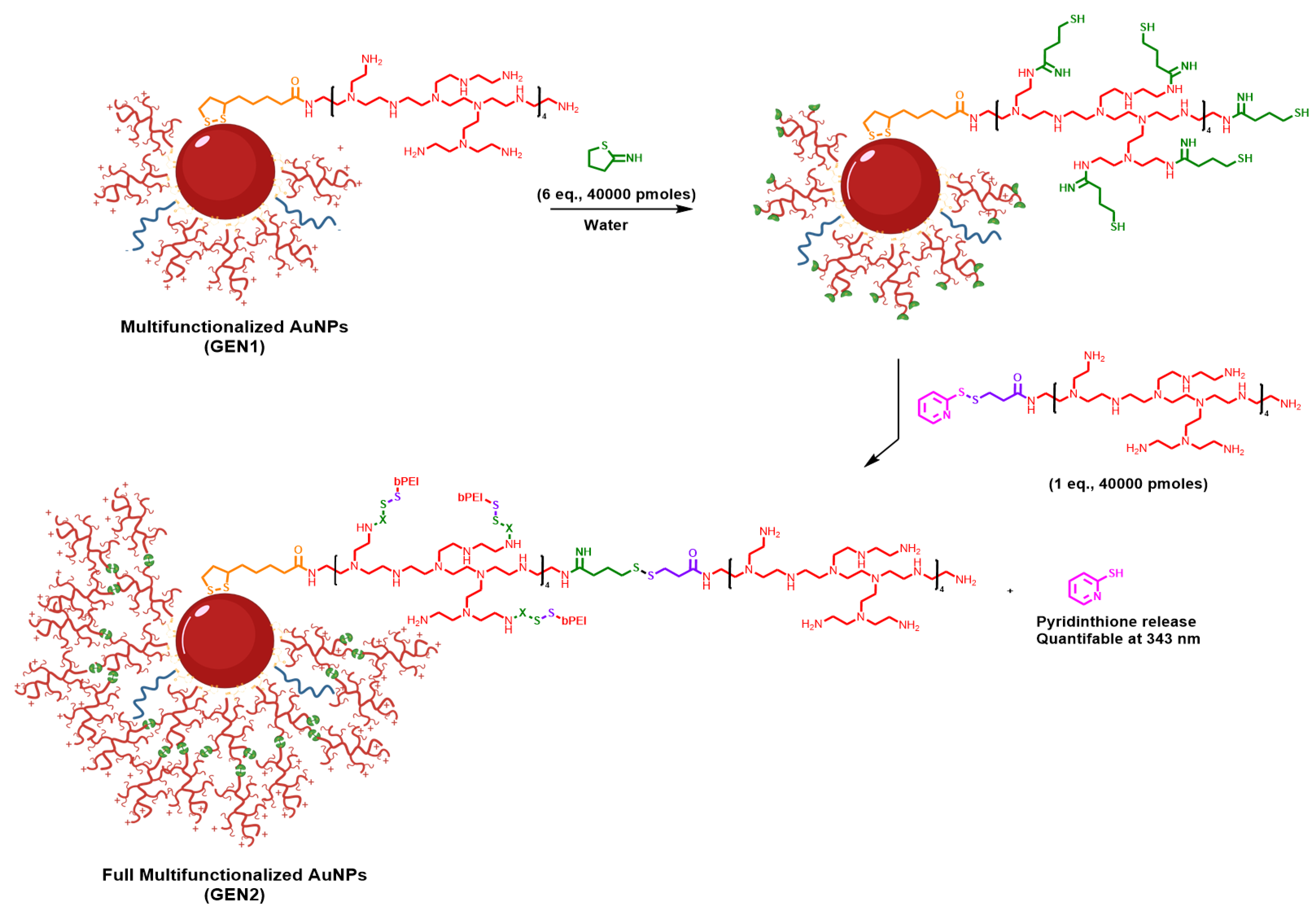
2.2.4. AuNPs GEN2
2.3. Oligonucleotides Experiments
2.3.1. Oligonucleotides Synthesis
2.3.2. Oligonucleotides Incubation
2.3.3. Oligonucleotides Transfection
2.4. Cell Lines and Culture Conditions
2.5. Chemotherapy
2.6. Nanoparticles Treatment
2.7. Combination Treatment
3. Results
3.1. Preparation of Multi Functionalized AuNPs
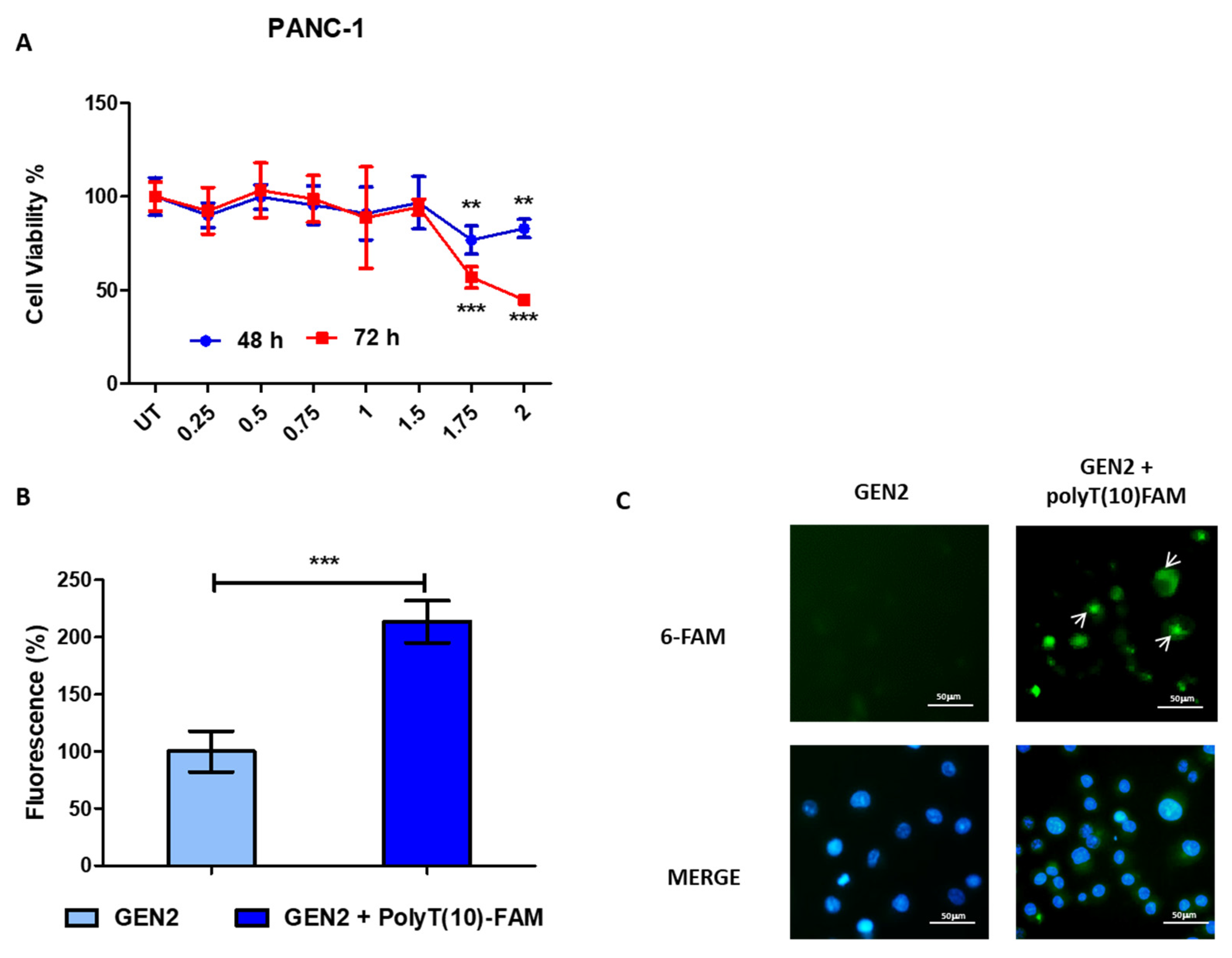
3.2. Transfection Efficacy of GEN2-AuNPs
3.3. Biological Activity of GEN2-AuNPs
3.4. Mutant and Wild Type p53 Cancer Cells Show Different Sensitivity to Chemotherapy
3.5. Modified Gold Nanoparticles Reduce Mutant p53 Cancer Cell Proliferation
3.6. Modified Gold Nanoparticles Reduce Chemoresistance in Mutant p53 Cancer Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xie, M.; Zhang, Q.; Mcmichael, J.F.; Wyczalkowski, M.A.; Wendl, M.C.; Ley, T.J.; Wilson, R.K.; Raphael, B.J. Mutational Landscape and Significance across 12 Major Cancer Types. Nature 2014, 502, 333–339. [Google Scholar] [CrossRef]
- Kim, M.P.; Lozano, G. Mutant P53 Partners in Crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Kim, T.J.; Lee, J.H.; Choi, J.H. Mutant P53 Stimulates Cell Invasion through an Interaction with Rad21 in Human Ovarian Cancer Cells. Sci. Rep. 2017, 7, 9076. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.J.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant P53 Drives Invasion by Promoting Integrin Recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Prokocimer, M.; Molchadsky, A.; Rotter, V. Dysfunctional Diversity of P53 Proteins in Adult Acute Myeloid Leukemia: Projections on Diagnostic Workup and Therapy. Blood 2017, 130, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Buller, R.E.; Lallas, T.A.; Shahin, M.S.; Sood, A.K.; Hatterman-Zogg, M.; Anderson, B.; Sorosky, J.I.; Kirby, P.A. The P53 Mutational Spectrum Associated with BRCA1 Mutant Ovarian Cancer. Clin. Cancer Res. 2001, 7, 831–838. [Google Scholar]
- Blandino, G.; Levine, A.J.; Oren, M. Mutant P53 Gain of Function: Differential Effects of Different P53 Mutants on Resistance of Cultured Cells to Chemotherapy. Oncogene 1999, 18, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Valenti, F.; Ganci, F.; Fontemaggi, G.; Sacconi, A.; Strano, S.; Blandino, G.; Di Agostino, S. Gain of Function Mutant P53 Proteins Cooperate with E2F4 to Transcriptionally Downregulate RAD17 and BRCA1 Gene Expression. Oncotarget 2015, 6, 5547–5566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Agostino, S.; Strano, S.; Emiliozzi, V.; Zerbini, V.; Mottolese, M.; Sacchi, A.; Blandino, G.; Piaggio, G. Gain of Function of Mutant P53: The Mutant P53/NF-Y Protein Complex Reveals an Aberrant Transcriptional Mechanism of Cell Cycle Regulation. Cancer Cell 2006, 10, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Fiorini, C.; Cordani, M.; Padroni, C.; Blandino, G.; Di Agostino, S.; Donadelli, M. Mutant P53 Stimulates Chemoresistance of Pancreatic Adenocarcinoma Cells to Gemcitabine. Biochim. Biophys. Acta-Mol. Cell Res. 2015, 1853, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonetto, G.; Koifman, G.; Silberman, A.; Attery, A.; Solomon, H.; Levin-Zaidman, S.; Goldfinger, N.; Porat, Z.; Erez, A.; Rotter, V. Mutant P53-Dependent Mitochondrial Metabolic Alterations in a Mesenchymal Stem Cell-Based Model of Progressive Malignancy. Cell Death Differ. 2019, 26, 1566–1581. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-Function Mutant P53 Promotes Cell Growth and Cancer Cell Metabolism via Inhibition of AMPK Activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordani, M.; Oppici, E.; Dando, I.; Butturini, E.; Dalla Pozza, E.; Nadal-Serrano, M.; Oliver, J.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant P53 Proteins Counteract Autophagic Mechanism Sensitizing Cancer Cells to MTOR Inhibition. Mol. Oncol. 2016, 10, 1008–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordani, M.; Pacchiana, R.; Butera, G.; D’Orazi, G.; Scarpa, A.; Donadelli, M. Mutant P53 Proteins Alter Cancer Cell Secretome and Tumour Microenvironment: Involvement in Cancer Invasion and Metastasis. Cancer Lett. 2016, 376, 303–309. [Google Scholar] [CrossRef]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 Reactivates Mutant P53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, H.C.; Yang, L.P.; Fitzgerald, A.L.; Osman, A.; Woo, S.H.; Myers, J.N.; Skinner, H.D. The P53-Reactivating Small Molecule RITA Induces Senescence in Head and Neck Cancer Cells. PLoS ONE 2014, 9, e104821. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhou, L.; Hong, B.; Van Den Heuvel, A.P.J.; Prabhu, V.V.; Warfel, N.A.; Kline, C.L.B.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Small-Molecule NSC59984 Restores P53 Pathway Signaling and Antitumor Effects against Colorectal Cancer via P73 Activation and Degradation of Mutant P53. Cancer Res. 2015, 75, 3842–3852. [Google Scholar] [CrossRef] [Green Version]
- Maan, M.; Pati, U. CHIP Promotes Autophagy-Mediated Degradation of Aggregating Mutant P53 in Hypoxic Conditions. FEBS J. 2018, 285, 3197–3214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Hiraki, M.; Namba, T.; Egawa, N.; Baba, K.; Tanaka, T.; Noshiro, H. Andrographolide Induces Degradation of Mutant P53 via Activation of Hsp70. Int. J. Oncol. 2018, 53, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Foggetti, G.; Ottaggio, L.; Russo, D.; Mazzitelli, C.; Monti, P.; Degan, P.; Miele, M.; Fronza, G.; Menichini, P. Autophagy Induced by SAHA Affects Mutant P53 Degradation and Cancer Cell Survival. Biosci. Rep. 2019, 39, BSR20181345. [Google Scholar] [CrossRef] [Green Version]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-Mediated Autophagy Degrades Mutant P53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [Green Version]
- Selivanova, G.; Wiman, K.G. Reactivation of Mutant P53: Molecular Mechanisms and Therapeutic Potential. Oncogene 2007, 26, 2243–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ubby, I.; Krueger, C.; Rosato, R.; Qian, W.; Chang, J.; Sabapathy, K. Cancer Therapeutic Targeting Using Mutant–P53-Specific SiRNAs. Oncogene 2019, 38, 3415–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu, A.K.; Iyer, S.; Chandra, S.; Adhikari, A.S.; Iwakuma, T.; Mandal, T.K. Novel siRNA formulation to effectively knockdown mutant p53 in osteosarcoma. PLoS ONE 2017, 12, e0179168. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; He, X.; Wang, K.; Xie, C.; Zhou, B.; Qing, Z. Ultrasmall Near-Infrared Gold Nanoclusters for Tumor Fluorescence Imaging in Vivo. Nanoscale 2010, 2, 2244–2249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chu, W.; Foroushani, A.D.; Wang, H.; Li, D.; Liu, J.; Barrow, C.J.; Wang, X.; Yang, W. New Gold Nanostructures for Sensor Applications: A Review. Materials 2014, 7, 5169–5201. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, P.; Han, G.; De, M.; Kim, C.K.; Rotello, V.M. Gold Nanoparticles in Delivery Applications. Adv. Drug Deliv. Rev. 2008, 60, 1307–1315. [Google Scholar] [CrossRef]
- Silva, F.; Zambre, A.; Campello, M.P.C.; Gano, L.; Santos, I.; Ferraria, A.M.; Ferreira, M.J.; Singh, A.; Upendran, A.; Paulo, A.; et al. Interrogating the Role of Receptor-Mediated Mechanisms: Biological Fate of Peptide-Functionalized Radiolabeled Gold Nanoparticles in Tumor Mice. Bioconjug. Chem. 2016, 27, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Schuemann, J.; Berbeco, R.; Chithrani, D.B.; Cho, S.H.; Kumar, R.; McMahon, S.J.; Sridhar, S.; Krishnan, S. Roadmap to Clinical Use of Gold Nanoparticles for Radiation Sensitization. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 189–205. [Google Scholar] [CrossRef] [Green Version]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled Drug Delivery Vehicles for Cancer Treatment and Their Performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Zhang, L.; Zhang, Y.; Wang, B.; Wang, Y.; Fan, Q.; Huang, W.; Wang, L. Plasmonic Nanobiosensor Based on Hairpin DNA for Detection of Trace Oligonucleotides Biomarker in Cancers. ACS Appl. Mater. Interfaces 2015, 7, 2459–2466. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat. Rev. Cancer 2016, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Adiseshaiah, P.P.; Crist, R.M.; Hook, S.S.; McNeil, S.E. Nanomedicine Strategies to Overcome the Pathophysiological Barriers of Pancreatic Cancer. Nat. Rev. Clin. Oncol. 2016, 13, 750–765. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Zhu, Y.; Oupický, D. Recent Advances in Delivery of Drug-Nucleic Acid Combinations for Cancer Treatment. J. Control. Release 2013, 172, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Tu, Y.; Wang, X.; Cao, C.; Hu, Y.; Shao, J.; Weng, L.; Mou, X.; Dong, X. A Photo-Triggered Antifungal Nanoplatform with Efflux Pump and Heat Shock Protein Reversal Activity for Enhanced Chemo-Photothermal Synergistic Therapy. Biomater. Sci. 2021, 9, 3293–3299. [Google Scholar] [CrossRef]
- Ramamoorth, M.; Narvekar, A. Non Viral Vectors in Gene Therapy—An Overview. J. Clin. Diagn. Res. 2015, 9, GE01–GE06. [Google Scholar] [CrossRef]
- Tang, G.P.; Zeng, J.M.; Gao, S.J.; Ma, Y.X.; Shi, L.; Li, Y.; Too, H.P.; Wang, S. Polyethylene Glycol Modified Polyethylenimine for Improved CNS Gene Transfer: Effects of PEGylation Extent. Biomaterials 2003, 24, 2351–2362. [Google Scholar] [CrossRef]
- Vijayakameswara Rao, N.; Ko, H.; Lee, J.; Park, J.H. Recent Progress and Advances in Stimuli-Responsive Polymers for Cancer Therapy. Front. Bioeng. Biotechnol. 2018, 6, 110. [Google Scholar] [CrossRef] [Green Version]
- Gosselin, M.A.; Guo, W.; Lee, R.J. Efficient Gene Transfer Using Reversibly Cross-Linked Low Molecular Weight Polyethylenimine. Bioconjug. Chem. 2001, 12, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Bauhuber, S.; Hozsa, C.; Breunig, M.; Göpferich, A. Delivery of Nucleic Acids via Disulfide-Based Carrier Systems. Adv. Mater. 2009, 21, 3286–3306. [Google Scholar] [CrossRef]
- Devos, S.L.; Miller, T.M. Antisense Oligonucleotides: Treating Neurodegeneration at the Level of RNA. J. Am. Soc. Exp. Neurother. 2013, 10, 486–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danielsen, M.B.; Lou, C.; Lisowiec-Wachnicka, J.; Pasternak, A.; Jørgensen, P.T.; Wengel, J. Gapmer Antisense Oligonucleotides Containing 2′,3′-Dideoxy-2′-Fluoro-3′-C-Hydroxymethyl-β-d-Lyxofuranosyl Nucleotides Display Site-Specific RNase H Cleavage and Induce Gene Silencing. Chem.-A Eur. J. 2020, 26, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.; Sciabola, S.; Salatto, C.; Weng, Y.; Moshinsky, D.; Little, J.; Walters, E.; Kreeger, J.; Dimattia, D.; Chen, T.; et al. Chemical Modification Study of Antisense Gapmers. Nucleic Acid Ther. 2012, 22, 344–359. [Google Scholar] [CrossRef]
- Wuithschick, M.; Birnbaum, A.; Witte, S.; Sztucki, M.; Vainio, U.; Pinna, N.; Rademann, K.; Emmerling, F.; Kraehnert, R.; Polte, J. Turkevich in New Robes: Key Questions Answered for the Most Common Gold Nanoparticle Synthesis. ACS Nano 2015, 9, 7052–7071. [Google Scholar] [CrossRef] [PubMed]
- Prigodich, A.E.; Seferos, D.S.; Massich, M.D.; Giljohann, D.A.; Lane, B.C.; Mirkin, C.A. Nano-Flares for MRNA Regulation and Detection. ACS Nano 2009, 3, 2147–2152. [Google Scholar] [CrossRef]
- Al-Johani, H.; Abou-Hamad, E.; Jedidi, A.; Widdifield, C.M.; Viger-Gravel, J.; Sangaru, S.S.; Gajan, D.; Anjum, D.H.; Ould-Chikh, S.; Hedhili, M.N.; et al. The Structure and Binding Mode of Citrate in the Stabilization of Gold Nanoparticles. Nat. Chem. 2017, 9, 890–895. [Google Scholar] [CrossRef]
- Pensa, E.; Cortés, E.; Corthey, G.; Carro, P.; Vericat, C.; Fonticelli, M.H.; Benítez, G.; Rubert, A.A.; Salvarezza, R.C. The Chemistry of the Sulfur-Gold Interface: In Search of a Unified Model. Acc. Chem. Res. 2012, 45, 1183–1192. [Google Scholar] [CrossRef]
- Zheng, N.; Fan, J.; Stucky, G.D. One-Step One-Phase Synthesis of Monodisperse Noble-Metallic Nanoparticles and Their Colloidal Crystals. J. Am. Chem. Soc. 2006, 128, 6550–6551. [Google Scholar] [CrossRef]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione Levels in Human Tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef]
- Carnerero, J.M.; Sánchez-Coronilla, A.; Martín, E.I.; Jimenez-Ruiz, A.; Prado-Gotor, R. Quantification of Nucleobases/Gold Nanoparticles Interactions: Energetics of the Interactions through Apparent Binding Constants Determination. Phys. Chem. Chem. Phys. 2017, 19, 22121–22128. [Google Scholar] [CrossRef]
- Kim, B.; Han, G.; Toley, B.J.; Kim, C.K.; Rotello, V.M.; Forbes, N.S. Tuning Payload Delivery in Tumour Cylindroids Using Gold Nanoparticles. Nat. Nanotechnol. 2010, 5, 465–472. [Google Scholar] [CrossRef]
- Curtis, K.A.; Miller, D.; Millard, P.; Basu, S.; Horkay, F.; Chandran, P.L. Unusual Salt and PH Induced Changes in Polyethylenimine Solutions. PLoS ONE 2016, 11, e0158147. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Liu, J. Methods for Preparing DNA-Functionalized Gold Nanoparticles, a Key Reagent of Bioanalytical Chemistry. Anal. Methods 2017, 9, 2633–2643. [Google Scholar] [CrossRef]
- Zhang, X.; Servos, M.R.; Liu, J. Instantaneous and Quantitative Functionalization of Gold Nanoparticles with Thiolated DNA Using a PH-Assisted and Surfactant-Free Route. J. Am. Chem. Soc. 2012, 134, 7266–7269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallops, C.E.; Yu, C.; Ziebarth, J.D.; Wang, Y. Effect of the Protonation Level and Ionic Strength on the Structure of Linear Polyethyleneimine. ACS Omega 2019, 4, 7255–7264. [Google Scholar] [CrossRef]
- Fukumoto, Y.; Obata, Y.; Ishibashi, K.; Tamura, N. Cost-Effective Gene Transfection by DNA Compaction at PH. Cytotechnology 2010, 62, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussif, O.T.; Lezoualc’h, F.; Zanta, M.A.; Mergnyt, M.D.; Scherman, D.; Demeneixt, B.; Behr, J.-P. A Versatile Vector for Gene and Oligonucleotide Transfer into Cells in Culture and in Vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [Green Version]
- Pezzoli, D.; Giupponi, E.; Mantovani, D.; Candiani, G. Size Matters for in Vitro Gene Delivery: Investigating the Relationships among Complexation Protocol, Transfection Medium, Size and Sedimentation. Sci. Rep. 2017, 7, 44134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malloggi, C.; Pezzoli, D.; Magagnin, L.; De Nardo, L.; Mantovani, D.; Tallarita, E.; Candiani, G. Comparative Evaluation and Optimization of Off-the-Shelf Cationic Polymers for Gene Delivery Purposes. Polym. Chem. 2015, 6, 6325–6339. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Q.; Carrier, F.; Fornace, A.J. Induction of Cellular P53 Activity by DNA-Damaging Agents and Growth Arrest. Mol. Cell. Biol. 1993, 13, 4242–4250. [Google Scholar] [CrossRef] [Green Version]
- Blattner, C.; Tobiasch, E.; Litfen, M.; Rahmsdorf, H.J.; Herrlich, P. DNA Damage Induced P53 Stabilization: No Indication for an Involvement of P53 Phosphorylation. Oncogene 1999, 18, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Lakin, N.D.; Jackson, S.P. Regulation of P53 in Response to DNA Damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [Green Version]
- Yip, K.W.; Reed, J.C. Bcl-2 Family Proteins and Cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.Q.; Bührer, E.D.; Berezowska, S.; Marti, T.M.; Xu, D.; Froment, L.; Yang, H.; Hall, S.R.R.; Vassella, E.; Yang, Z.; et al. MTOR Mediates a Mechanism of Resistance to Chemotherapy and Defines a Rational Combination Strategy to Treat KRAS-Mutant Lung Cancer. Oncogene 2019, 38, 622–636. [Google Scholar] [CrossRef]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.L.; Lin, F.J.; Gguo, Y.J.; Shao, Z.M.; Ou, Z.L. Gemcitabine Resistance in Breast Cancer Cells Regulated by PIi3K/AaKT-Mediated Cellular Proliferation Exerts Negative Feedback via the MEK/MAaPK and MTORr Pathways. Onco Targets Ther. 2014, 7, 1033–1042. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.H.; Lin, C.; Liu, M.M.; Zhang, J.; Tao, Z.H.; Hu, X.C. Src Inhibition Can Synergize with Gemcitabine and Reverse Resistance in Triple Negative Breast Cancer Cells via the AKT/c-Jun Pathway. PLoS ONE 2016, 11, e0169230. [Google Scholar] [CrossRef]
- Baron, B.; Wang, Y.; Maehara, S.I.; Maehara, Y.; Kuramitsu, Y.; Nakamura, K. Resistance to Gemcitabine in the Pancreatic Cancer Cell Line KLM1-R Reversed by Metformin Action. Anticancer Res. 2015, 35, 1941–1949. [Google Scholar]
- Chai, X.; Chu, H.; Yang, X.; Meng, Y.; Shi, P.; Gou, S. Metformin Increases Sensitivity of Pancreatic Cancer Cells to Gemcitabine by Reducing CD133+ Cell Populations and Suppressing ERK/P70S6K Signaling. Sci. Rep. 2015, 5, 14404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, M.C.; Lin, P.L.; Wang, Y.C.; He, T.Y.; Lee, M.C.; Yeh, S.D.; Chen, C.Y.; Lee, H. Mutant P53 Confers Chemoresistance in Non-Small Cell Lung Cancer by Upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Li, L.; Guan, X.; Xiong, L.; Miao, X. Mutant P53 Gain of Function and Chemoresistance: The Role of Mutant P53 in Response to Clinical Chemotherapy. Chemotherapy 2016, 62, 43–53. [Google Scholar] [CrossRef]
- Nakamura, M.; Sugimoto, H.; Ogata, T.; Hiraoka, K.; Yoda, H.; Sang, M.; Sang, M.; Zhu, Y.; Yu, M.; Shimozato, O.; et al. Improvement of Gemcitabine Sensitivity of P53-Mutated Pancreatic Cancer MiaPaCa-2 Cells by RUNX2 Depletion-Mediated Augmentation of TAp73-Dependent Cell Death. Oncogenesis 2016, 5, e233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordani, M.; Butera, G.; Dando, I.; Torrens-Mas, M.; Butturini, E.; Pacchiana, R.; Oppici, E.; Cavallini, C.; Gasperini, S.; Tamassia, N.; et al. Mutant P53 Blocks SESN1/AMPK/PGC-1α/UCP2 Axis Increasing Mitochondrial O2ˉ· Production in Cancer Cells. Br. J. Cancer 2018, 119, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Kazemi Oskuee, R.; Dabbaghi, M.; Gholami, L.; Taheri-Bojd, S.; Balali-Mood, M.; Mousavi, S.H.; Malaekeh-Nikouei, B. Investigating the Influence of Polyplex Size on Toxicity Properties of Polyethylenimine Mediated Gene Delivery. Life Sci. 2018, 197, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, N.; Liu, J.; Liu, Y.; Zhang, C.; Long, S.; Luo, G.; Zhang, L.; Zhang, Y. Mutant P53 Drives Cancer Chemotherapy Resistance Due to Loss of Function on Activating Transcription of PUMA. Cell Cycle 2019, 18, 3442–3455. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEN | LP-PEG | LP-PEI | Traut’s Reagent | PDP-PEI |
|---|---|---|---|---|
| 1 | 3 nmol | 7 nmol | - | - |
| 2 | 3 nmol | 7 nmol | 40 nmol | 40 nmol |
| Entry | Oligonucleotide | Sequence |
|---|---|---|
| 1 | Control 1 | 5′-ACGUGACACGTTCGGAGAAUU-3′ |
| 2 | Control 2 | 5′-UGCGCTCCTGGACGTAGCCU-3′ |
| 3 | Gapmer p53.1 | 5′-CAAAGCTGTTCCGTCCCAGU-3′ |
| 4 | Gapmer p53.2 | 5′-GACUCCAGTGGTAATCTAC-3′ |
| 5 | Gapmer p53.3 | 5′-GAAAUTTGCGTGTGGAGUA-3′ |
| 6 | Gapmer p53.4 | 5′-GGACATACCAGCTTAGAUUUU-3′ |
| Entry | Cell Line | Tumor Tissue | P53 Mutation |
|---|---|---|---|
| 1 | PANC-1 | Pancreas | R273H |
| 2 | MDA-MB-231 | Breast | R280K |
| 3 | MCF-7 | Breast | Wild Type |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Garrido, E.; Cordani, M.; Somoza, Á. Modified Gold Nanoparticles to Overcome the Chemoresistance to Gemcitabine in Mutant p53 Cancer Cells. Pharmaceutics 2021, 13, 2067. https://doi.org/10.3390/pharmaceutics13122067
García-Garrido E, Cordani M, Somoza Á. Modified Gold Nanoparticles to Overcome the Chemoresistance to Gemcitabine in Mutant p53 Cancer Cells. Pharmaceutics. 2021; 13(12):2067. https://doi.org/10.3390/pharmaceutics13122067
Chicago/Turabian StyleGarcía-Garrido, Eduardo, Marco Cordani, and Álvaro Somoza. 2021. "Modified Gold Nanoparticles to Overcome the Chemoresistance to Gemcitabine in Mutant p53 Cancer Cells" Pharmaceutics 13, no. 12: 2067. https://doi.org/10.3390/pharmaceutics13122067